
摘要
临床特点.
X连锁铁粒幼细胞性贫血伴共济失调(X-linked sideroblastic anemia and ataxia, XLSA/A)的特点是:中度贫血、男性早发性脊髓小脑综合征(主要表现为:行走延迟、童年早期明显共济失调、辨距不良和轮替运动障碍)。起病时,意向性震颤轻微,构音障碍为轻至中重度。共济失调被描述为无进展或缓慢进展。腿部上运动神经元(upper motor neuron, UMN)受损体征,表现为:深部腱反射活跃、非持续性踝阵挛、一些男性巴氏征阳性或可疑阳性。已有需要拐杖或轮椅的报道。斜视见于一些男性。可发生眼球震颤和长度异常性扫视。可见轻度学习障碍和抑郁。轻度小细胞低色素性贫血不引起症状。携带者(杂合) 女性神经系统检查正常,可表现出轻度血液系统异常。
诊断/检测.
具有特征性神经系统发现、存在轻度小细胞低色素性贫血、全血总红细胞原卟啉(total erythrocyte protoporphyrin, TEP)和锌原卟啉(zinc erythrocyte protoporphyrin, ZnEP)升高以及骨髓检查见环形铁粒幼细胞的男性应疑诊XLSA/A。外周血涂片见含铁小体。1名男性因ABCB7半合子致病性变异的鉴定而明确诊断。
女性神经系统检查正常,血涂片可同时见小细胞低色素红细胞和正常红细胞。她们的骨髓检查可见环形铁粒幼细胞。
管理.
对症治疗: XLSA/A男性可从早期物理治疗中获益,以便获得大肌肉运动技能。可能需要适应装置,如:踝关节固定矫形器和助行架。有利的饮食用具可帮助促进儿童期的独立技能。言语治疗可改善构音障碍的理解问题。书写困难可用计算机进行文字处理解决。
诊断
X连锁铁粒幼细胞性贫血伴共济失调(X-linked sideroblastic anemia and ataxia, XLSA/A)男性表现如下征象:
- 共济失调和不协调。注:Pagon et al [1985]描述了非进行性共济失调; Hellier et al [2001]描述共济失调可缓慢进展。
腿部上运动神经元(upper motor neuron, UMN)受损体征(如深部腱反射活跃、非持续性踝阵挛、巴氏征阳性或可疑阳性)(见于某些男性)
- 轻度、无症状的小细胞低色素性贫血
红细胞压积波动在从26%至35%。
平均红细胞体积 (Mean corpuscular volume, MCV fl)降低(表1)。
- 骨髓检查见环形铁粒幼细胞,示储存铁增多(图1)。注:血清铁参数正常。
- 全血总红细胞原卟啉(total erythrocyte protoporphyrin, TEP)和锌原卟啉(zinc erythrocyte protoporphyrin, ZnEP)高水平进一步支持诊断 [Pagon et al 1985, Bekri et al 2000, Hellier et al 2001, Maguire et al 2001, D’Hooghe et al 2012]。
注: (1) 全血TEP可作为筛查手段[Piomelli et al 1975, Hart & Piomelli 1981]。(2) 如未鉴定出ABCB7致病性变异(见后XLSA/A的诊断),但鉴定出至少一名患病存在特征性骨髓改变和高水平的TEP,而且家族史符合X连锁遗传,强烈提示XLSA/A诊断。
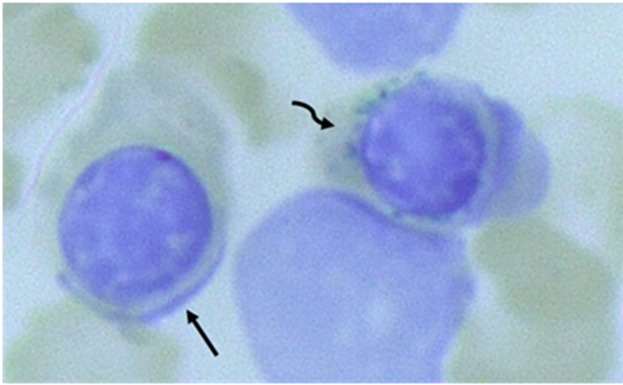
表1.
XLSA/A的平均红细胞体积(MCV fl)
| 组 | 平均红细胞体积 (MCV fl) |
|---|---|
| 正常男性 | 89.1 ± 5.01 |
| 正常女性e | 87.6 ± 5.5 |
| 患病男性 | 58至68 |
| 女性携带者 | 83至90 |
Pagon et al [1985] (4例 受累 男性和3例女性肯定携带者)
杂合女性 神经系统检查正常,可表现出轻度血液学异常,包括:血涂片含铁小体[Hellier et al 2001] 以及骨髓片见环形铁粒幼细胞[Pagon et al 1985]。一些杂合女性的全血TEP和ZnEP也升高[Pagon et al 1985]。
XLSA/A确诊依据是:男性存在ABCB7半合子。
表2.
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
临床特点
到目前为止, 已有4个无关 X连锁铁粒幼细胞性贫血伴共济失调(X-linked sideroblastic anemia and ataxia, XLSA/A)报道[Allikmets et al 1999, Bekri et al 2000, Hellier et al 2001, Maguire et al 2001, D'Hooghe et al 2012]。
Pagon et al [1985]报道了2个非进行性脊髓小脑综合征伴铁粒幼细胞性贫血家系,均按X连锁隐性遗传模式分离。2代4名男性和无关家系的另1名男性受累。 Bekri et al [2000]报道了患有XLSA/A的两兄弟。 Maguire et al [2001]报道了另一个家系中有患病的两兄弟和2名患病的舅舅。D’Hooghe et al [2012]报道了1名 XLSA/A男孩。
共济失调/神经系统发现.男性有早发性脊髓小脑综合征,主要表现为:行走延迟、儿童早期明显共济失调、辨距不良和轮替运动障碍。起病时,意向性震颤轻微,构音障碍为轻至中重度。
家系中一些(不是全部)受累成员的共济失调,看起来随时间推移改善,如躯干蹒跚减轻,行走变得越来越容易[Pagon et al 1985]。然而,五、六十岁的老人可见行走困难缓慢恶化[Pagon et al 1985, Bekri et al 2000, Hellier et al 2001]。
已有需要拐杖或轮椅的报道。
腿部上运动神经元(upper motor neuron, UMN)受损体征,表现为:深部腱反射活跃、非持续性踝阵挛、一些男性巴氏征阳性或可疑阳性。
斜视见于一些男性。眼球外转运动正常。然而,可发生眼球震颤和长度异常性扫视。
智力通常在正常范围。可见轻度学习障碍和抑郁 [Pagon & Bird, 个人交流], 还有1例患"精神分裂症"的报道 [Hellier et al 2001]。
无高弓足、脊柱侧弯和肌肉萎缩。
未见视神经萎缩或视网膜营养不良引起的视力损害。
大多数病例的脑MRI示小脑萎缩或发育不良[Raskind et al 1991]。
贫血. 贫血为轻度且不引起症状。
铁储存. 尽管发现铁储存增加、骨髓检查发现环形铁粒幼细胞,但无系统性铁过载的记述。在Pagon et al [1985]和Hellier et al [2001]报道的家系中,血清铁研究均正常,包括:血清铁浓度,总铁结合力(total iron binding capacity, TIBC)、TIBC饱和度和血清铁蛋白浓度。
外显率
目前已报道4个家系和1例男性单发病例(即, 家系中单个发生)[Pagon et al 1985, Bekri et al 2000, Hellier et al 2001, D’Hooghe et al 2012]。 由于忽视具有典型共济失调的轻度贫血男性,此病的患病率可能被低估。
遗传相关(等位基因)疾病
除了此GeneReview讨论的表型以外,无其它表型与ABCB7生殖细胞致病性变异有关。
在对难治性贫血伴环状铁粒幼细胞(RARS),一种以幼红细胞线粒体中过量铁积聚为特征的获得性骨髓增生异常综合征(MDS)个体的研究中,Boultwood et al [2008] ide已鉴定出骨髓环形铁粒幼细胞百分比增高ABCB7 表达水平降低的相关性,提示ABCB7 是RARS的候选基因。 SF3B1(一种RNA剪切机器组份)突变与大多数RARS相关[Yoshida et al 2011]。 Nikpour et al [2013]发现当SF3B1突变时,ABCB7表达下调。而且,ABCB7下调会减少K562细胞的红系分化、生长及克隆形成[Nikpour et al 2013]。
鉴别诊断
铁粒幼细胞性贫血. 铁粒幼细胞性贫血是以环形铁粒幼细胞(骨髓幼稚红细胞的线粒体中,病理性铁过载)为特征的一类异质性的获得性或遗传性贫血; 线粒体的核周定位导致特征性的环形外观[Camaschella 2008, Bergmann et al 2010]。 铁包涵体,被称为含铁小体,也可见于更成熟的红细胞[Sears & Udden 2004, Camaschella 2008] (图1)。
最常见的先天性铁粒幼细胞性贫血是ALAS2突变引起的X连锁铁粒幼细胞性贫血(X-linked sideroblastic anemia, XLSA)。XLSA以肝脏和系统性铁过载但无共济失调(因ALAS2表达限于红系组织)为特征[Napier et al 2005]。
其它引起先天性铁粒幼细胞性贫血的原因包括:编码影响线粒体代谢蛋白的基因突变 [Bergmann et al 2010, Fujiwara & Harigae 2013] 和/或影响铁硫(Fe-S)簇蛋白生物合成、组装及功能。铁硫蛋白对于基本代谢过程(如呼吸和基因表达)来说必不可少,其在线粒体合成,与线粒体载脂蛋白形成线粒体铁硫蛋白有关,或转运至胞质(通过ABCB7转运蛋白)协助胞质和胞核铁硫蛋白形成[Sheftel et al 2010]。这些基因如下:
- SLC25A38 (编码一种红系特异性线粒体转运蛋白) [Guernsey et al 2009]
- SLC19A2 (编码一种高亲和性硫胺素转运蛋白)
- PUS1 (RNA修饰酶假尿嘧啶核苷合成酶1) [Bykhovskaya et al 2004]
- ABCB7 (编码线粒体ATP结合盒转运蛋白), XLSA/A的病因
- GLRX5 (编码单巯基谷氧还蛋白5) [Camaschella et al 2007],与一种Fe-S簇生物合成缺乏有关(线粒体Fe/S蛋白组装)。线粒体铁代谢失调导致铁粒幼细胞性贫血。
- YARS2 (线粒体酪氨酰tRNA合成酶) [Riley et al 2010]
另一种与铁硫簇蛋白关联的共济失调是Friedreich共济失调 (Friedreich ataxia, FRDA),FXN突变引起,此基因编码线粒体蛋白,其主要作用是为Fe-S簇合成提供生物可利用形式的铁(Figure 2) [Ye & Rouault 2010b]。在FRDA患者的心肌细胞和神经元中发现了铁过量积聚 [Rouault & Tong 2008, Ye & Rouault 2010b];但 FRDA患者未见明显贫血,提示frataxin对于血红素和红系造血来说并非必不可少,或者frataxin缺乏不影响红系造血组织[Stemmler et al 2010, Ye & Rouault 2010a]。
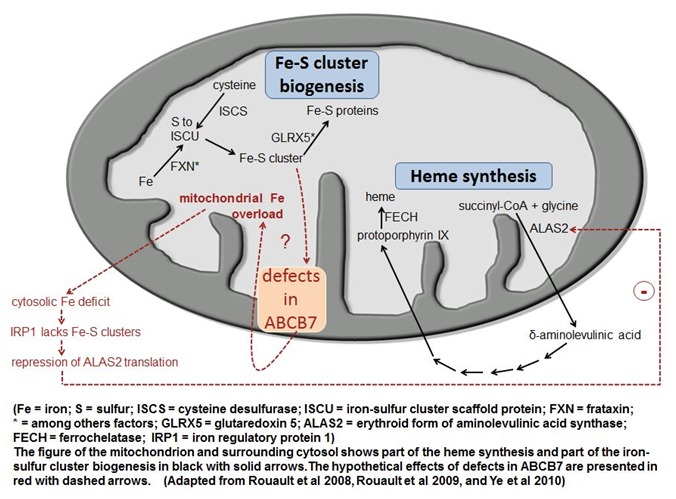
图2.
Fe -S簇生物合成、血红素合成和ABCB7缺乏的假想效应 经获准改编自D’Hooghe等 [2012]
值得注意的是,在frataxin水平降低的小鼠培养成纤维细胞中观察到:与人类疾病一样,一些铁硫簇蛋白缺乏,线粒体中铁堆积,并有氧化剂敏感性[Calmels et al 2009]。这些作用之间的因果联系还没有得到很好的解释[Stemmler et al 2010]。
共济失调.许多可遗传共济失调的诊断可能很困难。一些遗传性共济失调通常在3岁之前起病,包括共济失调性毛细血管扩张症、 婴儿起病的脊髓小脑性共济失调、 X连锁铁粒幼细胞性贫血伴共济失调 (XLSA/A)、 先天性糖基化障碍和小脑畸形 (如Dandy-Walker畸形) [Bernard & Shevell 2008]。
X连锁脊髓小脑性共济失调 已有报道,但罕见(见遗传性共济失调概述)。未见与贫血相关。在任何无法解释的X连锁脊髓小脑性共济失调中,即使无明显血液学改变,ABCB7突变应予考虑。
管理
初次诊断后的评估
为了确定被诊断为铁粒幼细胞性贫血伴共济失调(sideroblastic anemia and ataxia, XLSA/A)个体的疾病严重程度,建议行以下评估:
- 神经系统检查
- 脑CT或MRI
- 遵嘱行心理测试
- 医学遗传学咨询
对症治疗
XLSA/A无有效治疗。
共济失调男性可从早期物理治疗中获益,以便获得大肌肉运动技能。可能需要适应装置,如:踝关节固定矫形器和助行架。
有利的饮食用具可帮助促进儿童期的独立技能。
言语治疗可改善因构音障碍导致的理解问题。
书写困难可用计算机进行文字处理解决。
贫血常为轻度且无症状,不需治疗;未发生肝脏和系统性铁过载。
监管
是否通过常规筛查血清铁浓度、总铁结合力(total iron binding capacity, TIBC)和血清铁蛋白浓度,在老年个体中检测可能存在的铁过载,这是有争议的;铁过载理论上是可能的,但未被报道。
研究中的疗法
对于一些涉及铁分布异常的疾病,一种可能的治疗途径是药物介导的铁搬迁。去铁酮(Deferiprone, DFP),一种用于治疗铁过载的铁的螯合剂,当用于治疗区域性铁积聚性疾病时,有铁搬迁能力。由于它们可能的副作用,铁载体(微生物分泌的小高亲和性铁螯合物)和其它螯合剂必须谨慎用药;因此,铁再分布药物在一些铁分布异常疾病(如XLSA/A)中的潜在作用,有必要进行缜密的研究 [Boddaert et al 2007, Camaschella 2008, Kakhlon et al 2010]。
检索ClinicalTrials.gov可获得大量疾病的临床研究信息。注:可能没有关于本病的临床试验。
遗传咨询
遗传咨询是向个人和家庭提供关于遗传疾病的性质、遗传方式和后果信息的过程,以帮助他们做出明智的医疗和个人决策。以下部分涉及遗传风险评估以及利 用家族史和基因检测澄清家庭成员的遗传状态。本部分并不意味着解决个体可能面临的所有个人、文化或伦理问题,或代替遗传学专业人士的咨询。—ED.
携带者检测
杂合女性 (携带者) 无症状。
- 她们神经系统检查正常,无小脑功能障碍。
她们红细胞压积正常,血涂片可见小细胞低色素红细胞和正常红细胞二态性,骨髓检查见铁粒幼细胞。一些也可有总红细胞原卟啉(total erythrocyte protoporphyrin, TEP)和锌原卟啉(zinc erythrocyte protoporphyrin, ZnEP)水平增高 [Pagon et al 1985, D’Hooghe et al 2012]。
相关遗传咨询问题
家庭计划
- 判断遗传风险、明确携带者状态、探讨产前检测可用性的最佳时间是妊娠之前。
DNA库 储存DNA(通常从血白细胞中提取),以备将来可能使用。因为将来试验技术以及对基因、等位基因变异和疾病的认识可能会提高,所以应考虑储存受累个体的DNA。
产前检测
如果已鉴定出患病家系成员的致病性ABCB7变异,高风险妊娠的产前检测就可以在提供此疾病/基因检测或定制产前检测的临床实验室完成。
对于一些疾病,如XLSA,产前检测的需求不常见。 关于产前检测的应用,尤其是,如果此检测被看作是以终止妊娠为目的,而不是为了早期诊断,医疗专家之间以及家系内部可能存在不同的看法。虽然大多数中心认为产前检测的决定是父母的选择,但商讨这些问题是合适的。
胚胎植入前遗传学诊断(PGD) 可以是某些已鉴定出致病性ABCB7变异家系的一个选择。
资源
为了此病个体及其家庭的利益,GeneReviews员工挑选了以下疾病特定的和/或伞形支持组织和/或登记处。GeneReviews不为其它组织提供的信息负责。关于选择标准的信息,点击这里。
- euro-ATAXIA (European Federation of Hereditary Ataxias)Ataxia UKLincoln House, Kennington Park, 1-3 Brixton RoadLondon SW9 6DEUnited KingdomPhone: +44 (0) 207 582 1444Email: smillman@ataxia.org.uk
- Medline Plus
- National Ataxia Foundation2600 Fernbrook LaneSuite 119Minneapolis MN 55447Phone: 763-553-0020Email: naf@ataxia.org
分子遗传学
分子遗传学和OMIM表格中的信息可能与GeneReview其它部分的信息不一致:表格可能包含更新的信息。 —ED.
Table A.
表 B.
OMIM关于X-连锁铁粒幼细胞性贫血伴共济失调的入口(在OMIM查看全部)
分子遗传学发病机制
ABCB7编码一种参与铁稳态的线粒体三磷酸腺苷(adenosine triphosphate, ATP)结合盒(ATP-binding cassette, ABC)转运蛋白。ABC转运蛋白家族包括一大组ATP依赖跨膜蛋白,其特异性地跨细胞或细胞器膜转运多种底物[Holland 2011, Moitra & Dean 2011]。人类基因组包含49种ABC基因。与细菌相比,人类线粒体出人意料地含有很少数目的(≤4)ABC蛋白[Burke & Ardehali 2007, Zutz et al 2009]。线粒体ABC蛋白属于B亚家族,组装成半转运蛋白同源二聚体[Zutz et al 2009]。ABCB7见于线粒体内膜。
由ABCB7转运的底物未被充分描述。首先,ABCB7被认为参与血红素从线粒体向胞质的转运[Shimada et al 1998]。近期不同物种间的研究提示:血红素生物合成途径、铁硫(Fe–S)簇生物合成与线粒体铁稳态之间的关系。真核生物最常见的Fe–S簇是[2Fe–2S] 和[4Fe–4S]簇。Fe–S簇是古老的生物辅基,在许多生物学过程(包括:线粒体呼吸链活动和各种其它酶促和调节作用)中必不可少的。线粒体铁过载是人类Fe–S簇组装紊乱的突出特征[Rouault & Tong 2008, Sheftel et al 2010]。可能是ABCB7转运蛋白Fe–S簇和/或一种还不知道的调节分子从线粒体传递线粒体有足够铁的信号(图2)。那么,如果Fe–S簇合成或血红素合成被破坏,胞质/胞核区室会感知到线粒体铁缺乏,反应性显著增加线粒体铁储存。
见 发病机制的附加信息 (pdf).
基因结构.ABCB7含16 个外显子[Shimada et al 1998, Bekri et al 2000]. 关于基因和蛋白质信息的详细概述见 表A, 基因。
致病性等位基因变异. 见表2和 表3。 仅有4个无关 XLSA/A家系报道,每个均有完全不同的错义突变。值得注意的是,鉴定出的致病性变异是中等严重的错义突变。
- Maguire et al [2001]报道了有两兄弟受累和两名患病舅舅的家系。 在两兄弟和仍存活舅舅中,Maguire et al [2001]发现了位于第10号外显子的半合子错义致病性变异(c.1234G>C; p.Val412Leu),其导致ABCB7蛋白第6推定跨膜区末尾的1个替换。其母是此变异的杂合子。
表3.
本篇GeneReview讨论的ABCB7 致病性变异
| DNA核苷酸改变 | 蛋白质核苷酸改变 | 参考序列 |
|---|---|---|
| c.627A>T | p.Glu209Asp | NM_004299.3 NP_004290.2 |
| c.1203T>G | p.Ile401Met | |
| c.1234G>C | p.Val412Leu | |
| c.1300G>A | p.Glu434Lys |
变异分类注释: 表中变异由作者提供。GeneReview工作人员未独立核对变异分类。
命名法注释: GeneReviews遵循人类基因组变异协会的标准命名惯例(www.hgvs.org). 关于命名法的解释见快速参考。
正常基因产物. ATP结合盒,B亚家族,成员7蛋白(ABCB7) 属于三磷酸腺苷结合盒转运蛋白超家族;其酵母同源物,Atm1p,在胞质铁硫(Fe–S)簇包含蛋白的成熟过程中起核心作用[Bekri et al 2000]。ABCB7参与红系分化过程中血红素的生成[Taketani et al 2003],也被认为转运一种胞质Fe–S簇从线粒体到胞质成熟过程所需的组份[Napier et al 2005]。因而,线粒体似乎在血红素合成和Fe–S簇生物合成过程中都很重要。
ABCB7在骨髓和小脑中均高表达。这可能解释了为什么ABCB7缺乏男性有共济失调 [Allikmets et al 1999, Ye & Rouault 2010a, Ye & Rouault 2010b]。在XLSA/A观察到的共济失调可能与神经细胞中,线粒体铁负荷介导的损伤和/或线粒体铁稳态破坏有关[Napier et al 2005]。
异常基因产物. 酵母的互补研究提示:人类突变ATP结合盒,B亚家族,成员7蛋白(ABCB7) 由轻度部分功能缺失等位基因引起[Allikmets et al 1999, Bekri et al 2000],其导致胞质Fe–S簇蛋白减少。
Pondarre et al [2006]制造了条件性小鼠同源Abcb7 等位基因敲除,正式阐明了XLSA/A因部分功能缺失性突变引起,其直接或间接抑制血红素生物合成[Pondarré et al 2007]。实际上,如一种敲除小鼠模型所示,导致显著蛋白功能缺失的突变必定是致命的[Pondarre et al 2006]。
参考文献
引用文献
- Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet. 1999;8:743–9. [PubMed: 10196363]
- Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood. 2000;96:3256–64. [PubMed: 11050011]
- Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, Neufeld EJ. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54:273–8. [PMC free article: PMC2843911] [PubMed: 19731322]
- Bernard G, Shevell M. The wobbly child: an approach to inherited ataxias. Semin Pediatr Neurol. 2008;15:194–208. [PubMed: 19073328]
- Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–8. [PubMed: 17379741]
- Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, Littlewood TJ, Malcovati L, Della Porta MG, Jädersten M, Killick S, Giagounidis A, Bowen D, Hellström-Lindberg E, Cazzola M, Wainscoat JS (2008) The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS ONE 3:e1970. [PMC free article: PMC2276313] [PubMed: 18398482]
- Burke MA, Ardehali H. Mitochondrial ATP-binding cassette proteins. Transl Res. 2007;150:73–80. [PubMed: 17656326]
- Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004;74:1303–8. [PMC free article: PMC1182096] [PubMed: 15108122]
- Calmels N, Schmucker S, Wattenhofer-Donzé M, Martelli A, Vaucamps N, Reutenauer L, Messaddeq N, Bouton C, Koenig M, Puccio H. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS One. 2009;4:e6379. [PMC free article: PMC2710521] [PubMed: 19629184]
- Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia. Br J Haematol. 2008;143:27–38. [PubMed: 18637800]
- Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–8. [PubMed: 17485548]
- D'Hooghe M, Selleslag D, Mortier G, Van Coster R, Vermeersch P, Billiet J, Bekri S. X-linked sideroblastic anemia and ataxia: a new family with identification of a fourth ABCB7 gene mutation. Eur J Paediatr Neurol. 2012;16:730–5. [PubMed: 22398176]
- Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39:270–81. [PubMed: 12382202]
- Fujiwara T, Harigae H. Pathophysiology and genetic mutations in congenital sideroblastic anemia. Pediatr Int. 2013;55:675–9. [PubMed: 24003969]
- Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, Saint-Amant L, Schmidt PJ, Orr A, Bottomley SS, Fleming MD, Ludman M, Dyack S, Fernandez CV, Samuels ME. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651–3. [PubMed: 19412178]
- Hart D, Piomelli S. Simultaneous quantitation of zinc protoporphyrin and free protoporphyrin in erythrocytes by acetone extraction. Clin Chem. 1981;27:220–2. [PubMed: 7460270]
- Hellier KD, Hatchwell E, Duncombe AS, Kew J, Hammans SR. X-linked sideroblastic anaemia with ataxia: another mitochondrial disease? J Neurol Neurosurg Psychiatry. 2001;70:65–9. [PMC free article: PMC1763461] [PubMed: 11118249]
- Holland IB. ABC transporters, mechanisms and biology: an overview. Essays Biochem. 2011;50:1–17. [PubMed: 21967049]
- Kakhlon O, Breuer W, Munnich A, Cabantchik ZI. Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation. Can J Physiol Pharmacol. 2010;88:187–96. [PubMed: 20393584]
- Maguire A, Hellier K, Hammans S, May A. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L. Br J Haematol. 2001;115:910–7. [PubMed: 11843825]
- Moitra K, Dean M. Evolution of ABC transporters by gene duplication and their role in human disease. Biol Chem. 2011;392:29–37. [PubMed: 21194360]
- Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005;105:1867–74. [PubMed: 15528311]
- Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, Giai V, Fernandez-Mercado M, Papaemmanuil E, Högstrand K, Jansson M, Vedin I, Stephen Wainscoat J, Campbell P, Cazzola M, Boultwood J, Grandien A, Hellström-Lindberg E. The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia. 2013;27:889–96. [PMC free article: PMC3794445] [PubMed: 23070040]
- Pagon RA, Bird TD, Detter JC, Pierce I. Hereditary sideroblastic anaemia and ataxia: an X linked recessive disorder. J Med Genet. 1985;22:267–73. [PMC free article: PMC1049446] [PubMed: 4045952]
- Piomelli S, Lamola AA, Poh-Fitzpatrick MF, Seaman C, Harber LC. Erythropoietic protoporphyria and lead intoxication: the molecular basis for difference in cutaneous photosensitivity. I. Different rates of disappearance of protoporphyrin from the erythrocytes, both in vivo and in vitro. J Clin Invest. 1975;56:1519–27. [PMC free article: PMC333130] [PubMed: 1202082]
- Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, Anderson SA, Eisenstein RS, Fleming MD. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulphur cluster biogenesis. Hum Mol Genet. 2006;15:953–64. [PubMed: 16467350]
- Pondarré C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109:3567–9. [PMC free article: PMC1852240] [PubMed: 17192398]
- Raskind WH, Wijsman E, Pagon RA, Cox TC, Bawden MJ, May BK, Bird TD. X-linked sideroblastic anemia and ataxia: linkage to phosphoglycerate kinase at Xq13. Am J Hum Genet. 1991;48:335–41. [PMC free article: PMC1683027] [PubMed: 1671320]
- Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, McKenzie M, Compton A, Lim SC, Thorburn D, Ryan MT, Giegé R, Bahlo M, Christodoulou J. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome. Am J Hum Genet. 2010;87:52–9. [PMC free article: PMC2896778] [PubMed: 20598274]
- Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. [PMC free article: PMC2574672] [PubMed: 18606475]
- Sears DA, Udden MM. Pappenheimer bodies: a brief historical review. Am J Hematol. 2004;75:249–50. [PubMed: 15054821]
- Sheftel A, Stehling O, Lill R. Iron-sulfur proteins in health and disease. Trends Endocrinol Metab. 2010;21:302–14. [PubMed: 20060739]
- Shimada Y, Okuno S, Kawai A, Shinomiya H, Saito A, Suzuki M, Omori Y, Nishino N, Kanemoto N, Fujiwara T, Horie M, Takahashi E. Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia. J Hum Genet. 1998;43:115–22. [PubMed: 9621516]
- Stemmler TL, Lesuisse E, Pain D, Dancis A. Frataxin and mitochondrial FeS cluster biogenesis. J Biol Chem. 2010;285:26737–43. [PMC free article: PMC2930671] [PubMed: 20522547]
- Taketani S, Kakimoto K, Ueta H, Masaki R, Furukawa T. Involvement of ABC7 in the biosynthesis of heme in erythroid cells: interaction of ABC7 with ferrochelatase. Blood. 2003;101:3274–80. [PubMed: 12480705]
- Ye H, Rouault TA. Erythropoiesis and iron sulfur cluster biogenesis. Adv Hematol 2010a;2010. [PMC free article: PMC2939393] [PubMed: 20862391]
- Ye H, Rouault TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010b;49:4945–56. [PMC free article: PMC2885827] [PubMed: 20481466]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. [PubMed: 21909114]
- Zutz A, Gompf S, Schagger H, Tampe R. Mitochondrial ABC proteins in health and disease. Biochim Biophys Acta. 2009;1787:681-90. [PubMed: 19248758]
章节注释
作者履历
Soumeya Bekri (2014至今)
Marc D’Hooghe, MD (2014至今)
Thomas D Bird, MD; University of Washington, Seattle (1998-2014)
Roberta A Pagon, MD; University of Washington, Seattle (1998-2014)
Pieter Vermeersch (2014-至今)
修订沿革
- 2014年4月3日 (me) 全面更新实时发布
- 2009年4月7日 (me) 全面更新实时发布
- 2008年3月24日 (cd) 修订版: 无临床检测
- 2006年3月1日 (me) 综述发布到实时网站
- 1998年11月12日 (bp) 首稿