
摘要
临床特征.
Donnai-Barrow综合征(DBS)以典型的颅面部畸形(眼距过宽、囟门增大) 、眼部异常(高度近视、视网膜脱离、进行性视力下降和虹膜缺损)、感音神经性耳聋、胼胝体发育不全、智力障碍和先天性膈疝(CDH)和/或脐膨出为特征。家系间和家系内其表型都存在差异。
诊断/检测.
DBS的诊断基于其特征性的临床表现和其独特形式的低分子量蛋白尿相结合。LRP2,编码低密度脂蛋白受体相关蛋白2前体蛋白(巨蛋白),是已知的DBS相关的唯一基因。
处理.
对症治疗:手术修复脐膨出和/或膈疝;对于近视使用矫正镜片;视网膜脱离预防性治疗(例如:视网膜周边激光光凝治疗);对于耳聋使用助听器和/或人工耳蜗植入;对于不同程度的智力障碍、视力下降、听力损失给予特定的教育;对于痫性发作使用抗癫痫药物。
监测:连续的听力学检查;密切的眼部监测来检测视网膜脱离;包含血尿素氮(BUN)和肌酐浓度的连续的肾功能的血清学检查。
遗传咨询.
DBS以常染色体隐性遗传方式进行遗传。一般来说,一个受累儿童的父母必然为每人携带一个突变等位基因的杂合子;一个单亲二倍体的实例已被报导。当父母双方都确定为一个致病性变异的携带者,患者的每一个同胞是患者的概率为25%,是无症状携带者的概率为50%,还有25%的概率既不是患者也不是携带者。如果致病性变异在家系中被确定,通过实验室提供感兴趣基因的检测或定制化检测项目,对有风险的亲属进行携带者检测和对高危妊娠进行产前检测是合理的。
诊断
临床诊断
Donnai-Barrow综合征(DBS)的诊断基于其特征性临床特征的识别结合其独特形式的低分子量蛋白尿。它能通过LRP2致病性变异的检测确诊,LRP2编码低密度脂蛋白受体相关蛋白2前体蛋白(巨蛋白)。既没有一个单独的特征对DBS来说是能确定诊断的,也没有正式的诊断标准。然而,当以下结构和功能异常中的几个出现的时候需要考虑诊断:
颅面部.
显著的眼距增宽、大的前囟、宽额缝、前发际线的寡妇尖、低鼻梁、短鼻和后旋耳几乎见于100%的受累的个体。面部表现,虽然不粗陋,但是具有特征性(见图 1)。
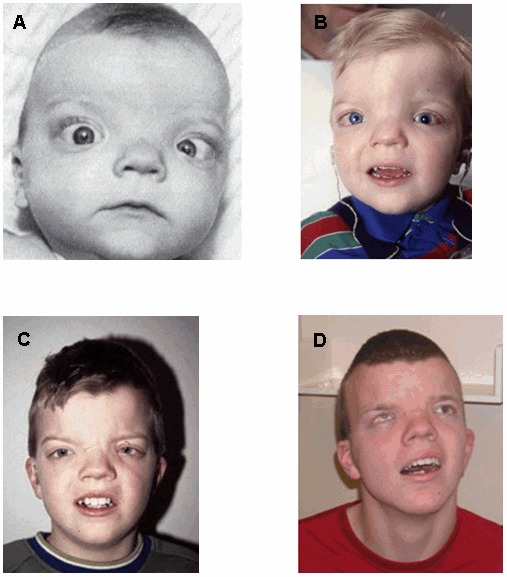
眼睛. 眼球增大(导致突眼的表现)和睑裂下斜在所有受累的个体中都能见到。虹膜缺损见于一小部分的患者。高度近视(>6个屈光度)、视网膜脱离、视网膜色素变性和进行性视力下降可见。感音神经性耳聋是普遍的表现。
膈疝 (或膈膨升)和/或脐膨出(或脐疝)都分别在大约50%的受累的个体中被报导。这两种缺陷同时发生大约见于1/3的病例。注意:这两种表现都缺乏或其中一个缺乏并不能排除诊断。
神经系统. 完全或部分胼胝体发育不全 (ACC)在几乎所有的临床诊断为DBS的患者都有报导(见图 2)。有趣的是,临床诊断为面-眼-声-肾(FOAR)综合征(包括Devriendt et al [1998]报导的病例,确定存在LRP2的突变,其表现与DBS/FOAR的特征存在重叠)。
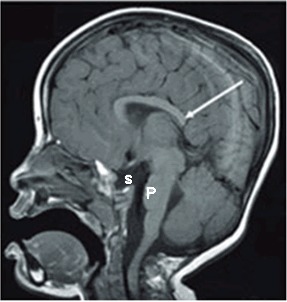
图 2.
一个3岁女性MRI显示出许多的异常,包括胼胝体发育不良(最显著的为胼胝体嘴和压部)(长白箭头),部分空蝶鞍和小脑桥。
注意:FOAR综合征,或面-眼-声-肾综合征,现在被认为是DBS的等位基因病。
发育延迟几乎总是出现。
检测
有功能的巨蛋白(由LRP2编码的蛋白)缺乏或异常,阻止正常的肾近曲小管重吸收巨蛋白配体,导致100%的DBS患者低分子量的蛋白在尿液中过量溢出。尿蛋白电泳确定的两个重要的低分子量蛋白是:
- 视黄醇结合蛋白 (RBP)
- 维生素D结合蛋白 (DBP)
注意:有限的数据显示这些物质在尿液中过量溢出会导致血浆中维生素A(视黄醇)和维生素D的浓度降低。注意:使用一种“油尺”(它能确定白蛋白)来评估尿蛋白不能作为尿蛋白电泳的合适的替代方法,因为DBS患者溢出的尿蛋白分子分子量低于白蛋白。
检测策略
为了在一个有DBS临床特征的先证者中确定/建立诊断:
- 进行脑MRI扫描来确定部分或完全胼胝体发育不全
- 进行尿蛋白电泳来确定低分子量蛋白尿和特征性的带型
- LRP2序列分析
有风险亲属的携带者检测 需要先在家系中确定致病性变异。注意:携带者是这种常染色体隐性遗传病的杂合子且没有发展成此病的风险。然而,从几个被证明为杂合子的人中收集的尿液发现比正常的总的蛋白要高[Kantarci et al 2007]。
临床特征
临床描述
因为许多受累的妊娠被终止或者继发于先天畸形的围产期的死亡,Donnai-Barrow综合征(DBS)患者比较少,其长期随访和自然史的信息有限。以下信息基于引文Holmes & Schepens [1972]、Donnai & Barrow [1993]、Schowalter et al [1997]、Devriendt et al [1998]、Avunduk et al [2000]、Chassaing et al [2003]、Chen [2007]、Kantarci et al [2007]和Patel et al [2007],除非另有说明,在Pober et al [2009]中都重新梳理了一遍。在一些病例中报导了相对较高的出生体重(~4 kg)。在出生后的前几周,有膈疝和/或脐膨出这些并发症的新生儿的紧急外科治疗和术后护理占据主导地位。进行性的视力下降导致失明常见,它似乎是高度近视的结果,伴随的风险为视网膜脱离,虽然及时治疗能增加视力保存的机会。在一些病例中,周围屏障激光凝固法的预防性治疗已经成功能地预防了视网膜脱离。进行性视网膜变性已经在少数患者中观察到;视网膜变性的确切性质和进展的概率仍然未知。感音神经性耳聋较普遍。一些患者用助听器可保留部分听力,少数患者接受了人工耳蜗植入。然而,2个长期存活者(15和21岁)有显著的听力损失(定义见耳聋和遗传性听力损失概述)。运动里程碑事件仅仅轻微延迟,大多数儿童有自制力。没有关于智力功能的正式研究存在,但是可得的数据显示所有DBS患者都有智力障碍,其轻重程度不一,介于轻度到中度的范围。因为严重的视力和听力缺陷,正式的评估是困难的。已知的年龄最大的DBS患者21岁,进入了一所对其听力和视力问题提供特殊支持的大学。他没有言语,但是使用英国手语进行沟通。他能自己穿衣服和做一些事情,例如保持房间整洁、整理床铺和操作微波炉和DVD播放机。他参与了支持就业活动,享受邮递和贴标签的事务。他喜欢社交活动和去餐厅,虽然切食物时他仍然需要一些帮助。其他的脑部异常的表现,例如脑室周围结节状异位和脑回结构异常,越来越多地被发现。其他器官系统的受累(例如心血管系统的室间隔缺损[VSD]和泌尿生殖系统的双角子宫)已被报导但是罕见。因为DBS胎儿和死于新生儿期的受累婴儿的有限检查,这样的缺陷的患病率可能被低估。一个伴局灶性节段性肾小球硬化的患者已被报导;是否这个表现是巧合或者DBS的一个罕见的表现目前未知[Shaheen et al 2010]。儿童和青春期的一般健康状况是好的,虽然一些患者在青春期出现了癫痫性疾病。在合适的时间会出现青春期发育。身高和体重处于正常范围之内;两个年龄较大的儿童身材高大,身高在第90百分位数。
病理生理学. 巨蛋白有许多配体。因为DBS患者巨蛋白缺乏或功能障碍,似乎有可能肾脏重吸收一些复合物,例如视黄醇或维生素D功能障碍,导致血浆和/或细胞内的缺乏。从几个受累个体的初步数据可以确定,与对照组相比,其循环视黄醇浓度降低[Kantarci et al 2007]。巨蛋白也定位于人类大脑,其在胚胎发生的神经元发育过程中起重要作用,它能转运配体,使其穿过脉络丛进入星形胶质细胞。这些过程的异常能导致可观察到的脑的畸形和智力障碍[Bento-Abreu et al 2008]。进一步的证据表明,在巨蛋白缺乏的情况下,细胞生长和眼内液体生成的调节异常是眼球增大和高度近视的可能原因[Veth et al 2011]。
基因型-表型相关性
目前,还不能通过基因型来预测表型。关于家系内表型差异的出现,其解释还未知,与家系间成员同一表型的差异相当[Kantarci et al 2007]。
命名
当提到DBS的时候以下名称不应该再被使用:
- 眼和面部异常、内眦距过宽和耳聋综合征
- Holmes-Schepens综合征
- 膈疝-脐膨出-眼距增宽综合征
- 膈疝-眼距增宽-近视-耳聋综合征
患病率
没有可获得的发病率或患病率数据。然而,受累的个体父母近亲婚配的富集提示突变的等位基因是罕见的。DBS在不同的种族中被报导,包括北欧和中欧、中东、欧洲裔美国人和非洲裔美国人。没有哪一个种族占主导地位。
遗传相关性(等位基因)疾病
DBS [Donnai & Barrow 1993]和FOAR综合征[Holmes & Schepens 1972, Schowalter et al 1997]曾经被作为两个不同的疾病被报导,虽然人们意识到它们的临床特征存在重叠[Devriendt et al 1998, Chassaing et al 2003]。现在人们知道这些综合征在本质上是相同的,都是由LRP2的致病性变异引起的[Kantarci et al 2007]。除了DBS/FOAR综合征,目前还未发现其他表型与LRP2的致病性变异相关。
鉴别诊断
Donnai-Barrow综合征(DBS)与先天性膈疝(CDH)相关(见先天性膈疝概述)。以下综合征包含DBS的一些特征:
处理
初步诊断后的评估
为了确定被诊断为Donnai-Barrow综合征患者疾病的严重程度,建议进行以下工作:
- 神经影像学检查(最好是MRI扫描)
- 包含青光眼筛查的详细的眼科学检查
- 听力学评估
- 尿蛋白定量测定
- 包含血尿素氮(BUN)和血清肌酐浓度的肾功能的血清学检测
- 循环维生素A、维生素D和其他钙状态调节因子水平的检测
- 如果症状提示癫痫性疾病,做EEG检查
- 临床遗传咨询
对症治疗
以下是合适的:
- 外科修复膈疝和术后评估呼吸功能以确定长期呼吸问题的风险是否增加
- 外科修复脐膨出
注意:脐膨出和/或膈疝的外科修复,与具有这些缺陷的其他遗传综合征的儿童相比,看起来风险并没有增加。 - 对于近视提供矫正镜片;治疗视网膜脱离或青光眼
- 提供合适的助听器和/或人工耳蜗植入(更详细的资料见耳聋和遗传性听力损失概述)
- 对不同程度的智力、视力和听力异常进行专门的教育
- 对于癫痫发作使用抗癫痫药物
监测
一旦考虑诊断DBS,就应该进行正规的眼科学检查。需要进行频繁和连续监测;应该根据呈现出的表现来确定安排检查。治疗(例如周围激光光凝治疗)能使高度近视导致的视网膜脱离的风险降到最低。一旦考虑诊断DBS,就应该进行听力评价。听力损失自然史和可能的进展的数据目前还不可得,因此还没有建立监测时间表。基于Shaheen et al [2010]的报导,定期肾功能的监测似乎是谨慎的。然而,DBS相关性肾功能不全的频率目前还未知。
正在研究中的治疗
想要获取各种不同疾病和情况的临床研究的信息,搜索ClinicalTrials.gov。注意:对于这种疾病可能没有临床试验。
遗传咨询
遗传咨询是向患者及其家庭提供关于遗传性疾病的特征、遗传方式和影响的信息的过程,以帮助他们做出明智的医疗和个人决策。以下部分涉及到遗传风险评估和使用家族史和遗传检测来明确家系成员的遗传状态。这一部分不是为了解决患者可能面临的所有个人的、文化的或伦理问题,也不是为了替代遗传学专业人员的咨询工作。—ED.
遗传方式
Donnai-Barrow综合征(DBS)以常染色体隐性遗传方式进行遗传。
家系成员风险
先证者的父母
- 杂合子(携带者)是无症状的,因为他们不表现出任何的结构性的出生缺陷或颅面部畸形。然而,从几个被证实为杂合子的个体收集的尿液中显示出比正常高的总蛋白[Kantarci et al 2007];对于确定这一发现的频率(和临床意义,如果有的话)需要进一步的研究。
先证者的同胞
- 由于在受累的同胞之间发生的主要结构性出生缺陷(例如脐膨出或CDH)有差别,在一个同胞中出现这些缺陷中的某一个并不能预测在另个同胞上也发生这些缺陷中的其中一个或都发生。
- 一旦一个有风险的同胞被确认未受累,他/她是携带者的概率为2/3。
先证者的后代. 关于DBS患者的生殖问题还没有发表报导。
产前检测和植入前遗传学诊断
分子遗传学检测. 一旦LRP2致病性变异在一个受累的家系成员中被确定,对Donnai-Barrow综合征风险增加的妊娠进行产前检测和植入前遗传学诊断是可能的选择。
胎儿超声检查. 对于具有DBS高危风险的妊娠可以使用超声检查或胎儿MRI扫描进行产前诊断,可以检测一些异常例如部分或完全性胼胝体发育不全、膈疝、脐膨出和包含眼距增宽和眼球突出的特征性的面部表现。
资源
GeneReviews工作人员选择了以下的疾病特异性和/或伞状支持组织和/或登记系统,为患有此病的个体和家庭提供帮助。对于其他组织提供的信息GeneReviews不予负责。关于选择标准的信息,点击这里。
- American Association on Intellectual and Developmental Disabilities (AAIDD)501 3rd Street NorthwestSuite 200Washington DC 20001Phone: 202-387-1968Fax: 202-387-2193Email: sis@aaidd.org
- American Society for Deaf Children (ASDC)800 Florida Avenue NortheastSuite 2047Washington DC 20002-3695Phone: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)Fax: 410-795-0965Email: info@deafchildren.org; asdc@deafchildren.org
- CHERUBSThe Association of Congenital Diaphragmatic Hernia Research, Awareness and Support3650 Rogers Road#290Wake Forest NC 27587Phone: 919-610-0129Fax: 815-425-9155Email: info@cdhsupport.org
- National Eye Institute31 Center DriveMSC 2510Bethesda MD 20892-2510Phone: 301-496-5248Email: 2020@nei.nih.gov
分子遗传学
分子遗传学和OMIM表中的信息可能与GeneReview中其他地方的信息有差异:表格中可能包含更新的信息。-ED.
表 A.
Donnai-Barrow综合征:基因和数据库
分子遗传学机制
Donnai-Barrow综合征(DBS)被假定是由低密度脂蛋白受体相关蛋白2基因(LRP2)的功能丧失性致病性变异引起的,此基因编码一种具有内吞作用的跨膜糖蛋白巨蛋白(LRP-2/gp330)[Kantarci et al 2007]。巨蛋白是在大鼠的Heymann肾炎中作为致病性自身抗原第一次被描述的。大多数巨蛋白敲除的老鼠(巨蛋白 -/-)由于呼吸功能不全在出生后不久死亡[Willnow et al 1996, Leheste et al 1999]。对巨蛋白缺乏的老鼠进行检查发现包含ACC和轻度前脑无裂畸形的前脑异常
[Willnow et al 1996, Leheste et al 1999]。对少数的DBS患者进行详细的神经影像学检查,普遍存在胼胝体异常和眼球异常,但是没有一个有前脑无裂畸形的证据。在一个受累个体中的脑室周围结节状异位和皮质脑回异常强调了巨蛋白早期人类大脑发育过程中的重要作用[Kantarci et al 2007]。巨蛋白在肾脏发育过程中不是必需的,但是在內吞过程中是需要的
[Willnow et al 1996, Leheste et al 1999, Anzenberger et al 2006, Fisher & Howie 2006]。巨蛋白缺乏老鼠分泌的低分子量血浆蛋白包含DBP和RBP,尿液中的α1-微球蛋白和气味结合蛋白[Leheste et al 1999]。与巨蛋白缺乏老鼠相似,所有受累的确定存在LRP2致病性变异的个体都显示出了低分子量蛋白尿伴DBP和RBP的排泄增加[Kantarci et al 2007]。
基因结构.LRP2 横跨大约236 kb的基因组DNA,包含79个编码外显子。 外显子 1的前半段和外显子79的后一部分不被翻译[Hjälm et al 1996]。关于基因和蛋白信息的更详细总结,见表 A,基因.
致病性等位基因变异. 在8个受累家系中的12个致病性变异已被记录[Kantarci et al 2007, Kantarci et al 2008]。每个家系都有其特有的致病性变异,分布于整个基因。在患者中发现的LRP2纯合或复合杂合变异有小的缺失或插入,或保守的剪接位点、无义和错义变异,遗传自其杂合携带者的父母。作为一个罕见事件,有一个患者是LRP2致病性变异的纯合子,其来自2号染色体的父源单亲二倍体。已经明确的是,其父亲是这个变异的杂合携带者,而母亲有两个良性的LRP2等位基因[Kantarci et al 2008]。
表 2.
选择的LRP2致病性变异
| DNA核苷酸改变 (别名 1) | 蛋白氨基酸改变 (别名 1) | 参考序列 |
|---|---|---|
| c.770-2A>G (IVS7-2A>G) | -- | NM_004525-.2 NP_004516-.2 |
| c.1093C>T | p.Arg365Ter | |
| c.1341+2T>G (IVS11+2T>G) | -- | |
| c.2840-1G>A (IVS18-1G>A) | -- | |
| c.7564T>C | p.Tyr2522His | |
| c.8452+1G>A (IVS44+1G>A) | -- | |
| c.8516_8519delTTTA | p.Tyr2840CysfsTer67 (Val2839ValfsTer67) | |
| c.9358_9359delAG | p.Ser3120TrypfsTer26 | |
| c.9484_9485delGT | p.Val3162LeufsTer2 | |
| c.10195C>T | p.Arg3399Ter | |
| c.11469_11472delTTTG 2 | p.Cys3823TrpfsTer159 | |
| c.13139dupC (13139insC) | p.Cys4381MetfsTer12 (Pro4380ProfsTer12) |
变异分类需注意的:表格中列出的变异由作者提供。GeneReviews工作人员没有再单独证实变异的分类。
命名需注意的:GeneReviews按照人类基因组变异协会(varnomen-.hgvs.org)规定的命名标准来命名。对于命名的解释见快速参考。
1.
不符合当前命名规定的变异名称
2. 在一个受累的DBS/FOAR个体中的由父源单亲二倍体导致的纯合性致病性变异[Kantarci et al 2008]
正常基因产物. 600-kd的巨蛋白包含4655个氨基酸。巨蛋白是低密度脂蛋白(LDL)受体基因家族的成员之一,其主要在吸收或分泌上皮的顶端表达。蛋白的大的细胞外结构域包含36个富含半胱氨酸的LDL受体A群(补充型)模体,37个LDL受体B群(包含YWTD)模体和一个EGF样的重复。蛋白的单个跨膜结构域后面跟着的是包含两个NpxY模体的一个短的胞质内(C末端)的结构域。各种组织中都能表达巨蛋白,包括大脑、眼睛、肾近端小管、肺、肠道、子宫、输卵管、男性生殖道和胚胎卵黄囊。巨蛋白结合50多个配体,包括脂蛋白、维生素结合蛋白、激素、酶和免疫和应激反应相关蛋白。有人提出巨蛋白与索尼克刺猬蛋白发生相互作用。在发育过程中,受体相关蛋白和巨蛋白在不同的组织中共表达。巨蛋白和一类膜受体cubilin(gp280)共享许多配体[Fisher & Howie 2006]。
异常基因产物. 导致DBS的LRP2致病性变异被预测为导致蛋白功能的缺失基于在所有受累的个体中都观察到了相似的表型,包括那些纯合移码变异导致的密码子提前终止。对于导致DBS特征性的先天畸形的突变蛋白,没有可获得的详细的病理生理学的描述或解释。
参考资料
引用文献
- Anzenberger U, Bit-Avragim N, Rohr S, Rudolph F, Dehmel B, Willnow TE, Abdelilah-Seyfried S. Elucidation of megalin/LRP2-dependent endocytic transport processes in the larval zebrafish pronephros. J Cell Sci. 2006;119:2127-37. [PubMed: 16638803]
- Avunduk AM, Aslan Y, Kapicioğlu Z, Elmas R. High myopia, hypertelorism, iris coloboma, exomphalos, absent corpus callosum, and sensorineural deafness: report of a case and further evidence for autosomal recessive inheritance. Acta Ophthalmol Scand. 2000;78:221-2. [PubMed: 10794262]
- Bento-Abreu A, Velasco A, Polo-Hernández E, Pérez-Reyes PL, Tabernero A, Medina JM. Megalin is a receptor for albumin in astrocytes and is required for the synthesis of the neurotrophic factor oleic acid. J Neurochem. 2008;106:1149-59. [PubMed: 18466341]
- Chassaing N, Lacombe D, Carles D, Calvas P, Saura R, Bieth E. Donnai-Barrow syndrome: four additional patients. Am J Med Genet A. 2003;121A:258-62. [PubMed: 12923867]
- Chen CP. Syndromes and disorders associated with omphalocele (III): single gene disorders, neural tube defects, diaphragmatic defects and others. Taiwan J Obstet Gynecol. 2007;46:111-20. [PubMed: 17638618]
- Devriendt K, Standaert L, Van Hole C, Devlieger H, Fryns JP. Proteinuria in a patient with the diaphragmatic hernia-hypertelorism-myopia-deafness syndrome: further evidence that the facio-oculo-acoustico-renal syndrome represents the same entity. J Med Genet. 1998;35:70-1. [PMC free article: PMC1051192] [PubMed: 9475100]
- Donnai D, Barrow M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: a newly recognized autosomal recessive disorder? Am J Med Genet. 1993;47:679-82. [PubMed: 8266995]
- Fisher CE, Howie SE. The role of megalin (LRP-2/Gp330) during development. Dev Biol. 2006;296:279-97. [PubMed: 16828734]
- Hjälm G, Murray E, Crumley G, Harazim W, Lundgren S, Onyango I, Ek B, Larsson M, Juhlin C, Hellman P, Davis H, Akerström G, Rask L, Morse B. Cloning and sequencing of human gp330, a Ca(2+)-binding receptor with potential intracellular signaling properties. Eur J Biochem. 1996;239:132-7. [PubMed: 8706697]
- Holmes LB, Schepens CL. Syndrome of ocular and facial anomalies, telecanthus, and deafness. J Pediatr. 1972;81:552-5. [PubMed: 4626128]
- Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GC, Bieth E, Chassaing N, Lacombe D, Devriendt K, Teebi A, Loscertales M, Robson C, Liu T, MacLaughlin DT, Noonan KM, Russell MK, Walsh CA, Donahoe PK, Pober BR. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957-9. [PMC free article: PMC2891728] [PubMed: 17632512]
- Kantarci S, Ragge NK, Thomas NS, Robinson DO, Noonan KM, Russell MK, Donnai D, Raymond FL, Walsh CA, Donahoe PK, Pober BR. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008;146A:1842-7. [PMC free article: PMC2891749] [PubMed: 18553518]
- Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, Aucouturier P, Moskaug JO, Otto A, Christensen EI, Willnow TE. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol. 1999;155:1361-70. [PMC free article: PMC1867027] [PubMed: 10514418]
- Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007;22:462-4. [PubMed: 17621530]
- Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birth Defects Res A Clin Mol Teratol. 2009;85:76-81. [PMC free article: PMC2882234] [PubMed: 19089858]
- Schowalter DB, Pagon RA, Kalina RE, McDonald R. Facio-oculo-acoustico-renal (FOAR) syndrome: case report and review. Am J Med Genet. 1997;69:45-9. [PubMed: 9066882]
- Shaheen IS, Finlay E, Prescott K, Russell M, Longoni M, Joss S. Focal segmental glomerulosclerosis in a female patient with Donnai-Barrow syndrome. Clin Dysmorphol. 2010;19:35-7. [PubMed: 19952924]
- Veth KN, Willer JR, Collery RF, Gray MP, Willer GB, Wagner DS, Mullins MC, Udvadia AJ, Smith RS, John SWM, Gregg RG, Link BA. Mutations in zebrafish lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLoS Genet. 2011;7:e1001310. [PMC free article: PMC3040661] [PubMed: 21379331]
- Willnow TE, Hilpert J, Armstrong SA, Rohlmann A, Hammer RE, Burns DK, Herz J. Defective forebrain development in mice lacking gp330/megalin. Proc Natl Acad Sci U S A. 1996;93:8460-4. [PMC free article: PMC38693] [PubMed: 8710893]
推荐阅读
- Kantarci S, Donahoe PK. Congenital diaphragmatic hernia (CDH) etiology as revealed by pathway genetics. Am J Med Genet C Semin Med Genet. 2007;145C:217-26. [PubMed: 17436295]
章注
致谢
感谢Dr Patricia K Donahoe的领导和持续的支持。也感谢许多把DBS/FOAR患者介绍给我们的医生。最后,我们由衷感谢所有参与到我们研究中的家庭,通过他们我们才能够发现造成DBS/FOAR的基因。
修订历史
- 2011年6月28日 在网页上发布实时全面更新
- 2008年8月28日 内容发布到实时网页上
- 2008年4月28日 原创投稿