
摘要
临床特征.
查尔综合征具有典型面部特征、动脉导管未闭(PDA)和小指中节指骨发育不全或发育低下三联征的特点。典型的面部特征为面中部扁平、鼻梁扁平、宽扁鼻尖、眼下垂、下斜睑裂、轻度上睑下垂、人中短导致三角嘴和嘴唇厚(或张开)。
诊断/实验室检查.
查尔综合征的诊断以临床表现为依据。TFAP2B基因的突变是唯一已知导致查尔综合征的突变。50%受累患者TFAP2B基因序列分析发现了突变。
处理.
对症处理: 新生儿期后动脉导管未闭的处理由主动脉向肺动脉分流的程度决定;选择手术结扎或导管插入阻塞导管。听力损失、视力问题和发育迟缓均按常规方式处理。
遗传咨询.
查尔综合征以常染色体显性方式遗传。由新生突变引起的病例比例尚不清楚。如果先证者的父母受累,那么同胞受累风险为50%.如果父母在临床上不受影响,先证者同胞的风险似乎很低。患有查尔综合征个体的每个小孩有50%的几率遗传这个变异并表现出这种疾病。如果在受影响家庭成员中发现这种致病变异 ,通过实验室提供的相关基因的产前检查或定制化检测项目对高危妊娠产妇进行产前诊断是有必要的.
诊断
临床诊断
查尔综合征的诊断依据以下临床特征的存在:
- 典型的面部特征:面中部扁平、鼻梁扁平、宽扁鼻尖、眼下垂、下斜睑裂、轻度上睑下垂和有短人中、突出的柱状人中、向上的朱红色边缘形成的三角形口以及厚的(张开的)外翻的嘴唇.[Char 1978]
- 动脉导管未闭(PDA)
- 小指中节指骨发育不全或发育低下
分子遗传学检测
基因Gene.TFAP2B是已知唯一突变导致查尔综合征的基因.
Table 1.
分子遗传学检测在查尔综合征中的应用摘要
基因 1 | 检测方法 | 基因突变检测 2 | 采用检测方法检测突变频率 3 |
|---|---|---|---|
| TFAP2B | 序列分析 Sequence analysis 4 | TFAP2B突变 TFAP2B mutations 5 | ~50% |
- 1.
染色体位点和蛋白质见基因和数据库的表A。
- 2.
等位基因变异信息见分子遗传学。
- 3.
- 4.
序列分析检测变异是良性、可能良性、意义未明、可能致病,或致病。致病变异可能包括小的基因内缺失或插入和错义、无义、剪接位点突变;典型地,检测不到外显子或整个基因的缺失或重复。有关序列分析结果解释的考虑问题,请点击这里。
- 5.
因为迄今为止证实了的致病突变都导致了具有显性负性影响的突变蛋白的产生,可能是关键区域的编码区的突变成为错义缺陷,尤其是基本结构域。剪接位点改变和产生单倍剂量不足的罕见突变也有报道。 [Mani et al 2005]
检测策略T
先证者的确诊. 查尔综合征先证者的诊断是通过临床症状确立的。大约50%受累患者可以检测到TFAP2B基因突变。
对高危妊娠进行产前诊断和植入前产前诊断(PGD)需要事先鉴定家庭中的致病突变。
临床特点
临床描述
查尔综合征具有典型的面部特征(见图1)、动脉导管未闭(PDA)和典型地手部异常(见诊断)的三联征。
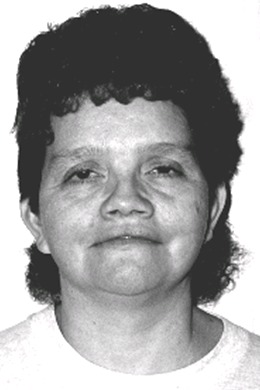
Figure 1.
经Satoda等人允许,复制了一个有典型面部特征的患有查尔综合征女性的图片。Typical [1999]
PDA. 动脉导管,即主动脉和肺动脉之间的动脉连接,它将血液从肺部分流出去,在出生后不久就会关闭。如果动脉导管持续开放,让左右分流(从体循环进入肺循环)发生,如果不纠正就会导致肺动脉高压。目前还没有得到有关查尔综合征患者在出生几周后动脉导管未闭自发关闭的可能性的信息,如果有也可能很低。
与查尔综合征相关的不太常见的特征
- 多乳头 [Zannolli et al 2000]
- 牙齿发育不全:四个象限都缺少第二和/或第三臼齿 [Mani et al 2005; Gelb, unpublished observation]
- 足畸形:指间关节融合或单侧指突 [Sweeney et al 2000], 间质多指趾畸形interstitial polydactyly [Slavotinek et al 1997], 并指畸形[Slavotinek et al 1997]
- 听力异常:受累患者严重双侧听力损失 [Gelb, 通过对查尔综合征一个原始家庭扩大范围的成员的未发表的研究]
- 发育迟缓:轻至中度
- 其他心脏缺陷(如,肌部多发室间隔缺损、复杂先天缺陷)
- 其他手部异常:间质多指趾畸形 [Slavotinek et al 1997];小指末节指状指(远端指间关节融合), 中指发育不全 [Babaoğlu et al 2012].
- 异常睡眠 [Mani et al 2005]
基因型-表型相关性
在Satoda et al [2000]、Zhao et al [2001]和Mani et al [2005] 讨论的8个TFAP2B基因突变中有5个影响DNA结合,而有1个突变即p.Pro62Arg影响反式激活结构域;另外2个突变是有关内含子的,预计导致单倍剂量不足。带有p.Pro62Arg突变的家庭始终有更轻微的面部畸形而且14个受累成员都没有手部缺陷。相反,PDA和其他心血管缺陷很高。心血管异常如此普遍的原因尚待解释,尤其是在面部特征轻微和手部正常的情况下,当基本区域突变导致明显面部畸形和手部异常时,而PDA的患病率仍然很低。
外显率
查尔综合征的外显率还没有正式确定。一个有TFAP2B基因致病性突变的无症状患者的情况已经说明。 [Mani et al 2005].
预期
查尔综合征的预期还没有被描述。
患病率
查尔综合征的患病率还没有确定,但被认为是相当低的。
遗传相关(等位基因)疾病
TFAP2B基因致病性突变已在PDA患者中发现,但没有查尔综合征的其他特征(非综合征PDA) [Khetyar et al 2008, Chen et al 2011].
鉴别诊断
面部特征. 与查尔综合征相关的典型面部特征通常是显著的,通常不会与其他疾病中观察到的面部特征相混淆。面部轮廓与上颌骨发育不良(宾德综合征)相似。
动脉导管未闭(PDA)Patent ductus arteriosus (PDA) 大约占所有先天性心脏病疾病的10%.足月儿中孤立PDA(没有其他先天性心脏缺陷)的发生率为1/200。PDA被认为在早产儿中更常见。它是先天性风疹综合征的心脏病变之一。PDA是常染色体显性遗传而且是非综合征型的隐匿疾病。 [Mani et al 2002].
筛选出的一组孤立PDA的患者很少发现TFAP2B基因突变的存在。[Khetyar et al 2008, Chen et al 2011].
胸主动脉瘤/主动脉夹层合并PDA是有关常染色体显性遗传的一种疾病,包括胸主动脉瘤(能进行解剖)和PDA。它与查尔综合征可以在基因上区别开来,它是由于编码肌球蛋白重链11的基因突变导致的[Zhu et al 2006]。可以参考 Thoracic Aortic Aneurysms and Aortic Dissections.
手异常. 与查尔综合征相关的手异常可以与小指弯曲一样小,这可以是一个正常发现,并与许多其他综合征重叠。
心-手综合征. 心-手综合征相关包括PDA、二尖瓣主动脉瓣和手异常(小指发育不全和短指),但面部正常 [Gelb et al 1999]。这种疾病可以与查尔综合征在基因上区别,对TFAP2B位点使用链接排除进行记录
其他需要考虑的心-手疾病:
- 霍尔特综合征
- Tabatznik syndrome
- 心-手三型
- 桡骨缺失综合征
- 尺骨-乳腺综合征
- 过度生长综合征
- 常染色体显性遗传罗宾诺综合征
- 埃利伟氏综合征
管理
初步诊断后的评估
推荐以下评估手段来确立查尔综合征个体的疾病状况和需求:
- 疑似有查尔综合征的婴儿和儿童:仔细的心脏评估,通常包括超声心电图
注:新生儿评估也许不完全是能提供有用信息的,因为动脉导管可能在任何新生儿体内持续开放数天。 - 医学遗传咨询
对症处理
对查尔综合征患者的管理主要关注心血管的介入。新生儿期后动脉导管未闭的处理取决于从主动脉到肺动脉的分流程度。可以选择手术结扎或导管阻塞。
查尔综合征最显著的外部症状,即畸形和手畸形,在生命早期不需要特别地护理。由于它们的缺陷的影响,当受累患者经历童年和青少年时期时,变形特征变得重要。资料表明,查尔综合征没有可用的成功的整形手术干预措施。
监测
患有查尔综合征的小孩在婴儿期和儿童期需要儿科关注。
尽管某些特定医疗问题包括听力损失、视力问题和发育迟缓在受累儿童中相对罕见,但他们的患病率比一般人群高。儿科医生对受影响儿童进行持续的发育评估可能是有益的,因此因根据需要提供早期干预。
亲属风险的评估
见遗传咨询中的有关风险的评估。
还在调研阶段的治疗方法
可通过搜索ClinicalTrials.gov 获取各种疾病表型的临床试验的信息。注:该疾病可能没有临床试验。
遗传咨询
遗传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识,以及符合个人情况的决定。接着几个段落是涉及遗传风险评估,根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的咨询工作。--作者ED
遗传方式
查尔综合征是常染色体显性遗传。
家属的风险
先证者的父母
- 一些诊断为查尔综合征的患者,父母也受累。
- 由于从头基因的突变,有查尔综合征的先证者也许会有这种疾病。由新生突变引起的病例比例暂不清楚。
- 建议评估有明显新生突变的先证者的父母包括关注面部外观的身体检查、心脏和四肢,如果检测到手或脚的异常,可以进行射线检查;如果心脏检查不正常,可以做超声心动图检查。
先证者的同胞
- 先证者同胞的风险取决于父母的遗传状态。
- 如果先证者父母受累,同胞风险为50%。
- 当父母没有临床症状时,先证者同胞的风险似乎很低。
- 尽管没有胚系嵌合病例的报道,但它仍然是可能的。
先证者的后代。每个患有查尔综合征的个体的孩子都有50%的几率遗传这种突变并患有这种疾病。
其他家庭成员. 其他家庭成员风险取决于先证者父母的状况。如果父母是受累的,那么他或她的家庭成员存在风险。
遗传咨询相关问题
明显新生突变家族的考虑。. 当有常染色体显性遗传情况的先证者的双亲之一有这种疾病的临床证据,先证者很可能有新生突变。然而,可能的非医学解释包括非生物学父亲或母亲(如辅助生殖),或秘密收养也应该考虑。
生育规划
- 确定遗传风险和讨论产前检测可用性的最佳时间是在怀孕前。
- 为受累的、是携带者或有携带风险的年轻人提供适当的遗传咨询(包括后代潜在风险和生育选择的讨论)。
DNA库是储存DNA(通常从白细胞提取)以备将来可能使用的。因为将来对有关基因、等位变异和疾病的理解和检测方法可能提高,应该考虑受累个体的DNA库。
产前检测
分子遗传检测。如果一个家庭成员的致病变异被确认,高危妊娠的产前检测可从提供产前的基因检测或定制化检测项目的临床实验室中得到。
超声检查。对于高风险孕妇来说,产前超声检查可以确定异常的手或脚以及复杂的先天性心脏缺陷。由于动脉导管未闭是胎儿的正常特征,所以不能再子宫内进行诊断。
复杂的先天性心脏疾病的产前发现可能会改变婴儿出生时的管理以及建议改变能为复杂心脏缺陷提供紧急干预的分娩中心的需要。
胚胎植入前遗传学诊断(PGD)可能成为致病突变已经确定的家庭的一种选择。
信息来源
为了该病患者及其家庭成员的方便,GeneReviews的员已经选择了下述的针对该病的,或者包括其他GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击获取详细信息。
- My46 Trait Profile
- Children's Heart FoundationPO Box 244Lincolnshire IL 60069-0244Phone: 888-248-8140 (toll-free); 847-634-6474Fax: 847-634-4988Email: info@childrensheartfoundation.org
- Congenital Heart Information Network (CHIN)101 North Washington AvenueSuite 1AMargate City NJ 08402-1195Phone: 609-822-1572Fax: 609-822-1574Email: mb@tchin.org
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其他部分的信息不一致:表格里信息可能更加新--ED
Table A.
查尔综合征:基因和数据库
| 基因 | 染色体位置 | 蛋白质 | 位点特异性 | HGMD |
|---|---|---|---|---|
| TFAP2B | 6p12 | Transcription factor AP-2-beta | TFAP2B database | TFAP2B |
数据从以下标准参考资料内整理:基因来自HGNC;染色体位置、位置名称、关键区域、基因互补群信息来自OMIM;蛋白信息来自UniProt。在此提供了超链接的相关数据库的相关描述,请点击这里。
基因结构. cDNA有1650bp和总体规模大约为2kb的编码区。有关基因和蛋白的详细信息,见表A,基因。
良性等位基因变异. SNP数据库中有两个TFAP2B编码变体: c.411C>A (p.Asp137Glu) and c.739T>G (p.Ser247Ala) (see Table 2).
致病性等位基因变异. 在7个查尔综合征不相关的个体和家族中报道了9个TFAP2B变异。 [Satoda et al 2000, Zhao et al 2001, Mani et al 2005, Babaoğlu et al 2012] (see Table 2). 6个突变影响基本结构域。第7个错义突变,p.Pro62Arg, 改变了活化域内的PY序列。所有这些错误的变化影响了高度保守的残基。在6个基本结构域变异中,有5个影响了精氨酸残基。这是由于这些残基通常对转录因子的DNA结合很重要,而且编码精氨酸残基的6个密码子中有4个含有CpG二核苷酸。另外两个突变影响内含子。 [Mani et al 2005]. (更多信息,见表A)
Table 2.
选择TFAP2B等位基因变体
| 变异等位基因种类 | DNA核苷酸变化 (Alias 1 ) | 蛋白质氨基酸变化 | 参考序列 |
|---|---|---|---|
| 正常 | c.411C>A | p.Asp137Glu | X95694 |
| c.739T>G | p.Ser247Ala | ||
| 致病性 | c.185C>G | p.Pro62Arg | |
| c.600+5G>A (IVS3+5G>A) | -- | ||
| c.673C>T | p.Arg225Cys 2 | ||
| c.673C>A | p.Arg225Ser 2 | ||
| c.706C>T | p.Arg236Cys 2 | ||
| c.791C>A | p.Ala264Asp 2 | ||
| c.821G>A | p.Arg274Gln 2 | ||
| c.821-1G>C (IVS4-1G>C) | -- | ||
| c.865C>T | p.Arg289Cys 2 |
变异分类注释:表中所列变异由作者提供。GeneReviews 工作人员还没有独立验证变异分类。
关于命名的说明:GeneReviews遵循人类基因组变异协会 (varnomen
- .hgvs.org )的标准命名规范. 有关术语的解释,请参阅“快速参考资料”。- 1.
不符合当前命名规范的变异命名。
- 2.
6个影响基本结构域的突变
正常 基因产物: 转录因子AP-2β。AP-2β转录因子蛋白质家族有一个高度保守的结构。一半的蛋白质氨基端包含反式激活结构域,这是这个蛋白质家族which is the least well-conserved domain among this family of proteins. 除了转录因子AP-2δ, 所有的AP-2蛋白在反式激活区域都包含有PY序列。一半的高度保守的蛋白质羧基端,包含一个基本结构域和螺旋- -螺旋结构域。前者对DNA结合至关重要,后者对二聚体作用至关重要。
异常 基因产物: 目前报道的7个转录因子AP-2β 错义突变中,有6个影响基本结构域。体外和细胞培养分析中记录了不同程度的DNA损伤,包括同型二聚体和异型二聚体以及反式激活[Satoda et al 2000, Zhao et al 2001]。二聚体似乎是正常的。因为这些突变干扰了与它们共同表达的正常的AP-2蛋白质的功能,因此它们的影响是显性负效的。第7个突变影响了反式激活域的PY序列。该突变保留了DNA结合功能,但是对反式激活有显性负效。内含子3突变(c.600+5G>A) 引起外显子3跳跃式的异常剪接,这导致了一种过早终止密码子的移码突变,并可能导致转录本的无意义的衰减[Mani et al 2005]。因此,这种分子缺陷导致单倍剂量不足。另外的内含子突变还没有正式检测,但应该也有类似的负面影响,导致单倍剂量不足。
参考资料
参考文献
- 1. Babaoğlu K, Oruç M, Günlemez A, Gelb BD. Char syndrome, a familial form of patent ductus arteriosus, with a new finding: hypoplasia of the 3rd finger. Anadolu Kardiyol Derg. 2012;12:523 - 4. [PubMed: 22728731]
- 2. Bertola DR, Kim CA, Sugayama SM, Utagawa CY, Albano LM, Gonzalez CH. Further delineation of Char syndrome. Pediatr Int. 2000;42:85 - 8. [PubMed: 10703243]
- 3. Char F. Peculiar facies with short philtrum, duck-bill lips, ptosis and low-set ears--a new syndrome? Birth Defects Orig Artic Ser. 1978;14:303 - 5. [PubMed: 728571]
- 4. Chen YW, Zhao W, Zhang ZF, Fu Q, Shen J, Zhang Z, Ji W, Wang J, Li F. Familial nonsyndromic patent ductus arteriosus caused by mutations in TFAP2B. Pediatr Cardiol. 2011;32:958 - 65. [PubMed: 21643846]
- 5. Gelb BD, Zhang J, Sommer RJ, Wasserman JM, Reitman MJ, Willner JP. Familial patent ductus arteriosus and bicuspid aortic valve with hand anomalies: a novel heart-hand syndrome. Am J Med Genet. 1999;87:175 - 9. [PubMed: 10533032]
- 6. Khetyar M, Syrris P, Tinworth L, Abushaban L, Carter N. Novel TFAP2B mutation in nonsyndromic patent ductus arteriosus. Genet Test. 2008;12:457 - 9. [PubMed: 18752453]
- 7. Mani A, Meraji SM, Houshyar R, Radhakrishnan J, Mani A, Ahangar M, Rezaie TM, Taghavinejad MA, Broumand B, Zhao H, Nelson-Williams C, Lifton RP. Finding genetic contributions to sporadic disease: a recessive locus at 12q24 commonly contributes to patent ductus arteriosus. Proc Natl Acad Sci U S A. 2002;99:15054 - 9. [PMC free article: PMC137543] [PubMed: 12409608]
- 9. Mani A, Radhakrishnan J, Farhi A, Carew KS, Warnes CA, Nelson-Williams C, Day RW, Pober B, State MW, Lifton RP. Syndromic patent ductus arteriosus: evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder. Proc Natl Acad Sci U S A. 2005;102:2975 - 9. [PMC free article: PMC549488] [PubMed: 15684060]
- 10. Satoda M, Gelb BD. Char syndrome and TFAP2B. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, eds. Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis. San Francisco, CA: Oxford University Press; 2003:798-803.
- 11. Satoda M, Pierpont ME, Diaz GA, Bornemeier RA, Gelb BD. Char syndrome, an inherited disorder with patent ductus arteriosus, maps to chromosome 6p12-p21. Circulation. 1999;99:3036 - 42. [PubMed: 10368122]
- 12. Satoda M, Zhao F, Diaz GA, Burn J, Goodship J, Davidson HR, Pierpont ME, Gelb BD. Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus. Nat Genet. 2000;25:42 - 6. [PubMed: 10802654]
- 13. Slavotinek A, Clayton-Smith J, Super M. Familial patent ductus arteriosus: a further case of CHAR syndrome. Am J Med Genet. 1997;71:229 - 32. [PubMed: 9217229]
- 14. Sweeney E, Fryer A, Walters M. Char syndrome: a new family and review of the literature emphasising the presence of symphalangism and the variable phenotype. Clin Dysmorphol. 2000;9:177 - 82. [PubMed: 10955477]
- 15. Zannolli R, Mostardini R, Matera M, Pucci L, Gelb BD, Morgese G. Char syndrome: an additional family with polythelia, a new finding. Am J Med Genet. 2000;95:201 - 3. [PubMed: 11102923]
- 16. Zhao F, Weismann CG, Satoda M, Pierpont ME, Sweeney E, Thompson EM, Gelb BD. Novel TFAP2B mutations that cause Char syndrome provide a genotype-phenotype correlation. Am J Hum Genet. 2001;69:695 - 703. [PMC free article: PMC1226056] [PubMed: 11505339]
- 17. Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343 - 9. [PubMed: 16444274]
推荐阅读
- Eckert D, Buhl S, Weber S, Jäger R, Schorle H. The AP-2 family of transcription factors. Genome Biol. 2005;6:246. [PMC free article: PMC1414101] [PubMed: 16420676]
本章节的注解
致谢
这项工作得到了美国国立卫生研究院(HL098123)到BDG的部分支持。
更新历史
- 24 January 2013 (me) Comprehensive update posted live
- 19 March 2008 (me) Comprehensive update posted live
- 17 June 2005 (me) Comprehensive update posted live
- 15 August 2003 (ca) Review posted live
- 18 April 2003 (bg) Original submission