
诊断
提示性发现
具有以下临床和影像学发现的个体,应怀疑遗传性纤维化性皮肌炎伴腱挛缩,肌病和肺纤维化(POIKTMP)。
临床
- 早发性硬皮病(以脸颊和脸部红斑,皮肤萎缩,毛细血管扩张和斑驳的色素沉着为特征),头皮头发稀疏,睫毛和/或眉毛稀疏或缺失的贫血
- 多汗症不耐高温
- 四肢轻度淋巴水肿
多发性挛缩,尤其是儿童三头肌肱三头肌挛缩
- 肌病伴弥漫性进行性肌无力,脊柱侧弯
- 限制性肺综合征和/或肺纤维化
- 外分泌胰腺功能不全
- 肝功能不全
- 血液学异常,如血小板减少,骨髓细胞减少
其他:
- 相对矮小的身材
- 白内障
- 指甲营养不良
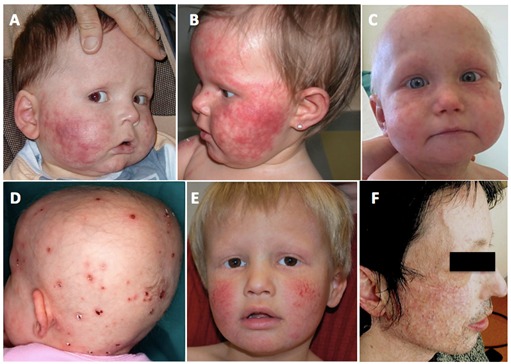
Figure 1.
图1:面部和头皮皮肤病变AE。小儿头皮,眉毛和睫毛的硬皮病和脱发
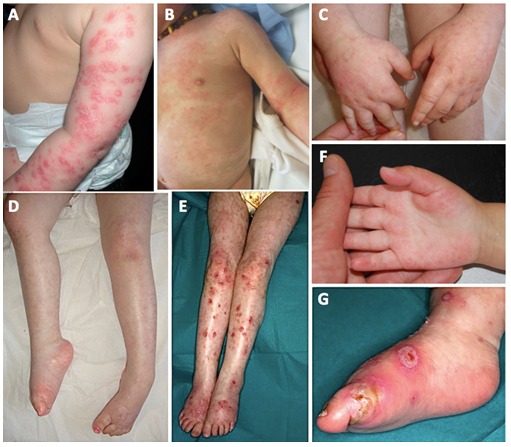
Figure 2.
图2 :下肢皮肤病变A.上肢湿疹样和牛皮癣样皮肤病
影像学
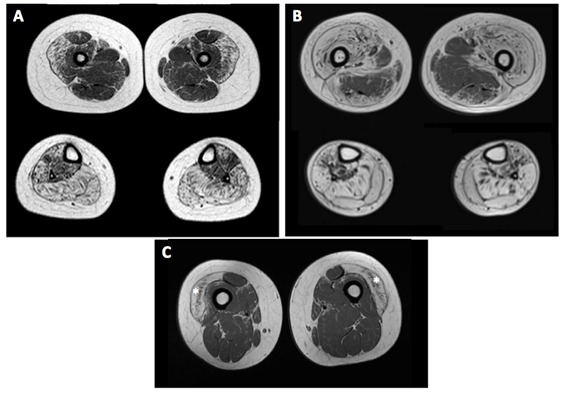
- 参见图3。肌肉MRI通常显示腿部严重弥漫性脂肪浸润,尤其是:
- 股外侧肌(胫骨后部相对保留);和
- 大腿前部(后部相对保留)。
- 这些发现可以证实无症状个体的肌肉受累
建立诊断
POIKTMP的诊断建立在早发性硬皮病伴有其他异常的先证者中,尤其是肌肉挛缩和/或肌肉无力,并通过分子遗传学检测鉴定FAM111B中的杂合致病性错义突变(表1)。
分子遗传学检测方法可以包括单基因检测,多基因组的使用以及更全面的 基因组检测:
- 单基因测试。首先执行FAM111B的序列分析。可以进行基因靶向的缺失/重复分析;但是,由于POIKTMP可能是由显性负遗传机制引起的,并且由于尚未报告大的基因内缺失或重复,因此测试基因内缺失或重复不太可能确定致病变异(请参见Molecular Genetics)。
- 更复杂的 基因组的 检测 如果单基因测序检测(和/或使用包含FAM111B的多基因检测方法)未能确认具有POIKTMP功能的个体的诊断,则可以考虑进行包括外显子组测序和基因组测序在内的更全面的基因组检测。这样的测试可以提供先前未考虑的诊断(例如,导致相似临床表现的一个或多个不同基因的突变)。
表格1。
分子遗传学检测用于肌腱挛缩症,肌病和肺纤维化的遗传性纤维化硬皮病
基因 1
测试方法
可通过这种方法检测到的具有致病变异体2的先证者所占的比例
FAM111B
序列分析 3
13/13 4
基因靶向的缺失/重复分析 5
未知6
2.有关在该基因中检测到的等位基因突变的信息,请参见《分子遗传学》。
3.序列分析可检测出各种变异,可能是致病性或良性,可能良性的变体。致病性变异可能包括小的基因内缺失/插入和错义,无义和剪接位点变异;通常,未检测到外显子或全基因缺失/重复。
4.Mercier等[2013],Mercier等[2015],Seo等[2016],Takeichi等[2017]
5.基因靶向的缺失/重复分析检测基因内缺失或重复。可以使用的方法包括:定量PCR,长距离PCR,多重连接依赖性探针扩增(MLPA)和设计用于检测单外显子缺失或重复的基因靶向微阵列。
6.在受影响的个体和功能研究中检测到的变异体谱[作者,未发表的观察结果]提示疾病为显性负性机制。因此,缺失 / 重复测试不太可能检测出致病变异。
able 1.
临床特征
临床描述
具有肌腱挛缩,肌病和肺纤维化(POIKTMP)的遗传性纤维化硬皮病的患者可以表现出很少或许多相关的临床特征。功能的严重性(例如,皮肤或肌肉异常)可能会有所不同。已经观察到家族内临床变异性[ Khumalo等人2006,Seo等人2016 ]。
值得注意的是,以下对临床特征的描述是基于迄今已发表的25 例经分子确诊的受影响个体的发现[ Mercier等,2013;Mercier等,2015;Seo等,2016;Takeichi等,2017 ]。
值得注意的是,据报道三人具有Rothmund-Thomson综合征样表型的个体可能具有FAM111B致病变异。然而,尚无法进行分子诊断[ Lessem et al 1980,Otsu et al 2008,Meier&Schwarz 2012 ]。
皮肤。皮肤异常是最早的发现。值得注意的是,随着时间的推移,皮肤病变-特别是面部皮肤恶性皮肤病-会改善。
- 皮肤异色病出现在婴儿早期,通常在头六个月。它主要定位在面部(图1)。暴露在阳光下会出现面部红斑的短暂加重。斑驳的色素沉着也是硬皮病的组成部分。
- 大多数人都患有多汗症。
- 下肢和/或上肢淋巴水肿,可能在儿童时期出现,通常较轻;蜂窝织炎可使其并发。
- 慢性红斑和鳞状皮肤病,经常在四肢观察到被描述为湿疹样,鱼鳞样病或牛皮癣样病
- 还可观察到手指硬化和轻度掌跖角化病。
- 头发/指甲。在几乎所有受累的个体中发现稀疏的头皮头发和稀疏或不存在的睫毛和/或不同严重程度的眉毛。三个受影响的个体患有轻度指甲发育不良。
肌肉
- 肌肉挛缩症通常出现在儿童时期,最早可在两岁时出现。受累最常见的肌肉是肱三头肌,导致跟腱缩短和足内翻畸形。还观察到上肢挛缩(肱二头肌和腕伸肌)。
- 肌病。大多数受影响的个体会发展出四肢的近端和远端肌肉的进行性肌无力。第一种表现(在下肢中观察到)是近端而不是远端。肌肉无力的变化范围从童年时期的走动丧失到成年后没有症状[ Mercier等,2013;Mercier等,2015;Seo等,2016;Takeichi等,2017 ]。
肌肉无力通常与肌肉萎缩有关,有时与胸腰椎侧弯有关。
血清肌酸激酶正常或轻度升高。执行时,肌电图可能显示正常或肌病模式。
肺.
- 可以观察到复发性支气管炎。肺功能异常伴有限制性肺疾病是常见的。
- 一些成年人发展为进行性间质性肺纤维化,表现为进行性呼吸困难和干咳。在出现首次呼吸道症状后三到四年内,它可能会危及生命。
消化道
其他
- 据报道,相对的矮小身材和/或体重增加与青春期延迟有关。
- 血液学发现包括嗜酸性粒细胞增多或轻度血小板减少。在多重家族中报道了骨髓细胞减少[ Seo et al 2016 ]。
- 甲状腺功能减退症的描述是在一个14岁的女孩中[ Takeichi et al 2017 ]。
- 眼睛。据报道,白内障是在13岁的女孩中出现的[ Mercier等,2015年 ]。
认知发展和功能正常。值得注意的是,一个人患有精神分裂症[ Mercier et al 2015 ],这可能是偶然的关联。
组织病理学
组织病理学
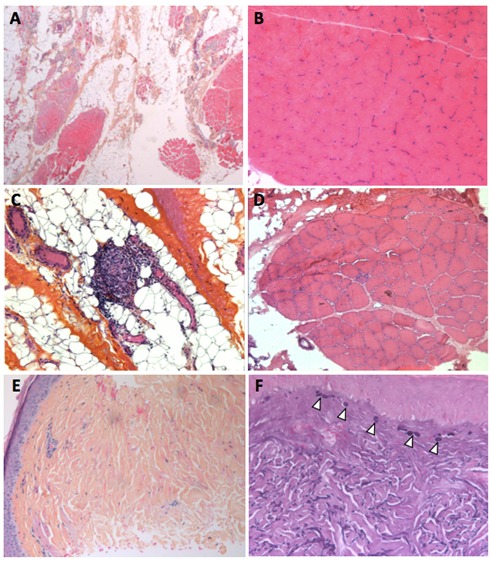
- 肌肉组织学显示大量脂肪浸润。残留的肌肉组织由具有正常纤维或具有中央核的萎缩纤维的碎裂的肌肉束组成(图4 AD)。在组织化学或免疫标记上未发现神经病变特征(即正常的ATPase模式)或线粒体网络异常。蛋白质印迹分析可以显示钙蛋白酶的第二次减少。
- 皮肤组织学显示表皮萎缩的特征性模式,在真皮的浅层和深层真皮中具有胶原蛋白硬化和弹性变性。乳头状真皮中的弹性球体与弥漫性轻微的胶原硬化有关(图4 EF)。
- 在一个南非家庭的一个受影响成员的尸检结果中发现,脂肪的弥散性扩散和包括肺,食道和胰腺在内的器官的纤维化[ Khumalo et al 2006 ]。
基因型-表型相关
POIKTMP最常见的原因是蛋白质的预测胰蛋白酶样半胱氨酸/丝氨酸肽酶结构域,特别是功能域环中的错义 FAM111B致病变体的杂合性。
- 位于环外的上游变异体(密码子421、430)似乎与较不严重的表型有关,特别是在肌肉特征方面[ Mercier et al 2015,Seo et al 2016,Takeichi et al 2017 ]。
- 该表型在密码子621致病变种似乎比在密码子625,627与致病变种观察到表型较不严重,和628 [ 库马洛等人2006,Mercier的等2013 ]。
需要进一步的研究来确认这些初步的基因型与表型的相关性。
外显率
据我们所知, POIKTMP的外显率是100%,在儿童早期的皮肤功能发生。
患病率
POIKTMP的患病率未知。迄今为止,仅报道了25例具有分子确证的POIKTMP的个体。这种情况被认为是普遍存在的,可能未得到充分诊断。
遗传相关(等位基因)疾病
此GeneReview中讨论的表型外,没有其他表型与FAM111B中的致病变异有关。
管理
初步诊断后的评估
为了确定诊断为遗传性纤维化鬼臼皮炎伴肌腱挛缩,肌病和肺纤维化(POIKTMP)的患者的疾病程度和需求,建议进行以下评估:
- 皮肤病学评估
- 物理疗法评估
- 由肺部专家进行的评估,包括肺功能测试,以评估限制性肺疾病和/或肺纤维化
- 肌肉MRI评估渐进性肌肉受累(可选)
- 验血:
- 肝功能: SGOT, SGPT, ALP, GGT
- 基线全血细胞计数与鉴别。如果有临床指征,则应通过CBC和骨髓活检对有贫血或血细胞减少症临床证据的个体进行评估
- 如果存在脂肪泻,粪便中的弹性蛋白酶水平
- 眼科检查以评估白内障或其他异常
- 咨询临床遗传学家和/或遗传咨询师
对症治疗
皮肤科的
- 避免过度日晒,并使用同时具有UVA和UVB防护的防晒霜
- 脉冲染料激光可以作为老年治疗皮疹的毛细血管扩张成分的一种治疗方法,在较年长的人群中进行讨论。但是,没有有关其使用的数据。
- 避免过多的热量暴露和控制发烧,尤其是在生命的最初几年中,以防止过热
- 淋巴水肿管理(手动淋巴引流,压迫)
- 局部治疗湿疹样病变(润肤剂,局部类固醇)
肌肉
- 物理疗法和运动以促进活动能力和预防挛缩
- 对于肌肉无力的人,还应补充钙和维生素D,以预防骨质减少。
肺
- 必要时使用自动充气的手动通气袋或机械吹气-吹气装置
- 必要时进行无创通气
胰腺。补充胰酶治疗胰腺外分泌功能不全
肝。熊去氧胆酸治疗胆汁淤积
成长。根据需要进行肠内喂养
白内障。手术切除视觉上明显的白内障
监管
从诊断时开始的年度监视(以适应患者的症状):
- 皮肤病学评估,用于皮肤病,四肢淋巴水肿,湿疹样病变,指甲和头发的变化
- 物理疗法评估肌肉无力或挛缩
- 肺功能检查
- 验血:
- 全血细胞计数与鉴别
- 肝功能测试:SGOT,SGPT,ALP,GGT
- 眼科检查
- 监测骨科并发症,尤其是挛缩(注意跟腱挛缩)和脊柱侧弯
需要避免的用药/情况
避免以下情况:
- 过度暴露于阳光下,可能加剧皮疹
- 由于多汗症继发的热不耐症而暴露于热
评估家庭及亲戚的发病风险
为亲属遗传风险检测的相关事项要做遗传咨询的,见遗传咨询( Genetic Counseling) 。
临床试验
对于该病及相关情况临床研究的信息,请搜索 ClinicalTrials.gov 美国和 www.ClinicalTrialsRegister.eu 欧洲
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。下一节讨论遗传风险评估以及家族史和遗传测试的使用,以阐明家族成员的遗传状况。本部分的目的不是要解决个人可能面临的所有个人,文化或道德问题,也不能替代与遗传学专业人员的咨询。
遗传方式
具有肌腱挛缩,肌病和肺纤维化(POIKTMP)的遗传性纤维化硬皮病以常染色体显性遗传 。
先证者的父母
先证者的后代. 患有POIKTMP的个体的每个孩子都有50%的机会遗传FAM111B致病性变异;观察到家族内临床变异性(见临床描述)。
有明显新发生 de novo 致病性变异的家庭需要考虑的:
当具有常染色体显性遗传 状况的先证者的父母均未检测到先证者的致病变体或该疾病的临床证据时,该致病变体很可能是新发生的。但是,也要考虑到非医学的解释,包括非生物学父亲 或母亲(例如,辅助生殖)和未公开的收养。
家庭计划
- 确定遗传风险和讨论产前检查可用性的最佳时间是在怀孕之前。
DNA库 是DNA(通常从白细胞中提取)的存储,以备将来使用。由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑到受累的个体的库DNA 。
Resources
GeneReviews staff has selected the following disease-specific and/or umbrellasupport organizations and/or registries for the benefit of individuals with this disorderand their families. GeneReviews is not responsible for the information provided by otherorganizations. For information on selection criteria, click here.
No specific resources for Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis have been identified by GeneReviews staff.
分子遗传学
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。
表A.
肌腱挛缩症,肌病和肺纤维化的遗传性纤维化硬皮病:基因和数据库Tab
| Gene | Chromosome Locus | Protein | HGMD | ClinVar |
|---|---|---|---|---|
| FAM111B | 11q12 | Protein FAM111B | FAM111B | FAM111B |
分子遗传病理
基因结构。FAM111B包含四个外显子。ATG转录起点位于外显子 3中。有关基因和蛋白质信息的详细摘要,请参见表A,基因。
致病变异。在报道的致病突变FAM111B几乎都是错义和公认的聚集半胱氨酸/丝氨酸蛋白酶样肽酶结构域的FAM111B蛋白。
已经报道了氨基酸残基p.Lys421,p.Gln430,p.Thr621,p.Thr625,p.Arg627和p.Ser628的变异:
- 在一个POIKTMP 家族病例中,在多个受影响的个体中报告了p.Thr621Asp 病原体变异 [ Mercier等人2015 ]。
- 致病性变体p.Arg627Gly和p.Ser628Asn已在多个受影响的个体中被鉴定为新发生[ Mercier等,2013;Mercier等,2015 ]。
据报道,该基因其他区域的两个致病变异p.Gln430Pro和p.Lys421del与较不严重的表型有关 [ Mercier等,2015;Seo等,2016;Takeichi等,2017 ]。
表3。
在此讨论该基因的 FAM111B致病变异Table 3.
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
| c.1261_1263delAAG | p.Lys421del | NM_198947 NP_945185 |
| c.1289A>C | p.Gln430Pro | |
| c.1861T>G | p.Tyr621Asp | |
| c.1874C>A | p.Thr625Asn | |
| c.1879A>G | p.Arg627Gly | |
| c.1883G>A | p.Ser628Asn |
变异分类说明:表中列出的变异体由作者提供。GeneReviews的工作人员尚未独立验证变异的分类。
命名说明:GeneReviews遵循人类基因组变异学会的标准命名惯例(varnomen
- .hgvs.org )网站). 有关术语的解释,请参阅快速参考Quick Reference。
正常基因产物。所述FAM111B蛋白质被预测为含有胰蛋白酶样半胱氨酸/丝氨酸肽酶结构域。有人建议在DNA复制中起作用[ Aviner et al 2015 ]。
异常基因产物。POIKTMP的疾病机制尚不清楚;但是,致病变体和功能研究的范围表明存在显性负效机制[ Mercier et al 2015 ; 作者,未发布的数据]。
References
Literature Cited
- Aviner R, Shenoy A, Elroy-Stein O, Geiger T. Uncovering hidden layers of cell cycle regulation through integrative multi-omic analysis. PLoS Genet. 2015;11:e1005554. [PMC free article: PMC4595013] [PubMed: 26439921]
- Khumalo NP, Pillay K, Beighton P, Wainwright H, Walker B, Saxe N, Mayosi BM, Bateman ED. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155:1057 - 61. [PubMed: 17034542]
- Lessem J, Bjerre I, Forslund M. Epilepsy and myopathy in a patient with Rothmund-Thomson's syndrome. Acta Med Scand. 1980;207:237 - 9. [PubMed: 6768225]
- Meier M, Schwarz A. Rothmund-Thomson syndrome--a single case report with systemic muscular atrophy, multiple organ fibrosis and pulmonary cachexia. Rheumatology (Oxford). 2012;51:2109 - 11. [PubMed: 22711845]
- Mercier S, Küry S, Salort-Campana E, Magot A, Agbim U, Besnard T, Bodak N, Bou-Hanna C, Breheret F, Brunelle P, Caillon F, Chabrol B, Cormier-Daire V, David A, Eymard B, Faivre L, Figarella-Branger D, Fleurence E, Ganapathi M, Gherardi R, Goldenberg A, Hamel A, Igual J, Irvine AD, Israel-Biet D, Kannengiesser C, Laboisse C, Le Caignec C, Mahe JY, Mallet S, MacGowan S, McAleer MA, McLean I, Meni C, Munnich A, Mussini JM, Nagy PL, Odel J, O'Regan GM, Pereon Y, Perrier J, Piard J, Puzenat E, Sampson JB, Smith F, Soufir N, Tanji K, Thauvin C, Ulane C, Watson RM, Khumalo NP, Mayosi BM, Barbarot S, Bezieau S. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. [PMC free article: PMC4608180] [PubMed: 26471370]
- Mercier S, Küry S, Shaboodien G, Houniet DT, Khumalo NP, Bou-Hanna C, Bodak N, Cormier-Daire V, David A, Faivre L, Figarella-Branger D, Gherardi RK, Glen E, Hamel A, Laboisse C, Le Caignec C, Lindenbaum P, Magot A, Munnich A, Mussini JM, Pillay K, Rahman T, Redon R, Salort-Campana E, Santibanez-Koref M, Thauvin C, Barbarot S, Keavney B, Bézieau S, Mayosi BM. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93:1100 - 7. [PMC free article: PMC3853004] [PubMed: 24268661]
- Otsu U, Moriwaki S, Iki M, Nozaki K, Horiguchi Y, Kiyokane K. Early blistering, poikiloderma, hypohidrosis, alopecia and exocrine pancreatic hypofunction: a peculiar variant of Rothmund-Thomson syndrome? Eur J Dermatol. 2008;18:632 - 4. [PubMed: 18952524]
- Seo A, Walsh T, Lee MK, Ho PA, Hsu EK, Sidbury R, King MC, Shimamura A. FAM111B Mutation Is Associated With Inherited Exocrine Pancreatic Dysfunction. Pancreas. 2016;45:858 - 62. [PMC free article: PMC4841754] [PubMed: 26495788]
- Takeichi T, Nanda A, Yang HS, Hsu CK, Lee JY, Al-Ajmi H, Akiyama M, Simpson MA, McGrath JA. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2017;176:534 - 6. [PubMed: 27406236]
Chapter Notes
Revision History
- 13 October 2016 (bp) Review posted live
- 1 February 2016 (sm) Original submission