
概述
临床特征.
非综合征型听力损失与耳聋 (DFNB1)以先天的非进行性轻度到极重度感音神经性听力受损为特征。无其他相关医学发现。
管理.
对症治疗:助听器; 参加合适的教育项目; 对极重度耳聋患者考虑人工耳蜗移植。
监控:监控包括年检和重测听力确定听力损失的稳定性。
有风险亲属评估:如果两个致病变异均已在受累的家庭成员中发现, 可用分子遗传学检测在有风险亲属幼年时来明确基因型从而可提供合适的早期支持与管理。
遗传咨询.
DFNB1循常染色体隐性遗传方式。先证者父母每次怀孕有25%几率生耳聋孩子,50%几率生携带者健听孩子以及25%几率生非携带者健听孩子。 当导致DFNB1的GJB2致病变异已在受累的家庭成员中检出,可对有风险的亲属做携带者检测,在孕期做产前检测,以及植入前遗传诊断。
诊断
提示性发现
病人若有以下表现当怀疑患有由GJB2双等位基因的致病变异造成的非综合征型听力损失与耳聋 (DFNB1):
- 由听觉脑干反应或纯音听力测试检出轻度到极重度的先天的一般非进行性感音神经性听力损失注:(1) 听力以分贝 (dB)衡量。每个频率阈值 0-dB 标志正常年轻成人50%的时间能听到发出音调的听力水平。听力阈值在25 dB之内认为听力正常。 (2) 听力损失程度分级为轻度(26-40 dB)、中度((1-55 dB)、中重度(56-70 dB)、重度(71-90 dB)、或极重度(>90 dB)。 听力损失频率分为低频(<500Hz)、中频(501-2000 Hz),或高频(>2000 Hz)(见遗传性听力损失与耳聋概论)。
- 病历和体检无相关系统发现
- 非综合征型听力损失家族史符合常染色体隐性遗传
建立诊断
患有轻度到极重度先天的一般非进行性感音神经性听力损失的先证者,若分子遗传学检测出GJB2(编码连接蛋白26)双等位基因的致病变异则DFNB1诊断成立(见表1).
DFNB1患者有以下两者之一:
- 纯合或复合杂合的GJB2致病变异(99%); 或
分子遗传检测方法包括目标基因检测(单基因检测和/或多基因包) 和基因组的检测(全面的基因组测序)。
目标基因检测要求临床医生根据表型数据决定可能牵涉到什么基因, 而全面的基因组的检测则不要求。因为造成遗传性听力损失与耳聋许多原因表型有交叉,多数遗传性听力损失与耳聋患者凭以下两种方法之一来确诊: 全面基因组测序(建议) 或目标基因检测(考虑)。
建议检测
建议最先进行全面耳聋基因包检测包括所有已知非综合征型耳聋基因和综合征型类非综合征型耳聋基因 (见鉴别诊断和遗传性听力损失与耳聋概论) (图1).
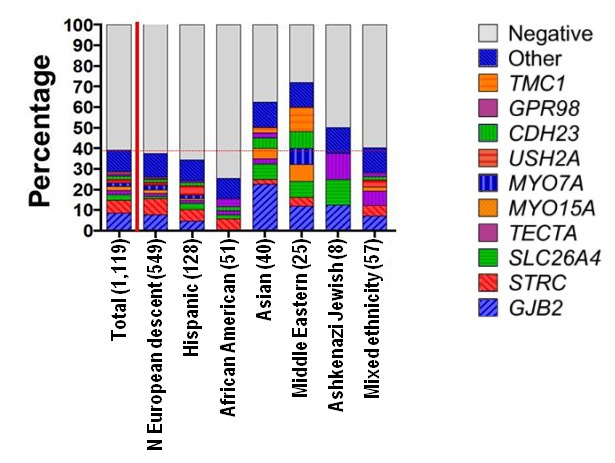
注: (1) 现有基因包包括的基因和对每个基因检测的诊断敏感性各个实验室和各时期各不相同[Shearer & Smith 2015]。(2) 一些多基因包可能包括和本GeneReview讨论病情无关的基因;所以,临床医生需要决定哪个多基因包在最经济实惠的情况下提供最佳诊断机会同时限制次要发现。(3) 基因包的方法可包括序列分析、缺失/重复分析、 和/或其他非基于测序的检测;检测拷贝数变异必须包括在听力损失基因包内[Shearer et al 2014]。关于更多基因包信息请点击这里。
考虑检测
如果无法进行听力损失基因包检测,可考虑单基因检测。但除非仅限于对先天的重度到极重度非综合征型听力损失患者,只进行GJB2序列分析并不经济有效。给每个先天听力损失患者无论严重程度都先做GJB2单基因检测,如果未检出再测其他基因的做法并非基于证据也不经济有效[Jayawardena et al 2015, Shearer & Smith 2015].
如果光凭表型不足以确定目标基因检测,可考虑包括外显子组测序和基因组测序的全面基因组的测序。外显子组测序和基因组测序均须有合适的检测前和检测后遗传咨询相伴。关于更多全面基因组检测信息请点击这里。
表 1.
用于诊断DFNB1的分子遗传检测
2.
3.
比例不同,取决于种族背景。表中数据反映主要为祖籍北欧的美国人群筛查结果。
4.
测序分析检测变异分为良性、疑似良性、意义不确定、疑似致病、或致病。致病变异可能包括基因内部小的缺失/插入和错义、无义、及剪接位点变异; 通常, 外显子或全基因缺失/重复不能被检测。关于解读序列分析结果考虑事项, 点击这里。
5.
目标基因缺失/重复分析能发现基因内缺失或重复。可用方法包括: 定量PCR、 长距离PCR、多重连接探针扩增(MLPA)、和专门设计检测目标基因单个外显子缺失或重复的芯片。
6.
目标基因缺失/重复检测将检出从单个外显子到全基因; 但这些方法可能无法检出大片断缺失和/或邻近多基因缺失 (如Feldmann et al [2009]所描述) 的断点。
临床特征
临床描述
非综合征型听力损失与耳聋(DFNB1) 以先天的(出生就有)非进行性感音神经性听力受损为特征。家系成员间耳聋程度差异不足为奇。
- 有报道有GJB2变异的儿童通过新生儿听力筛查但表现为迟发性听力损失[Norris et al 2006, Orzan & Murgia 2007].
在一项大规模对来自16个国家1531位常染色体隐性遗传轻度到中度非综合征型耳聋(年龄中位数8岁; 90%在0到26岁之间)患者GJB2基因型和听力测试数据进行的横断面分析,在所有研究对象和在部分特定基因型的研究对象中进行听力阈值对年龄的线性回归分析没有显示任何人听力损失显著加剧[Snoeckx et al 2005]。该发现和以前的研究相符[Orzan et al 1999, Löffler et al 2001]; 但是,鉴于回归横断面分析的本质,不能完全排除进行性听力损失。
尽管Snoeckx et al [2005]发现稍有不对称,在90%的患者中两耳0.5, 1.0, and 2.0 kHz纯音平均听力差别不超过15 dB。
前庭平衡功能正常; 受累的婴儿和幼童没有平衡问题,适龄时间学坐和学走。
除了听力受损,受累的患者健康;寿命正常。
基因型-表型相关性
大量研究显示可能基于基因型来预测表型。至今最大的研究是对来自16个国家1531位常染色体隐性遗传轻度到中度非综合征型耳聋患者的GJB2基因型和听力测试数据的横断面分析[Snoeckx et al 2005]。在找到的83个不同的变异中,47个预测为非截短(即错义变异)而36个预测为截短(即过早终止密码)。如此分类, 作者们定义了三种基因型类型: 双等位基因的截短(T/T)变异,双等位基因的非截短(NT/NT)变异, 和复合杂合截短/非截短(T/NT)变异(表2)。
表2.
| GJB2基因型类别 | 占所有DFNB1比例 | 听力损失 | |||
|---|---|---|---|---|---|
| 轻度 | 中度 | 重度 | 极重度 | ||
| T/T | 77.3% 1 | 0%-3% | 10%-12% | 25%-28% | 59%-64% |
| NT/NT | 6.2% 2 | 53% | 26% | 8% | 13% |
| T/NT | 16.5% 3 | 29%-37% | 24%-29% | 10%-17% | 24%-30% |
流行率
在美国、法国、英国、新西兰/澳大利亚,DFNB1 大约占先天的重度到极重度常染色体隐性遗传非综合征型听力损失与耳聋的50%[Angeli 2008, Azaiez et al 2004, Green et al 1999]. 基于以下计算,普通人群大致发病率为十万分之14: 先天的遗传性听力受损发病率 1:2000 新生儿, 其中70% 为非综合征型听力损失。75%至80%非综合征型听力损失病例为常染色体隐性; 其中, 50% 是由于GJB2双等位基因的致病变异。因此, 万分之五 x 0.7 x 0.8 x 0.5 = 十万分之14:
- 25%的巴勒斯坦家系中 [Shahin et al 2002]
- 估计24%的西伯利亚阿尔泰人中[Posukh et al 2005]
- 18%的汉族中国人[Duan et al 2015]
- 16%的伊朗聋人中[Bazazzadegan et al 2012]
- 15%的回族人中[Duan et al 2015]
- 11%的西藏家系中 [Duan et al 2015]
相关遗传疾病
等位基因相关其他疾病.下列常染色体显性遗传疾病由于GJB2杂合的致病变异引起:
- 角膜炎-鱼鳞病-耳聋(Keratitis-ichthyosis-deafness, KID)综合征(OMIM 148210)是一种外胚层发育不良疾病,受累的人患血管化角膜炎、进行性红斑角化病、极重度感音神经性耳聋、以及疤痕性脱发和易患鳞状细胞癌[Richard et al 2002, van Geel et al 2002, van Steensel et al 2002].
- 类豪猪鱼鳞病-耳聋(Hystrix-like ichthyosis-deafness, HID)综合征(OMIM 602540)是一种以感音神经性耳聋和皮肤角化过度为特征的角化病。出生不久到一岁即出现红皮病,整个皮肤表明出现刺状和鹅卵石突起的角化过度。有些病人出现严重掌跖角化和疤痕性脱发。HID综合征被认为和KID综合征的区别在于: (1) 皮肤症状涉及范围和发生时间; (2)角膜炎严重程度; 和(3) 电镜特征[van Geel et al 2002].
- Vohwinkel综合征(OMIM 124500)是一种"残害性"弥漫性角化病,因为周边角化过度导致手指脚趾自切断。轻度到中度感音神经性耳聋经常和该病相关联[Maestrini et al 1999].
其他.在一个以极重度语前耳聋、智力和身心发育迟缓、二趾弯曲、额顶绺为特征的邻近基因综合征患者身上检出与GJB2致病性变异在不同染色体上(反式)的包括GJB2, 相邻基因GJA3和GJB6, 及部分CRYL1的大片断缺失[Feldmann et al 2009].
鉴别诊断
见耳聋和遗传性听力损失概论 了解关于导致非综合征型听力损失和耳聋基因的信息。
表3.
涉及听力损失的常染色体隐性综合征
综合征 | 除听力损失外其他独特征状 | 听力损失 | 备注 | |
|---|---|---|---|---|
| Usher综合征I型 | 视网膜色素变性【注1】 | 见【注2】 | 先天的双侧极重度感音神经性听力损失 |
|
| Usher 综合征II型 | USH2A ADGRV1 DFNB31 | 先天的双侧感音神经性听力损失: 低频轻度到重度; 高频重度到极重度 |
| |
| Usher 综合征III型 | CLRN1 | 语后进行性感音神经性听力损失 |
| |
| Pendred综合征 | 甲状腺肥大 | SLC26A4 【注3】 | 听障通常为先天的 且经常为重到极重度(轻度到中度进行性听障也有) |
|
| Jervell和Lange-Nielsen综合征 | 心脏传导缺陷;长 QTc, 通常 >500毫秒 | KCNQ1 KCNE1 | 先天的极重度双侧感音神经性听力损失 |
|
视网膜色素变性是一种进行性双侧对称的视网膜杆细胞和锥细胞功能退化。
2.
至少在九个不同基因座里的致病变异导致Usher综合征I型。六个基因座里的基因–MYO7A (USH1B), USH1C, CDH23 (USH1D), PCDH15 (USH1F), USH1G, 和CIB2 (USH1J)–已找到。
3.
4.
亦称作增大前庭导水管(enlarged vestibular aqueduct,EVA)
5.
DVA伴有耳蜗发育不良称为Mondini畸形或发育不良。
6.
甲状腺肿大一般不在出生时表现,但在青春期(40%)或成年(60%)发生。
7.
治疗涉及用beta-肾上腺素阻断剂、心脏起搏器、和植入式除颤器, 以及避免导致加长QT间隔的药物和已知促使休克的活动。
常染色体隐性非综合征型听力损失没有找到GJB2变异和进行型听力损失:
- 颞骨薄切计算机辅助断层成像(CT)显示扩大的前庭导水管提示DFNB4 [Azaiez et al 2007];
其他导致先天的重度到极重度听力损失在单例聋儿家庭考虑:
- 先天性巨细胞病毒(cytomegalovirus, CMV), 最常见的非遗传性先天的听力损失的原因
- 早产、出生体重轻、Apgar新生儿评分低、感染、和任何需要到新生儿重症监护病房治疗的疾病
管理
初诊后随访评估
为确定确诊由GJB2 (DFNB1)双等位基因的致病变异引起的非综合征型听力损失与耳聋患者的波及程度和需求, 建议进行下列评估:
- 用适合年龄的测试,比如ABR测试、听觉稳态反应(Auditory Steady-State Response, ASSR)、纯音测听等,全面评定听力
- 眼科评估屈光不正
- 咨询遗传专科医生和/或遗传咨询师
对症治疗
指征如下:
- 配合适的助听器
- 参加合适的专为听障者的教育计划
- 考虑人造耳蜗移植(cochlear implantation,CI),给极重度耳聋患者的优良康复选择
另见遗传性听力损失与耳聋以了解更多管理细节的讨论。
监控
以下合适:
- 由熟悉遗传性听障的医生年检
- 重复测听确认听力稳定
要避免的事物/情况
听力损失患者须避免暴露于已知致聋的环境。对轻度到中度听力损失的DFNB1患者而言最重要的是避免反复过度暴露于很响的噪声。
遗传咨询
遗传咨询是向患者及其家庭提供遗传病的本质、遗传方式及其可能造成的影响等方面信息的过程,从而帮助他们做出知情的医疗和个人决策 。 以下诸节涉及遗传风险评估和利用家族史和基因检测来确定家庭成员的基因状态。这一章节并不能解决患者所有可能面临的个人、文化或伦理的问题,或取代向遗传专业人士咨询—编者按。
遗传咨询相关事宜
见管理, 有风险亲属评估以了解有关为早期诊断和治疗而对有风险亲属进行评估的信息
下列几点值得一提:
- 和打手语的聋文化团体成员交流需要技术高的翻译服务。
- 聋文化团体成员可能视耳聋为一种特征而非残疾、障碍、或需要“治疗”、“治愈”或“预防”的疾病。
- 许多聋人对获得他们自身耳聋原因,包括医疗、教育、社会服务等信息而不是关于预防、生殖、和计划生育的信息感兴趣。
- 一些措词首选: 概率或几率而不是风险;聋或听障而不是听力受损。诸如“异常”等措词当避免。
生育规划
库存DNA指为将来可用而储存DNA(一般从白细胞里提取)。因为很可能检测方法和我们对基因和变异的认知将来会有所改善,需要考虑库存受累的患者的DNA。
产前检测和植入前遗传诊断
一旦在DFNB1家系一员检出GJB2致病变异,就可能对有风险怀孕进行DFNB1的产前检测和植入前遗传诊断。
许多聋人对获得关于解释耳聋原因的信息比对生育风险信息更感兴趣。所以确定并解释家庭/患者的疑问和顾虑至关重要。"相对认为耳聋是病理现象的医学模式,许多聋人不认为他们有残疾而是认为他们自己是一独特文化群体,有自己的语言习俗信仰。对聋人有效的遗传咨询策略包括认识到感知风险的主观性和某些聋人可能更愿意生育聋儿。" — 引资Arnos et al [1991]
医学专业人士和不同家庭成员对产前检测可能有不同的看法,特别是为终止妊娠而不是为早期诊断而作的 。虽然大部分医疗中心认为产前检测的决定是父母的选择,和患者讨论这些事宜是合适的。
资源
为了使该病患者本人及其家庭受益,GeneReviews工作人员选择了以下疾病专司和/或联合支持机构和/或患者注册登记。GeneReviews对其他机构提供的信息概不负责。了解关于信息选择的标准,点击这里。
- 亚历山大•格雷厄姆•贝尔耳聋听障协会3417 Volta Place NorthwestWashington DC 20007电话: 866-337-5220 (toll-free); 202-337-5220; 202-337-5221 (TTY)传真: 202-337-8314电邮: info@agbell.org
- 美国聋儿协会(American Society for Deaf Children, ASDC)800 Florida Avenue NortheastSuite 2047Washington DC 20002-3695电话: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)传真: 410-795-0965电邮: info@deafchildren.org; asdc@deafchildren.org
- 宝宝的听力该网页由美国国家耳聋和其他交流疾病研究所(National Institute on Deafness and Other Communication Disorders)支持和开发, 提供新生儿听力筛查和听力损失的相关信息。
- 美国国家聋人协会 (National Association of the Deaf, NAD)8630 Fenton StreetSuite 820Silver Spring MD 20910电话: 301-587-1788; 301-587-1789 (TTY)传真: 301-587-1791电邮: nad.info@nad.org
- 美国国家图书馆医学遗传参考主页
分子遗传
分子遗传信息和OMIM 表格可能不同于GeneReview其他地方的描述: 表格可能包括更新的信息。-编者按
表A.
非综合征型听力损失与耳聋DFNB1: 基因和数据库
| 基因座名 | 基因 | 染色体位置 | 蛋白 | 基因座专门数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|---|
| DFNB1 | GJB2 | 13q12-.11 | 间隙连接beta-2蛋白 | 连接蛋白-耳聋主页(GJB2) 遗传性听力损失主页 (GJB2) 辛辛那提儿童医院医学中心-人类遗传突变数据库(GJB2) | GJB2 | GJB2 |
表B.
OMIM非综合征型听力损失与耳聋条目,DFNB1 (见OMIM全文)
引言
间隙连接通道能让离子和不超过大约1.2 kd的相对小分子量代谢产物通过[Harris & Bevans 2001]。不同间隙连接通道之间离子选择性和开关机制的差异反映了人体内多于20种不同连接蛋白异型体的存在。大多连接蛋白有共同的基因构建,包含整个编码区的一个大外显子和另一个非编码外显子被不同大小的内含子隔开。GJB2编码连接蛋白26。
基因结构.GJB2编码序列(2号外显子)编码226-氨基酸的蛋白。关于基因和蛋白的详细概论,见表A, 基因。
致病变异.见表4。 导致常染色体隐性遗传非综合征型听力损失的大量GJB2致病变异列在连接蛋白-耳聋主页.
常见GJB2致病变异:
- c.35delG
- 北欧后裔携带者频率2%-4%[Snoeckx et al 2005]
- 也在阿拉伯人、贝都因人、印度人、和巴基斯坦人后裔中被报道过
- p.Val37Ile, 泰国人中最常见(携带者频率: 11.6%) [Hwa et al 2003]; 和轻度听力损失相关联[Snoeckx et al 2005, Abe et al 2000, Wilcox et al 2000, Kenna et al 2001, Lin et al 2001, Marlin et al 2001]
- p.Met34Thr, 等位基因频率1%-2%和轻度听力损失相关联[Snoeckx et al 2005]
下列三个包括包含GJB2上游序列和GJB6一部分的大片段缺失导致降 低GJB2表达, 想必是由于缺失同染色体上的cis-调控元件。
- ~309-kb 缺失 (称作∆GJB6-D13S1830)* 在西班牙人中常见 (占GJB2序列分析未检出原因的西班牙聋人里DFNB1等位基因的67%) [Wu et al 2002, Stevenson et al 2003, Del Castillo et al 2003]
- ~232-kb 缺失 (称作∆GJB6-D13S1854)* 在西班牙人中也相对常见 (占GJB2序列分析或309-kb缺失筛查未检出原因的西班牙聋人里DFNB1等位基因的25%) [del Castillo et al 2005]
*注: 这两个缺失也在祖籍欧洲的其他人群中以较低频率被检出[Wu et al 2002, Del Castillo et al 2003, Stevenson et al 2003].
表4.
列选GJB2致病变异
| DNA碱基变化 (别名【注1】) | 预测蛋白变化 | 参考序列 |
|---|---|---|
| c.35delG | p.Gly12ValfsTer1 | NM_004004-.4 NP_003995-.2 |
| c.35G>T | p.Gly12Val | |
| c.-3179G>A 2 (IVS1+1G>A) | -- | |
| c.56G>C | p.Ser19Thr | |
| c.101T>C | p.Met34Thr 3 | |
| c.109G>A | p.Val37Ile 3 | |
| c.167delT | p.Leu56ArgfsTer26 | |
| c.235delC | p.Leu79CysfsTer3 | |
| c.231G>A | p.Trp77Arg | |
| c.269T>C | p.Leu90Pro | |
| c.339T>G | p.Ser113Arg | |
| c.358_360delGAG | p.Glu120del | |
| c.427C>T | p.Arg143Trp | |
| c.487A>G | p.Met163Val | |
| c.551G>C | p.Arg184Pro | |
| ∆GJB6-D13S1830 | ||
| ∆GJB6-D13S1854 | ||
| del(chr13:19,837,344-19,968,698) |
变异分类备注: 表中所列变异为作者所提供。GeneReviews工作人员尚未独立核实变异分类。
变异命名备注: GeneReviews按人类基因组变异协会(Human Genome Variation Society,www-.hgvs.org)的标准命名约定。命名解释见快捷参考 。
1.
变异命名未遵循当前规范
2.
IVS1+1G>A在基因组的序列(参考序列NC_000013-.9)2号外显子起始上游-3179碱基处。
3.
p.Met34Thr and p.Val37Ile与正常或轻度听力损失相关联。见致病变异讨论。
正常基因产物.GJB2编码连接蛋白26, 即beta-2间隙连接蛋白,由226 氨基酸构成。六个连接蛋白聚合成大约中央2.3-nm的孔形成连接子。相邻细胞的连接子通过共价键形成细胞间通道。大量连接子聚集称作斑,是间隙连接的组分。间隙连接允许细胞间直接通过其中央水相孔洞交换离子和分子,从而使可电兴奋组织的活性同步,以及使不可电兴奋组织交换代谢产物和信号分子。连接蛋白26与自身、连接蛋白32、连接蛋白46、及连接蛋白50组合形成有功能的间隙连接通道。
异常基因产物.GJB2致病变异导致下列之一:
- 失去连接蛋白26 的功能(p.Gly12Val, p.Ser19Thr, c.35delG, p.Leu90Pro)
- 无法形成同型间隙连接通道(p.Val37Ile, p.Trp77Arg, p.Ser113Arg, p.Glu120del, p.Met163Val, p.Arg184Pro, and c.235delC)
- 干扰蛋白翻译(p.Arg184Pro) [Snoeckx et al 2005]
参考资料
发表的指南/共识声明
- American College of Medical Genetics. Statement on universal newborn hearing screening. Available online. 2000. Accessed 11-14-17.
- Genetics Evaluation of Congenital Hearing Loss Expert Panel, American College of Medical Genetics. Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Available online. 2002. Accessed 11-14-17.
引用文献
- Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:41-3. [PMC free article: PMC1734448] [PubMed: 10633133]
- Arnos KS, Israel J, Cunningham M. Genetic counseling of the deaf. Medical and cultural considerations. Ann N Y Acad Sci. 1991;630:212-22. [PubMed: 1952592]
- Angeli SI. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope. 2008;118:2014-23. [PubMed: 18758381]
- Azaiez H, Chamberlin GP, Fischer SM, Welp CL, Prasad SD, Taggart RT, del Castillo I, Van Camp G, Smith RJ. GJB2: the spectrum of deafness-causing allele variants and their phenotype. Hum Mutat. 2004;24:305-11. [PubMed: 15365987]
- Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122:451-7. [PubMed: 17690912]
- Bazazzadegan N, Nikzat N, Fattahi Z, Nishimura C, Meyer N, Sahraian S, Jamali P, Babanejad M, Kashef A, Yazdan H, Sabbagh Kermani F, Taghdiri M, Azadeh B, Mojahedi F, Khoshaeen A, Habibi H, Reyhanifar F, Nouri N, Smith RJ, Kahrizi K, Najmabadi H. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss--a twelve year study. Int J Pediatr Otorhinolaryngol. 2012;76:1164-74. [PubMed: 22695344]
- del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martin Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl HH, Kanaan M, Nance WE, Petit C, Smith RJ, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005;42:588-94. [PMC free article: PMC1736094] [PubMed: 15994881]
- Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, Cockburn DJ, Pandya A, Siemering KR, Chamberlin GP, Ballana E, Wuyts W, Maciel-Guerra AT, Alvarez A, Villamar M, Shohat M, Abeliovich D, Dahl HH, Estivill X, Gasparini P, Hutchin T, Nance WE, Sartorato EL, Smith RJ, Van Camp G, Avraham KB, Petit C, Moreno F. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73:1452-8. [PMC free article: PMC1180408] [PubMed: 14571368]
- Duan SH, Zhu YM, Wang YL, Guo YF. Common molecular etiology of nonsyndromic hearing loss in 484 patients of 3 ethnicities in northwest China. Acta Otolaryngol. 2015;135:586-91. [PubMed: 25761933]
- Feldmann D, Le Maréchal C, Jonard L, Thierry P, Czajka C, Couderc R, Ferec C, Denoyelle F, Marlin S, Fellmann F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur J Med Genet. 2009;52:195-200. [PubMed: 19101659]
- Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211-6. [PubMed: 10376574]
- Harris AL, Bevans CG. Exploring hemichannel permeability in vitro. Methods Mol Biol. 2001;154:357-77. [PubMed: 11218659]
- Heathcote K, Syrris P, Carter ND, Patton MA. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J Med Genet. 2000;37:50-1. [PMC free article: PMC1734451] [PubMed: 10633135]
- Hwa HL, Ko TM, Hsu CJ, Huang CH, Chiang YL, Oong JL, Chen CC, Hsu CK. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet Med. 2003;5:161-5. [PubMed: 12792423]
- Jayawardena AD, Shearer AE, Smith RJH. Sensorineural hearing loss – a changing paradigm for its evaluation. Otolaryngol Head Neck Surg. 2015;153:843-50. [PMC free article: PMC4730883] [PubMed: 26216887]
- Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patients with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 2001;127:1037-42. [PubMed: 11556849]
- Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, Narisawa K, Takasaka T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet. 2000;90:141-5. [PubMed: 10607953]
- Kutkowska-Kaźmierczak A, Niepokój K, Wertheim-Tysarowska K, Giza A, Mordasewicz-Goliszewska M, Bal J, Obersztyn E. Phenotypic variability in gap junction syndromic skin disorders: experience from KID and Clouston syndromes' clinical diagnostics. J Appl Genet. 2015;56:329-37. [PMC free article: PMC4543413] [PubMed: 25575739]
- Lin D, Goldstein JA, Mhatre AN, Lustig LR, Pfister M, Lalwani AK. Assessment of denaturing high-performance liquid chromatography (DHPLC) in screening for mutations in connexin 26 (GJB2). Hum Mutat. 2001;18:42-51. [PubMed: 11438992]
- Löffler J, Nekahm D, Hirst-Stadlmann A, Gunther B, Menzel HJ, Utermann G, Janecke AR. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur J Hum Genet. 2001;9:226-30. [PubMed: 11313763]
- Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237-43. [PubMed: 10369869]
- Marlin S, Garabedian EN, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg. 2001;127:927-33. [PubMed: 11493200]
- Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H, Keats B, Friedman TB. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500-5. [PubMed: 9819448]
- Norris VW, Arnos KS, Hanks WD, Xia X, Nance WE, Pandya A. Does universal newborn hearing screening identify all children with GJB2 (Connexin 26) deafness? Ear Hear. 2006;27:732-41. [PubMed: 17086082]
- Orzan E, Murgia A. Connexin 26 deafness is not always congenital. Int J Pediatr Otorhinolaryngol. 2007;71:501-7. [PubMed: 17222463]
- Orzan E, Polli R, Martella M, Vinanzi C, Leonardi M, Murgia A. Molecular genetics applied to clinical practice: the Cx26 hearing impairment. Br J Audiol. 1999;33:291-5. [PubMed: 10890143]
- Posukh O, Pallares-Ruiz N, Tadinova V, Osipova L, Claustres M, Roux AF. First molecular screening of deafness in the Altai Republic population. BMC Med Genet. 2005;6:12. [PMC free article: PMC1079841] [PubMed: 15790391]
- Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341-8. [PMC free article: PMC447609] [PubMed: 11912510]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393-9. [PubMed: 9856479]
- Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, Kanaan M. Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet. 2002;110:284-9. [PubMed: 11935342]
- Shearer AE, Kolbe DL, Azaiez H, Sloan CM, Frees KL, Weaver AE, Clark ET, Nishimura CJ, Black-Ziegelbein EA, Smith RJ. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6:37. [PMC free article: PMC4067994] [PubMed: 24963352]
- Shearer AE, Smith RJ. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg. 2015;153:175-82. [PMC free article: PMC4743024] [PubMed: 26084827]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441-50. [PMC free article: PMC4796320] [PubMed: 26969326]
- Smith RJ, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879-90. [PubMed: 15752533]
- Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, Pollak A, Ploski R, Murgia A, Orzan E, Castorina P, Ambrosetti U, Nowakowska-Szyrwinska E, Bal J, Wiszniewski W, Janecke AR, Nekahm-Heis D, Seeman P, Bendova O, Kenna MA, Frangulov A, Rehm HL, Tekin M, Incesulu A, Dahl HH, du Sart D, Jenkins L, Lucas D, Bitner-Glindzicz M, Avraham KB, Brownstein Z, Del Castillo I, Moreno F, Blin N, Pfister M, Sziklai I, Toth T, Kelley PM, Cohn ES, Van Maldergem L, Hilbert P, Roux AF, Mondain M, Hoefsloot LH, Cremers CW, Lopponen T, Lopponen H, Parving A, Gronskov K, Schrijver I, Roberson J, Gualandi F, Martini A, Lina-Granade G, Pallares-Ruiz N, Correia C, Fialho G, Cryns K, Hilgert N, Van de Heyning P, Nishimura CJ, Smith RJ, Van Camp G. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am J Hum Genet. 2005;77:945-57. [PMC free article: PMC1285178] [PubMed: 16380907]
- Stevenson VA, Ito M, Milunsky JM. Connexin-30 deletion analysis in connexin-26 heterozygotes. Genet Test. 2003;7:151-4. [PubMed: 12885339]
- Tennessee Department of Health. Pediatric Audiology Guidelines – Tennessee Genetic Centers Referral Pattern for Hearing Loss. Appendix 4. Available online. 2005. Accessed 11-14-17.
- van Geel M, van Steensel MA, Kuster W, Hennies HC, Happle R, Steijlen PM, Konig A. HID and KID syndromes are associated with the same connexin 26 mutation. Br J Dermatol. 2002;146:938-42. [PubMed: 12072059]
- van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis- ichthyosis-deafness syndrome. J Invest Dermatol. 2002;118:724-7. [PubMed: 11918723]
- Wilch E, Azaiez H, Fisher RA, Elfenbein J, Murgia A, Birkenhäger R, Bolz H, Da Silva-Costa SM, Del Castillo I, Haaf T, Hoefsloot L, Kremer H, Kubisch C, Le Marechal C, Pandya A, Sartorato EL, Schneider E, Van Camp G, Wuyts W, Smith RJ, Friderici KH. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin Genet. 2010;78:267-74. [PMC free article: PMC2919588] [PubMed: 20236118]
- Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, Collins V, Wilcox LJ, McKinlay Gardner RJ, Kamarinos M, Cone-Wesson B, Williamson R, Dahl HH. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399-405. [PubMed: 10830906]
- Wu BL, Lindeman N, Lip V, Adams A, Amato RS, Cox G, Irons M, Kenna M, Korf B, Raisen J, Platt O. Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet Med. 2002;4:279-88. [PubMed: 12172394]
提议阅读
- Petit C, Levilliers J, Marlin S, Hardelin J. Hereditary hearing loss. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 254. New York, NY: McGraw-Hill. Available online.
章节备注
鸣谢
由RO1-DC02842 from the NIDCD (RJHS)基金资助
既往作者
Mary-Kayt N Jones, BA (2016-present)
Daryl A Scott, MD, PhD; University of Iowa (1998-2001)
Val C Sheffield, MD, PhD; University of Iowa (1998-2001)
Richard JH Smith, MD (1998-present)
Guy Van Camp, PhD; University of Antwerp (1998-2016)
改版历史
- 18 August 2016 (bp) Comprehensive update posted live
- 2 January 2014 (me) Comprehensive update posted live
- 14 July 2011 (me) Comprehensive update posted live
- 11 July 2008 (me) Comprehensive update posted to live Web site
- 21 December 2005 (me) Comprehensive update posted to live Web site
- 14 March 2005 (rjs) Revision: information on GJB6 deletions
- 15 July 2004 (rjs) Revision: use of an interpreter
- 27 October 2003 (me) Comprehensive update posted to live Web site
- 24 April 2001 (me) Comprehensive update posted to live Web site
- 28 September 1998 (pb) Review posted to live Web site
- 4 April 1998 (rjs) Original submission