
概述
临床特征.
SCN1A-相关癫痫疾病包含疾病谱系范围,从轻型的简单的高热惊厥 (FS) 和全面性癫痫伴热性惊厥附加症 (GEFS+),到严重型的Dravet综合征和难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC) 。 伴难治性癫痫的表型包括Dravet综合征(也称为婴儿严重肌阵挛癫痫 [SMEI] 或婴儿期多形态肌阵挛癫痫 [PMEI]),通常与进展性的痴呆相关。较少见的表型包括肌阵挛-站立不能性发作(MAE或Doose综合征),Lennox-Gastaut综合征 (LGS),婴儿痉挛症和疫苗相关的脑病和癫痫发作。SCN1A相关癫痫疾病的 表型 在同一个家族中可能存在差异。
管理.
对症治疗: 抗癫痫药物 (AEDs) 包括苯二氮卓类(地西泮和氯硝西泮),司替戊醇(欧洲使用;目前美国FDA未批准),托吡酯和丙戊酸。氯巴占可用于治疗Lennox-Gastaut综合征的癫痫发作。苯巴比妥是有效的但耐受性差,因为它对认知有影响。采用生酮饮食降低癫痫发作频率已让一部分 受累的 的个体受益。
预防并发症: 失张力发作或肌阵挛-站立不能性发作的患者,应使用保护头盔。
监控: 对神经系统、认知和行为恶化进行系列的神经心理学评估;对新的或不同的癫痫发作类型进行EEG监测。
应避免的媒介/环境: AEDs: 卡马西平、拉莫三嗪和氨己烯酸,可能会诱发或加重肌阵挛性发作;苯妥英可能会诱发舞蹈手足徐动症。某皮些活动中,突然丧失意识可能导致受伤或死亡(如,洗澡、游泳、潜水,或高空作业/高空运动)。
孕期管理: 孕妇应接受咨询了解妊娠期使用抗癫痫药物的风险和益处,增加孕妇的叶酸补充剂至4000 µg/天的利弊,怀孕对抗癫痫代谢的影响,以及怀孕对癫痫发作的影响。
其他: 用于治疗SMEI的抗癫痫药物氯巴占和司替戊醇,未被美国FDA批准。睡眠剥夺和疾病可加剧癫痫发作。癫痫患者应了解机动车驾驶相关法律。
遗传咨询.
SCN1A-相关癫痫疾病为 常染色体显性遗传 。SCN1A相关癫痫疾病的 先证者 可能有一个遗传的或 de novo 致病性变异. de novo致病性变异引起的病例的百分比在各 表型中不同:SCN1A相关癫痫疾病的先证者存在 受累的 父亲或母亲,该百分比随着先证者表型严重程度的增加而下降;因此,大部分SCN1A-相关SMEI 和 ICE-GTC都是de novo变异导致。SCN1A相关癫痫疾病患者的每个孩子都有50%概率遗传到该致病性变异;然而,由于 外显率下降,癫痫发作的风险小于100%。若已知家族中存在致病性变异,可对高风险的孕妇进行产前诊断。
GeneReview Scope
| SCN1A-相关癫痫疾病:包含表型 1 |
|---|
|
同义词、曾用名,见 Nomenclature.
- 1.
上述表型的其他遗传因素,见 Differential Diagnosis.
诊断
SCN1A相关癫痫疾病包含从轻微到严重的表型的疾病谱系范围。由于仅依据临床症状无法确诊,必需对SCN1A基因 杂合的 致病性变异进行检测。检出SCN1A 致病性变异 对 受累的 患者的癫痫疾病的临床管理也可能有所提示 (see Treatment of Manifestations and Agents/Circumstances to Avoid).
SCN1A相关癫痫疾病的表型包括*:
- 高热惊厥 (FS), 可能有或没有提示SCN1A相关癫痫疾病的指征
- 全面性癫痫伴热性惊厥附加症 (GEFS+)
- Dravet 综合征, 也称为 难治性儿童癫痫伴全面性强直-阵挛性发作 (SMEI) 或婴儿期多形态肌阵挛癫痫 (PMEI)
注: 认为用"Dravet 综合征" 比较好,因为描述性疾病名所提示的肌阵挛性发作可能在一些症状类似的儿童中是没有发现的。 - 严重肌阵挛性癫痫,边界性 (SMEB)
- 难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC), 该类别在ILAE中未被定义,与ILAE分类系统中的迟发型Dravet综合征最为相似。
注: 该分类在SCN1A 相关文献中广泛应用,因此为考虑完整性包括在内。 - 婴儿期部分性癫痫发作伴可变病灶,也称为婴儿游走性部分性癫痫, 或 严重婴儿多灶性癫痫 Harkin et al [2007]
*注: 文献中用于描述表型的术语,有时与International League Against Epilepsy (ILAE) 定义的癫痫综合征标准术语不同.
较少见的关联表型
- 肌阵挛-站立不能性发作 (MAE, Doose 综合征), 最初在概念上定义为具有普遍的癫痫遗传倾向的群体。在ILAE分类系统中,是包括Dravet综合征、良性肌阵挛性癫痫,以及儿童期起病的癫痫伴全面性癫痫。
- Lennox-Gastaut综合征 (LGS), 相关表现有EEG慢棘波、全面性发作、以及智力障碍. Selmer et al [2009] 报道了在22个LGS患者群体中发现一名成人患者检出SCN1A 致病性变异.
- 婴儿痉挛症
- 疫苗相关性脑病和癫痫发作
SCN1A相关癫痫疾病临床拟诊复杂的原因有以下三方面:
- 表型涵盖了广泛的不同严重程度,甚至在同一个家族中也存在不同的严重程度.
- 癫痫表型不完全特异 (也就是, 在其它疾病中也能见到).
- 有些癫痫表型为家族中观察到的表型,而非某一个家族成员个体的表型。
SCN1A相关癫痫疾病特异性的家族特征包括以下:
- 一个或以上家族成员具有癫痫,尤其是发作形式大于一种
- 高热惊厥:
- 1岁以内 [Bonanni et al 2004]
- 6岁以后 [Scheffer & Berkovic 1997]
- 伴异常严重程度 (包括癫痫持续状态) [Baulac et al 1999]
- 无诱因 (如,无发热) 发作 (可能为全身性强直-阵挛,肌阵挛,肌阵挛-不稳定,或失神发作) [Scheffer & Berkovic 1997]
- 接种疫苗后发作史
- 偏侧抽搐发作
- 环境刺激诱发发作,包括热、温度变化、强光、或忙碌嘈杂的环境
注: 因为这些暗示特征可能发生在一些家庭成员而不发生在另一些成员身上,必须要有完整的家庭病史。
SCN1A相关癫痫疾病需检测到SCN1A 杂合的 致病性变异. 见 Table 1.
- 另一种方法是利用 表型靶向检测 ,对包含SCN1A 及其它感兴趣基因进行序列分析(见 Differential Diagnosis). 注:包含的基因及多基因包检测的技术方法在各实验室有差异并随时间改变。
Table 1.
SCN1A相关癫痫疾病的分子遗传学检测总结
| 基因 1 | 检测方法 | 该技术检出致病性变异先证者所占比例 |
|---|---|---|
| SCN1A | 序列分析 2 | 73%-92% 3 |
| 缺失/重复 分析 4 | 8%-27% 5, 6, 7, 8 |
- 1.
见 Table A. Genes and Databases 染色体 位点 和蛋白质. 见 Molecular Genetics 等位基因变异信息。
- 2.
- 3.
基于从100%中减去8%-27% 缺失 频率实验值。
- 4.
- 5.
Using a variety of methods to identify deletions encompassing the对采用 序列分析未检测到SCN1A 致病性变异 的SMEI患者,采用各种不同的方法检测包含 SCN1A 位点 的缺失,Madia et al [2006] 在39名患者中的3名(8%)找到4个缺失, Mulley et al [2006] 在13名中的2名(15%)找到缺失, Suls et al [2006] 在11名中的3名 (27%)找到缺失。在这三个研究中,共在序列分析未检出变异的63名SMEI患者中的8名 (12%) 中检出SCN1A 缺失.
- 6.
Marini et al [2009] 发现 序列分析 未检出 致病性变异 的Dravet综合征患者中,12.5%具有MLPA检出的拷贝数变异。
- 7.
- 8.
邻近基因缺失综合征 表现为严重癫痫、智力障碍和 变形的 特征的,包含SCN1A 基因和 SCN2A 基因的染色体 位点 2q23-q24 缺失。已有一名 受累的 个体被报道 [Pereira et al 2004].
SCN1A 致病变异个体的次要或调节基因:
- SCN9A. SCN9A 致病变异被认为在SCN1A 变异引发的疾病中起调节作用;在有些病例中,孤立存在的SCN9A 致病变异(无SCN1A 变异)被认为与Dravet综合征相关。 [Singh et al 2009, Mulley et al 2013] (见 Differential Diagnosis).
- CACNB4. 一名Dravet 表型患者在 de novoSCN1A nonsense 变异(Arg568Ter)之外,有一个CACNB4 错义 变异(Arg468Gln)。Dravet 综合征的表型被认为至少部分是由于增加的Ca(v)2.1电流介导的兴奋性神经递质释放的增加引起的。[Ohmori et al 2008b].
- CACNA1A. 一个针对48名检出 SCN1A 致病变异的Dravet综合征患者的研究发现,21/48 患者具有似乎是常见变异(多态性)的 CACNA1A 改变。文章作者们对检出已知SCN1Ah致病变异并有 CACNA1A 多态性 变异的 受累的 个体的临床特点,与没有 CACNA1A 多态性变异的受累的个体进行比较。研究中有40名患者检出已知SCN1Ah致病变异并表现Dravet综合征;在这40名患者中,20名有 CACNA1A 多态性变异的个体与20名无 CACNA1A 多态性变异的个体的临床特征进行比较。[Ohmori et al 2013]. 有 CACNA1A 多态性变异的受累的个体的癫痫发作发作较早、一岁以内的癫痫发作时间延长的次数较频繁,且失神发作的次数较频繁。[Ohmori et al 2013].
注: 考虑到与给定 致病性变异 相关的Dravet综合征的表型差异,必须谨慎解释所谓的调节基因的作用。也就是说,只有在已编制了可能的修饰因子的综合列表时才能解释Dravet综合症的表型变异性。
临床特征
临床描述
SCN1A相关癫痫疾病的自然史受到癫痫 表型 的强烈影响,从轻微一端的单纯型热性惊厥 (FS) 和全面性癫痫伴热性惊厥附加症 (GEFS+),到严重一端的婴儿严重肌阵挛癫痫 (SMEI) 和难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC) 。 [Kimura et al 2005, Mantegazza et al 2005, Fujiwara 2006, Gennaro et al 2006].
表型 即使在具有相关 致病性变异 的家族成员中也存在变化。 Figure 1 显示了一个 家系 证明了 可变的表现度. 由于这种可变的表达性,很难预测长期预后。
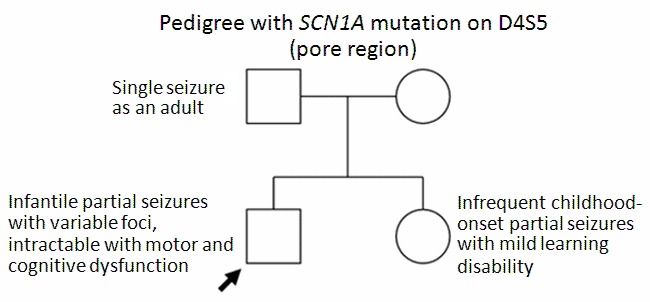
与认知结果差的相关特征包括早期肌阵挛和失神癫痫发作 [Ragona et al 2011].
青春期后癫痫发作的严重程度会降低; 但是,他们很少完全痊愈。只有大约16%的Dravet综合征患者的癫痫发作完全消退,这意味着抗惊厥治疗通常是终生的。完全痊愈倾向于发生在生命早期癫痫发作较轻的个体中。Akiyama et al [2010] 发现发作完全消失与一生中惊厥性癫痫持续状态发作少于3次呈相关性。
难治性癫痫发作(例如,Dravet综合征)的表型通常引起癫痫性脑病,一种进行性痴呆。脑病的根本原因尚不清楚:癫痫发作的影响,最明显的解释,不能与药物的作用或SCN1A突变对认知的影响区分开 [Riva et al 2009].
除了响应强烈的环境刺激而发作癫痫发作外,具有SCN1A突变的个体通常具有以冲动性、注意力不集中和注意力分散为特征的ADHD样 表型 。 可能与GABA系统无法“过滤掉”不重要的感觉输入有关,这些症状往往对常规兴奋剂药物反应迟钝。
患有严重癫痫表型的个体通常会出现姿势改变的运动表现(臀部,膝盖和躯干屈曲,出现“驼背”外观)和共济失调。尽管步态通常被描述为“共济失调”,但 受累的 个体的蹲伏方式来看似乎比人们的预期要熟练得多。步态变化往往在年龄较大的儿童中更为普遍。在一项研究中,这些变化在5岁之前不存在,但存在于10名6-12岁儿童中的5名和9名13岁或以上儿童中的8名 [Rodda et al 2012]. 在一项队列研究中,10名Dravet综合征成人中有5名具有中屈膝步态 [Rilstone et al 2012]. 随着年龄增长而恶化的模式是:被动膝关节伸展和髋关节伸展的减少;外部胫骨扭转增加;;和扁平足 [Rodda et al 2012]. 髋部内旋未显示与年龄相关的变化。步态变化通常在儿童期开始,但通常在癫痫发作后发展。受影响个体的共济失调程度大于单独使用抗惊厥药物所预期的程度。
SCN1A-相关癫痫疾病的表型, 在 Table 2 在总结,包括以下:
高热惊厥 (FS). 这些儿童期癫痫发作仅与发烧有关。 流行病学定义要求如下:
- 六个月或之后发病
- 五岁前消失
- 体温高于 38° C (无其它 CNS 感染的证据)
- 无其他可识别的原因
发热性癫痫发作分为单纯性热性惊厥和复杂的热性惊厥。 如果存在以下任何一种情况,则发热性惊厥被认为是复杂的:
- 持续起过15分钟
- 24小时内发作次数超过一次
- 发作期间表现任一种部分性(局灶性)特征
高热惊厥附加症 (FS+). 这组热性惊厥(简单或复杂)具有以下任何特征:
- 1岁以内起病
- 持续至6岁以后
- 异常严重程度(包括癫痫持续状态)
- 无诱因 (如, 无热) 出现的任何发作类型
全面性癫痫. 否则,该 表型 与儿童期或青春期发病的特发性全身性癫痫无法区分。由SCN1A突变引起的全身性癫痫通常是强直性,阵挛性,强直 - 阵挛性,肌阵挛性或失神。
全面性癫痫伴高热惊厥附加症 (GEFS+). 该术语是指家族性而非个人的发现 [Arzimanoglou et al 2004].
在 GEFS+家族中,具有 可变的表现度 和不完全 外显率 的癫痫以 常染色体显性遗传 模式遗传。尽管可以在任一家族中看到完整范围的相关表型,但癫痫发作表型倾向于疾病谱中表型轻微的一端 [Scheffer & Berkovic 1997] ,由于发作严重的类型具有生殖劣势,因此不太可能呈 家族性 [Claes et al 2001].
患有GEFS +的家族中受影响的个体通常在儿童早期患有热性惊厥(或FS +),随后是偶然的强直,阵挛性,肌阵挛或失神发作,其对儿童晚期或青春期早期的药物有反应和缓解。在免疫接种的情况下首次发作的GEFS +儿童的比例似乎高于与FS +和GEFS +无关的热性惊厥儿童的比例。
Dravet 综合征. 该 表型 定义为癫痫发作在出生后的第一年(通常在6个月左右;在某些情况下在3个月之前),不会缓解,并且通常演变为包括肌阵挛性癫痫发作。
- 早期癫痫发作往往是长期发热性癫痫发作。 癫痫发作有时可以由适度高热(例如,热浴,体力活动)诱发。
- 任何癫痫发作类型都是可能的; 全身强直 - 阵挛性,肌阵挛性和半身性癫痫发作是最常见的。
- 肌阵挛性癫痫发作往往出现在疗程的后期,通常与认知功能障碍,共济失调和精神运动性退化的出现相吻合。
- 癫痫持续状态很常见,药物控制很困难。
- 最初的脑电图通常是正常的,但随着时间的推移会出现癫痫样活动。模式可以包括广泛尖峰和波放电,多个尖峰和波(也称为多尖峰和波)放电,以及多局灶尖峰。
严重肌阵挛性癫痫, 交界性 (SMEB).该描述有时用于具有一些但不是全部SMEI特征的儿童 [Fukuma et al 2004].
难治性儿童癫痫伴全面性强直-阵挛性发作(ICE-GTC). 该 表型 定义为全身性癫痫发作,包括失神发作和在婴儿期或儿童期发病的全身性强直 - 阵挛性发作。然而,部分性癫痫发作可发生在高达13%受累的 个体 [Bonanni et al 2004]. 也可以看到局部癫痫,交替的半身性或复杂的部分性癫痫发作。 频繁全身性强直 - 阵挛性发作的儿童常发生认知功能障碍。ICE-GTC和Dravet综合征之间的区别尚不清楚,前者不包括在ILAE分类系统中。
婴儿部分性癫痫发作伴有可变灶. 该 表型 定义为在婴儿期开始的局灶性癫痫发作,其具有涉及两个脑半球的多个独立的癫痫发作区域。 多灶性部分性癫痫发作通常是首发表现; 然而,在一些儿童中,首发症状是热性惊厥。严重程度各不相同,药物耐药性很常见,但不是绝对的。 肌阵挛性癫痫发作很少见,但可通过给予使钠通道失活的药物(包括苯妥英,卡马西平或拉莫三嗪)来促使癫痫发作。可能发生认知恶化,尤其是当癫痫发作控制不完全时。脑电图显示多灶性独立尖峰; 可以看到广泛的尖峰和波放电。
Table 2.
SCN1A相关癫痫疾病中癫痫发作表型的分布情况
| 疾病 | 分布 |
|---|---|
| 单纯性高热惊厥 (FS) | 未知 |
| 高热惊厥附加症 (FS+) | 未知 |
| 全面性癫痫伴热性惊厥附加症 (GEFS+) | 5%-10% 1 |
| 婴儿严重肌阵挛癫痫 (SMEI) | 33%-90% 2 |
| 难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC) | 70% 3 |
与SCN1A突变相关的不常见表型的特征和过程包括:
肌阵挛-站立不能性发作 (MAE, 也称 Doose综合征). 该 表型 定义为肌阵挛,失张力和非典型失神发作的组合。发病通常在两岁后(范围:7个月 - 8年)。
虽然可以发生孤立的肌阵挛性癫痫发作以及强直性癫痫发作,但它们并不是这种综合征的特征(这使它们与Lennox-Gastaut综合征区别开来)。癫痫发作前的发展通常是正常的。该病程范围可以从不伴认知障碍的自发性癫痫发作,到伴严重智力残疾的难治性癫痫发作 [Arzimanoglou et al 2004].
Ebach et al [2005] 比较两个队列,以确定MAE 表型 是否比严重的特发性全身性婴儿癫痫(SIGEI)表型对SCN1A 致病性变异 的存在更具特异性。他们在20名患有MAE的儿童中发现了一个致病变异,在18名患有SIGEI的儿童中发现了两个致病变异; 小样本量排除了统计学上显着的结果。
Lennox-Gastaut综合征 (LGS). 该 表型 被定义为EEG上的缓慢棘波,发育迟缓和多种类型的全身性癫痫发作(特别是非典型性失神,强直性和失张力性癫痫发作)。LGS通常在童年时期(2-14岁)开始。在这种综合征中可以看到任何类型的癫痫发作; 癫痫持续状态很常见 [Arzimanoglou et al 2004]. 只有少数具有LGS表型的人具有SCN1A 致病性变异, 他们通常发生在Dravet综合征家庭中 [Singh et al 2001]。该子类型的特征描述仍然很少。尚不清楚SCN1A相关的LGS是否与其他病因的LGS表型不同。
婴儿痉挛症. 该 表型 定义为聚集性癫痫发作,显示与脑电图上的慢波瞬态相关的短暂(<1秒)轴向收缩,通常随后是背景的广泛衰减。 这两项发现可能与快速活动相混合。静息EEG(癫痫发作之间)显示高压减慢和称为低血球性心律失常的多灶性尖峰态 [Arzimanoglou et al 2004]. 已经报道过一次SCN1A致病性 错义 变异与婴儿痉挛的关联 [Wallace et al 2003]. 单一案例占报告案例的比例不到1%,尽管发表偏好使得难以估计实际比例。
疫苗相关的脑病和癫痫发作. 该 表型 定义为免疫后48小时婴儿癫痫发作和脑病的突然发作。Berkovic et al [2006] 在14名被诊断患有疫苗后脑病的儿童中发现11名具有SCN1A 致病性变异。 Tro-Baumann et al [2011] 报道,70名具有SCN1A致病变异体和Dravet表型的个体中,有19名在接种疫苗后有癫痫发作史。
脑MRI在疾病早期通常是正常的; 然而,它经常演变为显示皮质萎缩,小脑萎缩,白质高信号,脑室扩大,海马硬化或皮质发育不良[Striano et al 2007]. 在生命早期具有更严重 表型 的个体通常在生命后期的MRI上看到更多的萎缩性变化。
基因型-表型关联
Mulley et al [2005] 发现大多数SCN1A致病变异聚集在C-末端和连接S5和S6的孔环中,特别是在蛋白质的前三个结构域中 (Figure 2).
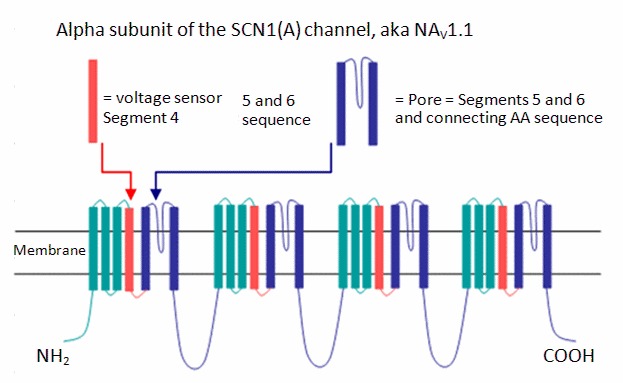
位于电压传感器或孔道区域的致病性的 nonsense 变异和 错义 变异通常导致更严重的 表型 [Zuberi et al 2011]; 然而,截短的变异,并不一定会导致严重的表型 [Suls et al 2010, Yu et al 2010].
具有位于形成孔道区域的 错义 变异和SCN1A蛋白截短的患者更倾向于有步态变化[Kanai et al 2004, Rilstone et al 2012]. 这些变化可能是由于 SCN1A 致病性变异 对小脑浦肯野细胞的直接作用 [Catterall et al 2010].
分子水平上确诊 SMEI 的患者中估计有5%具有 家族性 错义 SCN1A ,且其他亲属具有较轻微的 表型 (如, GEFS+) [Mulley et al 2005].
命名法
全面性癫痫伴热性惊厥附加症,曾称为GEFS+2型。
难治性婴儿部分性癫痫发作,曾称为 ICEGTC.
外显率
SCN1A相关癫痫疾病表现为不完全的 外显率 和 可变的表现度.
外显率根据不同 表型有所不同。 例如, Bonanni et al [2004] 估计GEFS+ 表型的 外显率 t约为70% ,而 Mantegazza et al [2005] 报道 家族性 单纯性热性惊厥的外显率为90%
预期
SCN1A相关癫痫疾病,未进行预期观察。
发病率
SCN1A相关癫痫疾病的发病率未知。
遗传相关(等位基因)疾病
与SCN1A致病性相关的其它表型:
- Panayiotopolous综合征 [Grosso et al 2007]
- 家族性自闭症 [Weiss et al 2003]
- 拉斯穆森脑炎与 致病性变异 p.Arg1575Cys 相关的拉斯穆森脑炎 [Ohmori et al 2008a]
鉴别诊断
具有SCN1A 变异的典型表型,不是诊断 SCN1A相关癫痫疾病的必要条件也不是充分条件。其它疾病(包括由其它基因导致的)可能关联相同的表型。
将 SCN1A相关癫痫疾病从其它潜在可治疗疾病中识别出来是最为重要的,包括 [Arzimanoglou et al 2004, Roger et al 2006]:
- Pyridoxine-dependent seizures 及B6-相关癫痫
- "亚叶酸反应性" 癫痫,一种罕见的癫痫病,它可以随着日常的叶酸管理而改善。
- 先天性代谢紊乱,包括线粒体功能障碍,可通过乳酸,酮,氨,氨基酸的异常血清浓度和/或尿液有机酸异常浓度来诊断 (见 Mitochondrial Diseases Overview)
- Biotinidase deficiency, 通常在 新生儿筛查 中发现。
- Glucose transporter type 1 deficiency, 可通过低CSF 葡萄糖浓度诊断,并对生酮饮食有反应。
- 肝卟啉症,通常表现为尿液中的光敏卟啉和红细胞中降低的单倍体胆色素原(PBG)脱氨酶 (见 Acute Intermittent Porphyria, Porphyria Cutanea Tarda, Hepatic Coproporphyria, and Variegate Porphyria.)
若家族史为阴性或未知,散发性 癫痫 (如,那些没有遗传因素的癫痫) 应被考虑在鉴别诊断之内,与影像学发现非特异性的任何癫痫病因一样。一些需考虑的一般伤害类别如下 [Arzimanoglou et al 2004, Roger et al 2006]:
- 外伤
- 缺氧
- 脑膜炎或出血的后遗症
- 传染性或自身免疫性脑炎
- 血管炎
- 副肿瘤综合征
- 毒素 (包括戒毒)
- 内分泌失调
若家族史为阳性,有其他亲属患癫痫,鉴别诊断包括以下遗传性癫痫综合征 [Arzimanoglou et al 2004, Roger et al 2006]:
- 良性 家族性 婴儿发作 (见 KCNQ3-Related Disorders)
- 良性儿童癫痫伴中央颞区棘波(外侧裂性癫痫)(见 KCNQ2-Related Disorders, KCNQ3-Related Disorders)
- 儿童枕部癫痫
- 失神癫痫
- 家族性颞叶癫痫
- 具有可变灶的家族性局灶性癫痫
- 全面性癫痫伴热性惊厥附加症 (GEFS+)
迄今为止,已鉴定出至少12个与 家族性热性惊厥相关的基因座和至少8个与全面性癫痫伴热性惊厥附加症(GEFS +)相关的基因座。单纯热性惊厥的 表型 通常比 GEFS+相关热性惊厥的表型严重程度低。 (见 Clinical Description) [Nakayama & Arinami 2006].
见 OMIM 系列表型: Seizures, familial febrile 和 Epilepsy, generalized, with febrile seizures ,查看OMIM中该 表型 相关的基因。
已知与GEFS +相关的遗传基因座包括:
- 电压门控钠离子通道基因
- SCN1B (位点名称: GEFSP1) [Wallace et al 1998, Wallace et al 2002, Audenaert et al 2003], 该基因变异可能为 常染色体隐性遗传 导致Dravet综合征 [Patino et al 2009], 起病期比SCN1A 或 GABRG2 相关疾病晚 [Sijben et al 2009]
- SCN2A [Sugawara et al 2001], 该基因变异也可能导致伴严重肌阵挛癫痫、共济失调和疼痛的 表型 [Liao et al 2010]. 在Dravet 综合征患儿中,SCN1A 致病性变异比 SCN2A 致病性变异发生频率更高,比例约9:1 [Shi et al 2009].
- 偶尔,孤立的SCN9A 致病性变异(无 SCN1A 变异)与Dravet 综合征相关 [Singh et al 2009]; 发生概率未知。家系研究显示,在患有Dravet综合征的尚未发现SCN1A 致病性变 的个体,SCN9A致病变异体的发生率相对较高。
- GABAA 受体基因
- GABRG2 (位点名称: GEFSP3) [Baulac et al 2001, Wallace et al 2001a], 比 SCN1A相关不常见。 Shi et al [2010] 发现140位Dravet-GEFS+ 谱系的儿童癫痫患者中有1位具有 GABRG2 致病性变异.
- GABRD (位点名称: GEFSP5) [Dibbens et al 2004]
- PCDH19, 编码原钙粘附因子 [Depienne et al 2009, Marini et al 2010] ,位于 X 染色体. PCDH19 基因可能导致 Dravet 综合征 表型; 大多数有症状患者为女性。该综合征特征为早期发育正常,和倾向于发生在人群中的发热和温度诱发的癫痫发作。发作的起病时间倾向于稍晚 (年龄 ≥12 个月). 尽管相较于SCN1A 致病性变异的患者,这些患者可能较少发生肌跳跃型抽搐和失神癫痫,其表型可能与 Dravet 综合征相当类似;此外,这些患者通常对相同的药物有反应(如,司替戊醇) [Nikanorova et al 2011].
- GEFSP6 (8p23-p21), 没有已知离子通道基因的区域 [Baulac et al 2008]
具有 2q24.2 缺失 的癫痫患者,可能有 SCN1A 或候选基因 SLC4A10的缺失 [Krepischi et al 2010].
管理
对以下初步诊断的评估
为了确定被诊断患有SCN1A相关癫痫疾病的个体的疾病和需求程度,建议进行以下评估:
- 神经病学检查
- 认知神经心理学评估
- 行为神经心理评估
- 脑电图 (EEG), 包括当发作或症状学不清楚时使用视频脑电图遥测术
- 临床遗传学评估 [Pal et al 2010]
对症治疗
最好由熟悉该病症的药物疗法的医生(例如,儿科癫痫病学家)提供护理。 癫痫控制至关重要,因为患有SCN1A相关癫疾病的儿童癫痫发作突然不明原因死亡(SUDEP)的风险很高。 此外,长期急性癫痫发作可能导致永久性损伤 [Chipaux et al 2010, Takayanagi et al 2010].
药物治疗侧重于异常的SCN1A通道不成比例地影响GABA神经元的观察结果 [Yu et al 2006] 以及相关的癫痫发作对与GABA受体结合的抗癫痫药物(AEDs)的最佳反应:
- 氯巴占Clobazam (0.2-1 mg mg/kg/day), 欧洲标准护理的一部分,现已获得美国FDA的批准。 氯巴占被FDA批准用于治疗Lennox-Gastaut综合征的癫痫发作 [Selmer et al 2009].
- 司替戊醇Stiripentol (30-100 mg/kg/day) 被癫痫学家接受为SCN1A相关癫痫疾病的有效治疗药物。它是欧洲早期护理标准的一部分,在其他常规抗惊厥药失败后在美国使用。FDA未批准其在美国使用,但其对SCN1A相关癫痫疾病的有效性证据比任何其他药物更具特异性(基于对婴儿期严重肌阵挛性癫痫(SMEI)患者癫痫发作减少的双盲评估)[Chiron et al 2000]). 因此,司替戊醇不被视为“研究性”疗法。
Thanh et al [2002] 证明了该药物相比于安慰剂的功效; 仅观察到婴儿和幼儿的中度副作用,包括嗜睡,食欲不振和偶然的中性粒细胞减少。 在美国最近对82名患有Dravet综合征的儿童进行的一项调查中,发现司替戊醇可有效减少癫痫持续发作 [Wirrell et al 2013].
司替戊醇直接作用于GABAA 受体 [Quilichini et al 2006], 也是肝酶CYP3A4, CYP1A2, 和 CYP2C19的一种潜在抑制剂。因此,它会使一些常见AEDs的血清浓度升高,包括丙戊酸,氯巴占及其非氯巴占代谢物 [Thanh et al 2002]. 当与丙戊酸和氯巴占一起使用时,通常不允许超过50毫克/公斤/天的剂量。
12岁以上的儿童可能因消化道副作用和恶心而不能耐受司替戊醇 [Thanh et al 2002]. - 苯二氮平类Benzodiazepines. 服用司替戊醇的患者必需谨慎使用苯二氮平类 [Thanh et al 2002]. 单次输注地西泮和氯硝西泮似乎是安全的[Thanh et al 2002].
- 托吡酯Topiramate [Coppola et al 2002]
- 丙戊酸Valproic acid (10-30 mg/kg/day) [Thanh et al 2002]
- 乙琥胺Ethosuximide. 可能对失神癫痫有效。剂量通常受胃肠道副作用的限制,可以通过更频繁的给药来最小化副作用。
- 左乙拉西坦Levetiracetam (20-80 mg/kg/day). 通常有效,但可能使一些患者的癫痫发作恶化 [Caraballo et al 2010].
- 溴化钾Potassium bromide. 美国FDA未批准,但在日本广泛使用并具合理效果。[Tanabe et al 2008].
- 苯巴比妥Phenobarbital. 苯巴比妥虽然有效,但由于其对认知的影响而耐受性差。 当与司替戊醇联合使用时,苯巴比妥的血清浓度增加,因为司替戊醇减缓了巴比妥类药物的代谢和排泄。
- 生酮饮食Ketogenic diet.Dressler et al [2010] 报道,62.5%的Dravet综合征患者保持生酮饮食6个月而癫痫发作减少了50%以上。Nabbout et al [2011] 在15名患者中的研究发现也支持在Dravet综合征使用生酮饮食.
睡眠剥夺和疾病会加剧与SCN1A相关癫痫疾病发作; 因此,应鼓励良好的睡眠卫生。睡眠呼吸暂停的合并症也可能在患有癫痫的个体中频繁发生 [Malow et al 2000], 并且可影响癫痫控制、行为和认知。如果怀疑有阻塞性或中枢性睡眠呼吸暂停,应考虑多导睡眠图。
癫痫发作并不总是对传统的AEDs有反应。坊间证据表明,以下药物/治疗方式可能对SCN1A相关的SMEI癫痫发作有效 [Dravet et al 2002]:
- 乙琥胺Ethosuximide和高剂量吡拉西坦piracetam用于肌阵挛性癫痫发作
- 皮质类固醇
- 免疫球蛋白
多个家庭报告有帮助的非医疗干预包括以下内容 [Nolan et al 2008]:
- 放置静脉通路装置
- 创建便携式微环境
- 有书面的急诊科协议
- 为家族建立紧急路线
- 在呼叫中分配给父母以减轻对兄弟姐妹的影响
- 创造个人时间以减少父母压力
- 寻找喘息护理
- 联系互联网支持小组
二次并发症的预防
应建议患有失张力性癫痫发作或肌阵挛性癫痫的个体佩戴防护头盔。
虽然免疫可能引发癫痫发作,但它不会影响疾病的自然过程。 McIntosh等[2010]回顾性地观察了14名患有Dravet综合征的人群,发现免疫对认知结果没有影响。 这些作者提出,免疫计划不会改变,并且通过提供预定的长效NSAID(例如萘普生)可以降低免疫后发烧的风险。 治疗神经病学家还可以考虑在免疫接种时间附近暂时增加抗惊厥剂量。
应建议患有失张力性癫痫发作或肌阵挛性癫痫的个体佩戴防护头盔。
尽管免疫可能会引发癫痫,但它不会影响疾病的自然过程。McIntosh et al [2010] 回顾性地观察了14名患有Dravet综合征的人群,发现免疫对认知结果没有影响。 这些作者提出,不改变免疫计划并且通过提供预定的长效NSAID(例如萘普生)可以降低免疫后发烧的风险。 治疗神经病学家还可以考虑在免疫接种期间暂时增加抗惊厥剂量。
监测
神经,认知和行为恶化的连续神经心理学评估是合适的。
当怀疑新的或不同的癫痫发作类型时,脑电图监测是适当的。
应避免的媒介/环境
对于大多数形式的癫痫有效的几种抗癫痫药物(AEDs),可能由于杂合的SCN1A致病变异而导致癫痫发作恶化:
- 卡马西平、拉莫三嗪和氨己烯酸Carbamazepine, lamotrigine, and vigabatrin, [Horn et al 1986, Guerrini et al 1998, Ceulemans et al 2004a]
- 苯妥英Phenytoin, 可能会加重癫痫发作并导致舞蹈病 [Saito et al 2001]
- 卢非酰胺Rufinamide, 有一种类似于卡马平和苯妥英的药理学机制,也可能会加剧癫痫发作
- 对乙酰氨基酚Acetaminophen, 是过量的肝毒素。考虑到与抗惊厥药物,特别是丙戊酸盐和托吡酯相互作用的可能性 [Nicolai et al 2008],应避免使用对乙酰氨基酚。 任何NSAIDs都可以作为退热药使用,并且风险更低。
应避免突然意识丧失导致伤害或死亡的活动(例如,洗澡,游泳,开车或在高处工作/运动)。
有风险亲属的评估
见 Genetic Counseling 有关为 遗传咨询 目的检测有风险亲属的问题。
孕期管理
除了 Genetic Counseling部分建议考虑的内容外,其他孕期相关考虑如下:
- 畸形风险 (尤其是子宫中丙戊酸暴露引起) [Samrén et al 1997]) 和微小畸形
- 增加孕妇的叶酸补充剂至4000 μg/天的利弊,尤其是孕期使用丙戊酸或卡马西平的女性
- 子宫内接触抗惊厥药物对未来认知发育的影响 [Meador et al 2009]
- 抗惊厥剂对激素避孕方法的影响
- 抗惊厥剂对受孕的影响; 服用抗惊厥药的母亲患并发症的风险
- 妊娠对抗惊厥代谢的影响
- 妊娠对母亲癫痫发作的影响
怀孕、计划生育和避孕是每一个患有癫痫的育龄妇女都应该提出的问题。这些考虑并不是SCN1A相关癫痫疾病特有的或(除药物选用外)SCN1A相关癫痫疾病所能影响。
正在研究中的疗法
大麻衍生化合物 (包括大麻二酚 [CBD], 四氢大麻酚 [THC], 和大麻油), 统称为“大麻素”,基于坊间传闻得到了大量媒体关注;然而,目前并无科学证据表明它们的疗效。因此,它们不应该被用来代替已知有效的治疗方法。需要进行随机临床试验,以确定大麻素治疗是否有效。在开始这一试验方面取得了快速进展,预计将于2014年开始。大麻素是具有生物活性的,可能具有精神和/或全身的副作用;它们也可以作为一种免疫抑制剂和动物模型中的抗炎药,因此在考虑广泛使用之前需要在未成年人类中进行仔细研究 [Rieder et al 2010, Bergamaschi et al 2011].
丘脑深部电刺激 (DBS) 被 Andrade et al [2010] 报道用于两名 Dravet 综合征儿童患者并随访10年。其中一名在植入后表现出“显著改善”,而另一名未受益。
拉克酰胺Lacosamide has not been studied in SCN1A-related seizure disorders; however, there are theoretic reasons why it may be effective [Curia et al 2009].
维拉帕米Verapamil 被报道对两名由SCN1A 变异导致的患严重癫痫的女孩有帮助 [Iannetti et al 2009]; 然而,它在之前还未被正式研究过。
搜索 ClinicalTrials.gov 获取有关各种疾病和病症的临床研究信息。
其他
应使癫痫患者了解当地机动车驾驶法和医师报告法。
海马硬化可能是SCN1A相关癫痫疾病的第二特征 [Livingston et al 2009], 但由于这种疾病中普遍存在癫痫发作潜力,因此没有被证实的手术作用.值得注意的是, Scheffer et al [2007] 报道了两例SCN1B突变患者颞叶手术后的良好预后。
遗传咨询
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
遗传模式
SCN1A相关癫痫疾病为 常染色体显性遗传 模式.
注: 大多数SCN1A相关婴儿严重肌阵挛癫痫(SMEI) 和难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC) 均由 de novo 杂合的 致病性变异导致。
家族成员风险
先证者 的父母
- 患有SCN1A相关癫痫疾病的 先证者 可能是由 de novo 致病性变异 导致。案例中 de novo 变异致病的比例,根据 表型 不同而有所差异。随着先证者表型严重程度的增加,患有SCN1A相关癫痫疾病是患者有 受累的 父母的百分比下降。
- 大于95%的GEFS+ 患者有一位父/母具有相同的 SCN1A 致病性变异.
- 只有大约 5% SMEI 先证者有一位父/母具有相同的SCN1A 致病性变异 [Wallace et al 2003, Ceulemans et al 2004b, Fukuma et al 2004].
- 对患有 SMEI 并确认具有 SCN1A 致病性变异 的儿童的父母进行检测发现,95% (76/80) 儿童的致病变异为 先证者 的 de novo 变异。在4名父/母具有致病变异的儿童中,2名具有 错义 变异,2名具有截短变异;父母无症状或仅有轻微癫痫 [Gennaro et al 2003, Nabbout et al 2003].
- 若在 先证者 发现的 致病性变异 在父母的白细胞提取DNA中均未检测到,则父母具有该致病性变异的风险均低,但由于存在 胚系嵌合 的可能性,仍高于普通人群。胚系嵌合已被记录过 [Gennaro et al 2006, Selmer et al 2009, Azmanov et al 2010], 在Dravet综合征家族中可能发生率高达7% [Depienne et al 2010].
- 在进行适当的评估之前,无法确认显然的阴性的家族史。尽管95%SCN1A相关的 GEFS+ 患者有一位 受累的 父母,其家族史可能显示为阴性,原因为未能识别家族成员的疾病或症状出现之前早期死亡。若父/母是最先发生 致病性变异 , 她/他可能具有该致病性变异的 体细胞嵌合 并仅有轻微或最轻度的受累 [Gennaro et al 2006].
先证者 的同胞
先证者 的后代
- SCN1A相关癫痫疾病患者的每一个孩子都有50% 概率遗传到 致病性变异.
- 外显率是不完全的 (见 Penetrance) 并随 表型 而有所不同。
- GEFS+ 患者的下一代可能具有比他们更严重 受累的 情况。比如,他们的孩子可能患有Dravet综合征。
先证者 的其它亲属。家族成员的风险决定于先证者父母的状态:若父母一方是 受累的 或具有一个 致病性变异, 则其他家族成员的患病风险高于普通人群。
相关的遗传咨询问题
解释有风险的无症状亲属的检测结果. 对检出家族特异 致病性变异 的无症状亲属的遗传咨询应包括 外显率 下降和仅依据 分子遗传学检测 预测 表型 能力有限的信息。
具有明显 de novo 致病性变异 的家族的一些考虑:若 常染色体显性遗传 症状的 先证者 的父母均没有该致病性变异或疾病临床表现,则该变异可能为 de novo. 然而,可能的非医学解释包括: 非生物学父亲 或母亲 (如,辅助生殖) 或也可以调查一下未公开的收养的可能性。
家庭计划
- 确定遗传风险的最佳时间和产前检测可用性的讨论是在怀孕前。
DNA 银行 是将DNA (通常从白细胞中提取)存储以备将来使用。因为检测方法学及我们对于基因、等位基因变异和疾病的理解可能进步更新,需考虑将 受累的 个体的DNA进行储存。
资源
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- Dravet Syndrome FoundationPhone: 203-392-1950Fax: 203-907-1940Email: info@dravetfoundation.org
- My46 Trait Profile
- American Epilepsy Society (AES)
- Canadian Epilepsy AllianceCanadaPhone: 1-866-EPILEPSY (1-866-374-5377)
- Epilepsy Foundation8301 Professional Place EastSuite 200Landover MD 20785-7223Phone: 800-332-1000 (toll-free)Email: ContactUs@efa.org
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
- International Ion Channel Epilepsy Patient Registry
分子遗传学
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. - ED.
Table A.
SCN1A相关癫痫疾病: 基因和数据库
Table B.
SCN1A相关癫痫疾病的OMIM条目 (View All in OMIM)
分子遗传学致病机制:
SCN1A 编码神经元电压门控钠离子通道alpha亚基(也称为 Nav1.1) 。SCN1A相关癫痫疾病因此在概念化为“离子通道病”,以癫痫发作(及其后遗症)为主要表现。 分子异常导致神经元功能障碍,并最终导致皮质网络水平上的过度兴奋:癫痫的必要条件。
SCN1A 是 染色体 2q24 上编码钠离子通道基因簇的一部分,该基因簇还包括 SCN2A和SCN3A [Mulley et al 2005]. 钠离子通道的 alpha亚基组成的膜通道。每个alpha 亚基蛋白有4个结构域,由环连接的6个跨膜区段。(Figure 2). 孔道-线氨基酸位于 S5, S6, 以及 P-loop, 后者连接了 S5 和 S6。电压感应器位于 S4, 带正电的氨基酸使得感应到膜电势变化 [Catterall 2000]. 尽管癫痫相关的致病性变异被发现于 Nav1.1的所有区域,但在C-末端发生更频繁,在某种程序上发生于N-末端,D1-D5的P-环,以及电压感应器 [Ceulemans et al 2004b, Mulley et al 2006].
基因结构.SCN1A在 基因组的 DNA中大约占84 M,转录组为 8,100 bp (reference sequence NM_006920.4). 该 基因 包含26个外显子,编码1,998 个氨基酸 (参考序列 NP_008851.3). 剪接变异已被报道过 [Wallace et al 2001b]. 关于基因和蛋白的详细概述信息,见 Table A, 基因.
致病性等位基因变异
- Dravet综合征. 婴儿严重肌阵挛癫痫(SMEI) 表型 相关的致病性变异,几乎有一半是截短变异 [Mulley et al 2006]. 其余的包括 错义 变异 (39%-43%; 比 GEFS+ 表型更少, 但具有相似的 Nav1.1 拓扑分布), 剪接位点 变异 (7%), 和缺失 (3%) [参考 Mulley et al 2005].
- 难治性儿童癫痫伴全面性强直-阵挛性发作 (ICE-GTC). 10名ICE-GTC 患者中7名具有 SCN1A 错义 致病性变异 [Mulley et al 2005]. 在两个遗传性致病性变异案例中,患者父母患有GEFS+.
- 婴儿部分性癫痫发作伴多灶性. 在作者的经验中,这类案例经常具有影响Nav1.1孔道区域或羧基端的 错义 致病性变异。
- 疫苗相关性脑病和癫痫发作. 有5个关于 Nav1.1 截短致病性变异的报道,6个关于 Nav1.1 保守区域 错义 致病性变异的报道 [Berkovic et al 2006].
正常 基因产物. 见 Molecular Genetic Pathogenesis.
异常 基因产物. SCN1A相关疾病的分子致病机制可能取决于特定的 致病性变异 类型 (功能丧失 vs. 活性改变). 致病机制是一个活跃的研究领域;似乎主要影响是GABA能神经元兴奋抑制的丧失。[Escayg & Goldin 2010].
参考文献
Literature Cited
- Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia. 2010;51:1043 - 52. [PubMed: 20041943]
- Andrade DM, Hamani C, Lozano AM, Wennberg RA. Dravet syndrome and deep brain stimulation: seizure control after 10 years of treatment. Epilepsia. 2010;51:1314 - 6. [PubMed: 19919661]
- Arzimanoglou A, Guerrini R, Aicardi J. Aicardi's Epilepsy in Children. 3 ed. Philadephia, PA: Lippincott Williams & Wilkins; 2004.
- Audenaert D, Claes L, Ceulemans B, Lofgren A, Van Broeckhoven C, De Jonghe P. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology. 2003;61:854 - 6. [PubMed: 14504340]
- Azmanov DN, Zhelyazkova S, Dimova PS, Radionova M, Bojinova V, Florez L, Smith SJ, Tournev I, Jablensky A, Mulley J, Scheffer I, Kalaydjieva L, Sander JW. Mosaicism of a missense SCN1A mutation and Dravet syndrome in a Roma/Gypsy family. Epileptic Disord. 2010;12:117 - 24. [PubMed: 20562086]
- Baulac S, Gourfinkel-An I, Couarch P, Depienne C, Kaminska A, Dulac O, Baulac M, LeGuern E, Nabbout R. A novel locus for generalized epilepsy with febrile seizures plus in French families. Arch Neurol. 2008;65:943 - 51. [PubMed: 18625863]
- Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud'homme JF, Baulac M, Brice A, LeGuern E. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet. 1999;65:1078 - 85. [PMC free article: PMC1288241] [PubMed: 10486327]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46 - 8. [PubMed: 11326274]
- Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. 2011;6:237 - 49. [PubMed: 22129319]
- Berkovic SF, Harkin L, McMahon JM, Pelekanos JT, Zuberi SM, Wirrell EC, Gill DS, Iona X, Mulley JC, Scheffer IE. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006;5:488 - 92. [PubMed: 16713920]
- Bonanni P, Malcarne M, Moro F, Veggiotti P, Buti D, Ferrari AR, Parrini E, Mei D, Volzone A, Zara F, Heron SE, Bordo L, Marini C, Guerrini R. Generalized epilepsy with febrile seizures plus (GEFS+): clinical spectrum in seven Italian families unrelated to SCN1A, SCN1B, and GABRG2 gene mutations. Epilepsia. 2004;45:149 - 58. [PubMed: 14738422]
- Caraballo RH, Cersósimo R, De los Santos C. Levetiracetam-induced seizure aggravation associated with continuous spikes and waves during slow sleep in children with refractory epilepsies. Epileptic Disord. 2010;12:146 - 50. [PubMed: 20483715]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13 - 25. [PubMed: 10798388]
- Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849 - 59. [PMC free article: PMC2901973] [PubMed: 20194124]
- Ceulemans B, Boel M, Claes L, Dom L, Willekens H, Thiry P, Lagae L. Severe myoclonic epilepsy in infancy: toward an optimal treatment. J Child Neurol. 2004a;19:516 - 21. [PubMed: 15526956]
- Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol. 2004b;30:236 - 43. [PubMed: 15087100]
- Chipaux M, Villeneuve N, Sabouraud P, Desguerre I, Boddaert N, Depienne C, Chiron C, Dulac O, Nabbout R. Unusual consequences of status epilepticus in Dravet syndrome. Seizure. 2010;19:190 - 4. [PubMed: 20172746]
- Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, Dulac O, Pons G. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356:1638 - 42. [PubMed: 11089822]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327 - 32. [PMC free article: PMC1226119] [PubMed: 11359211]
- Coppola G, Capovilla G, Montagnini A, Romeo A, Spano M, Tortorella G, Veggiotti P, Viri M, Pascotto A. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res. 2002;49:45 - 8. [PubMed: 11948006]
- Curia G, Biagini G, Perucca E, Avoli M. Lacosamide: a new approach to target voltage-gated sodium currents in epileptic disorders. CNS Drugs. 2009;23:555 - 68. [PMC free article: PMC4878900] [PubMed: 19552484]
- Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, Benyahia B, Quelin C, Carpentier W, Julia S, Afenjar A, Gautier A, Rivier F, Meyer S, Berquin P, Hélias M, Py I, Rivera S, Bahi-Buisson N, Gourfinkel-An I, Cazeneuve C, Ruberg M, Brice A, Nabbout R, Leguern E. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009;5:e1000381. [PMC free article: PMC2633044] [PubMed: 19214208]
- Depienne C, Trouillard O, Gourfinkel-An I, Saint-Martin C, Bouteiller D, Graber D, Barthez-Carpentier MA, Gautier A, Villeneuve N, Dravet C, Livet MO, Rivier-Ringenbach C, Adam C, Dupont S, Baulac S, Héron D, Nabbout R, Leguern E. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet. 2010;47:404 - 10. [PubMed: 20522430]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315 - 9. [PubMed: 15115768]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371 - 7. [PubMed: 16054936]
- Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy. In: Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3 ed. Eastleigh, UK: John Libbey; 2002:81-103.
- Dressler A, Stöcklin B, Reithofer E, Benninger F, Freilinger M, Hauser E, Reiter-Fink E, Seidl R, Trimmel-Schwahofer P, Feucht M. Long-term outcome and tolerability of the ketogenic diet in drug-resistant childhood epilepsy--the Austrian experience. Seizure. 2010;19:404 - 8. [PubMed: 20598586]
- Ebach K, Joos H, Doose H, Stephani U, Kurlemann G, Fiedler B, Hahn A, Hauser E, Hundt K, Holthausen H, Muller U, Neubauer BA. SCN1A mutation analysis in myoclonic astatic epilepsy and severe idiopathic generalized epilepsy of infancy with generalized tonic-clonic seizures. Neuropediatrics. 2005;36:210 - 3. [PubMed: 15944908]
- Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51:1650 - 8. [PMC free article: PMC2937162] [PubMed: 20831750]
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus--and prevalence of variants in patients with epilepsy. Am J Hum Genet. 2001;68:866 - 73. [PMC free article: PMC1275640] [PubMed: 11254445]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343 - 5. [PubMed: 10742094]
- Fujiwara T. Clinical spectrum of mutations in SCN1A gene: severe myoclonic epilepsy in infancy and related epilepsies. Epilepsy Res. 2006;70 Suppl 1:S223 - 30. [PubMed: 16806826]
- Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain. 2003;126:531 - 46. [PubMed: 12566275]
- Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, Ohfu M, Inoue T, Watanachai A, Kira R, Matsuo M, Muranaka H, Sofue F, Zhang B, Kaneko S, Mitsudome A, Hirose S. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia. 2004;45:140 - 8. [PubMed: 14738421]
- Gaily E, Anttonen AK, Valanne L, Liukkonen E, Träskelin AL, Polvi A, Lommi M, Muona M, Eriksson K, Lehesjoki AE. Dravet syndrome: new potential genetic modifiers, imaging abnormalities, and ictal findings. Epilepsia. 2013;54:1577 - 85. [PubMed: 23808377]
- Gennaro E, Santorelli FM, Bertini E, Buti D, Gaggero R, Gobbi G, Lini M, Granata T, Freri E, Parmeggiani A, Striano P, Veggiotti P, Cardinali S, Bricarelli FD, Minetti C, Zara F. Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem Biophys Res Commun. 2006;341:489 - 93. [PubMed: 16430863]
- Gennaro E, Veggiotti P, Malacarne M, Madia F, Cecconi M, Cardinali S, Cassetti A, Cecconi I, Bertini E, Bianchi A, Gobbi G, Zara F. Familial severe myoclonic epilepsy of infancy: truncation of Nav1.1 and genetic heterogeneity. Epileptic Disord. 2003;5:21 - 5. [PubMed: 12773292]
- Grosso S, Orrico A, Galli L, Di Bartolo R, Sorrentino V, Balestri P. SCN1A mutation associated with atypical Panayiotopoulos syndrome. Neurology. 2007;69:609 - 11. [PubMed: 17679682]
- Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39:508 - 12. [PubMed: 9596203]
- Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM, Sadleir LG, Andermann E, Gill D, Farrell K, Connolly M, Stanley T, Harbord M, Andermann F, Wang J, Batish SD, Jones JG, Seltzer WK, Gardner A, Sutherland G, Berkovic SF, Mulley JC, Scheffer IE. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843 - 52. [PubMed: 17347258]
- Heron SE, Scheffer IE, Iona X, Zuberi SM, Birch R, McMahon JM, Bruce CM, Berkovic SF, Mulley JC. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin. J Med Genet. 2010;47:137 - 41. [PubMed: 19589774]
- Horn CS, Ater SB, Hurst DL. Carbamazepine-exacerbated epilepsy in children and adolescents. Pediatr Neurol. 1986;2:340 - 5. [PubMed: 3508708]
- Iannetti P, Parisi P, Spalice A, Ruggieri M, Zara F. Addition of verapamil in the treatment of severe myoclonic epilepsy in infancy. Epilepsy Res. 2009;85:89 - 95. [PubMed: 19303743]
- Kanai K, Hirose S, Oguni H, Fukuma G, Shirasaka Y, Miyajima T, Wada K, Iwasa H, Yasumoto S, Matsuo M, Ito M, Mitsudome A, Kaneko S. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology. 2004;63:329 - 34. [PubMed: 15277629]
- Kimura K, Sugawara T, Mazaki-Miyazaki E, Hoshino K, Nomura Y, Tateno A, Hachimori K, Yamakawa K, Segawa M. A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev. 2005;27:424 - 30. [PubMed: 16122630]
- Krepischi AC, Knijnenburg J, Bertola DR, Kim CA, Pearson PL, Bijlsma E, Szuhai K, Kok F, Vianna-Morgante AM, Rosenberg C. Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy. Epilepsia. 2010;51:2457 - 60. [PubMed: 21204806]
- Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, Bellan-Koch A, Petrou S, Ahonen VE, Lerche H, Lehesjoki AE. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. 2010;75:1454 - 8. [PubMed: 20956790]
- Livingston JH, Cross JH, Mclellan A, Birch R, Zuberi SM. A novel inherited mutation in the voltage sensor region of SCN1A is associated with Panayiotopoulos syndrome in siblings and generalized epilepsy with febrile seizures plus. J Child Neurol. 2009;24:503 - 8. [PubMed: 19339291]
- Madia F, Striano P, Gennaro E, Malacarne M, Paravidino R, Biancheri R, Budetta M, Cilio MR, Gaggero R, Pierluigi M, Minetti C, Zara F. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology. 2006;67:1230 - 5. [PubMed: 17030758]
- Malow BA, Levy K, Maturen K, Bowes R. Obstructive sleep apnea is common in medically refractory epilepsy patients. Neurology. 2000;55:1002 - 7. [PubMed: 11061259]
- Mantegazza M, Gambardella A, Rusconi R, Schiavon E, Annesi F, Cassulini RR, Labate A, Carrideo S, Chifari R, Canevini MP, Canger R, Franceschetti S, Annesi G, Wanke E, Quattrone A. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci U S A. 2005;102:18177 - 82. [PMC free article: PMC1312393] [PubMed: 16326807]
- Marini C, Mei D, Parmeggiani L, Norci V, Calado E, Ferrari A, Moreira A, Pisano T, Specchio N, Vigevano F, Battaglia D, Guerrini R. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology. 2010;75:646 - 53. [PubMed: 20713952]
- Marini C, Mei D, Temudo T, Ferrari AR, Buti D, Dravet C, Dias AI, Moreira A, Calado E, Seri S, Neville B, Narbona J, Reid E, Michelucci R, Sicca F, Cross HJ, Guerrini R. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007;48:1678 - 85. [PubMed: 17561957]
- Marini C, Scheffer IE, Nabbout R, Mei D, Cox K, Dibbens LM, McMahon JM, Iona X, Carpintero RS, Elia M, Cilio MR, Specchio N, Giordano L, Striano P, Gennaro E, Cross JH, Kivity S, Neufeld MY, Afawi Z, Andermann E, Keene D, Dulac O, Zara F, Berkovic SF, Guerrini R, Mulley JC. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia. 2009;50:1670 - 8. [PubMed: 19400878]
- McIntosh AM, McMahon J, Dibbens LM, Iona X, Mulley JC, Scheffer IE, Berkovic SF. Effects of vaccination on onset and outcome of Dravet syndrome: a retrospective study. Lancet Neurol. 2010;9:592 - 8. [PubMed: 20447868]
- Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW., NEAD Study Group. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med. 2009;360:1597 - 605. [PMC free article: PMC2737185] [PubMed: 19369666]
- Mulley JC, Nelson P, Guerrero S, Dibbens L, Iona X, McMahon JM, Harkin L, Schouten J, Yu S, Berkovic SF, Scheffer IE. A new molecular mechanism for severe myoclonic epilepsy of infancy: exonic deletions in SCN1A. Neurology. 2006;67:1094 - 5. [PubMed: 17000989]
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat. 2005;25:535 - 42. [PubMed: 15880351]
- Mulley JC, Hodgson B, McMahon JM, Iona X, Bellows S, Mullen SA, Farrell K, Mackay M, Sadleir L, Bleasel A, Gill D, Webster R, Wirrell EC, Harbord M, Sisodiya S, Andermann E, Kivity S, Berkovic SF, Scheffer IE, Dibbens LM. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. 2013;54:e122 - 6. [PubMed: 23895530]
- Nabbout R, Copioli C, Chipaux M, Chemaly N, Desguerre I, Dulac O, Chiron C. Ketogenic diet also benefits Dravet syndrome patients receiving stiripentol: a prospective pilot study. Epilepsia. 2011;52:e54 - 7. [PubMed: 21569025]
- Nabbout R, Gennaro E, Dalla Bernardina B, Dulac O, Madia F, Bertini E, Capovilla G, Chiron C, Cristofori G, Elia M, Fontana E, Gaggero R, Granata T, Guerrini R, Loi M, La Selva L, Lispi ML, Matricardi A, Romeo A, Tzolas V, Valseriati D, Veggiotti P, Vigevano F, Vallee L, Dagna Bricarelli F, Bianchi A, Zara F. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 2003;60:1961 - 7. [PubMed: 12821740]
- Nakayama J, Arinami T. Molecular genetics of febrile seizures. Epilepsy Res. 2006;70:S190 - 8. [PubMed: 16887333]
- Nicolai J, Gunning B, Leroy PL, Ceulemans B, Vles JS. Acute hepatic injury in four children with Dravet syndrome: valproic acid, topiramate or acetaminophen? Seizure. 2008;17:92 - 7. [PubMed: 17697789]
- Nikanorova M, Genton P, Johannessen SI, Johannessen Landmark C. Stiripentol. In Nikanorova M, Genton P, Johannessen SI, Johannessen Landmark C. Topics in Epilepsy Volume 4: Orphan Drugs in Epilepsy. Esher, UK: John Libbey Eurotext; 2011:39-47.
- Nolan K, Camfield CS, Camfield PR. Coping with a child with Dravet syndrome: insights from families. J Child Neurol. 2008;23:690 - 4. [PubMed: 18344453]
- Ohmori I, Ouchida M, Kobayashi K, Jitsumori Y, Inoue T, Shimizu K, Matsui H, Ohtsuka Y, Maegaki Y. Rasmussen encephalitis associated with SCN 1 A mutation. Epilepsia. 2008a;49:521 - 6. [PubMed: 18031552]
- Ohmori I, Ouchida M, Kobayashi K, Jitsumori Y, Mori A, Michiue H, Nishiki T, Ohtsuka Y, Matsui H. CACNA1A variants may modify the epileptic phenotype of Dravet syndrome. Neurobiol Dis. 2013;50:209 - 17. [PubMed: 23103419]
- Ohmori I, Ouchida M, Miki T, Mimaki N, Kiyonaka S, Nishiki T, Tomizawa K, Mori Y, Matsui H. A. CACNB4 mutation shows that altered Ca(v)2.1 function may be a genetic modifier of severe myoclonic epilepsy in infancy. Neurobiol Dis. 2008b;32:349 - 54. [PubMed: 18755274]
- Pal DK, Pong AW, Chung WK. Genetic evaluation and counseling for epilepsy. Nat Rev Neurol. 2010;6:445 - 53. [PubMed: 20647993]
- Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O'Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764 - 78. [PMC free article: PMC2749953] [PubMed: 19710327]
- Pereira S, Vieira JP, Barroca F, Roll P, Carvalhas R, Cau P, Sequeira S, Genton P, Szepetowski P. Severe epilepsy, retardation, and dysmorphic features with a 2q deletion including SCN1A and SCN2A. Neurology. 2004;63:191 - 2. [PubMed: 15249644]
- Quilichini PP, Chiron C, Ben-Ari Y, Gozlan H. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABA-A receptor channels. Epilepsia. 2006;47:704 - 16. [PubMed: 16650136]
- Ragona F, Granata T, Dalla Bernardina B, Offredi F, Darra F, Battaglia D, Morbi M, Brazzo D, Cappelletti S, Chieffo D, De Giorgi I, Fontana E, Freri E, Marini C, Toraldo A, Specchio N, Veggiotti P, Vigevano F, Guerrini R, Guzzetta F, Dravet C. Cognitive development in Dravet syndrome: a retrospective, multicenter study of 26 patients. Epilepsia. 2011;52:386 - 92. [PubMed: 21269283]
- Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P. Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology. 2010;215:598 - 605. [PMC free article: PMC3005548] [PubMed: 19457575]
- Rilstone JJ, Coelho FM, Minassian BA, Andrade DM. Dravet syndrome: seizure control and gait in adults with different SCN1A mutations. Epilepsia. 2012;53:1421 - 8. [PubMed: 22780858]
- Riva D, Vago C, Pantaleoni C, Bulgheroni S, Mantegazza M, Franceschetti S. Progressive neurocognitive decline in two children with Dravet syndrome, de novo SCN1A truncations and different epileptic phenotypes. Am J Med Genet A. 2009;149A:2339 - 45. [PubMed: 19764027]
- Rodda JM, Scheffer IE, McMahon JM, Berkovic SF, Graham HK. Progressive gait deterioration in adolescents with Dravet syndrome. Arch Neurol. 2012;69:873 - 8. [PubMed: 22409937]
- Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3 ed. Eastleigh, UK: John Libbey; 2006.
- Saito Y, Oguni H, Awaya Y, Hayashi K, Osawa M. Phenytoin-induced choreoathetosis in patients with severe myoclonic epilepsy in infancy. Neuropediatrics. 2001;32:231 - 5. [PubMed: 11748493]
- Samrén EB, van Duijn CM, Koch S, Hiilesmaa VK, Klepel H, Bardy AH, Mannagetta GB, Deichl AW, Gaily E, Granström ML, Meinardi H, Grobbee DE, Hofman A, Janz D, Lindhout D. Maternal use of antiepileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38:981 - 90. [PubMed: 9579936]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479 - 90. [PubMed: 9126059]
- Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, Xu R, Jackson G, Adams J, Connellan M, Petrou S, Wellard RM, Briellmann RS, Wallace RH, Mulley JC, Berkovic SF. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. 2007;130:100 - 9. [PubMed: 17020904]
- Selmer KK, Lund C, Brandal K, Undlien DE, Brodtkorb E. SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy Behav. 2009;16:555 - 7. [PubMed: 19782004]
- Shi X, Huang MC, Ishii A, Yoshida S, Okada M, Morita K, Nagafuji H, Yasumoto S, Kaneko S, Kojima T, Hirose S. Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J Hum Genet. 2010;55:375 - 8. [PubMed: 20485450]
- Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 2009;31:758 - 62. [PubMed: 19783390]
- Sijben AE, Sithinamsuwan P, Radhakrishnan A, Badawy RA, Dibbens L, Mazarib A, Lev D, Lerman-Sagie T, Straussberg R, Berkovic SF, Scheffer IE. Does a SCN1A gene mutation confer earlier age of onset of febrile seizures in GEFS+? Epilepsia. 2009;50:953 - 6. [PubMed: 19292758]
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, White HS, Filloux F, Leppert MF. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649. [PMC free article: PMC2730533] [PubMed: 19763161]
- Singh R, Andermann E, Whitehouse WP, Harvey AS, Keene DL, Seni MH, Crossland KM, Andermann F, Berkovic SF, Scheffer IE. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia. 2001;42:837 - 44. [PubMed: 11488881]
- Striano P, Mancardi MM, Biancheri R, Madia F, Gennaro E, Paravidino R, Beccaria F, Capovilla G, Dalla Bernardina B, Darra F, Elia M, Giordano L, Gobbi G, Granata T, Ragona F, Guerrini R, Marini C, Mei D, Longaretti F, Romeo A, Siri L, Specchio N, Vigevano F, Striano S, Tortora F, Rossi A, Minetti C, Dravet C, Gaggero R, Zara F. Brain MRI findings in severe myoclonic epilepsy in infancy and genotype-phenotype correlations. Epilepsia. 2007;48:1092 - 6. [PubMed: 17381446]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384 - 9. [PMC free article: PMC33477] [PubMed: 11371648]
- Suls A, Claeys KG, Goossens D, Harding B, Van Luijk R, Scheers S, Deprez L, Audenaert D, Van Dyck T, Beeckmans S, Smouts I, Ceulemans B, Lagae L, Buyse G, Barisic N, Misson JP, Wauters J, Del-Favero J, De Jonghe P, Claes LR. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat. 2006;27:914 - 20. [PubMed: 16865694]
- Suls A, Velizarova R, Yordanova I, Deprez L, Van Dyck T, Wauters J, Guergueltcheva V, Claes LR, Kremensky I, Jordanova A, De Jonghe P. Four generations of epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology. 2010;75:72 - 6. [PubMed: 20484682]
- Takayanagi M, Haginoya K, Umehara N, Kitamura T, Numata Y, Wakusawa K, Hino-Fukuyo N, Mazaki E, Yamakawa K, Ohura T, Ohtake M. Acute encephalopathy with a truncation mutation in the SCN1A gene: a case report. Epilepsia. 2010;51:1886 - 8. [PubMed: 20491869]
- Tanabe T, Awaya Y, Matsuishi T, Iyoda K, Nagai T, Kurihara M, Yamamoto K, Minagawa K, Maekawa K. Management of and prophylaxis against status epilepticus in children with severe myoclonic epilepsy in infancy (SMEI; Dravet syndrome)--a nationwide questionnaire survey in Japan. Brain Dev. 2008;30:629 - 35. [PubMed: 18424028]
- Thanh TN, Chiron C, Dellatolas G, Rey E, Pons G, Vincent J, Dulac O. Arch Pediatr. 2002;9:1120 - 7. [Long-term efficacy and tolerance of stiripentaol in severe myoclonic epilepsy of infancy (Dravet's syndrome)] [PubMed: 12503502]
- Tro-Baumann B, von Spiczak S, Lotte J, Bast T, Haberlandt E, Sassen R, Freund A, Leiz S, Stephani U, Boor R, Holthausen H, Helbig I, Kluger G. A retrospective study of the relation between vaccination and occurrence of seizures in Dravet syndrome. Epilepsia. 2011;52:175 - 8. [PubMed: 21219303]
- Wallace RH, Hodgson BL, Grinton BE, Gardiner RM, Robinson R, Rodriguez-Casero V, Sadleir L, Morgan J, Harkin LA, Dibbens LM, Yamamoto T, Andermann E, Mulley JC, Berkovic SF, Scheffer IE. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;61:765 - 9. [PubMed: 14504318]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001a;28:49 - 52. [PubMed: 11326275]
- Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL Jr, Mulley JC, Berkovic SF. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001b;68:859 - 65. [PMC free article: PMC1275639] [PubMed: 11254444]
- Wallace RH, Scheffer IE, Parasivam G, Barnett S, Wallace GB, Sutherland GR, Berkovic SF, Mulley JC. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology. 2002;58:1426 - 9. [PubMed: 12011299]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366 - 70. [PubMed: 9697698]
- Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8:186 - 94. [PubMed: 12610651]
- Wirrell EC, Laux L, Franz DN, Sullivan J, Saneto RP, Morse RP, Devinsky O, Chugani H, Hernandez A, Hamiwka L, Mikati MA, Valencia I, Le Guern ME, Chancharme L, de Menezes MS. Stiripentol in Dravet syndrome: results of a retrospective U.S. study. Epilepsia. 2013;54:1595 - 604. [PubMed: 23848835]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142 - 9. [PubMed: 16921370]
- Yu MJ, Shi YW, Gao MM, Deng WY, Liu XR, Chen L, Long YS, Yi YH, Liao WP. Milder phenotype with SCN1A truncation mutation other than SMEI. Seizure. 2010;19:443 - 5. [PubMed: 20630778]
- Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011;76:594 - 600. [PubMed: 21248271]
章节说明
修订记录
- 15 May 2014 (me) Comprehensive update posted live
- 10 November 2011 (me) Comprehensive update posted live
- 29 November 2007 (me) Review posted to live Web site
- 13 October 2006 (msm) Original submission