
概要
临床表现.
在成人中,X连锁迟发性脊椎骨骺发育不良(X连锁SEDT)的特征在于具有显著短躯干和臂展长度长于身高的不成比例的身材矮小。出生时,患病男性的高度正常且有正常的身体比例。患病男性从大约六至八岁开始出现身高增高迟缓。成年的最终身高通常为137-163厘米。伴随骨关节炎的进展致关节和背部疼痛。髋关节、膝关节和肩关节通常受累但程度可不同。髋关节置换术通常早在40岁就被需要。指间关节通常是幸免的。运动和认知能力正常。
诊断:
诊断X连锁SEDT,这依赖于临床和影像学特征的联合诊断,通常在童年可能多见。青少年和成年男性躯干相对短,呈现不成比例的身材矮小和桶状胸。上下身比例通常为0.8左右。臂展长度通常超过身高的10-20厘米。通常在青春期之前出现的特征性影像学检查结果包括:多发性骨骺异常,扁平椎(椎体平坦扁平)侧视上下椎体呈驼峰状,成年后椎间盘间隙狭窄,脊柱侧弯,齿状突异常,股骨颈短,髋内翻,成年初期就有早发性关节炎的迹象。TRAPPC2(SEDL)是已知的唯一导致X连锁SEDT的基因。分子遗传学检测在临床诊断为X连锁SEDT男性患者中揭示了超过80%TRAPPC2的突变。
疾病管理:
对症治疗:手术治疗包括关节置换术(髋,膝,肩)或脊柱外科(脊柱侧弯或后凸畸形)。经常需要在外科手术之前或之后进行标准化慢性疼痛管理。
监测:每年对关节疼痛和脊柱侧弯评估进行随访,学龄前颈椎影像学检查,任何涉及全身麻醉外科手术要先评估临床上十分重要的齿状突异常情况。
需避免的情况:齿状突发育不全的个体应当避免过度颈部屈伸。对脊柱和承重关节施加过度压力的活动和职业也应当避免。
亲属风险评估:风险的男性的症状前检测可避免对身材矮小和/或骨关节炎等原因造成不必要的诊断测试。
遗传咨询:
X连锁SEDT是在X连锁方式遗传的。无论是否有家族史,对生育有患病儿子的母亲进行分子遗传学检测都能发现他们是TRAPPC2致病突变的携带者。女性携带者在每一次受孕过程中均有50%的几率将致病突变传递下去,若后代是男性则成为患者,若后代是女性则成为不患病的携带者。患病男性的儿子都不会受到影响(不会患病),所有的女儿都将是TRAPPC2致病突变的携带者。如果确定了家系中的致病性突变,那么对高风险女性亲属进行致病突变的携带者检测和高危胎儿产前检测是可行的。
诊断
疑似诊断
当在男性中发现以下异常应怀疑为X连锁迟发性脊椎骨骺发育不良(X连锁SEDT):
- 在青春期或成年期不成比例的身材矮小,相对短的躯干和桶状胸。上下身体比例通常为0.8左右。臂展通常超过身高的10-20厘米。颈短,驼背和腰椎前凸过度可能在青春期表现较为明显。
- 早发性骨关节炎,尤其在髋关节。
- 家族史(家系图)一致指向X连锁隐性遗传病。阳性家族史有一定的作用,但不是必需的。
- 腭裂和视网膜脱落的情况下(通常存在于先天性SED,见鉴别诊断)
确切诊断
诊断X连锁SEDT应建立在先证者具有特征X线检查结果或X线检查仍未有定论但分子遗传学检测已证实(表1)。
影像学检查
下面的影像学表现可能不会在患病男孩的童年早期表现,但通常出现在青春期前(图1):
{kind=link}
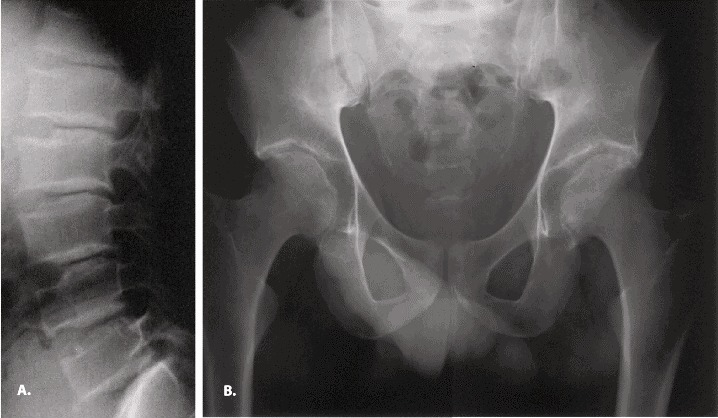
31岁男性SEDT患者的射线照片。椎体上下呈现驼峰样的扁平椎,两髋关节严重退行性改变。
- 多发性骨骺异常
- 扁平椎(椎体平坦扁平)侧视上下椎体呈驼峰状,成年后椎间盘间隙狭窄
- 脊柱侧弯
- 齿状突异常
- 股骨颈短
- 髋内翻
- 成年初期有早发性关节炎的迹象
- 有症状男性的影像学检查应由有骨骼结构异常放射检查经验的放射科医师进行检查。
- 单个基因检测首先包括TRAPPC2(以前称为SEDL)的测序分析,如果没有发现致病性突变再进行缺失/重复分析。
- 多个基因的芯片检测,其包括TRAPPC2和其他可以被考虑的感兴趣(有关)的基因(见鉴别诊断)。如果专家放射学解读不可用,此方法可能特别有用。注:基因检测需要考虑来自不同实验室的多个基因的芯片检测和随时间变化的灵敏度。
表1
X连锁SEDT的分子遗传学检测
| 基因 1 | 检测方法 | 部分先证者 通过以下方法检测致病性突变 2 | |
|---|---|---|---|
| 男性患者 | 女性携带者 | ||
| TRAPPC2 | 测序分析 3 | 80% 4 | 80% 5 |
|
基因靶向的缺失/重复分析 6 |
10% 7 | 10% | |
| Unknown 8 | NA | 10% | 10% |
1.染色体定位和蛋白质信息请参阅表A.基因和数据库。
2.有关该等位基因突变检测的信息见分子遗传学。
3.序列分析检测到的突变结果有意义不明确的(良性的,可能是良性的)或者致病的。致病性突变可能包括基因内的微缺失/插入、无义突变、剪接位点突变。而整个外显子或整个基因的缺失或重复没有被检测到过。对于关于序列分析问题的解释,请点击这里。
4.患病男性样品在序列分析之前的PCR扩增失败/缺失可能可以提示是一个假(多重)外显子或整个在X染色体上的基因缺失。需要通过额外的缺失/重复测试来进一步确认。
5.在女性杂合子的基因组DNA序列分析中并不能检测到一个或多个外显子或整个X连锁基因的缺失。
6.鉴定外显子或整个基因的缺失/重复不能通过对编码区和侧翼内含子区域的基因组DNA的序列分析来完成。需要通过以下方法:定量PCR、长片段PCR、多重连接探针扩增(MLPA)、以及染色体微阵列(CMA),其中包括该基因/染色体的片段。
7.疑似诊断的男性患者序列分析中发现的缺失需要通过后续的缺失/重复检测来进一步确认。
8.到目前为止没有被发现。很难确定分子诊断结果是否为阴性,这反应了基因异质性和临床误诊的可能性。
临床特征
临床描述
男性.出生时,男性患者的长度正常并有正常的身体比例。男性患者从上小学(6-8岁)开始出现身高增长的迟缓。最终成年身高通常是137-163厘米 [Whyte et al 1999, Unger et al 2007, Jones et al 2013].
成人X连锁SEDT患者有短躯干和臂展明显高于身高的不成比例的身材矮小。
伴随骨关节炎的进行性关节和背部疼痛。髋关节,膝关节和肩关节通常不同程度受累。髋关节置换通常在40岁前就需要。指间关节通常是幸免的。
男性患者有正常的运动和认知能力。寿命和智力正常。
女性.女性携带者通常没有明显的表型变化,但较轻的骨关节炎已有报道[Whyte et al 1999].
基因型与表现型的关联
数据不足以可靠地去确定临床表现程度的与特定TRAPPC2突变的关联性。所有迄今为止的致病突变,不论其分子基础是否相同,都导致几乎相同的表型,包括 突变。
命名与术语
脊椎骨骺发育不良是描述一类在影像学检测中有异常的严重骨骼发育不良,包括假性软骨发育不全。先天性型患者在出生时就有明显的异常,而迟发型患者通常在学龄期才有典型症状。
迟发型SED通常是以X连锁隐性形式遗传,尽管也有相当少见的常染色体显性或常染色体隐性“迟发型”被报道。
流行
患病率为1/150000-1/200000 [Wynne-Davies & Gormley 1985].
TRAPPC2致病性突变在几个种族中均被发现,包括欧洲 [Gedeon et al 2001],日本[Matsui et al 2001]和中国 [Shu et al 2002],观测表明没有具体的高风险人人群或种族。
遗传相关 (等位基因) 疾病
除了GeneReview上讨论的部分没有其他表型是已知且与TRAPPC2突变相关。
鉴别诊断
X连锁迟发性脊椎骨骺发育不良(X连锁SEDT)是根据它的延迟发病和X连锁的遗传方式与其他形式的脊椎骨骺发育不良(SED)区别开来。其他形式的SED包括:
- 先天性SED(OMIM 183900), 在出生时不成比例的身材矮小通常很明显,有特征的影像学改变。患病个体通常有中线腭裂,伴视网膜脱落的 高度近视和听力损失的风险。先天性SED是SED最常见的形式。它是由COL2A1的杂合致病性突变导致的,该基因编码Ⅱ型胶原蛋白,以常染色体显性方式遗传。
- 迟发性SED,常染色体形式遗传(罕见)。常染色体显性遗传由COL2A1致病性突变导致;隐性遗传形式已经在临床上被发现,但没有找到致病因。
- 儿童进行性假性风湿关节病(OMIM208230)。一种在三岁到八岁之间发病的常染色体隐性遗传病。与X-连锁SEDT不同,关节肿胀和手部受累是这种疾病的特点。
- Morquio综合征(粘多糖贮积症Ⅳ型)的特点是轻度的多发性成骨不全、齿突发育不全、身材矮小,和角膜云翳。(见粘多糖贮积病IVA型和GLB1 -相关疾病)。Morquio综合征是由于以下两种酶之一的缺乏引起的:半乳糖胺-6-硫酸酯酶或β-半乳糖苷酶。它是一种常染色体隐性遗传病。
- 常染色体显性多发性骨骺发育不良(MED)在幼年早期就有表现,通常在运动后臀部和/或膝部疼痛。受影响的孩子抱怨长途步行很疲劳。蹒 跚步态可能存在。成年身高在正常的较低范围或轻度矮小。与躯干相比,四肢相对较短。有进行性的疼痛和关节畸形,导致早发性骨关节炎,特别是大负重关节受累。根据定义,脊柱是正常的,尽管可以观察到Schmorl体和不规则的椎体终板。五种基因的致病变种可导致显性MED:COMP, COL9A1, COL9A2, COL9A3和MATN3。
- 休门氏病(OMIM 181440)一个用于无论何种病因导致的脊柱早发性关节炎的术语。
- 脊柱外周发育不良(OMIM 271700)以常染色体显性方式遗传,手部脚部和尺骨短小。一个被报道的家系携带有COL2A1的致病性突变。
- 斯蒂克勒综合征。表型有多种,可包括:近视,白内障和视网膜脱落;传导性与神经性耳聋;面中部不发达和腭裂(单独或作为罗宾序列的一部分);和轻度脊椎骨骺发育不良和/或早发性关节炎。斯蒂克勒综合征可由COL2A1、COL11A1或COL11A2致病性突变以常染色体显性方式遗传; 而斯蒂克勒综合征在COL9A1、COL9A2或COL9A3的致病性突变则以常染色体隐性方式遗传。
管理
初步诊断后的评估
为了确立疾病范围或程度和X-连锁性脊椎骨骺发育不良(X连锁SEDT)个体诊断,以下应注意:
- 准确诊断所需的影像学检测资料用于记录在疾病呈现时的程度。
- X连锁SEDT个体需要评估有临床意义的齿状突发育不全的可能性。
- 医学遗传学咨询是合适的。
对症治疗
外科手术包括关节置换术(髋关节、膝关节,肩关节)或脊柱手术(矫正脊柱侧弯或脊柱后凸畸形)。
矫形外科手术前后的慢性疼痛管理是常规的,经常需要。
并发症的预防
在涉及全身麻醉的任何外科手术之前,应该对颈椎椎管影像学结果进行鉴定以评估有临床意义的齿状突发育不全。
监控
受影响的个体每年应接受关节疼痛和脊柱侧弯的随访。
应在学龄对颈椎椎管影像学结果进行鉴定,以评估有临床意义的齿状突发育不全。
应避免的情况
以下情况应当要避免:
- 齿状突发育不全的患者,极度的颈部屈曲和伸展
- 对脊柱和承重关节施加过度压力的活动和职业
亲属风险评估
如果家系中TRAPPC2致病性突变是已知的,高风险的男性症状前检测可早期诊断和排除其他原因造成身材矮小和/或骨关节炎从而避免不必要的检测。
在研究阶段的治疗方式
搜索ClinicalTrials.gov获取更加广泛的关于疾病和症状的临床研究信息。注意:这种疾病可能还没有临床试验。
遗传咨询
遗传咨询是向个人和家庭提供关于遗传疾病的致病实质,遗传性和基因异常情况的相关信息,以帮助他们获得充分的医疗知情和对疾病的情况做出各种个人决定过程。以下部分涉及遗传风险评估,家族史和遗传诊断的应用,以阐明家庭成员的遗传状况。本节不是为了解决个人可能面临的所有个人、文化或伦理问题,或代替与遗传学专业人士的咨询。
遗传模式
X-连锁性脊椎骨骺发育不良(X连锁SEDT)是X连锁的遗传病。
家庭成员风险
先证者父母
- 男患者的父亲既不是这种病的患者,也不会是TRAPPC2致病突变的携带者。
- 在一个家系中有超过一个以上的患者,则男患者的母亲肯定是该病的携带者。
- 被报道个体中科研实验室分子遗传学检测报告是可用的,所有的患病儿子的母亲都是TRAPPC2致病性突变的携带者,无论是否有家族史[Gedeon et al 2001]。患病儿子的母亲不是携带者的病例,迄今尚未有报道。
先证者同胞
- 同胞的发病风险取决于母亲(携带者)的情况。
- 如果先证者的母亲具有TRAPPC2致病性突变,在怀孕时传递致病突变的可能性是50%。继承致病突变的男性会成为患者;而继承致病性突变的女性将是携带者,通常不会受到影响。
- 如果先证者是散发病例(即,在一个家系只有单一个体患病),并且如果TRAPPC2致病性突变不能在其母亲的白细胞的DNA中检测到,那么该同胞的再发风险较低但由于可能存在孕妇生殖细胞嵌合体的情况而大于群体发病率。虽然没有生殖细胞嵌合体的报道,但这种情况仍然有可能。
男性先证者后代 患病男性将把自己的致病性突变传递给其所有的女儿而不会传递给其儿子。
先证者的其他亲属 先证者的姨母/小姨可能是携带者。姨母/小姨后代的风险取决于他们的性别,有可能是患者也可能会成为携带者。
携带者筛查
女性杂合子鉴定需要以下:
注:女性携带者是X连锁遗传病的杂合子,有可能在临床表现上存在轻微的与这种疾病相关的临床症状。
遗传咨询的相关问题
请参阅疾病管理。对先证者亲属进行风险评估是为了出于对高风险亲属早期诊断和治疗的目的。
家庭计划
- 确定遗传风险和讨论产前诊断有效性的最佳时间是怀孕前。
- 为年轻的患者、携带者或高风险携带者提供适当的遗传咨询(包括后代潜在患病风险的讨论和生殖选择)是合适的。
DNA银行
用于DNA(通常从白细胞中提取)的储存,以备将来使用。因为诊断方法和我们对基因,等位基因突变和疾病的理解可能会在未来得到提升,所以应该考虑对受影响个体的DNA进行储存。
产前诊断
如果TRAPPC2致病性突变已在患者家系中得到明确鉴定,对于高风险妊娠的产前诊断从临床实验室中获得TRAPPC2基因检测或定制的产前诊断都是可行的。
针对一些情况(如X连锁SEDT)产前诊断的请求要求不影响智力,并有一些可供选择的治疗是不常见的。在医疗专业人员和家庭内可能存在使用产前诊断观念方面的差异,特别是如果为了妊娠终止而不是早期诊断而进行的检查。虽然大多数中心会认为产前检测的决定是父母的选择,但是对这些问题的适当讨论是必要的。
植入前遗传学诊断 可以用于在其中一些已经明确TRAPPC2致病性突变的家系中。
资源
GeneReviews的工作人员选择了以下特定疾病和/或相关工作组织和/或为患有此障碍的个人及其家人的利益的一些注册机构。GeneReviews不对其他组织提供的信息负责。有关选择标准的信息,请单击此处 here.
-
Human Growth Foundation (HGF)997 Glen Cove AvenueSuite 5Glen Head NY 11545Phone: 800-451-6434 (toll-free)Fax: 516-671-4055Email: hgf1@hgfound.org
-
Little People of America, Inc. (LPA)250 El Camino RealSuite 201Tustin CA 92780Phone: 888-572-2001 (toll-free); 714-368-3689Fax: 714-368-3367Email: info@lpaonline.org
-
MAGIC Foundation6645 West North AvenueOak Park IL 60302Phone: 800-362-4423 (Toll-free Parent Help Line); 708-383-0808Fax: 708-383-0899Email: ContactUs@magicfoundation.org
-
International Skeletal Dysplasia RegistryUCLA615 Charles E. Young DriveSouth Room 410Los Angeles CA 90095-7358Phone: 310-825-8998Email: AZargaryan@mednet.ucla.edu
分子遗传学
分子遗传学部分和OMIM表格中的信息可能与GeneReview中的其他地方不同,表格可能包含更多更新信息-ED。
表A
X连锁迟发性脊柱骨骺发育不良:基因和数据库信息
| 基因 | 染色体定位 | 蛋白质 | 位点特异性 | HGMD |
|---|---|---|---|---|
| TRAPPC2 | Xp22 |
Trafficking protein particle complex subunit 2 | TRAPPC2 database | TRAPPC2 |
数据来自以下标准的参考文献:来自HGNC的基因信息; 染色体位点、基因座名称,临界区,互补群信息来源于OMIM; 蛋白来自UniProt。有关提供链接的数据库(Locus Specific,HGMD)的描述,请单击此处here。
表B
在OMIM中有关X连锁迟发性脊柱骨后发育不良的词目(View All in OMIM)
| 300202 | 运输蛋白粒子复合体, 亚组2; TRAPPC2 |
| 313400 | X连锁迟发性脊柱骨骺发育不良; SEDT |
基因结构. TRAPPC2(之前又称作SEDL)共有为6个外显子,转录起始点位于外显子3中。关于基因和蛋白质信息的详细概述,参见表A,基因。
致病性的等位基因突变.
导致X连锁SEDT的TRAPPC2致病性突变包括:剪接位点的变体、无义突变、缺失和少有的错义突变。实例:外显子3、4和5中的二核苷酸缺失;外显子6中的四核苷酸缺失;外显子5中的五核苷酸缺失;外显子3和4中3'端剪切位点的突变;外显子2、3、4、5和6中5'端剪切位点的突变;外显子3、4、5和6中的无义突变;错义突变(具体见表3);外显子3,6和4-6的缺失。表2总结了多见的致病性突变。
注意:外显子和多个外显子的缺失(详见HGMD,表A)没有在杂合子女性的测序分析中检测到(见表1)。
表2
TRAPPC2常见的致病性突变
| 在所有患病个体中所占比例(%) | 致病性突变位点 1 |
|---|---|
| ~18% | c.93+5G>A |
| ~5% | c.157_158delAT |
| ~4% | c.191_192delTG |
| ~13% | c.271_275delCAAGA |
| ~9% | 其他致病性突变 |
参考文献:Gedeon et al [2001],Shaw et al [2003],Fiedler et al [2004].
1.每个致病性突变的详细信息见表3。
表3
部分选择性的等位基因突变信息
| DNA 核酸改变 (Alias 1) |
蛋白质/氨基酸改变 | 参考序列 |
|---|---|---|
| c.93+5G>A (IVS3+5G>A) |
-- | NM_001011658 NP_001011658 |
| c.139G>T | p.Asp47Tyr | |
| c.157_158delAT | p.Met53ValfsTer35 | |
| c.191_192delTG | p.Val64GlyfsTer24 | |
| c.218C>T | p.Ser73Leu | |
| c.248T>C | p.Phe83Ser | |
| c.271_275delCAAGA | p.Gln91ArgfsTer9 | |
| c.389T>A | p.Val13 |
命名与术语的注释:GeneReviews遵循Human Genome Variation Society (www.hgvs.org)中标准化的命名原则。命名与术语的解释,请参见Quick Reference。
突变分类的注释:在表格中列出的突变来源于文献。GeneReviews的工作人员没有单独核实这些突变分类。
1.不符合当前命名原则的突变名称。
正常基因产物。TRAPPC2(“Sedlin”)编码一种似乎被广泛表达的由140个氨基酸组成的蛋白质[Gedeon et al 1999, Gécz et al 2000]。Sedlin是TRAPP(运输蛋白粒子)复合物的重要组成部分,能够将原胶原三聚体(例如Ⅱ型胶原)从内质网运输到高尔基体,最终将这些蛋白质运输到细胞外基质中 [Venditti et al 2012]。
异常基因产物。几乎所有的TRAPPC2致病性突变都被预测将产生一个无效的等位基因或截短的蛋白质产物。
参考文献
Literature Cited
-
Fiedler J, Le Merrer M, Mortier G, Heuertz S, Faivre L, Brenner RE. X-linked spondyloepiphyseal dysplasia tarda: Novel and recurrent mutations in 13 European families. Hum Mutat. 2004;24:103. [PubMed: 15221797]
-
Gécz J, Hillman MA, Gedeon AK, Cox TC, Baker E, Mulley JC. Gene structure and expression study of the SEDL gene for spondyloepiphyseal dysplasia tarda. Genomics. 2000;69:242 - 51. [PubMed: 11031107]
-
Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gecz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet. 1999;22:400 - 4. [PubMed: 10431248]
-
Gedeon AK, Tiller GE, Le Merrer M, Heuertz S, Tranebjaerg L, Chitayat D, Robertson S, Glass IA, Savarirayan R, Cole WG, Rimoin DL, Kousseff BG, Ohashi H, Zabel B, Munnich A, Gecz J, Mulley JC. The molecular basis of X-linked spondyloepiphyseal dysplasia tarda. Am J Hum Genet. 2001;68:1386 - 97. [PMC free article: PMC1226125] [PubMed: 11349230]
-
Jones KL, Jones MC, del Campo M. Smith's Recognizable Patterns of Human Malformation. 7th ed. Philadelphia, PA: WB Saunders; 2013.
-
Matsui Y, Yasui N, Ozono K, Yamagata M, Kawabata H, Yoshikawa H. Loss of the SEDL gene product (Sedlin) causes X-linked spondyloepiphyseal dysplasia tarda: Identification of a molecular defect in a Japanese family. Am J Med Genet. 2001;99:328 - 30. [PubMed: 11252002]
-
Savarirayan R, Thompson E, Gecz J. Spondyloepiphyseal dysplasia tarda (SEDL, MIM #313400). Eur J Hum Genet. 2003;11:639 - 42. [PubMed: 12939648]
-
Shaw MA, Brunetti-Pierri N, Kadasi L, Kovacova V, Van Maldergem L, De Brasi D, Salerno M, Gecz J. Identification of three novel SEDL mutations, including mutation in the rare, non-canonical splice site of exon 4. Clin Genet. 2003;64:235 - 42. [PubMed: 12919139]
-
Shu SG, Tsai CR, Chi CS. Spondyloepiphyseal dysplasia tarda: report of one case. Acta Paediatr Taiwan. 2002;43:106 - 8. [PubMed: 12041616]
-
Unger S, Lachman RS, Rimoin DL: Chondrodysplasias. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, eds. Emery & Rimoin’s Principles and Practice of Medical Genetics. 5 ed. New York, NY: Churchill Livingstone; 2007:3709-53.
-
Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, Beznoussenko GV, Mironov AA, Mironov A Jr, Zelante L, Piemontese MR, Notarangelo A, Malhotra V, Vertel BM, Wilson C, De Matteis MA. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. 2012;337:1668 - 72. [PMC free article: PMC3471527] [PubMed: 23019651]
-
Whyte MP, Gottesman GS, Eddy MC, McAlister WH. X-linked recessive spondyloepiphyseal dysplasia tarda. Clinical and radiographic evolution in a 6-generation kindred and review of the literature. Medicine (Baltimore) 1999;78:9 - 25. [PubMed: 9990351]
-
Wynne-Davies R, Gormley J. The prevalence of skeletal dysplasias. An estimate of their minimum frequency and the number of patients requiring orthopaedic care. J Bone Joint Surg Br. 1985;67:133 - 7. [PubMed: 3155744]
章节注释
历史版本
- 11 June 2015 (me) Comprehensive update posted live
- 15 February 2011 (me) Comprehensive update posted live
- 5 April 2006 (me) Comprehensive update posted to live Web site
- 10 February 2004 (me) Comprehensive update posted to live Web site
- 30 December 2003 (cd) Revision: change in test availability
- 1 November 2001 (me) Review posted to live Web site
- 16 May 2001 (gt) Original submission