
简介
临床特征 营养不良型大疱性表皮松解症(DEB)是一种遗传性皮肤病,通常会在出生时影响皮肤和指甲。 DEB根据异常方式分为两种主要类型:隐性营养不良性大疱性表皮松解症(RDEB)和显性营养不良性大疱性表皮松解症(DDEB)。每种类型进一步分为多种临床亚型。缺乏 DEB家族史并不排除诊断。
严重全身性RDEB的临床发现包括皮肤起泡,表现为水疱,创伤小,可留下栗疹和疤痕。新生儿时期可能会出现水泡和糜烂,影响全身。口腔受累可能导致口腔起泡,舌头融合到口腔底部以及口腔大小逐渐缩小。食道糜烂可导致网状狭窄,导致严重吞咽困难。因此,营养不良以及维生素和矿物质的缺乏可能导致幼儿生长受限。角膜侵蚀会导致疤痕和视力丧失。手脚起泡,然后留下疤痕,使手指融合成“手套状”的手脚,并有挛缩和假性滑脱。侵袭性鳞状细胞癌的终生风险高于90%。
相反,不严重的RDEB形式的水疱可局限在手,脚,膝盖和肘部,有或无弯曲区域和躯干受累,在严重全身性RDEB中没有破坏性瘢痕。
在DDEB中,水疱通常很轻,仅限于手,脚,膝盖和肘部,但有疤痕的情况下可以治愈。营养不良的指甲,尤其是脚趾甲很常见,可能是DDEB的唯一表现。
诊断/测试。DEB的诊断建立在具有特征性临床发现的先证者中,并通过分子遗传学检测鉴定COL7A1中双等位基因的致病变异(RDEB)或 杂合的 致病性变异(DDEB)。已知致病性变异导致DEB的唯一 基因是COL7A1。如果 分子遗传学检测不能诊断,则可能需要用直接免疫荧光(IF)检查皮肤活检中的特定皮肤标志物和/或电子显微镜(EM)以进行诊断。
管理。表现的治疗:应将新的水疱刺破,干燥,并且在大多数情况下,应使用不粘的材料穿着,并用填充物覆盖以保持稳定性和保护作用,并用弹性包裹物固定以保持完整性。 RDEB严重且生长缓慢的婴儿和儿童需要注意体液和电解质的平衡,并可能需要营养支持,包括喂养胃造口术。根据需要用铁补充剂和输血治疗贫血。其他营养补品可能包括钙,维生素D,硒,肉碱和锌。职业疗法可能有助于预防手挛缩。通常需要重复手术松解手指。
预防主要表现:如果已知受累的胎儿患有任何形式的DEB,则剖宫产可以减少分娩过程中对皮肤的伤害。鼓励与年龄相称的游戏,包括对皮肤造成最小伤害的活动;需要敷料和填充物以保护骨骼突出面免引起水泡。
监控:从生命的第二个十年开始,对于没有出现鳞状细胞癌或出现疤痕组织愈合异常伤口进行活检。建议的定期测试包括每6至12个月筛查贫血和铁,锌,维生素D,硒和肉碱的缺乏症。建议每年进行超声心动图检查以鉴别扩张型心肌病,并建议进行骨矿物质密度检查以鉴别骨质疏松症。
应避免的药物/情况:衣服和鞋类穿着不合适或质地较粗;伤害皮肤的活动/绷带。
有风险的亲属的评估:对有风险的新生儿评估是否有水疱迹象,这样可以尽可能避免对皮肤的伤害。
遗传咨询。营养不良性大疱性表皮松解以 常染色体显性遗传(DDEB)或常染色体隐性遗传(RDEB)方式遗传。致病变异的分子表征是确定 遗传模式和再发风险的唯一准确方法。仅表型严重程度和IF / EM结果不足以诊断。
GeneReview 范围
| 营养不良性大疱性表皮松解性(DEB):包括表型 |
|---|
|
有关同义词和过时的名称,请参见 Nomenclature.
诊断
营养不良性大疱性表皮松解症(DEB)是一种遗传性疾病,通常在出生时就影响皮肤和指甲。 目前,DEB的分类基于2013年共识会议的发布[Fine et al 2014]。 诊断基于临床怀疑:皮肤脆弱,DEB家族史和诊断测试。 分子遗传分析是最确定的测试,但是直接免疫荧光(IF)和/或透射电子显微镜(EM)可能会有所帮助,尤其是在对亚型进行分类时。
DEB根据遗传方式分为两种主要类型:隐性营养不良性大疱性表皮松解症(RDEB)和显性营养不良性大疱性表皮松解症(DDEB)。 每种类型进一步分为多种临床亚型(请参阅 Nomenclature)。 缺乏已知的DEB家族史并不排除诊断。
提示性发现
具有以下临床表现的个体应怀疑营养不良性大疱性表皮松解症(DEB):
- 皮肤的脆弱性表现为水疱,创伤小,可愈合为纤毛和疤痕
- 水泡和腐蚀:
- 出生时导致皮肤先天性发育不全(缺少皮肤,尤其是四肢)
- 影响整个身体,包括粘膜(最严重的形式)或主要是手,脚,膝盖和肘部(较轻的形式)
- 导致手脚假性融合(严重形式)
- 导致口腔和/或食道瘢痕形成和狭窄
- 导致角膜糜烂,形成疤痕,导致视力下降
- 易患鳞状细胞癌
- 营养不良或缺乏指甲,尤其是脚趾甲
建立诊断
DEB的诊断建立在具有特征性临床发现的先证者中,并在 分子遗传学检测中鉴定出COL7A1中双等位基因的致病变异(RDEB)或杂合的致病性变异(DDEB)(请参阅Table 1)。如果分子遗传学检测不能诊断,则可能需要使用直接IF检查皮肤活检(请参阅Skin Biopsy)以检查特定的皮肤标志物和/或EM。常规组织学无用。
应当指出,并非所有临床医生都可以使用本节中描述的诊断工具(分子遗传学检测,皮肤活检的专门检查)。最近的一项研究将74例临床研究结果与遗传学确认结果进行了比较,发现与EB的类型和亚型高度一致。该技术在发展中国家可能有用[Yenamandra et al 2017].
分子遗传学检测方法可以包括靶向基因的检测(单基因检测,混合或系列单基因检测, multigene panel)和综合基因组的检测(染色体芯片分析, 外显子组测序, exome array, 基因组测序)的组合,方法取决于 表型。
以基因为目标的测试要求临床医生确定可能涉及哪些基因,而基因组的测试则不需要。由于DEB的表型很宽,因此具有可能在Suggestive Findings中描述的独特发现的个体可以使用基因靶向检测(见 Option 1)进行诊断,而具有与许多其他遗传性表皮松解性疾病(EB)无明显区别的表型的个体),或者出现在疾病更严重的进展之前的出生时或新生儿时期,更可能使用基因组测试来诊断(请参阅 Option 2)。
选项1 当表型和实验室发现提示诊断DEB 分子遗传学检测方法时,可包括单基因检测或 multigene panel使用:
- 包含COL7A1和其他感兴趣基因的大疱表皮松解性multigene panel (见Differential Diagnosis) 最有可能以最合理的成本鉴定出该病的遗传原因,同时限制了对确证基因的致病性变异和 意义不确定的鉴定没有解释底层的表型。注意:(1)套餐中包含的基因和每个基因所用测试的诊断敏感性因实验室而异,并可能随时间变化。 (2)一些多基因套餐可能包含与本GeneReviews中讨论的病症无关的基因。 (3)在某些实验室中,套餐选项可能包括定制的实验室设计套餐和/或定制的针对表型的外显子组分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括序列分析,删除/重复分析和/或其他非基于序列的测试。对于这种疾病,建议同时进行多基因分析(包括删除/重复分析)(见 Table 1)。
有关多基因套餐的介绍,请单击 here。有关订购基因检测的临床医生的更多详细信息,请参见here.
Table 1.
选项2 当该表型与以大疱性表皮松解为特征的许多其他遗传性疾病没有区别时,综合基因组的检测(不需要临床医生确定可能涉及哪些基因)是最佳选择。 外显子组测序是最常用的方法。基因组测序也是可以的。
如果外显子组测序不能诊断,可以考虑外显子组阵列(临床上可用时)。
有关全面的 基因组的测试的介绍,请单击here。 可在here找到订购基因组测试的临床医生的更多详细信息。
营养不良性大疱性表皮松解症的分子遗传学检测
| 基因 1 | 测试方法 | 用此方法检出致病性变异在先症者重占比 2 |
|---|---|---|
| COL7A1 3 | Sequence analysis 4 | 95% 5 |
| Gene-targeted deletion/duplication analysis 6 | <2% 7 |
- 1.
染色体 位点 和蛋白见Table A. Genes and Databases for.
- 2.
有关在该基因中检测到的等位基因变异的信息,请参见 Molecular Genetics。
- 3.
已经以隐性和显性遗传模式描述了COL7A1中的一些致病变异[Almaani et al 2011]。 如果在COL7A1中发现了两个变异,则可能需要进行父母亲测试以确定该变异是 双等位基因的。
- 4.
- 5.
在活检确诊DEB个人通过 序列分析致病变异的检出率是95% [Kern et al 2006, Bale & Pfendner 2014, Pfendner et al 2017].
- 6.
基因靶向的 deletion/duplication analysis检测基因内缺失或重复。 所使用的方法可能包括quantitative PCR,远程PCR,多重连接依赖探针扩增(MLPA),以及旨在检测单外显子缺失或重复的基因靶向微阵列。
- 7.
通过基因靶向的 deletion/duplication analysis可检测到具有 致病性变异的先证者的比例显性DEB为<1%, 隐性DEB为<2%[Pfendner et al 2017].
皮肤活检 EB的明确诊断最直接地是通过分子遗传学分析。过去,只有单基因测序可用时,必须首先进行活检以确定要分析的单个基因。但是,由于现在可以使用多基因套餐检测 [Lucky et al 2018],因此某些临床医生更愿意避免进行活检,除非基因分析无法做出诊断[Pfendner 2015, Tenedini et al 2015].
如果确定活检对诊断是必要的,则应从新鲜水疱(<12小时大)或机械性水疱的前缘进行活检,并应包括一些正常的邻近皮肤;较老的水泡发生变化,可能会掩盖诊断形态。通常使用椭圆形或刮胡切除。尽管打孔器活检会引入令人困惑的伪影,但谨慎使用打孔器可避免表皮的丢失[Intong & Murrell 2010].
光学显微镜不足以准确诊断大疱性表皮松解。
免疫荧光(IF)。通过皮肤活检IF抗体/抗原谱检查是建立DEB诊断的合适方法。直接IF可以揭示皮肤的层面,并有助于建立广泛的EB类型。即使没有分裂,皮肤中是否存在特定蛋白质也可能决定EB的类型。 IF还具有快速的优势 [Pohla-Gubo et al 2010, Meester et al 2018].
特征性发现:
- 使用抗体染色胶原蛋白VII减少或不存在。
- 在较温和的RDEB和DDEB中,胶原蛋白VII的染色可能看起来正常,但切割平面低于层板.
- 对其他抗原(例如层粘连蛋白332,胶原XVII,凝集素,α6β4整联蛋白和角蛋白5和14)进行正常染色有助于确认DEB的诊断。
透射电子显微镜(TEM)。 有时,尤其是在较温和形式的EB中,直接IF研究不足以做出诊断,因为可能检测到接近正常的抗原水平并且未观察到切割平面。 在这种情况下,皮肤活检的TEM检查可有助于检查细胞结构[Eady & Dopping-Hepenstal 2010].
特征性发现:
- 所有DEB. 在基底膜区域的层状骨膜下方观察到裂损。
- 隐性DEB (RDEB) 全身重型. 锚定纤维的明显减少,缺失或形态异常。
- 显性DEB (DDEB), RDEB-全身和局部
- 锚定原纤维的数量可能减少和/或形态改变。
- 在某些个体中可以观察到细胞内残留胶原蛋白VII。
- 在新生阶段出现短暂性水疱的某些个体中,VII型胶原可能会保留在基底角质形成细胞内,而不是被运输到基底膜区域。
临床特征
临床表现
在了解营养不良性大疱性表皮松解症(DEB)的分子基础之前,主要根据临床特征, 遗传模式以及在皮肤活检中检测到的胶原VII和锚定纤维的存在与否来确定类型和亚型。 当前的分类系统基于遗传模式(常染色体显性遗传DEB [DDEB]与 常染色体隐性遗传DEB [RDEB]),并通过胶原蛋白VII染色和在中鉴定出的特定COL7A1 致病性变异进一步分型。 给定受累的(参见Nomenclature)[Fine et al 2014]. 在本基因综述,术语“隐性DEB严重全身性”(RDEB-sev gen),“隐性DEB全身性和局部化”(包括其他几种亚型)和“显性DEB”(DDEB)(也包括其他亚型) )已被使用,下面将进行讨论。
见 Figure 1.
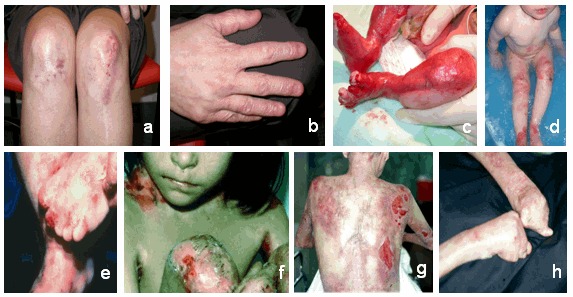
Figure 1.
营养不良性大疱性表皮松解的常见发现:a,b。 在成年人的主要DEB中发现膝盖和手的疤痕和营养不良的指甲
隐性DEB严重全身性(RDEB-sev gen)在这种经典的RDEB严重形式中,水疱在出生时即出现或在新生儿期变得明显。 最近对RDEB-sev gen的预后进行了综述 [Fine & Mellerio 2009a, Fine & Mellerio 2009b, Murrell 2010, Li et al 2017].
皮肤和皮肤癌的风险
- 在新生儿期可能会发现先天性皮肤发育不良,特别是四肢发育不良。
- 早在新生儿期,水泡就会影响整个皮肤,包括皮肤,口腔粘膜,食道粘膜和角膜。 慢性不愈合伤口和继发感染很常见,常为葡萄球菌,假单胞菌和链球菌感染。
- 在整个生命中水泡持续,疤痕可能会导致毁容和整形外科问题(请参阅下面的整形外科)。
- 许多个体长出大的不规则棕色斑块,其组织学上包括痣细胞的集合,被称为EB痣[Lanschuetzer et al 2010]。 迄今为止,尚未报道在这些痣中出现黑色素瘤的情况。
- 侵袭性鳞状细胞癌(SCC)的终生风险大于90%,且具有明显的转移趋势[Fine et al 2009]。 SCC通常出现在第三个十年,但最早可以出现在第二个十年 [Ayman et al 2002]。 受影响的个体通常会患侵袭性转移性SCC [Mellerio et al 2016].
口腔,胃肠道,生长和营养问题
- 口腔受累可能导致舌头融合到口腔底部(强直觉缺乏),并逐渐缩小口腔和张口的大小(口气),这与不良的牙齿卫生和龋齿一起影响进食量,最终营养 [Krämer et al 2012].
- 食道水疱和糜烂以及网状狭窄可引起严重的吞咽困难,并导致营养不良[Azizkhan et al 2006, Mortell & Azizkhan 2010]。 受累的人会患有食道疾病,很少或没有皮肤表现。 胃食管反流病也很常见。
- 肛门糜烂,液体和纤维摄入不足以及使用阿片类镇痛药会导致频繁的严重便秘。
- 由摄入不足引起的营养不良和对组织愈合的营养需求增加,可能导致年幼儿童的生长受限以及年长儿童的青春期缺乏或延迟。
- 维生素和矿物质缺乏症尤其是铁,锌,肉碱,硒和维生素D可能会发生 [Haynes 2010].
- 贫血归因于铁摄入不足和骨髓抑制导致的慢性疾病性贫血。
- 锌缺乏症可能会阻碍皮肤伤口的正常愈合。
- 肉碱和硒缺乏症在其他情况下也与心肌病有关,可能有助于DEB的发现。
- 骨质减少和骨质疏松症通常与维生素D缺乏症有关,其原因是营养不良,缺乏足够的阳光照射以及缺乏运动 [Martinez & Mellerio 2010, Rodari et al 2017].
眼 角膜侵蚀可导致疤痕和视力丧失 [Matsumoto et al 2005].
心脏 RDEB已报道扩张型心肌病,有时与硒和肉碱缺乏有关,在某些情况下可能致命 [Lara-Corrales et al 2010, Ryan et al 2016].
泌尿科/肾脏科 会发生尿道糜烂,狭窄,膀胱功能障碍和肾小球肾炎,有时会导致肾衰竭 [Fine et al 2004].
骨科 患有RDEB的人往往会出现手指挛缩和假性指突,导致“手套”或“茧”的手,从而导致功能受损和生活质量下降[Eismann et al 2014]。 尽管脚趾融合不会损害功能,但脚和脚踝以及较大的关节(膝盖,臀部,颈部)的疼痛性水疱和进行性挛缩会干扰移动和功能。
社会心理的 由于这种疾病的并发症以及大多数EB受累的患者所忍受的慢性疼痛,严重的压力可能会影响受累者的个人和家庭。 生活质量可能下降,老年人可能会发生心理社会障碍,包括焦虑,抑郁和药物依赖性/滥用 [Frew & Murrell 2010]–尽管最近的一项研究表明,DEB患者的疼痛与焦虑或抑郁无关 [Fortuna et al 2016]。
隐性DEB全身性和局部化 多种临床表型构成了RDEB的谱,其中许多不如RDEB-全身严重型。 该表型可能是轻度的,起泡仅限于手,脚,膝盖和肘部以及营养不良的指甲,或者相对较广泛,包括弯曲区域和躯干,但没有在RDEB-全身严重型中见到严重的破坏性疤痕。 根据类型的不同,水疱的发作从出生到儿童时期不等。
不太常见的RDEB-全身和-局部变异的一些鲜明特征:
- RDEB inversa. 躯干,脖子,大腿和腿出现水疱和皮肤萎缩,而手,脚,肘或膝盖几乎没有变化。另外, 表型类似于DEB类型,起泡并产生疤痕。 婴儿期可能出现手和水泡。
- RDEB 胫前瘙痒症常影响胫骨。 胫前水泡发展成瘙痒样角化过度病变。 病变主要发生在胫骨前部区域,不累及膝盖和皮肤的其他部位。 其他发现包括指甲营养不良,小而白色的疤痕(类异倍体皮肤病变)和肥大性疤痕,无胫前表现。
- RDEB 全身中间型 表现出广泛的水疱,有疤痕,粟丘疹和痣。 假性鼻甲可能与口腔病变以及指甲受损或缺失一起发生。 可能有发育迟缓,但不如RDEB全身严重。 鳞状细胞癌也发生在一些受累的个体中。
- RDEB 局部型 表现出严重的水泡和疤痕,但仅限于手和脚。 其他部位不受影响。 指甲通常不存在。 也没有生长受限和全身性疾病。 在具有这种亚型的个体中尚未报道鳞状细胞癌。
- RDEB 向心型(RDEB-CE)在出生时很明显,仅涉及手,脚和胫前区,指甲缺如。 尚未报道发育迟缓和全身性疾病。 尚未报道这种亚型患者的鳞状细胞癌。
- 新生儿的大疱性皮肤溶解通常仅限于新生儿期的一过性水疱[Fassihi et al 2005]。 这些个体的分子遗传学测试显示 杂合的COL7A1致病变异或(很少) 双等位基因的COL7A1致病变异[Frew et al 2011, Boccaletti et al 2015, Diociaiuti et al 2016].
显性DEB(DDEB)在这种较温和的DEB形式中,水泡通常仅限于手,脚,膝盖和肘部,可能相对较温和,可治愈但仍有疤痕。 营养不良的指甲,尤其是脚趾甲很常见,可能会造成指甲脱落。 在最温和的形式中,营养不良的指甲可能是唯一提到的特征[Dharma et al 2001, Sato-Matsumura et al 2002, Tosti et al 2003]。 DDEB的水泡通常会随着年龄的增长而有所改善,这可能是由于体育锻炼减少所致。 DDEB的亚型与RDEB的亚型相似,但可能以较轻的表现形式出现。 同一家族成员之间的临床差异可能很大。
- DDEB 全身型 (DDEB-gen) 是一种较温和的EB形式,其中COL7A1中的单个致病性变异导致广泛的水疱性疾病,这种疾病会影响婴儿期大多数摩擦部位,但通常会发展为成年后较轻的疾病。 水疱形成疤痕,指甲通常缺如。 其他系统通常不受影响,很少报告生长迟缓和鳞状细胞癌。
- DDEB 局部,指甲型只会影响指甲营养不良和脆弱。 没有皮肤异常。 但是,其他家庭成员可能会有更严重的表现。
基因型-表型相关
隐性DEB(RDEB)
- 最严重的形式是由COL7A1中双等位基因的致病性变异引起的,这些致病性变异是由于无效 或框架外变异如插入/缺失,单碱基改变和剪接连接而导致 [Mellerio et al 1999a, Gardella et al 2002a, Gardella et al 2002b, Mallipeddi et al 2003]。严重程度可能与终止密码子的位置有关[Tamai et al 1999]。然而,某些功能蛋白的存在或缺乏似乎是确定疾病严重程度的最重要因素。
- 中度严重形式通常是由一个等位基因上Gly-X-Y结构域内的甘氨酸取代和另一等位基因上的终止密码子引起的; 仅产生少量的部分功能蛋白[Murata et al 2000, Dharma et al 2001, Varki et al 2007].
- 不太严重的形式通常是由其他(非甘氨酸)氨基酸取代和剪接连接变异引起的。有广泛的表型变异性,并且在文献中已经报道了700多种致病变异 [Ashton et al 1999, Mellerio et al 1999b, Whittock et al 1999, Gardella et al 2002a, Murata et al 2004, Sawamura et al 2005, Varki et al 2007].
显性DEB(DDEB)。 DDEB的大多数产生是由于 显性负效氨基酸取代胶原VII的胶原三螺旋 结构域中的甘氨酸,尽管已经报道了一些剪接连接变异和其他氨基酸取代。相同的 致病性变异但表型可能表现出个体或家系内差异 [Murata et al 2000, Vaccaro et al 2000, Mallipeddi et al 2003, Nakamura et al 2004, Wessagowit et al 2005].
外显率
直到最近,在评估家庭成员的轻度疾病特征时,COL7A1的致病变异被认为是100%外显的。但是,在几个家庭中,患有DDEB和已知的COL7A1致病性变异的个体的亲属具有相同的变异,但没有这种疾病的迹象。因此,至少在DDEB中,渗透率似乎小于100%
[Almaani et al 2011; 作者,未发表的评论].
命名法
隐性DEB全身重度(RDEB-sev gen)最初称为Hallopeau-Siemens型(RDEB-HS)。
隐性DEB全身中度(RDEB-gen intermed )和RDEB局部化(RDEB-loc)最初称为非Halopau-Siemens型(RDEB-non-HS)。
在过去的15年中,DEB的命名已发生了四次更改。 最新的分类系统,称为“洋葱皮”术语,来自国际共识会议,其建议于2014年6月发布 [Fine et al 2014].。 这种分类系统首先将DEB划分为遗传模式,然后是胶原蛋白VII染色的组织学描述, 受累的特定COL7A1 致病性变异描述(参见 Table 2)。
有关与大疱表皮松解和交界性大疱表皮松解有关的最新命名建议的信息,请参见 Table 3(pdf)。
Table 2.
2008年DEB术语与提议的“洋葱皮”术语的比较-代表性示例
| 旧称 1 | 2014命名 |
|---|---|
| RDEB, severe generalized | RDEB generalized severe, collagen VII absent, COL7A1 pathogenic variants (specify type) |
| RDEB, generalized other | RDEB generalized intermediate, collagen VII reduced staining, COL7A1 pathogenic variants (specify type) |
| DEB-BDN | DEB-BDN, granular intraepidermal collagen VII staining, COL7A1 AD or AR pathogenic variants (specify) |
| DDEB generalized | DDEB generalized, normal collagen VII staining, COL7A1 致病性变异 (specify) |
BDN = 新生儿大疱性皮肤溶解; DDEB = 显性遗传营养不良型大疱性表皮松解症; RDEB = 隐性遗传营养不良型大疱性表皮松解症
- 1.
Per 2008 recommendations
患病率
根据国家EB注册中心的数据,EB的总体患病率为每百万活产11.07 [Fine 2016]。 DDEB和RDEB的患病率分别为每百万活产1.49和1.35。
据估计,美国人群中RDEB的携带者频率为370分之一 [Pfendner et al 2001].
遗传(等位基因)相关疾病
除此GeneReview中讨论的表型外,没有其他表型与COL7A1中的致病变异相关。
鉴别诊断
由20种不同基因的致病变异引起的大疱性表皮松解症(EB) 的四种主要类型是EB simplex(EBS),交界性EB(JEB),营养不良性EB(DEB)和Kindler syndrome(参见 Table 4)。尽管就某些类型的大疱性表皮松解症的诊断标准达成了共识,但罕见亚型的有效性及其诊断标准存在争议。参见 Murrell [2010]获得了出色的临床评价,Fine et al [2014] (full text;尤其是表I和VII)参见修订后的分类系统。
EB的四种主要类型具有皮肤脆弱性,表现为水疱和/或糜烂,几乎没有或没有创伤。所有类型的EB都有一个阳性的Nikolsky征(摩擦后未受累的皮肤起水泡)。没有针对特定类型的临床发现;因此,确定EB类型需要进一步的实验室评估。分子遗传学检测可用于建立诊断(请参阅Establishing the Diagnosis)。或者,可以对新诱发的水泡进行新鲜的皮肤活检,然后通过间接免疫荧光对关键的基底膜蛋白成分进行染色。通过确定裂面以及这些蛋白质成分的存在/不存在和分布来建立诊断。电子显微镜也是诊断性的,通常在较温和的EB类型中更有用。
临床检查可用于确定水泡程度,口腔和其他粘膜损伤的存在以及疤痕的存在和程度。
建立EB类型的临床发现存在以下局限性:
- 在幼儿和新生儿中,水泡和瘢痕形成的程度和严重性无法确定诊断,或者其程度不足以识别EB类型。
- 粘膜和指甲受累以及存在或不存在粟丘疹可能无助于判别。
- 炎症后的变化(例如在EBS,Dowling-Meara型(EBS-DM)中看到的变化)通常被误认为是瘢痕形成或斑驳色素沉着。
- 由于侵蚀或刮擦的感染,单纯性EB和交界性EB可能会出现疤痕,从而进一步损坏裸露的表面。
- 在EB的三种主要类型中都可以看到先天性皮肤缺失(即EBS,JEB,DEB),这并不是诊断的特征。
Table 3. 大疱表皮松解
| 临床特征 1 | EB类型 | EB 亚型 | 基因 | MOI | 皮肤活检结果 (切割水平) | |||
|---|---|---|---|---|---|---|---|---|
| 水泡范围 | 口腔/其他黏膜 病变 | 在疤痕存在/范围 | 其他(相关基因) 1 | |||||
| 根据基因&/或变异而轻到重 | 粘膜受累严重 |
|
| EBS 3 | EBS 基底层上 | TGM5 | AR | 在所有EBS中:在超微结构水平上,真皮-表皮交界处有裂口 |
| DSP | ||||||||
| PKP1 | ||||||||
| JUP | ||||||||
| EBS 基底层 | KRT5 | AD (AR 2) | ||||||
| KRT14 | ||||||||
| CD151 | ||||||||
| EXPH5 | AR | |||||||
| PLEC | ||||||||
| DST | ||||||||
| KLHL24 | ||||||||
| 中度至严重,取决于致病性变异的类型 |
|
在口腔 &鼻腔,手指&脚趾周围以及内部上呼吸道&指甲周围的皮肤上形成肉芽组织提示JEB-GS |
| JEB | JEB 泛发重型 | LAMA3 | AR | 在所有JEB中:在lucina lucida水平上进行拆分,或者:
|
| LAMB3 | ||||||||
| LAMC2 | ||||||||
| JEB 泛发 &局部 | LAMA3 | |||||||
| LAMB3 | ||||||||
| LAMC2 | ||||||||
| COL17A1 | ||||||||
| ITGB4 | ||||||||
| JEB 晚发型 | COL17A1 | |||||||
| JEB w/幽门闭锁 | ITGB4 | |||||||
| ITGA6 | ||||||||
| JEB w/呼吸&肾脏受累 | ITGA3 | |||||||
| JEB-LOC 综合征4 | LAMA3A | |||||||
| 广泛; 主要在手&脚上 | 在RDEB食管狭窄 |
RDEB中较大的儿童&成年人的手&脚疤痕引起的假性畸形(手套畸形) |
| DEB | RDEB 泛发重型 | COL7A1 | AR | 在基底膜下形成(在浅层真皮中) |
| RDEB 泛发 & 局部 | ||||||||
| DDEB (所有亚型) | AD | |||||||
| 易碎的皮肤薄弱w/急性起泡 |
| 带薄纸外观的皮肤组织萎缩 |
| Kindler syndrome | NA | FERMT1 | AR | 多重分裂平面; 特别是表皮,lucina层&densa层(多裂平面是机械性球囊疾病所特有的) |
部分采用自 Fine et al [2014]
- 1.
对于给定的EB类型,没有临床发现是特异的; 但是,某些临床发现更可能与单一类型的EB相关。
- 2.
曾有因KRT5和KRT14引起的常染色体隐性遗传EBS罕见病例报道 [Yasukawa et al 2002, Yiasemides et al 2008].
- 3.
EBS根据开裂的程度分为上基底和基底EBS。
- 4.
Punjabi印第安人描述了喉癌(LOC)综合征或JEB-LOC(OMIM 245660)。 JEB-LOC具有许多表型特征,类似于non-Herlitz junctional epidermolysis bullosa (NH-JEB) [Figueira et al 2007, Pfendner et al 2007]。 皮肤脆弱表现为轻度的水疱和手和面部的糜烂,并扩散到身体的其他部位,并伴有结lesions性病变。 新生儿可能会嘶哑,然后出现喉咙异常和生长异常,结膜疾病,指甲异常和牙釉质增生。 最终,结膜疾病可能导致失明,喉部疾病可能导致需要气管切开的危及生命的气道阻塞[Cohn & Murrell 2010].
管理
初步诊断后的评估
为了确定诊断为营养不良性大疱性表皮松解症(DEB)的患者的疾病程度和需求,建议进行以下评估(如果尚未完成)。
皮肤和皮肤癌的风险
- 全面评估整个皮肤表面是否有水泡,糜烂和感染
- 老年人鳞状细胞癌(SCC)的硬皮结,不愈合或疼痛性病变的评估
口腔,胃肠道和营养
- 检查口腔,包括粘膜起泡和糜烂,龋齿
- 如果出现吞咽困难症状,可吞咽钡剂以食道狭窄
- 测量身高,体重和BMI以评估营养状况和胃造口术喂养的需要
- 贫血和营养状况的基本实验室检查(请参阅 Surveillance)
- 如果诊断发生在年龄较大的个体中,则进行基本DEXA扫描以检查骨质疏松症
眼 眼科检查以评估角膜擦伤和疤痕
心脏 基本超声心动图
泌尿科/肾脏科 基本尿液分析以评估血尿和蛋白尿
骨科 物理或职业治疗师对手功能和活动/敏捷状态的评估
妇科的 如果患者在诊断中是青春期,则评估青春期状态
社会心理(心理学和社会工作)
- 由专业人员进行评估,以评估焦虑,抑郁,药物依赖和滥用
- 协助解决与学校有关的问题
- 协助获得所需的服务,保险和残障人士住宿
遗传学 咨询临床遗传学家和/或遗传咨询师
表现治疗
皮肤科 家庭必须确定许多正确有效的包扎方法中哪些适合他们:没有“最佳”方法。 但是,总的来说,应刺破并排出水疱以防止流体压力扩散 [Denyer 2010, Pope et al 2012, El Hachem et al 2014].
在大多数情况下,水泡和侵蚀的敷料涉及三层:
- 主(基础)层。 一种不粘在皮肤上的主要非粘性敷料。 对不同的主层的耐受是不同的。 主层可以包括以下任何一项:
- 带有或不带有不粘硅表面的不粘产品
- 浸有润肤剂(如凡士林)或局部防腐剂(如银,医用级蜂蜜或局部抗生素,如果有感染)的敷料
- 第二层 该层为主要层提供了稳定性,并增加了用于保护的填充。 通常使用柔软的纱布卷。
- 第三层 该层通常具有一定的弹性,可确保第一层和第二层敷料的完整性。
不愈合的伤口可能需要用生物皮肤替代品或临时猪或人尸体皮肤移植物覆盖。目前,涉及基因校正的自体细胞的新产品正在临床试验中(请参见Therapies Under Investigation)。
疼痛和瘙痒是影响DEB患者生活质量的主要因素 [Goldschneider et al 2014, Danial et al 2015]. 提倡多种局部,口服和心理疗法。
皮肤感染 在DEB中很常见。 EB中所有开放性伤口最终都被细菌定植。临床判断必须确定何时存在需要治疗的重大感染。许多受累的人感染了耐药菌,最常见的是耐甲氧西林的葡萄球菌(MRSA),铜绿假单胞菌和链球菌。抗生素和防腐剂都需要使用。
鳞状细胞癌(SCC)。 2016年出版物中总结了多种治疗SCC的方法[Mellerio et al 2016]. 没有公认的护理标准。
口服,胃肠道和营养。对于更严重受累的RDEB婴幼儿,生长不良可能是个问题,需要额外的营养支持,包括必要时进行饲喂胃造口术,以确保摄入足够的热量 [Stehr et al 2008, Mortell & Azizkhan 2010, Hubbard 2016].
食道狭窄和网状组织可反复扩张以改善吞咽[Castillo et al 2002, Kay & Wyllie 2002, Azizkhan et al 2006].
液体和电解质问题在新生儿期和患广泛疾病的婴儿中可能非常严重,甚至危及生命,需要谨慎处理。
贫血是RDEB的慢性问题,可以通过口服铁补充剂,静脉铁剂输注和/或红细胞输注来治疗。
其他营养缺乏症的治疗包括:
- 补充钙和维生素D以及静脉注射双膦酸盐治疗骨质减少和骨质疏松症;
- 硒和肉碱水平低时进行补充,可能有助于预防扩张型心肌病;
- 含量低时可以补充锌,以促进伤口愈合。
良好的牙齿保健对于确保进食能力和摄取足够的热量至关重要 [Harris et al 2001]。可能需要提取龋齿和拥挤。
眼 预防性使用眼部润滑剂,在某些情况下还应使用防护性隐形眼镜,可防止角膜擦伤。
心脏 如果在超声心动图上检测到心肌病,请咨询心脏病专家。使用β受体阻滞剂和/或ACE抑制剂的药物治疗可能能够控制或逆转它。
泌尿科/肾脏科 如果存在血尿或蛋白尿,请咨询泌尿科医生以解决尿道糜烂,狭窄或膀胱功能障碍,或咨询肾科医生,因为可能发生肾小球肾炎和肾衰竭 [Almaani & Mellerio 2010].
骨科
- 职业疗法可能有助于预防进行性手部挛缩。由于皮肤脆弱,手夹板可能会出现问题。
- 已经描述了通过几种方法手术松解手指。它通常需要重复执行[Marín-Bertolín et al 1999, Glicenstein et al 2000]。一些中心主张保留功能拇指,而不是全手释放。
妇科的 营养不良的女孩常见青春期延迟。
- 雌激素替代将启动青春期的迹象。
- 通常会抑制月经期,以防止贫血恶化。
社会心理的 包括社会服务和心理咨询在内的社会心理支持对于管理受累的及其家人至关重要。
监视
Table 4.
患有DEB的个人的建议监视
| 器官系统 | 评估 | 频率 |
|---|---|---|
| 血液 |
| 每6到12个月进行一次贫血评估 |
| 肾脏 |
| 每6-12个月一次评估肾功能和膀胱炎 |
| 肝脏 | 肝功能检查 | 每6-12个月进行一次肝功能评估 1 |
| 皮肤 | 锌 | 每6至12个月一次用于伤口愈合 |
| 骨骼 | 25-OH 维生素 D3 | 由于骨质疏松症的风险,每6-12个月一次 |
| 骨密度扫描 | 一年一次用于骨质疏松 | |
| 心脏 | 硒&肉碱 | 由于存在/低水平的心肌病风险,每6-12个月出现一次 |
| 超声心动图 | 一年一次心脏病监护 | |
| 免疫 |
| 每6到12个月进行一次炎症评估 |
- 1.
RDEB患者很少发生暴发性甚至致命的肝衰竭。 在重度RDEB中使用的一些用于止痛,止痒和/或感染的药物可能会对肝功能产生不利影响。
由于RDEB患者的一生中转移性鳞状细胞癌的风险大于90%,因此必须在生命的第二个十年中至少每年进行一次不愈合,疤痕组织旺盛或外观异常的伤口监测。可能需要对可疑病变进行频繁的活检,然后进行局部切除。受影响的人通常不愿意在诊所环境中完全脱衣服,换药期间的家庭照片可能就足够了。
避免的药物/情况
应避免穿着不合适或质地较粗的衣服和鞋类,因为它们可能导致外伤。
通常,应避免伤害皮肤的活动(例如远足,骑山地自行车,接触运动);应鼓励致力于参与此类活动的 受累的个人设计保护皮肤的方法。
大多数患有DEB的人不能忍受使用普通医用胶带或Band-Aids®。
评估亲戚处于危险中
对高危新生儿评估是否有水疱迹象 , 尽可能避免对皮肤的伤害。
鉴于家族内临床变异性,应该通过家庭中的COL7A1 致病性变异的 分子遗传学检测来阐明受累的明显无症状的高危亲戚的遗传状况,以便尽早发现。尽可能从迅速开始治疗和预防措施中受益的人。
有关与 遗传咨询目的有关的高危亲属测试的问题,请参见 Genetic Counseling。
怀孕管理
妊娠和EB的数据有限,但最近的一项调查未发现EB女性的妊娠相关并发症风险增加 [Intong et al 2017].
正在调查的疗法
当前的研究中有几种有希望的疗法,包括基因校正的自体表皮移植物[Siprashvili et al 2016],各种干细胞疗法包括骨髓移植,间充质干细胞和IPS细胞 [Tamai & Uitto 2016],,以及基因-校正的成纤维细胞 [Jacków et al 2016].
最近的一项加速临床试验表明,自体转基因角质形成细胞培养物可在患有严重JEB的7岁儿童中再生整个功能齐全的表皮 [Hirsch et al 2017]. DEB正在进行类似的研究[Siprashvili et al 2016].
使用RDEB的鼠模型和鼠的伤口模型,已证明可以将培养的真皮成纤维细胞(来自未受影响的人类受试者或来自RDEB的个体,经工程改造以表达胶原蛋白7蛋白[C7])可以注入鼠皮或移植的RDEB中皮肤等效物,然后注入的细胞将C7分泌到乳头状真皮中。在那里,C7并入真皮-表皮连接处(DEJ),形成新的锚定原纤维(AFs),并逆转了表皮-真皮依从性差的RDEB表型。还显示可以在家中静脉内(IV)施用细胞以打开皮肤伤口,促进愈合。这表明,将这种静脉注射入具有RDEB的个体的细胞可以定位在愈合伤口内,并持续分泌C7,然后可以将其掺入DEJ中并形成促进愈合的新AFs[Woodley et al 2013].
在美国搜索ClinicalTrials.gov,在EU Clinical Trials Register,以获取有关各种疾病和状况的临床研究信息。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分介绍了遗传风险评估以及家族史和基因检测的使用,以阐明家族成员的遗传状况。 本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —ED。
遗传方式
营养不良性大疱性表皮松解症(DEB)以 常染色体显性遗传或常染色体隐性遗传的方式遗传。
确定单发的情况下的遗传模式(即,家庭中的单发事件)。致病变异的分子表征是确定遗传模式和 再发风险的唯一准确方法。据报道,有七个个体在一个等位基因上具有隐性 致病性变异而在另一个等位基因上具有显性遗传的氨基酸取代,这提示在单独基于父母 表型(即 分子遗传学检测)预测复发风险时要谨慎 ) [Varki et al 2007].
仅表型严重性和EM / IF发现不足以确定遗传模式和 再发风险,因为隐性DEB的表型变异性极高[Hashimoto et al 1999, Vaccaro et al 2000, Mallipeddi et al 2003]。 表型较轻且无家族史的个体可能具有 常染色体显性遗传或常染色体隐性遗传DEB; RDEB中许多表型的描述了一些非常温和的DDEB [Hashimoto et al 1999, Vaccaro et al 2000, Mallipeddi et al 2003, Varki et al 2007].
常染色体显性遗传–对家庭成员的风险
先证者的父母
- 据报告,约70%的被诊断患有显性DEB(DDEB)的人有一个受累的父母。
- 大约30%的先证者可能患有 de novoCOL7A1 致病性变异 [Varki et al 2007];但是,由于偏误,这些数字可能无法反映新发突变的真实比例。
- 建议对具有明显 de novo 致病性变异的 先证者的父母进行分子遗传学检测。
- 如果在任一亲本的白细胞DNA中都无法检测到先证者中发现的 致病性变异,则可能的解释包括亲本的先证者中的de novo致病变异或亲本中的 胚系嵌合。母体胚系嵌合已有报道 [Cserhalmi-Friedman et al 2001, Fassihi et al 2006].
- 尽管诊断为DDEB的70%个体有受累的父母,仍有部分家族史阴性或由于外显率降低 ,除非对先证者的父母进行了分子遗传学检测,否则无法否定阴性的家族史。
- 注意:如果父母是首次发生致病性变异的个体,则他/她可能具有该变异的 体细胞嵌合,并且可能是轻度/轻度受累的。
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况:
- 如果先证者的父母是 受累的和/或已知是COL7A1 杂合的 致病性变异,同胞继承该变异的风险为50%。已观察到家族内临床变异性和外显率降低。
- 如果在任一亲本的白细胞DNA中均无法检测到 先证者中发现的 致病性变异,则同胞的风险较低(约等于106分之一),但比一般人群的同胞风险要大,因为 胚系嵌合已报道[Cserhalmi-Friedman et al 1999, Cserhalmi-Friedman et al 2001, Fassihi et al 2006].
- 如果父母未进行 分子遗传学检测但似乎在临床上未受影响,则由于父母的 胚系嵌合或外显率降低 , 先证者的同胞仍被认为罹患DDEB的风险增加。
先证者的后代。 DDEB个体的每个孩子都有50%的机会遗传COL7A1致病性变异。
其他家庭成员。对其他家庭成员的风险取决于先证者'父母的状态:如果父母患有COL7A1致病性变异,则他或她的家庭成员可能处于危险之中。
常染色体隐性遗传
家庭成员的风险
先证者的父母
患有隐性营养不良性大疱性表皮松解症(RDEB)的孩子的父母是杂合子(即一种COL7A1致病性变异的携带者)。
先证者的后代 具有RDEB的个体的后代是COL7A1中致病性变异的杂合子(携带者)。
其他家庭成员 先证者'父母的每个同胞都有可能成为COL7A1 致病性变异的携带者。
携带者(杂合子)检测 对高危亲属进行携带者测试需要事先确定该家族中的COL7A1致病变异。
相关的遗传咨询问题
有关以早期诊断和治疗为目的的评估高风险亲戚的信息,请参阅《管理,Evaluation of Relatives at Risk》。
具有明显de novo 致病性变异家庭的注意事项。当具有 常染色体显性遗传的先证者的父母均无先证者中鉴定出致病变体或该疾病的临床证据时,该致病变体很可能是新生的。但是,也可以探索非医学解释,包括 非生物学父亲或产妇(例如,辅助生殖)和未公开的收养。
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑受累的银行DNA。
产前检查和植入前遗传学诊断
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或登记册,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- DEBRA InternationalAm Heumarkt 27/3Vienna 1030AustriaPhone: +43 1 876 40 30-0Fax: +43 1 876 40 30-30Email: office@debra-international.org
- DebRA of America, Inc. (Dystrophic Epidermolysis Bullosa Research Association)16 East 41st Street3rd FloorNew York NY 10017Phone: 866-332-7276 (toll-free); 212-868-1573Email: staff@debra.org
- DebRA UKDebRA House13 Wellington Business ParkCrowthorne Berkshire RG45 6LSUnited KingdomPhone: +44 01344 771961Fax: +44 01344 762661Email: debra@debra.org.uk
- My46 Trait Profile
- DEB RegisterInternational registry of dystrophic epidermolysis bullosa (DEB) patients and associated COL7A1 mutations
- EBCare RegistryThe EBCare Registry is a resource for individuals and families 受累的 by all forms of epidermolysis bullosa (EB) and qualified researchers working on approved EB research projects.Phone: 866-332-7276Fax: 888-363-0790Email: coordinator@EBCare.org
分子遗传
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。 —编者。
Table A.
营养不良性表皮松解性大疱:基因和数据库
| 基因 | 染色体定位 | 蛋白质 | 特别位点数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| COL7A1 | 3p21 | Collagen alpha-1(VII) chain | COL7A1 database | COL7A1 | COL7A1 |
Table B.
营养不良性表皮松解的OMIM条目(View All in OMIM)
| 120120 | COLLAGEN, TYPE VII, ALPHA-1; COL7A1 |
| 131705 | TRANSIENT BULLOUS DERMOLYSIS OF THE NEWBORN; TBDN |
| 131750 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, AUTOSOMAL DOMINANT; DDEB |
| 131850 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, PRETIBIAL |
| 132000 | EPIDERMOLYSIS BULLOSA WITH CONGENITAL LOCALIZED ABSENCE OF SKIN AND DEFORMITY OF NAILS |
| 226600 | EPIDERMOLYSIS BULLOSA DYSTROPHICA, AUTOSOMAL RECESSIVE; RDEB |
| 226650 | EPIDERMOLYSIS BULLOSA, JUNCTIONAL, NON-HERLITZ TYPE |
| 604129 | EPIDERMOLYSIS BULLOSA PRURIGINOSA |
分子发病机理
COL7A1在包括表皮的基底角质形成细胞的角质形成细胞中表达,其中蛋白质产物被组装成具有螺旋三重胶原结构域的同源三聚体分子。然后,同三聚体通过二硫键结合成在层状牙本质下面的细胞外基质中的同二聚体结构,并形成将基膜锚定在下面的真皮上的锚定原纤维。锚定原纤维通过连接至层粘连蛋白5和直接位于其上方的角质形成细胞半桥粒而与基底膜相连。细胞内角蛋白中间丝网络直接与将角质形成细胞锚定在基底层上的半脂质体和导致角质形成细胞彼此牢固结合的桥粒直接连接。这些联系以及网络本身提供了稳定性和对压力的抵抗力,使角质形成细胞在轻度创伤期间能够保持其结构完整性,并锚定在基底膜和真皮上 [Bruckner-Tuderman 1999].
COL7A1中的致病变异可导致对轻微创伤的抵抗力降低,并导致水泡,这是DEB的标志。 致病性变异的类型,取代氨基酸的生化特性及其在蛋白质中的位置决定了水泡 表型的严重程度(请参见 Genotype-Phenotype Correlations)和遗传模式。优势DEB的家族内表型变异表明,其他因素也可能影响细胞对摩擦的抵抗力[Anton-Lamprecht & Gedde-Dahl 2002, Ortiz-Urda et al 2005].
基因结构。正常的cDNA包含9.2 kb,具有118个外显子中的8,833个核苷酸的 开放阅读框架,编码2,944个氨基酸,跨越32 kb。有关 基因 和蛋白质信息的详细摘要,请参见Table A,基因。
致病变异。在DDEB中,三重螺旋结构域 (Gly-X-Y;尤其是第73、74和75号外显子)中的甘氨酸取代变异占主导地位(> 75%)。 p.Gly2034Arg和p.Gly2043Arg是最常见的引起DDEB的致病变异,占美国最大人群中报告的主要致病变异的50%[Varki et al 2007]。在该区域外还可以发现甘氨酸取代以及其他氨基酸取代和剪接连接变异。然而,通常情况下,如果不确定亲本表型和相应的 基因型,就无法预测遗传模式。
在所有形式的DEB,已经描述了超过700种全基因DEC的隐性DEB变异[Ashton et al 1999, Mellerio et al 1999b, Whittock et al 1999, Gardella et al 2002a, Murata et al 2004, Sawamura et al 2005, Varki et al 2007].。在某些种族背景中已经描述了常见的致病变异,包括c.497dupA[Ashton et al 1999, Gardella et al 2002a],c.2470dupG [Mellerio et al 1999b],,p.Arg578Ter [Whittock et al 1999],c.3840delC[Whittock et al 1999]和c.4919delG [Whittock et al 1999] –在美国人群中多发。但是,每种占不超过所描述的致病变异总数的1%-2%。尽管已经描述了甘氨酸取代和其他氨基酸取代,但在RDEB中无意变异占主导。 RDEB的较轻形式通常是由剪切变异或其他错义变异引起的。
Table 5.
本GeneReviews中讨论的COL7A1变异
| DNA 核苷酸改变 (Alias 1) | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.497dupA (c.497insA) | p.Val168GlyfsTer12 | NM_000094 NP_000085 |
| c.1732C>T | p.Arg578Ter | |
| c.2470dupG (c.2470insG) | p.Asn825LysfsTer41 | |
| c.3840delC | p.Gly1281ValfsTer44 | |
| c.4919delG | p.Gly1640ValfsTer70 | |
| c.6100G>A | p.Gly2034Arg | |
| c.6127G>A | p.Gly2043Arg |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen- .hgvs.org ).的标准命名约定。 有关命名法的说明,请参见Quick Reference。- 1.
不符合当前命名约定的变异名称
正常基因产物。胶原VII是2,944个氨基酸的单体,可与三螺旋胶原结构域缔合为同型三聚体。然后,同型三聚体通过二硫键缔合成形成锚定原纤维的同型二聚体结构。
异常基因产物。隐性DEB通常是由于两个等位基因上的密码子过早终止变异或大片段缺失或框架外基因产物的剪接变异导致的COL7A1基因产物缺失而导致-尽管已证明某些甘氨酸替代变异具有常染色体隐性遗传以及 常染色体显性遗传,可能导致基因产物缺失(隐性)或缺陷基因产物(显性)。
在显性DEB中,在胶原 结构域(Gly-X-Y)中VII胶原被甘氨酸取代,可能导致异常的三重螺旋卷曲和部分无功能的蛋白质产物。这些蛋白质在电子显微镜下可能会表现出形态改变,而免疫荧光染色的强度可能正常或略有降低,因此除非存在切割平面,否则很难通过皮肤活检的免疫荧光染色进行诊断。另外, 非移码 外显子可以用来调节隐性疾病中的疾病严重程度并产生部分功能性的 基因产物 [McGrath et al 1999, Varki et al 2007].
患有隐性全身严重DEB的个体(RDEB-sev gen)的一生中,发生侵袭性转移性鳞状细胞癌的风险大于90%,机理不清楚直到Ortiz-Urda et al [2005] 研究RDEB角质形成细胞中Ras驱动肿瘤发生,并发现缺乏胶原蛋白VII的小鼠不形成肿瘤,而保留了特定VII的小鼠胶原蛋白VII片段(氨基末端非胶原 结构域NC1)具有致瘤性。恢复NC1表达可恢复胶原VII缺乏细胞的致瘤性。他们得出的结论是,胶原蛋白VII介导的肿瘤-基质相互作用促进了肿瘤的形成,而NC1序列在RDEB个体中的保留可能是其增加对鳞状细胞癌敏感性的因素。
参考文献
Literature Cited
- Almaani N, Liu L, Dopping-Hepenstal PJ, Lai-Cheong JE, Wong A, Nanda A, Moss C, Martinéz AE, Mellerio JE, McGrath JA. Identical glycine substitution mutations in type VII collagen may underlie both dominant and recessive forms of dystrophic epidermolysis bullosa. Acta Derm Venereol. 2011;91:262 - 6. [PubMed: 21448560]
- Almaani N, Mellerio JE. Genitourinary tract involvement in epidermolysis bullosa. Dermatol Clin. 2010;28:343 - 6. [PubMed: 20447500]
- Anton-Lamprecht I, Gedde-Dahl T. Epidermolysis bullosa. In: Rimoin DL, Connor MJ, Pyeritz RE, Korf BR, Emery AEH, eds. Principles and Practice of Medical Genetics. 4 ed. New York, NY: Churchill Livingstone Publishers; 2002.
- Ashton GH, Mellerio JE, Dunnill MG, Milana G, Mayou BJ, Carrera J, McGrath JA, Eady RA. Recurrent molecular abnormalities in type VII collagen in Southern Italian patients with recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol. 1999;24:232 - 5. [PubMed: 10354186]
- Ayman T, Yerebakan O, Ciftcioglu MA, Alpsoy E. A 13-year-old girl with recessive dystrophic epidermolysis bullosa presenting with squamous cell carcinoma. Pediatr Dermatol. 2002;19:436 - 8. [PubMed: 12383103]
- Azizkhan RG, Stehr W, Cohen AP, Wittkugel E, Farrell MK, Lucky AW, Hammelman BD, Johnson ND, Racadio JM. Esophageal strictures in children with recessive dystrophic epidermolysis bullosa: an 11-year experience with fluoroscopically guided balloon dilatation. J Pediatr Surg. 2006;41:55 - 60. [PubMed: 16410108]
- Bale S, Pfendner EG. Exome "slice" sequencing: an alternative to sequential gene sequencing in epidermolysis bullosa (EB). Abstract 485. Nashville, TN: American College of Medical Genetics Annual Clinical Genetics Meeting. 2014.
- Boccaletti V, Zambruno G, Castiglia D, Magnani C, Tognetti E, Fabrizi G, Cortelazzi C, Pagliarello C, Di Nuzzo S. Recessive bullous dermolysis of the newborn in preterm siblings with a missense mutation in type VII collagen. Pediatr Dermatol. 2015;32:e42 - 7. [PubMed: 25639640]
- Bruckner-Tuderman L. Hereditary skin diseases of anchoring fibrils. J Dermatol Sci. 1999;20:122 - 33. [PubMed: 10379704]
- Castillo RO, Davies YK, Lin YC, Garcia M, Young H. Management of esophageal strictures in children with recessive dystrophic epidermolysis bullosa. J Pediatr Gastroenterol Nutr. 2002;34:535 - 41. [PubMed: 12050581]
- Cohn HI, Murrell DF. Laryngo-onycho-cutaneous syndrome. Dermatol Clin. 2010;28:89 - 92. [PubMed: 19945620]
- Cserhalmi-Friedman PB, Garzon MC, Guzman E, Martinez-Mir A, Chung WK, Anyane-Yeboa K, Christiano AM. Maternal germline mosaicism in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2001;117:1327 - 8. [PubMed: 11710955]
- Cserhalmi-Friedman PB, Grossman J, Karpati S, Ahmad W, Horvath A, Christiano AM. Identification of a de novo glycine substitution in the type VII collagen gene in a proband with mild dystrophic epidermolysis bullosa. Exp Dermatol. 1999;8:143 - 5. [PubMed: 10232407]
- Danial C, Adeduntan R, Gorell ES, Lucky AW, Paller AS, Bruckner A, Pope E, Morel KD, Levy ML, Li S, Gilmore ES, Lane AT. Prevalence and characterization of pruritus in epidermolysis bullosa. Pediatr Dermatol. 2015;32:53 - 9. [PMC free article: PMC4315706] [PubMed: 25236506]
- Denyer JE. Wound management for children with epidermolysis bullosa. Dermatol Clin. 2010;28:257 - 64. viii-ix. [PubMed: 20447488]
- Dharma B, Moss C, McGrath JA, Mellerio JE, Ilchyshyn A. Dominant dystrophic epidermolysis bullosa presenting as familial nail dystrophy. Clin Exp Dermatol. 2001;26:93 - 6. [PubMed: 11260188]
- Diociaiuti A, Castiglia D, Giancristoforo S, Guerra L, Proto V, Dotta A, Boldrini R, Zambruno G, El Hachem M. Frequent occurrence of aplasia cutis congenita in bullous dermolysis of the newborn. Acta Derm Venereol. 2016;96:784 - 7. [PubMed: 26864810]
- Eady RA, Dopping-Hepenstal PJ. Transmission electron microscopy for the diagnosis of epidermolysis bullosa. Dermatol Clin. 2010;28:211 - 22. [PubMed: 20447483]
- Eismann EA, Lucky AW, Cornwall R. Hand function and quality of life in children with epidermolysis bullosa. Pediatr Dermatol. 2014;31:176 - 82. [PubMed: 24274904]
- El Hachem M, Zambruno G, Bourdon-Lanoy E, Ciasulli A, Buisson C, Hadj-Rabia S, Diociaiuti A, Gouveia CF, Hernández-Martín A, de Lucas Laguna R, Dolenc-Voljč M, Tadini G, Salvatori G, De Ranieri C, Leclerc-Mercier S, Bodemer C. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. 2014;9:76. [PMC free article: PMC4110526] [PubMed: 24884811]
- Fassihi H, Diba VC, Wessagowit V, Dopping-Hepenstal PJ, Jones CA, Burrows NP, McGrath JA. Transient bullous dermolysis of the newborn in three generations. Br J Dermatol. 2005;153:1058 - 63. [PubMed: 16225626]
- Fassihi H, Lu L, Wessagowit V, Ozoemena LC, Jones CA, Dopping-Hepenstal PJ, Foster L, Atherton DJ, Mellerio JE, McGrath JA. Complete maternal isodisomy of chromosome 3 in a child with recessive dystrophic epidermolysis bullosa but no other phenotypic abnormalities. J Invest Dermatol. 2006;126:2039 - 43. [PubMed: 16710310]
- Figueira EC, Crotty A, Challinor CJ, Coroneo MT, Murrell DF. Granulation tissue in the eyelid margin and conjunctiva in junctional epidermolysis bullosa with features of laryngo-onycho-cutaneous syndrome. Clin Exp Ophthalmol. 2007;35:163 - 6. [PubMed: 17362460]
- Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis bullosa registry. JAMA Dermatol. 2016;152:1231-8. [PubMed: 27463098]
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, Marinkovich MP, Martinez AE, McGrath JA, Mellerio JE, Moss C, Murrell DF, Shimizu H, Uitto J, Woodley D, Zambruno G. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70:1103 - 26. [PubMed: 24690439]
- Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009;60:203 - 11. [PubMed: 19026465]
- Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, Devries DT, Suchindran C., National Epidermolysis Bullosa Registry. Inherited epidermolysis bullosa and the risk of death from renal disease: experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis. 2004;44:651 - 60. [PubMed: 15384016]
- Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009a;61:367 - 84. [PubMed: 19700010]
- Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009b;61:387 - 402. [PubMed: 19700011]
- Fortuna G, Aria M, Cepeda-Valdes R, Garcia-Garcia SC, Moreno Trevino MG, Salas-Alanís JC. Role of dystrophic epidermolysis bullosa in anxiety, depression and self-esteem: a controlled cross-sectional study. J Dermatol. 2016;43:70 - 8. [PubMed: 26183725]
- Frew J, Lim SW, Klausseger A, Chow CW, Tran K, Su J, Orchard D, Varigos G, Sawamura D, Nishie W, Shimizu H, Murrell DF. Autosomal dominant bullous dermolysis of the newborn associated with a heterozygous missense mutation p.G1673R in type VII collagen. Australas J Dermatol. 2011;52:e1 - 4. [PubMed: 22070715]
- Frew JW, Murrell DF. Quality of life measurements in epidermolysis bullosa: tools for clinical research and patient care. Dermatol Clin. 2010;28:185 - 90. [PubMed: 19945634]
- Gardella R, Castiglia D, Posteraro P, Bernardini S, Zoppi N, Paradisi M, Tadini G, Barlati S, McGrath JA, Zambruno G, Colombi M. Genotype-phenotype correlation in italian patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2002a;119:1456 - 62. [PubMed: 12485454]
- Gardella R, Zoppi N, Zambruno G, Barlati S, Colombi M. Different phenotypes in recessive dystrophic epidermolysis bullosa patients sharing the same mutation in compound heterozygosity with two novel mutations in the type VII collagen gene. Br J Dermatol. 2002b;147:450 - 7. [PubMed: 12207583]
- Glicenstein J, Mariani D, Haddad R. The hand in recessive dystrophic epidermolysis bullosa. Hand Clin. 2000;16:637 - 45. [PubMed: 11117053]
- Goldschneider KR, Good J, Harrop E, Liossi C, Lynch-Jordan A, Martinez AE, Maxwell LG, Stanko-Lopp D., Dystrophic Epidermolysis Bullosa Research Association International. (DEBRA International). Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12:178. [PMC free article: PMC4190576] [PubMed: 25603875]
- Harris JC, Bryan RA, Lucas VS, Roberts GJ. Dental disease and caries related microflora in children with dystrophic epidermolysis bullosa. Pediatr Dent. 2001;23:438 - 43. [PubMed: 11699172]
- Hashimoto I, Kon A, Tamai K, Uitto J. Diagnostic dilemma of "sporadic" cases of dystrophic epidermolysis bullosa: a new dominant or mitis recessive mutation? Exp Dermatol. 1999;8:140 - 2. [PubMed: 10232406]
- Haynes L. Nutrition for children with epidermolysis bullosa. Dermatol Clin. 2010;28:289 - 301. [PubMed: 20447494]
- Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, Scaglione D, Reichelt J, Klausegger A, Kneisz D, Romano O, Secone Seconetti A, Contin R, Enzo E, Jurman I, Carulli S, Jacobsen F, Luecke T, Lehnhardt M, Fischer M, Kueckelhaus M, Quaglino D, Morgante M, Bicciato S, Bondanza S, De Luca M. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551:327 - 32. [PMC free article: PMC6283270] [PubMed: 29144448]
- Hubbard LD. Long-term outcomes in adults with recessive dystrophic epidermolysis bullosa fed by a gastrostomy tube in situ. Int J Dermatol. 2016;55:181 - 6. [PubMed: 26340599]
- Intong LR, Murrell DF. How to take skin biopsies for epidermolysis bullosa. Dermatol Clin. 2010;28:197 - 200. [PubMed: 20447481]
- Intong LRA, Choi SD, Shipman A, Kho YC, Hwang SJE, Rhodes LM, Walton JR, Chapman MG, Murrell DF. Retrospective evidence on outcomes and experiences of pregnancy and childbirth in epidermolysis bullosa in Australia and New Zealand. Int J Womens Dermatol. 2017;3:S1鈥揝5. [PMC free article: PMC5418959] [PubMed: 28492031]
- Jacków J, Titeux M, Portier S, Charbonnier S, Ganier C, Gaucher S, Hovnanian A. Gene-corrected fibroblast therapy for recessive dystrophic epidermolysis bullosa using a self-inactivating COL7A1 retroviral vector. J Invest Dermatol. 2016;136:1346 - 54. [PubMed: 26994967]
- Kay M, Wyllie R. Endoscopic dilatation of esophageal strictures in recessive dystrophic epidermolysis bullosa: new equipment, new techniques. J Pediatr Gastroenterol Nutr. 2002;34:515 - 8. [PubMed: 12050577]
- Kern JS, Kohlhase J, Bruckner-Tuderman L, Has C. Expanding the COL7A1 mutation database: novel and recurrent mutations and unusual genotype-phenotype constellations in 41 patients with dystrophic epidermolysis bullosa. J Invest Dermatol. 2006;126:1006 - 12. [PubMed: 16484981]
- Krämer SM, Serrano MC, Zillmann G, Gálvez P, Araya I, Yanine N, Carrasco-Labra A, Oliva P, Brignardello-Petersen R, Villanueva J. DEBRA International. Oral health care for patients with epidermolysis bullosa--best clinical practice guidelines. Int J Paediatr Dent. 2012;22 Suppl 1:1 - 35. [PubMed: 22937908]
- Lanschuetzer CM, Laimer M, Nischler E, Hintner H. Epidermolysis bullosa nevi. Dermatol Clin. 2010;28:179 - 83. [PubMed: 19945633]
- Lara-Corrales I, Mellerio JE, Martinez AE, Green A, Lucky AW, Azizkhan RG, Murrell DF, Agero AL, Kantor PF, Pope E. Dilated cardiomyopathy in epidermolysis bullosa: a retrospective, multicenter study. Pediatr Dermatol. 2010;27:238 - 43. [PubMed: 20609141]
- Li AW, Prindaville B, Gibson TE, Wiss K. Inpatient management of children with recessive dystrophic epidermolysis bullosa: a review. Pediatr Dermatol. 2017;34:647 - 55. [PubMed: 28944966]
- Lucky AW, Dagaonkar N, Lammers K, Husami A, Kissell D, Zhang K. A comprehensive next-generation sequencing assay for the diagnosis of epidermolysis bullosa. Pediatric Dermatology. 2018;35:188 - 97. [PubMed: 29334134]
- Mallipeddi R, Bleck O, Mellerio JE, Ashton GH, Eady RA, McGrath JA. Dilemmas in distinguishing between dominant and recessive forms of dystrophic epidermolysis bullosa. Br J Dermatol. 2003;149:810 - 8. [PubMed: 14616374]
- Marín-Bertolín S, Amaya Valero JV, Neira Gimenez C, Marquina Vila P, Amorrortu-Velayos J. Surgical management of hand contractures and pseudosyndactyly in dystrophic epidermolysis bullosa. Ann Plast Surg. 1999;43:555 - 9. [PubMed: 10560876]
- Martinez AE, Mellerio JE. Osteopenia and osteoporosis in epidermolysis bullosa. Dermatol Clin. 2010;28:353 - 5. [PubMed: 20447502]
- Matsumoto Y, Dogru M, Tsubota K. Ocular surface findings in Hallopeau-Siemens subtype of dystrophic epidermolysis bullosa: report of a case and literature review. Cornea. 2005;24:474 - 9. [PubMed: 15829808]
- McGrath JA, Ashton GH, Mellerio JE, Salas-Alanis JC, Swensson O, McMillan JR, Eady RA. Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. J Invest Dermatol. 1999;113:314 - 21. [PubMed: 10469327]
- Meester I, Igoucheva O, Alexeev V, South A, Moreno-Treviño MG, Salas-Alanis JC. High concordance between clinical diagnosis of epidermolysis bullosa and immunofluorescence with a small, well-matched antibody panel. Australas J Dermatol. 2018;59:73 - 6. [PubMed: 28707324]
- Mellerio JE, Ashton GH, Mohammedi R, Lyon CC, Kirby B, Harman KE, Salas-Alanis JC, Atherton DJ, Harrison PV, Griffiths WA, Black MM, Eady RA, McGrath JA. Allelic heterogeneity of dominant and recessive COL7A1 mutations underlying epidermolysis bullosa pruriginosa. J Invest Dermatol. 1999a;112:984 - 7. [PubMed: 10383749]
- Mellerio JE, Robertson SJ, Bernardis C, Diem A, Fine JD, George R, Goldberg D, Halmos GB, Harries M, Jonkman MF, Lucky A, Martinez AE, Maubec E, Morris S, Murrell DF, Palisson F, Pillay EI, Robson A, Salas-Alanis JC, McGrath JA. Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: best clinical practice guidelines. Br J Dermatol. 2016;174:56 - 67. [PubMed: 26302137]
- Mellerio JE, Salas-Alanis JC, Amaya-Guerra M, Tamez E, Ashton GH, Mohammedi R, Eady RA, McGrath JA. A recurrent frameshift mutation in exon 19 of the type VII collagen gene (COL7A1) in Mexican patients with recessive dystrophic epidermolysis bullosa. Exp Dermatol. 1999b;8:22 - 9. [PubMed: 10206718]
- Mortell AE, Azizkhan RG. Epidermolysis bullosas: management of esophageal strictures and enteric access by gastrostomy. Dermatol Clin. 2010;28:311 - 8. [PubMed: 20447496]
- Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res. 2004;295:442 - 7. [PubMed: 14727126]
- Murata T, Masunaga T, Shimizu H, Takizawa Y, Ishiko A, Hatta N, Nishikawa T. Glycine substitution mutations by different amino acids in the same codon of COL7A1 lead to heterogeneous clinical phenotypes of dominant dystrophic epidermolysis bullosa. Arch Dermatol Res. 2000;292:477 - 81. [PubMed: 11142768]
- Murrell D. Epidermolysis Bullosa: Part I – Pathogenesis and Clinical Features. 1 ed. Vol 28-1. Dermatologic Clinics, Elsevier; 2010.
- Nakamura H, Sawamura D, Goto M, Sato-Matsumura KC, LaDuca J, Lee JY, Masunaga T, Shimizu H. The G2028R glycine substitution mutation in COL7A1 leads to marked inter-familiar clinical heterogeneity in dominant dystrophic epidermolysis bullosa. J Dermatol Sci. 2004;34:195 - 200. [PubMed: 15113589]
- Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, Sakai LY, Lee H, Marinkovich MP, Khavari PA. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773 - 6. [PubMed: 15774758]
- Pfendner EG. Next-generation sequencing: comprehensive genetic testing for epidermolysis bullosa. Br J Dermatol. 2015;173:638 - 9. [PubMed: 26404574]
- Pfendner EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Basic science of epidermolysis bullosa and diagnostic and molecular characterization: proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007;46:781 - 94. [PubMed: 17651158]
- Pfendner E, Lewis T, Bale S. Exome slice testing for epidermolysis bullosa in a cohort of 260 individuals. Pediatr Dermatol. 2017;34:S1.
- Pfendner E, Uitto J, Fine JD. Epidermolysis bullosa carrier frequencies in the US population. J Invest Dermatol. 2001;116:483 - 4. [PubMed: 11231335]
- Pohla-Gubo G, Cepeda-Valdes R, Hintner H. Immunofluorescence mapping for the diagnosis of epidermolysis bullosa. Dermatol Clin. 2010;28:201 - 10. [PubMed: 20447482]
- Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, Goodman L, Coutts P, Wagner J, Allen U, Sibbald G. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67:904 - 17. [PMC free article: PMC3655403] [PubMed: 22387035]
- Ryan TD, Lucky AW, King EC, Huang G, Towbin JA, Jefferies JL. Ventricular dysfunction and aortic dilation in patients with recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2016;174:671 - 3. [PubMed: 26370777]
- Rodari G, Guez S, Manzoni F, Chalouhi KK, Profka E, Bergamaschi S, Salera S, Tadini G, Ulivieri FM, Spada A, Giavoli C, Esposito S. Birmingham epidermolysis severity score and vitamin D status are associated with low BMD in children with epidermolysis bullosa. Osteoporos Int. 2017;28:1385 - 92. [PubMed: 28012019]
- Sato-Matsumura KC, Yasukawa K, Tomita Y, Shimizu H. Toenail dystrophy with COL7A1 glycine substitution mutations segregates as an autosomal dominant trait in 2 families with dystrophic epidermolysis bullosa. Arch Dermatol. 2002;138:269 - 71. [PubMed: 11843659]
- Sawamura D, Goto M, Yasukawa K, Sato-Matsumura K, Nakamura H, Ito K, Nakamura H, Tomita Y, Shimizu H. Genetic studies of 20 Japanese families of dystrophic epidermolysis bullosa. J Hum Genet. 2005;50:543 - 6. [PubMed: 16189623]
- Siprashvili Z, Nguyen NT, Gorell ES, Loutit K, Khuu P, Furukawa LK, Lorenz HP, Leung TH, Keene DR, Rieger KE, Khavari P, Lane AT, Tang JY, Marinkovich MP. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316:1808 - 17. [PubMed: 27802546]
- Stehr W, Farrell MK, Lucky AW, Johnson ND, Racadio JM, Azizkhan RG. Non-endoscopic percutaneous gastrostomy placement in children with recessive dystrophic epidermolysis bullosa. Pediatr Surg Int. 2008;24:349 - 54. [PubMed: 18094979]
- Tamai K, Murai T, Mayama M, Kon A, Nomura K, Sawamura D, Hanada K, Hashimoto I, Shimizu H, Masunaga T, Nishikawa T, Mitsuhashi Y, Ishida-Yamamoto A, Ikeda S, Ogawa H, McGrath JA, Pulkkinen L, Uitto J. Recurrent COL7A1 mutations in Japanese patients with dystrophic epidermolysis bullosa: positional effects of premature termination codon mutations on clinical severity. Japanese Collaborative Study Group on Epidermolysis Bullosa. J Invest Dermatol. 1999;112:991 - 3. [PubMed: 10383751]
- Tamai K, Uitto J. Stem cell therapy for epidermolysis bullosa-does it work? J Invest Dermatol. 2016;136:2119 - 21. [PubMed: 27772543]
- Tenedini E, Artuso L, Bernardis I, Artusi V, Percesepe A, De Rosa L, Contin R, Manfredini R, Pellacani G, Giannetti A, Pagani J, De Luca M, Tagliafico E. Amplicon-based next-generation sequencing: an effective approach for the molecular diagnosis of epidermolysis bullosa. Br J Dermatol. 2015;173:731 - 8. [PubMed: 25913354]
- Tosti A, Piraccini BM, Scher RK. Isolated nail dystrophy suggestive of dominant dystrophic epidermolysis bullosa. Pediatr Dermatol. 2003;20:456 - 7. [PubMed: 14521572]
- Vaccaro M, Moretti G, Guarneri F, Cannavo S, Magaudda L. "Sporadic" dystrophic epidermolysis bullosa: a new dominant or mitis recessive mutation? Eur J Dermatol. 2000;10:436 - 8. [PubMed: 10980463]
- Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44:181 - 92. [PMC free article: PMC2598021] [PubMed: 16971478]
- Wessagowit V, Kim SC, Woong Oh S, McGrath JA. Genotype-phenotype correlation in recessive dystrophic epidermolysis bullosa: when missense doesn't make sense. J Invest Dermatol. 2005;124:863 - 6. [PubMed: 15816848]
- Whittock NV, Ashton GH, Mohammedi R, Mellerio JE, Mathew CG, Abbs SJ, Eady RA, McGrath JA. Comparative mutation detection screening of the type VII collagen gene (COL7A1) using the protein truncation test, fluorescent chemical cleavage of mismatch, and conformation sensitive gel electrophoresis. J Invest Dermatol. 1999;113:673 - 86. [PubMed: 10504458]
- Woodley DT, Wang X, Amir M, Hwang B, Remington J, Hou Y, Uitto J, Keene D, Chen M. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133:1910 - 3. [PMC free article: PMC3890237] [PubMed: 23321924]
- Yenamandra VK, Moss C, Sreenivas V, Khan M, Sivasubbu S, Sharma VK, Sethuraman G. Development of a clinical diagnostic matrix for characterizing inherited epidermolysis bullosa. Br J Dermatol. 2017;176:1624 - 32. [PubMed: 27925151]
- Yiasemides E, Trisnowati N, Su J, Dang N, Klingberg S, Marr P, Melbourne W, Tran K, Chow CW, Orchard D, Varigos G, Murrell DF. Clinical heterogeneity in recessive epidermolysis bullosa due to mutations in the keratin 14 gene, KRT14. Clin Exp Dermatol. 2008;33:689 - 97. [PubMed: 18713255]
- Yasukawa K, Sawamura D, McMillan JR, Nakamura H, Shimizu H. Dominant and recessive compound heterozygous mutations in epidermolysis bullosa simplex demonstrate the role of the stutter region in keratin intermediate filament assembly. J Biol Chem. 2002;277:23670 - 4. [PubMed: 11973334]
本章节的注解
更新历史
- 13 September 2018 (ha) 系统性更新发布到公开网页上
- 26 February 2015 (me) 系统性更新发布到公开网页上
- 4 November 2010 (me) 系统性更新发布到公开网页上
- 4 October 2007 (cd) Revision: deletion/duplication analysis 可在临床上使用
- 21 September 2007 (cd) Revision: deletion/duplication analysis 不可在临床上使用
- 17 October 2006 (cd) Revision: deletion/duplication analysis可在临床上使用
- 21 August 2006 (me) 内容发布到公开网页上
- 27 December 2005 (ep) 最早稿件