
简介
临床特征 肝静脉闭塞伴免疫缺陷疾病(VODI)的特征在于:(1)原发性免疫缺陷; (2)终末肝小叶血管闭塞和肝纤维化表现为肝肿大和/或肝衰竭。发病通常在六个月大之前。免疫缺陷包括严重的低丙种球蛋白血症,正常水平的循环T细胞,缺乏淋巴结生发中心和组织浆细胞缺乏的T细胞免疫缺陷的临床证据。发生细菌和机会性感染,包括耶氏肺孢子菌感染,皮肤粘膜念珠菌病和肠病毒或巨细胞病毒感染。过去,受累的患者预后很差,如果未诊断或未静脉注射免疫球蛋白(IVIG)和耶氏肺孢子菌的预防,则其生命的第一年死亡率为100%。然而,通过早期识别和治疗,预后得到了显著改善。
诊断/测试 VODI的诊断是根据以下临床诊断标准在先证者中建立的:
如果临床特征尚不确定,则通过分子遗传学检测鉴定SP110中双等位基因的致病变异。
管理 表现的治疗:一旦确诊为VODI,就应预防性使用IVIG和预防耶氏肺孢子菌; 及时地治疗感染;考虑肝移植,尽管并发症发生率可能很高;合适的预处理可以有效地进行骨髓移植。
预防主要表现:IVIG和耶氏肺孢子菌的预防。
监测:定期监测肝功能,血小板计数和血红蛋白水平;常规监测血清和尿液电解质,因为可能会出现抗利尿激素不适当的综合征(SIADH);在输注IVIG之前测量免疫球蛋白的浓度;支气管肺泡灌洗以诊断耶氏肺孢子菌感染;根据需要进行病毒培养或肺功能研究;临床有提示时可通过脑脊髓成像诊断脑白质营养不良。
应避免的药物/情况:已知易导致肝静脉闭塞性疾病(hVOD)的药物,包括环磷酰胺和生物碱/灌木茶。
对有风险的亲属的评估:如果家族中的两个致病变异都已知,则对年龄小于12个月的先证者同胞进行分子遗传学检测,以便进行早期诊断和治疗。
遗传咨询 VODI以常染色体隐性遗传的方式遗传。受累的孩子的父母都是杂合子(携带者),因此携带一种致病性变异。杂合子是无症状的。 受影响个体的每个同胞都有25%的机会发病,有50%的机会成为无症状携带者,有25%的机会不受影响 。如果已知一个家庭的两个致病性变异,可以对高危亲属进行携带者检测,对高危妊娠进行产前诊断。
诊断
提示性发现
以下个体应怀疑肝静脉闭塞伴免疫缺陷(VODI) :
临床诊断标准
- 具有细菌和机会性感染的免疫缺陷的临床证据,包括耶氏肺孢子菌感染,粘膜皮肤念珠菌病和肠病毒或巨细胞病毒感染
- 受累的个人或一级亲属的肝肿大或肝功能衰竭的证据,不能用其他因素解释(通常存在肝静脉闭塞性疾病,可确诊,但可能未发现或已解决)
- 发病通常在六个月大之前
- 家族史与常染色体隐性遗传一致
实验室特点
- IgA,IgM和IgG的血清浓度低
注意:免疫球蛋白水平是特定于年龄和实验室的,因此应与当地参考范围进行比较。
- 正常淋巴细胞数量以及CD4和CD8百分比
- 正常淋巴细胞对有丝分裂原的增殖反应
- 产生低细胞因子
放射学特点
- 肝超声检查。与肝静脉闭塞性疾病(HVOD)一致的特征可能包括肝脾肿大,胆囊壁增厚,门静脉直径增加,肝静脉直径减少,腹水和韧带十二指肠再狭窄。
- 多普勒超声检查。与HVOD一致的特征可能包括门静脉血流,脐旁静脉血流减少,肝动脉阻力增加。
组织病理学特征
HVOD(也称为肝窦阻塞综合征)可能包括3区末端肝小静脉内膜肿胀,管腔狭窄,小叶肝细胞坏死和肝窦充血(见 Figure 1)。
注意:如果肝活检禁忌,则肝超声检查和多普勒超声检查可提供 HVOD的支持证据。
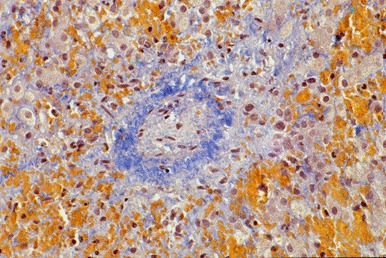
Figure 1.
肝活检显示一个女孩在五个月大时出现肝肿大和腹水,表现为血管闭塞,静脉周围纤维化,3区纤维化和肝细胞脱落(Picro-Mallory染色100x)
建立诊断
VODI的诊断是根据上述临床诊断标准在先证者中建立的。 如果临床特征尚无定论,则通过分子遗传学检测鉴定SP110中双等位基因的致病变异,可以进行诊断(参见Table 1)。
分子检测方法可以包括单基因检测, multigene panel的使用以及更全面的基因组的检测。
- 单基因测试。 如果仅发现一个或没有致病性变异 ,则首先进行SP110的序列分析,然后进行基因靶向的deletion/duplication analysis。
- 也可以考虑包括SP110和其他感兴趣的基因的multigene panel (请参阅 Differential Diagnosis)。注意:(1)基因套餐中包含的基因和每个基因 测试的诊断敏感性因实验室而异,并且可能随时间而变化。 (2)一些基因套餐可能包括与本《基因综述》中讨论的病症无关的基因;因此,临床医生需要确定哪个基因套餐最有可能以最合理的成本既能确定该病的遗传原因,同时减少意义不确定基因的和无法解释表型的致病性变异。 (3)在某些实验室中,基因套餐选项可能包括定制的实验室设计套餐和/或针对表型的外显子组定制分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括序列分析,deletion/duplication analysis和/或其他非基于序列的测试。
Table 1 1.
分子遗传学检测在肝静脉闭塞伴免疫缺陷的应用
| 基因 1 | 测试方法 | 通过该方法可检测出具有致病性变异 2的先证者的比例 |
|---|---|---|
| SP110 | Sequence analysis 3 | 13/13 (100%) 4 |
| Gene-targeted deletion/duplication analysis 5 | Unknown 6 |
- 1.
查染色体 位点 和蛋白质,见Table A. Genes and Databases.
- 2.
有关该 基因中检测到的等位基因变异的信息见Molecular Genetics .
- 3.
- 4.
迄今为止确定的所有致病变异均存在于外显子2、4、5和8中,最常见的是在黎巴嫩人群中发现的c.642delC [Roscioli et al 2006, Ruga et al 2006, Cliffe et al 2012, Wang et al 2012].
- 5.
基因靶向的 deletion/duplication analysis检测基因内的缺失或重复。 可以使用的方法包括:quantitative PCR,远程PCR,依赖多重连接的探针扩增(MLPA)和设计用于检测单外显子缺失或重复的基因靶向微阵列。
- 6.
没有有关基因靶向的deletion/duplication analysis检测率的数据。
临床特征
临床表现
肝静脉闭塞伴免疫缺陷(VODI)是与末梢肝小叶血管闭塞和肝小叶3区(位于中央静脉周围)纤维化相关的原发免疫缺陷。来自澳大利亚悉尼研究的所有儿童均在六个月大以前出现表现,大多数为单独或同时具有肝静脉闭塞性疾病(HVOD)特征的免疫缺陷后遗症(见Table 2)。
免疫缺陷的特征是严重的低球蛋白球蛋白血症,T细胞免疫缺陷的临床证据以及正常数量的循环T和B细胞,淋巴结生发中心缺失以及组织浆细胞缺失 [Roscioli et al 2006]。发生细菌和机会性感染,包括耶氏肺孢子菌肺炎感染,皮肤粘膜念珠菌病和肠病毒或巨细胞病毒感染。
肝静脉闭塞性疾病(hVOD) 90%的VODI儿童从出生开始出现肝肿大(宫内感染为83%)或肝功能衰竭(宫内感染为53%)。通常情况下,就诊婴儿已经病了数周,已经变得呼吸急促,生长障碍,被耶氏肺孢子菌引起的间质性肺炎收治,被发现患有腹水黄疸,患有低血球蛋白血症和血小板减少症,并有hVOD的证据。
神经病学 总体而言,VODI儿童中有30%的神经系统受累。据报道,没有人患有脑静脉阻塞性疾病。四名个体(B II.1和B II.2的姐妹[Table 3]和3名无关儿童)在验尸后发现多器官功能衰竭与广泛的脑坏死有关。一个惊人的发现是三名VODI患者(20%)存在脑脊髓白质营养不良。 5名个体患有病因不明的白质营养不良,而6名个体在CMV相关性胃肠炎后发展为这种并发症。在个体A II.1中,将最初诊断为脑血管意外伴右侧脑白质病变(可能为弓形虫或孔脑囊肿),后修订为脑脊髓白质营养不良。 B II.2个体在静脉免疫球蛋白(IVIG)和cotrimoxazole预防基础上保持良好状态,直到18岁时突然出现与脑部病变相关的轻瘫和尿储留。广泛调查未发现感染原因或静脉闭塞性疾病的证据。对大剂量IVIG和类固醇疗法有部分反应,提示有炎症病因[MW Lin和M Wong,个人交流]。
其他特性 VODI婴儿和儿童没有变形的能力。血小板减少症经常出现,但可能随着hVOD的使用而改善。一个孩子患有ADH分泌不当的综合症,大概是他的脑白质营养不良的表现。
发病率/死亡率 如果无法诊断和未经IVIG或皮下免疫球蛋白(SCIG)替代和吉氏肺孢子菌的治疗,则VODI在生命的第一年死亡率为100%,到青少年期总体死亡率为90% [Roscioli et al 2006]。但是,在最近确定的1岁以上的8位受累的患者中,只有三例死亡,且通过早期识别和治疗,预后明显改善[Cliffe et al 2012]。如果hVOD反复,可以通过继续进行IVIG和预防肺囊虫预防hVOD复发。但是,可能存在神经炎性并发症的持续风险(B II.2)。 1名儿童患者(A II.1,Table 3)在6岁时骨髓移植后hVOD复发后死亡。 Table 2 总结了悉尼20例经VODI临床诊断的个体的临床和免疫学特征(包括11例能够通过分子分析调查证实SP110致病性变异存在的患者)和8例新近确诊的患者[Cliffe et al 2012]。
Table 2
肝静脉闭塞性伴免疫缺陷病的临床和免疫学特征
| 临床表现 | 悉尼患病个体 w/VODI 1 | 备注 | 确诊个人 w/VODI w/Novel 致病变异 2 |
|---|---|---|---|
| 表现症状 <6 months | 20/20 (100%) | 7/8 | |
| 初次出现肝功能衰竭 | 4/20 (20%) | 1/12 HSCT后 3/12 无明显诱因 | 0/8 |
| 初次出现肝肿大 | 9/20 (45%) | 3/6 耶氏肺孢子菌 2/6 肝肿大w/out SOS | 6/8 1/8 肠病毒&弥散性巨细胞病毒 cytomegalovirus 3 |
| 耶氏肺孢子菌肺炎感染 | 12/20 (60%) | 7/12 确诊 5/12 可疑 | 1/8 可疑 3 1/8 确诊 4 |
| 皮肤粘膜念珠菌病 | 2/20 (10%) | 1/8 | |
| 其他表现 | 1/20 (5%) | 19 岁 | 1/8 肺纤维化 5 |
| 死亡 | 19/20 (95%) | 3/8 (38%) | |
| 从初始肝窦阻塞综合征恢复 | 4/20 (20%) | 1 完全康复 1 需肝移植的慢性肝病 1 SOS post-HSCT 1 发育障碍,反复误吸 | 4/8 |
| 神经系统异常 | 6/20 (35%) | 4/7 脑梗塞 2/7 白质营养不良 | 1/8白质营养不良 |
| 全丙种球蛋白过少血症 | 19/19 (100%) | 1/18 4个月大时正常免疫球蛋白的缺失 | 5/5 测试 1/5 开始IVIG后IgA&IgM的水平较低 |
| 正常淋巴细胞数 | 10/11 (92%) | 8/8 | |
| 正常NK细胞 | 12/12 (100%) | 3/3 3, 4, 5 | |
| 胞浆内 IFN-γ, IL-2, IL-4, IL-10减少 | 4/5 (80%) | 4小时低水平,48小时正常/升高水平 | 1/1 3 |
| 记忆T & B cells细胞数量减少 | 3/4 (75%) | 2/3 6 |
Table 3 总结了纯合性SP110致病性变异个体的临床特征 [Roscioli et al 2006, Cliffe et al 2012, Wang et al 2012, Ganaiem et al 2013].
Table 3
SP110纯合子致病变异个体的临床特征
| 患者 | SP110 (LRG_109) 病理变异 | 表现 | 血清Igs | 记忆 T/B 细胞 | T Cell 细胞因子 | 临床发现 | 死亡 |
|---|---|---|---|---|---|---|---|
| A II.1 1 黎巴嫩人 | c.642delC in 外显子 5 | 5个月大:免疫缺陷,血小板减少,SOS | ↓ | N/A | N/A | 左偏瘫 2, 反复性hVOD w/HSCT后伴GVHD | 是 |
| B II.1 1 黎巴嫩人 | 年龄7个月:免疫缺陷 | ↓ | N/A | N/A | 反复吸入继发的慢性肺疾病 | 是 (19岁) | |
| B II.2 1 黎巴嫩人 | 6个月:肝脾肿大,腹水,SOS | ↓ | ↓ | ↓ | 直到18岁(轻瘫,尿储留,脑部病变) | 未 | |
| C II.1 1 黎巴嫩人 | 年龄4个月:肝脾肿大,腹水,SOS,血小板减少症,皮肤粘膜念珠菌病 | ↓ | ↓ | ↓ | 慢性肝病,肝移植后门静脉高压症 | 是 | |
| D II.1 1 黎巴嫩人 | 年龄3个月:肝脾肿大,腹水,SOS | ↓ 3 | ↓ | ↓ | 肝移植后的噬血细胞综合征 | 是 | |
| 16 4 黎巴嫩人 | c.642delC in 外显子 5 | 年龄3个月:肝脾肿大,腹水,SOS | ↓ | ↓ | ↓ | 肺出血,多器官衰竭 | 是 |
| 5 4 黎巴嫩人 | c.642delC in 外显子 5 | 3个月大,呼吸窘迫 | ↓ | ↓ | N/A | SIADH, idiopathic 脑脊髓白质营养不良 | 未 |
| 6 4 黎巴嫩人 | c.642delC in 外显子 5 | 3个月大:慢性咳嗽,腹泻 8岁:肝脾肿大 年龄≥12岁:hVOD | ↓ | N/A | N/A | 特发性左额叶钙化囊肿,癫痫,CMV结肠炎,腹泻后脑脊髓炎伴下肢瘫痪,脑脊髓白质营养不良,食管念珠菌病,十二指肠淋巴细胞浸润 | 未 |
| 7 4 黎巴嫩人 | c.642delC 推定 5 | 年龄2个月:慢性腹泻、生长缓慢,中耳和呼吸道感染,肝脾肿大,血小板减少 | N/A | N/A | N/A | 小头畸形,肝活检与 w/SOS一致 | 是的,在11 个月大时腹泻导致败血性休克 |
| 8 4 黎巴嫩人 | c.642delC 推定5 | 年龄5个月大:上呼吸道疾病 年龄8个月:慢性腹泻,肝肿大,血小板减少症 | N/A | N/A | N/A | 肝活检与 w/SOS一致 | 是的,在3.5岁腹泻导致败血症性休克 |
| 9 4 黎巴嫩人 | c.642delC 推定 5 | 年龄2个月:腹水,肝肿大,贫血,血小板减少 | N/A | N/A | N/A | 肝活检与 w/SOS一致 | 是的,2.5个月中耳炎,腹泻,肺炎 |
| E I.1 1 黎巴嫩人 | c.40delC in 外显子 2 | 年龄3个月:免疫缺陷,血小板减少,肝脾肿大,有/无SOS证据 | ↓ | N/A | N/A | 肠病毒和耶氏肺孢子菌感染 | 是 |
| 4 4 西班牙裔 | c.78_79delinsAT (p.Ile27Leu) in 外显子 2 | 5岁以上:肝脾肿大,发烧,呼吸窘迫,全血细胞减少 | ↓ | ↓ | ↓ | 稳定&好 | 未 |
| 1 4 意大利人 | c.319_325dup GGTGCTT in 外显子 4 | 11个月:肝脾肿大,弥散性巨细胞病毒,轮状病毒肠胃炎,外阴脓肿,SOS | ↓ 开始 | ↓ | N/A | 从hVOD中恢复 | 未 |
| 2 4 意大利人 | c.667+1dup 外显子 5 剪接位点 | 3个月大的患者:肝脾肿大,生长障碍,呼吸窘迫/肺纤维化,腹泻 | ↓ | ↓ | N/A | 肝活检肝窦扩张,中度中央静脉&静脉窦窦底纤维化; 稳定 w/改进 | 未 |
| 3 4 巴勒斯坦阿拉伯人 | c.373del in 外显子 4 | 年龄3个月:发病前先进行级联检测,后确诊VODI。 无肝肿大或肝功能异常 | N/A | N/A | N/A | 稳定&好 | 未 |
Modified from Roscioli et al [2006]
CMV = cytomegalovirus,巨细胞病毒
GVHD = graft-versus-host disease,移植物抗宿主病
HSCT = hematopoietic stem cell transplantation,造血干细胞移植
hVOD = hepatic veno-occlusive disease,肝静脉闭塞性疾病
SOS = sinusoidal obstruction syndrome,肝窦梗阻综合征
SIADH = syndrome of inappropriate antidiuretic hormone secretion,抗利尿激素分泌异常综合征
注意:虽然不知道A,B和C家族是否有关联,但它们被认为具有共同的祖先。 初始纯合性作图分析中包括的个体:A II.1,B II.1,B II.2,C II.1、16(初始分析中为“ G”)和5(初始分析中为“ J”)。
受影响的个人名称是引用的文章中使用的名称。
- 1.
- 2.
继发于脑白质异常
- 3.
使用IVIG时,IgA和IgM血清浓度增加至正常下限。
- 4.
- 5.
基因型-表型相关
在具有不同致病变异的个体之间,未观察到VODI的临床表现有显著差异。
在外显子4中出现框外重复的孩子(个人1, Table 3)在11个月大时(晚于平均水平)出现了弥散性CMV感染,其他VODI儿童中并未发现。此外,记忆性T和B细胞数量正常,细胞因子生成正常-在其他VODI儿童中未观察到发现。
外显率
确认患有由SP110中的致病性变异引起的VODI的个体中, B细胞和T细胞免疫缺陷的外显率为100%。 在所有先证者或其 受累的患者的同胞中都表现出了hVOD。
大约10%的VODI儿童由于 受累的患者同胞而在年轻时确定,并在疾病过程的早期用IVIG治疗,可能仅在出现时表现出免疫缺陷。
命名法
由于饮食和地理上的联系,肝静脉闭塞性疾病以前被称为牙买加丛林茶疾病。现在,该术语已被肝静脉闭塞性疾病(hVOD)或肝窦阻塞综合征(SOS)取代,鉴于在全球范围内出现hVOD,该术语的限制较少。 hVOD和综合免疫缺陷的组合称为VODI。
患病率
VODI最初是由Mellis & Bale [1976]在黎巴嫩裔的澳大利亚人中发现的。随后,大多数报告患有VODI的儿童都是黎巴嫩裔。据计算,澳大利亚悉尼的黎巴嫩人中VODI的患病率为每2500人中就有1人 [Roscioli et al 2006]。来自澳大利亚和黎巴嫩的未公开数据表明,在黎巴嫩背景的家庭中很少发现与SP110外显子5致病性变异c.642delC相关的VODI,特别是与近亲婚配相关的家庭。这反过来表明,在移居黎巴嫩的外籍人士中漏诊了该诊断。
在非黎巴嫩血统的儿童中,VODI的患病率尚不清楚。但是,以下报告表明在其他人群中也观察到VODI表型。其他报告VODI:
- 西班牙裔的单发的VODI病例(即家庭中的单发),具有体液和细胞免疫缺陷
- 两名意大利儿童患有VODI[Ruga et al 2006, Cliffe et al 2012]
- 来自美国的一名患有VODI的西班牙裔儿童 [Cliffe et al 2012, Wang et al 2012]
遗传相关(等位基因)疾病
尚未发现SP110的等位基因相关孟德尔疾病或包括hVOD或免疫缺陷的SP110区域在内的相邻基因疾病。
鉴别诊断
尽管Washington et al [1993]报道的个体中以及在Buckley & Hutchins [1995]报道的一组艾滋病尸检中描述了与严重的联合免疫缺陷症(SCID)相关的肝窦阻塞综合征。在其他类别的免疫缺陷中,伴有肝静脉闭塞性疾病(hVOD)的免疫缺陷的研究表明,hVOD可能是VODI的主要特征,而不是免疫缺陷本身的继发特征。
hVOD的主要鉴别诊断是环境生物碱或细胞毒性相关肝静脉闭塞。然而,hVOD也与酒精性肝硬化 [Kishi et al 1999], ataxia-telangiectasia [Srisirirojanakorn et al 1999],骨质疏松[Corbacioglu et al 2006] (参见CLCN7-Related Osteopetrosis)和高嗜酸性粒细胞增多症(OMIM 607685)相关。 在鉴别免疫性表型时也应考虑HIV。
既往使用单核苷酸多态性(SNP)进行的病例对照研究还报告了hVOD和SNPs 在氨基甲酰磷酸合成酶1(CPS1,见Urea Cycle Disorders Overview),factor V Leiden(FVL),HFE(参见HFE-Associated Hereditary Hemochromatosis)和谷胱甘肽S-转移酶(GSTM1和GSTT1)基因的相关性。据报道,纯合性HFE Cys282Tyr 等位基因的相对风险为8.6,而GSTM1无效等位基因的相对风险为4.12 [Srivastava et al 2004, Kallianpur 2005, Kallianpur et al 2005]。没有对这些发现进行独立的验证。
没有报道称在没有免疫缺陷的情况下具有hVOD的个体中存在SP110致病变异。
管理
初步诊断后的评估
为了确定患有肝静脉闭塞伴免疫缺陷性疾病(VODI)的个体的疾病程度和需求,建议进行以下评估:
- 评估免疫功能,包括血清免疫球蛋白水平,T细胞和B细胞数量和百分比以及对有丝分裂原的T细胞增殖反应
- 如果可以的话,对记忆B和T细胞的数目以及细胞因子(IL2,IL4,IL6和IFNγ)的数量进行更广泛的免疫测试
- 全血细胞计数
- 评估肝功能(包括转氨酶,胆红素和白蛋白的血清浓度)和门静脉高压症(包括贫血和血小板减少症)的后遗症
- 咨询临床遗传学家和/或遗传咨询师
在考虑进行肝活检以进行肝静脉闭塞性疾病(hVOD)的组织学诊断之前,应先进行凝血分析和肝多普勒超声检查。凝血功能障碍和/或门脉高压禁止肝活检。
表现治疗
低球蛋白血症可通过静脉免疫球蛋白(IVIG)进行治疗,应从VODI诊断开始或在确诊具有纯合性SP110致病变异的症状前同胞中开始。合适的剂量为每四周一次0.4 g / kg,调整剂量以维持血IgG水平大于6 g / L。
预防肺孢子菌 VODI儿童应继续使用cotrimoxazole儿科混悬液(5 mL =甲氧苄氨嘧啶40 mg和磺胺甲恶唑200 mg,如果耐受该药物 )预防肺孢子菌 。这可以每日单剂或每周三天的单剂给药。推荐剂量为每公斤5 mg甲氧苄啶(0.625 mL / kg)或150 mg / M2 (3.75 mL / M2 )。
特定病原体的感染 应给予适当的支持,并使用抗菌药或抗病毒药。
可以考虑进行肝移植,但在迄今为止研究的VODI队列中,并发症的发生率很高(请参见Other)。
骨髓移植 Ganaiem et al [2013] 报告说,采用适当的预处理可能是一种有效的治疗方式(请参阅 Other)。
预防主要表现
在诊断时应开始常规IVIG预防与严重低血球蛋白血症相关的感染, cotrimoxazole预防耶氏肺孢子菌感染 (请参见Treatment of Manifestations)。
预防继发并发症
一些证据表明,在VODI早期进行免疫缺陷治疗可以降低hVOD发生或复发的风险。
监测
以下是适当的:
- 定期监测VODI儿童的肝功能,血小板计数和血红蛋白水平,因为可能发生肝衰竭和门脉高压
- 可能会发生抗利尿激素(SIADH),进行血清和尿液电解质监测
- 在IVIG输注之前测量免疫球蛋白浓度
- 支气管肺泡灌洗以诊断耶氏肺孢子菌感染;根据需要进行病毒培养或肺功能研究
- 临床指证时可进行脑脊髓成像诊断白质营养不良
避免的药物/情况
应避免易产生hVOD的物质(例如环磷酰胺和生物碱/灌木茶)。
评估处于危险中亲戚
评估未满12个月的先证者同胞 ,以便尽早确定将从治疗和预防措施中受益的人。大多数VODI儿童在6个月大之前就已经出现症状;但是,如果一个孩子在11个月大时出现症状,则应考虑在12个月以下的先证者同胞中进行测试。
评估包括:
- 如果家族中的致病变异已知,行分子遗传学检测;
- 如果不知道该家族的致病变异,则在出生时进行血清免疫球蛋白,全血细胞计数和肝功能检查,并在六个月时重复进行。
迄今为止描述的VODI个体的外显率为完全外显(即100%);因此,不建议对年龄超过12个月的先证者的健康高风险同胞进行 分子遗传学检测。
有关为遗传咨询目的而对高危亲戚进行测试的相关问题,请参见Genetic Counseling 。
孕期管理
对于一名受累的孕妇,在怀孕期间进行静脉内免疫球蛋白预防与严重低血球蛋白血症相关的感染,以及cotrimoxazole预防耶氏肺孢子菌感染是适当的。有证据表明,对因SP110纯合性致病性变异的婴儿进行早期治疗可能会改善长期预后。
正在调查的疗法
在美国搜索ClinicalTrials.gov ,在欧洲搜索www.ClinicalTrialsRegister.eu,以获取有关各种疾病和状况的临床研究信息。注意:可能没有针对该疾病的临床试验。
其他
在澳大利亚人群中,报道的hVOD是 VODI在造血干细胞移植(HSCT)后发生 。因此,HSCT后,VODI患者可能有hVOD的风险。 Ganaiem et al [2013]报道了首例成功的HSCT,表明这可能是采用适当预处理方案的有效治疗方式。 Ganaiem等人描述了一个近亲婚配的阿拉伯家庭,有八个受累的人。五人获得了HSCT,其中三人获得了成功。所有五个患者都接受了匹配的同胞或家庭成员骨髓(4位患者)或外周干细胞(1位患者)。所有五个人都接受了包含氟达拉滨和血清疗法以及烷化剂(环磷酰胺,白消安或曲奥舒凡)的预处理 。死亡的两个人也接受了塞替派,移植后死于hVOD和多器官衰竭。一个孩子在2014年完成单纯性HSCT后一直保持良好状态两年。她来自黎巴嫩血统,在四个月大时出现了可能的疱疹性肝炎,最初对阿昔洛韦产生临床反应,但在输血后发展为hVOD。她对IVIG,预防性Bactrim™和去纤蛋白反应良好。她的神经学正常,但是尽管凝血研究和血小板已经正常化,但在一周前的头部超声检查正常后,常规的移植前MRI发现无症状的双侧硬脑膜下血肿。在六个月大的时候,她接受了无关的脐带血移植,并使用了阿仑单抗,氟达拉滨,海藻糖和抗胸腺细胞球蛋白调节剂。在患者移植后两个月,预防性地将去纤罗肽继续使用。她的神经系统正常,两岁时的MRI扫描未显示解剖异常[M Wong,个人交流]。
2016年,一名澳大利亚近亲婚配的背景的澳大利亚婴儿在四个月大时出现肺囊肿,CMV感染和hVOD伴低血红蛋白血症。在分子遗传学检测. 上鉴定出纯合性SP110 致病性变异 (c.642delC)。在控制肺炎和hVOD的五个月大时,他接受了去除α/βTCR和CD19 +细胞后单倍父本HSCT。他接受了氟达拉滨,海藻糖和抗胸腺细胞球蛋白的预处理方案。给予去纤罗肽预防hVOD。移植后两个月,他状况良好,并且移植后无GVHD或hVOD复发,但患有中度CMV肝炎,在第60天停用Defibrotide。受累的患者和供体在移植前均为CMV阳性。 HSCT后70天,他发生了癫痫发作。 MRI显示整个大脑和脊髓广泛异常,包括水肿和环状增强病变区域。 CT显示广泛的钙化。 CMV视网膜炎很明显,CSF和玻璃体均为CMV PCR阳性[T Cole,J Smart和T Soosay Raj,个人交流]。
在移植方案中加入去纤蛋白预防可以提高生存率。预处理方案中去除第二种烷基化剂可能会降低与移植相关的hVOD的风险。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 下面介绍遗传风险评估以及家族史和基因检测的使用,以阐明家族成员的遗传状况。 本节的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —编者。
遗传模式
肝静脉闭塞伴免疫缺陷性疾病(VODI)以常染色体隐性遗传的方式遗传,完全外显。
家庭成员的风险
先证者的父母
先证者的同胞
- 受累的患者的每个同胞都有25%的机会受到感染,有50%的机会成为无症状的携带者,有25%的机会不受影响。
- 年龄大于12个月且已知未受影响的同胞有2/3的风险成为携带者。
- 杂合子(携带者)无症状,没有患病的风险。
先证者的后代 患有免疫缺陷的肝静脉闭塞性疾病的个体的后代是SP110中致病性变异的杂合子(携带者)。
先证者的其他家庭成员。先证者父母的每个同胞都有SP110致病性变异的携带者风险。
携带者(杂合子)检测
对高危亲属进行携带者测试需要事先确定该家族中SP110的致病变异。
相关的遗传咨询问题
有关评估12个月以下的高风险亲戚以进行早期诊断和治疗的信息,请参阅管理,Evaluation of Relatives at Risk。
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。 由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑建立受累的患者DNA银行。
产前检查和植入前遗传学诊断
一旦在一个受累的家庭成员中发现了SP110的致病变异,就可以对风险增加的妊娠进行产前检测,并可以对VODI进行植入前遗传诊断 。
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或登记册,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- International Patient Organisation for Primary Immunodeficiencies (IPOPI)FirsideMain RoadDownderry Cornwall PL11 3LEUnited KingdomPhone: +44 01503 250 668Fax: +44 01503 250 668Email: info@ipopi.org
- Jeffrey Modell Foundation/National Primary Immunodeficiency Resource Center747 Third AvenueNew York NY 10017Phone: 866-463-6474 (toll-free); 212-819-0200Fax: 212-764-4180Email: info@jmfworld.org
- European Society for Immunodeficiencies (ESID) RegistryDr. Gerhard KindleUniversity Medical Center Freiburg Centre of Chronic ImmunodeficiencyEngesserstr. 479106 FreiburgGermanyPhone: 49-761-270-34450Email: esid-registry@uniklinik-freiburg.de
分子遗传
分子遗传学和OMIM表中的信息可能与GeneReview中其他地方的信息不同:表可能包含最新信息。 —编者
Table A
肝静脉闭塞伴免疫缺陷性疾病:基因和数据库
| 基因 | 染色体定位 | 蛋白 | 位点数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| SP110 | 2q37 | Sp110 nuclear body protein | SP110 database SP110base | SP110 | SP110 |
Table B
肝静脉闭塞伴免疫缺陷性疾病的OMIM条目 (View All in OMIM)
基因结构 SP110编码三种主要的异型体:
- Sp110异型体A,NM_004509.3 (平均质量78.438 kd;转录本不包括 外显子17)
- 异型体B,NM_004510.3(平均质量61.940 kd;转录本包含一个替代的 外显子15,终止于外显子15内)
- 异型体C,NM_080424.2(平均质量81.211 kd;包括外显子17并终止于外显子19的全长转录本)
致病变异 参见Table 4.这些致病变中的大多数会导致移码变异,从而导致蛋白质截短。 c.642delC致病性变异已在黎巴嫩血统的多个先证者中得到鉴定[Roscioli et al 2006, Cliffe et al 2012].
Table 4
本基因综述中讨论的SP110致病变异
| DNA 核酸改变 (Alias 1) | 外显子 | 蛋白改变 | 参考文献 | 参考序列 |
|---|---|---|---|---|
| c.40delC | 2 | p.Gln14SerfsTer25 | Roscioli et al [2006] | NM_080424 NP_536349 |
| c.78_79delinsAT (78_79CA>AT) | 2 | p.Ile27Leu | Cliffe et al [2012], Wang et al [2012] | |
| c.319_325dupGGTGCTT | 4 | p.Ser109TrpfsTer5 | Ruga et al [2006], Cliffe et al [2012] | |
| c.373delA | 4 | p.Thr125LeufsTer3 | Cliffe et al [2012], Ganaiem et al [2013] | |
| c.642delC | 5 | p.Pro214ProfsTer14 | Roscioli et al [2006] | |
| c.667+1dupG | 5 | NA | Cliffe et al [2012] |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变体的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会(varnomen
- .hgvs.org )的标准命名约定。 有关命名法的说明,请参见 Quick Reference 。NA =不适用
- 1.
不符合当前命名约定的变异名称
正常基因产物. Sp110主要在白细胞和脾脏中表达。它是由干扰素γ和全反式维甲酸(ATRA)诱导的。据描述,SP110同工型B显示出作为视黄酸受体α(RARα)的有效转录共阻遏物的活性,可能是通过竞争性排斥受体上的活化剂 [Watashi et al 2003].
Sp110核体蛋白是Sp100 / Sp140早幼粒细胞白血病核体(PML NB)蛋白家族的成员。该蛋白具有一个Sp100结构域(氨基酸6-159),可与其他Sp100家族蛋白进行二聚化,一个核定位信号(氨基酸288-306)和一个核激素相互作用域(LXXLL型),充当ATRA响应元素。参与染色质介导的 基因转录的模块化蛋白质的共同特征的其他 结构域包括SAND结构域(氨基酸452-532),植物同源框结构域(氨基酸537-577)和溴结构域(氨基酸606-674)[Bloch et al 2000].。
Sp110核体蛋白与PML NB(一种核大分子复合物)相关,其被部署到活性宿主或参与到病毒DNA复制,转录和修复的区域,据报道与细胞凋亡,细胞周期控制和免疫反应有关。
基因产物异常。来自具有VODI和 纯合性 失活SP110变异的个体的EBV转染的B细胞显示,在正常数量的PML核体中,没有核Sp100特异性免疫标记。该发现与Sp110蛋白在免疫应答中具有重要作用而不是PML核体形成必不可少的一致[Roscioli et al 2006].
与VODI相关的大多数致病变异是功能丧失性变异。 c.78_79delinsAT(p.Ile27Leu)致病性变异发生在高度保守的Sp100 结构域内的高度保守的p.Ile27处(请参见正常基因产物)。 c.78_79delinsAT(p.Ile27Leu)致病变异大大降低了Sp110的稳定性[Cliffe et al 2012].
病理生理学 目前尚不清楚hVOD是SP110序列变异的直接表现,还是与肝窦的凋亡改变有关,还是继发于感染。然而,hVOD似乎在感染发生后发展。
参考文献
Literature Cited
- Bloch DB, Nakajima A, Gulick T, Chiche JD, Orth D, de La Monte SM, Bloch KD. Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Mol Cell Biol. 2000;20:6138 - 46. [PMC free article: PMC86089] [PubMed: 10913195]
- Buckley JA, Hutchins GM. Association of hepatic veno-occlusive disease with the acquired immunodeficiency syndrome. Mod Pathol. 1995;8:398 - 401. [PubMed: 7567938]
- Cliffe ST, Bloch DB, Suryani S, Kamsteeg EJ, Avery DT, Palendira U, Church JA, Wainstein BK, Trizzino A, Lefranc G, Akatcherian C, Megarbané A, Gilissen C, Moshous D, Reichenbach J, Misbah S, Salzer U, Abinun M, Ong PY, Stepensky P, Ruga E, Ziegler JB, Wong M, Tangye SG, Lindeman R, Buckley MF, Roscioli T. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol. 2012;130:735 - 742.e6. [PubMed: 22621957]
- Corbacioglu S, Honig M, Lahr G, Stohr S, Berry G, Friedrich W, Schulz AS. Stem cell transplantation in children with infantile osteopetrosis is associated with a high incidence of VOD, which could be prevented with defibrotide. Bone Marrow Transplant. 2006;38:547 - 53. [PubMed: 16953210]
- Ganaiem H, Eisenstein EM, Tenenbaum A, Somech R, Simanovsky N, Roscioli T, Weintraub M, Stepensky P. The role of hematopoietic stem cell transplantation in SP110 associated veno-occlusive disease with immunodeficiency syndrome. Pediatr Allergy Immunol. 2013;24:250 - 6. [PubMed: 23448538]
- Kallianpur AR. Genomic screening and complications of hematopoietic stem cell transplantation: has the time come? Bone Marrow Transplant. 2005;35:1 - 16. [PubMed: 15489868]
- Kallianpur AR, Hall LD, Yadav M, Byrne DW, Speroff T, Dittus RS, Haines JL, Christman BW, Summar ML. The hemochromatosis C282Y allele: a risk factor for hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2005;35:1155 - 64. [PubMed: 15834437]
- Kishi M, Maeyama S, Ogata S, Koike J, Uchikoshi T. Hepatic veno-occlusive lesions in severe alcoholic hepatitis and alcoholic liver cirrhosis: a comparative histopathological study in autopsy cases. Alcohol Clin Exp Res. 1999;23:47S - 51S. [PubMed: 10235278]
- Mellis C, Bale PM. Familial hepatic venoocclusive disease with probable immune deficiency. J Pediatr. 1976;88:236 - 42. [PubMed: 1249685]
- Roscioli T, Cliffe ST, Bloch DB, Bell CG, Mullan G, Taylor PJ, Sarris M, Wang J, Donald JA, Kirk EP, Ziegler JB, Salzer U, McDonald GB, Wong M, Lindeman R, Buckley MF. Mutations in the gene encoding the PML nuclear body protein Sp110 are associated with immunodeficiency and hepatic veno-occlusive disease. Nat Genet. 2006;38:620 - 2. [PubMed: 16648851]
- Ruga EM, Guariso G, Antiga LD, Guido M, Fassan M, Elia RD, Ziegler JB, Roscioli T, Cliffe ST, Buckley MF, Zancan L. Hepatic veno-occlusive disease with immunodeficiency syndrome: case report. Budapest, Hungary: 12th Meeting of the European Society for Immunodeficiencies. 2006.
- Srisirirojanakorn N, Finegold MJ, Gopalakrishna GS, Klish WJ. Hepatic veno-occlusive disease in ataxia-telangiectasia. J Pediatr. 1999;134:786 - 8. [PubMed: 10356154]
- Srivastava A, Poonkuzhali B, Shaji RV, George B, Mathews V, Chandy M, Krishnamoorthy R. Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood. 2004;104:1574 - 7. [PubMed: 15142875]
- Wang T, Ong P, Roscioli T, Cliffe ST, Church JA. Hepatic veno-occlusive disease with immunodeficiency (VODI): first reported case in the U.S. and identification of a unique mutation in Sp110. Clin Immunol. 2012;145:102 - 7. [PubMed: 22982295]
- Washington K, Gossage DL, Gottfried MR. Pathology of the liver in severe combined immunodeficiency and DiGeorge syndrome. Pediatr Pathol. 1993;13:485 - 504. [PubMed: 8372033]
- Watashi K, Hijikata M, Tagawa A, Doi T, Marusawa H, Shimotohno K. Modulation of retinoid signaling by a cytoplasmic viral protein via sequestration of Sp110b, a potent transcriptional corepressor of retinoic acid receptor, from the nucleus. Mol Cell Biol. 2003;23:7498 - 509. [PMC free article: PMC207568] [PubMed: 14559998]
本章节注解
更新历史
- 12 January 2017 (sw) 系统性更新发布到公开网页上
- 3 July 2013 (me)系统性更新发布到公开网页上
- 15 September 2009 (me) 系统性更新发布到公开网页上
- 21 February 2007 (me) 内容发布到公开网页上
- 29 November 2006 (mb) 初稿提交