
概述
临床特征 .
PIK3CA相关过度生长谱 (PROS) 包括一系列临床发现,其中核心特征是先天的或儿童早期出现的节段性/局灶性过度生长,伴有或不伴有细胞发育不良。在将 PIK3CA 确定为致病基因之前,PROS 根据所涉及的组织和/或器官分为不同的临床综合征(例如,MCAP [巨脑-毛细血管畸形] 综合征和 CLOVES [先天性脂肪瘤性躯干不对称过度生长、淋巴管、毛细血管、静脉和混合型血管畸形、表皮痣、骨骼和脊柱异常]综合征)。过度生长的主要区域包括大脑、四肢(包括手指和脚趾)、躯干(包括腹部和胸部)和面部,通常都呈不对称分布。脑过度生长可能伴有特定脑结构的继发性过度生长,导致脑室扩大、胼胝体明显增厚和小脑扁桃体异位伴后颅窝拥挤。血管畸形可能包括毛细血管畸形、静脉畸形和不太常见的动脉或混合(毛细血管-淋巴-静脉或动静脉)畸形。淋巴畸形可能出现在不同的位置(内部和/或外部),并可能导致各种临床问题,包括肿胀、疼痛和偶尔继发于外伤的局部出血。脂肪瘤过度生长可能发生在血管畸形的同侧或对侧(如果存在)。智力障碍的程度似乎主要与癫痫发作、皮质发育不良(例如多小脑回)和脑积水的存在和严重程度有关。许多孩子有喂养困难,这些困难通常是多因素的。内分泌问题影响少数个体,最常见的包括低血糖症(主要是低胰岛素性低酮症低血糖症)、甲状腺功能减退和生长激素缺乏症。
诊断/测试.
PROS 的诊断建立在先证者具有提示性发现和杂合的嵌合(或很少纯合的)激活 PIK3CA 中的致病性变异。来自临床受影响组织样本的 DNA 序列分析——最好是来自覆盖受影响区域的新获得的皮肤活检、来自过度生长组织的手术切除或来自未培养的组织(如皮肤成纤维细胞或其他组织)——应优先用于基因检测.对于体细胞变异检测,建议对整个 PIK3CA 编码区进行靶向捕获,然后在非常深的覆盖范围内进行 next-generation sequencing,因为它可以检测整个基因中非常低水平的嵌合。
管理.
症状的治疗:明显或脂肪瘤节段过度生长可能需要减瘤;脊柱侧弯和腿长差异可能需要骨科护理和手术干预。神经系统并发症(例如,阻塞性脑积水、颅内压升高、进行性和/或有症状的小脑扁桃体异位或 Chiari 畸形,以及脑过度生长/畸形患者的癫痫)可能需要神经外科干预。根据血管畸形的类型,可以使用硬化疗法、激光疗法或口服药物(如西罗莫司)。同样,淋巴畸形可以通过口服药物或仔细的手术减瘤来治疗,最好由血管异常小组来治疗。对于有疼痛的人,建议评估疼痛的来源并治疗根本原因。对于那些生长激素缺乏症,可以考虑试用生长激素治疗,仔细监测生长和过度生长。如果存在以下情况,则需要常规治疗:心脏和肾脏异常;智力障碍和行为问题;多指和足部畸形;凝血病或血栓形成;肾母细胞瘤;甲状腺功能减退症;和低血糖。
监测:每次访视:测量生长参数,包括头围、臂长、手、腿和脚;评估新的神经系统表现(癫痫发作、语气变化和 Chiari 畸形的其他体征/症状);监测发育进程和行为;评估运动技能;脊柱侧弯的临床评估和器官肿大和/或腹部肿块的腹部检查。建议进行连续头部 MRI 成像,频率取决于初始评估结果的严重程度和大脑成熟程度。对于中枢神经系统过度生长或发育不良的患者,每六个月进行一次脑部 MRI 检查,直到 2 岁,然后每年进行一次,直到 8 岁,以专门监测进行性脑积水和 Chiari 畸形。根据临床指示:对任何血管和/或淋巴管畸形进行临床评估和监测;肢体或肢体部分过度生长者的四肢 X 光片;对躯干过度生长的患者进行超声或 MRI 随访;脊柱侧弯或影响脊柱的畸形患者的脊柱 MRI。血液学咨询,并提供任何手术干预后评估血栓形成和凝血病风险的建议,特别是对于那些有 CLOVES表型和/或血管畸形的人。考虑每三个月进行一次肾脏超声检查,直到 8 岁(肾母细胞瘤的肿瘤筛查存在争议)。
遗传咨询.
PROS 疾病是不遗传的,因为大多数已确定的致病变异是体细胞性(嵌合)。迄今为止,尚未报告确认的垂直传播或同胞发生。 PIK3CA 致病性变异对先证者同胞的风险预计与普通人群相同。除了少数受影响的 PROS 个体外,所有受累个体都具有 PIK3CA 致病性变异的体细胞嵌合体,这表明突变发生在多细胞胚胎的一个细胞中受精后。因此,遗传给后代的风险预计低于 50%。
GeneReview
| PIK3CA 相关过度生长谱 (PROS):包括表型 1, 2 |
|---|
|
有关同义词和既往的名称,请参见 Nomenclature.
- 1.
对于这些表型的其他遗传原因,请参见 Differential Diagnosis.
- 2.
诊断
PIK3CA 相关过度生长谱 (PROS) 包含一系列临床发现,其中核心特征是在没有类似受影响个体的家族史的情况下, 先天的或儿童早期出现节段性/局灶性过度生长伴或不伴细胞发育不良(即在一个家庭中单次出现)。 在将 PIK3CA 确定为致病基因之前,PROS 根据所涉及的组织和/或器官分为不同的临床综合征(见GeneReview Scope).
提示性发现
具有以下临床、脑部 MRI 和家族史发现的个体应考虑 PROS [Keppler-Noreuil et al 2015, Mirzaa et al 2016, Kuentz et al 2017].
临床表现
- 任何多种组织的过度生长,包括(但不限于)脑、脂肪、血管、肌肉、骨骼、神经
- 血管畸形,包括(但不限于)毛细血管、静脉、动静脉或混合畸形
- 淋巴畸形
- 皮肤发现,包括表皮痣和色素沉着过度斑
- 手或脚的单个或多个异常(例如,大指、并指、多指、凉趾间隙)
- 肾脏畸形
- 良性肿瘤,肾母细胞瘤和肾母细胞瘤病除外(即弥漫性或多灶性持续胚胎细胞簇)
脑MRI检查结果。局灶性脑过度生长(伴有或不伴有皮质发育不良)包括:
- 半巨脑畸形(HMEG)
- 局灶性皮质发育不良(FCD)
- 发育不良性巨脑畸形(DMEG)
家族史。因为 PIK3CA 通常是由de novo嵌合致病性变异引起的,所以大多数先证者代表单发的病例(即在一个家族中单发)。极少数情况下,家族史可能与常染色体显性遗传一致(例如,多代受累的男性和女性)。
建立诊断
PROS 的诊断建立在先证者具有提示性发现和杂合的镶嵌(或很少纯合)激活 PIK3CA 中的致病性变异 [Keppler-Noreuil et al 2015](见Table 1和Molecular Genetics)。
分子诊断。分子基因检测包括靶向检测(单基因组基因检测或 multigene panel检测),虽然可以使用全基因组的检测(外显子组测序, 基因组测序)。由于大多数报告的 PIK3CA 致病变异是受精后的(因此是镶嵌的),因此可能需要测试一种以上的组织(在大多数情况下不包括血液):
- 经验表明,对来自临床受影响组织样本的 DNA 进行序列分析——最好是来自覆盖受影响区域的新获得的皮肤活检,来自过度生长组织的手术切除,或来自未培养的组织(例如,皮肤成纤维细胞或其他组织)——应该优先考虑用于基因检测。
- 受影响组织或培养细胞中激活变体的嵌合水平变化很大[Keppler-Noreuil et al 2014, Kuentz et al 2017].
- 基于目前的技术,不建议对血液或血液中的DNA 孤立的进行检测,因为除了 24 名患有巨脑-毛细血管畸形 (MCAP) 综合征的个体中的两人外,尚未在血液中发现 PIK3CA 致病性变异,他们有明显的 PIK3CA de novo 胚系 致病性变异[Rivière et al 2012].
基因靶向测试要求临床医生确定可能涉及哪些 基因,而基因组的测试则不需要。 具有Suggestive Findings中描述的独特发现的个体很可能使用基因靶向检测进行诊断(参见Option 1),而未考虑诊断为 PROS 的个体可以使用基因组检测进行诊断(参见 Option 2),如果使用适当的样品。
选项1
当表型发现提示诊断为 PROS 时,分子遗传学检测可以包括单基因检测或使用包括PIK3CA的multigene panel 。
- 注:(1)PROS中观察到的致病变异均与功能获得有关;因此,不推荐以目标基因的deletion/duplication analysis 。 (2) 未能检测到激活的 PIK3CA致病性变异并不排除在具有提示性发现的个体中诊断为 PROS,因为在许多受影响的个体中观察到低水平的嵌合 [Kurek et al 2012, Lee et al 2012, Lindhurst et al 2012, Jansen et al 2015, Mirzaa et al 2016].
- 可以考虑在适当的样本(见above)上包含 PIK3CA 和其他感兴趣的基因(见Differential Diagnosis)的multigene panel。注:(1)panel 中包含的基因和用于每个基因的检测的诊断工具敏感性因实验室而异,并且可能随时间而变化。 (2) 一些多基因 panel 可能包含与本 GeneReview 中讨论的疾病无关的基因。 (3) 在一些实验室,panel 选项可能包括定制的实验室设计的panel 和/或定制的以表型为重点的外显子组分析,其中包括临床医生指定的基因。 (4) panel 中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。
选项 2
当由于个体具有非典型表型特征而未考虑 PROS 的诊断时,可以考虑对适当样本进行基因组的测试(见上文)。
全基因组的测试不需要临床医生确定可能涉及哪些基因。 外显子组测序是最常用的;基因组测序也是可能的。
有关全面 基因组的测试的介绍,请单击here。 有关订购基因组检测的临床医生的更多详细信息,请参见here。
Table 1
PIK3CA相关过度生长谱中使用的分子遗传学检测
| 基因1 | 方法 |
| |||
|---|---|---|---|---|---|
| PIK3CA | 测序 3, 4 | 100% 5 | |||
| 靶向基因 deletion/duplication analysis 6 | 无报道7 | ||||
- 1.
查染色体 位点 和蛋白质,见Table A. Genes and Databases.
- 2.
有关该 基因中检测到的等位基因变异的信息见Molecular Genetics .
- 3.
- 4.
体细胞 PIK3CA 变异检测方法的选择(见Molecular Genetics)取决于几个因素。一些制造商已经开发了基于 PCR 的检测方法,用于检测癌症中常见的特定 PIK3CA 变异,其中许多变异也在 PROS 个体中发现。数据表明,虽然存在突变热点(例如,PIK3CA 密码子 542、545 和 1047),但仍有大量罕见的致病变异。
- 5.
- 6.
基因靶向deletion/duplication analysis 检测基因内缺失或重复。使用的方法可能包括 quantitative PCR、长程 PCR、多重连接依赖性探针扩增 (MLPA) 和设计用于检测单外显子缺失或重复的基因靶向微阵列。
- 7.
在 PROS 中观察到的致病变异都与功能获得有关。因此,部分或全部 PIK3CA 缺失和重复不太可能导致这种表型
临床特征
临床表现
PIK3CA 相关过度生长谱 (PROS) 包括可能伴有或不伴有细胞发育不良的广泛组织的过度生长。 在了解 PROS 的分子性质之前,临床上描述了许多不同但重叠的表型并命名(参见 GeneReviews Scope)。
一般而言,PROS 可分为 孤立的(当一个人的局灶性病变仅影响一个组织或身体部位;见 Table 2)和一个 综合征性(即过度生长加上至少两个其他特征) 两个系统;见 Table 3)。
Table 2
孤立的 PIK3CA 相关过度生长表型受影响器官或组织
| 器官或组织 | 表型 | 注解 |
|---|---|---|
| 脑/头部 | HMEG: 大脑过度生长影响 1 半球 w/或 w/o 皮质发育不良 |
|
| 局灶性皮质发育不良 1 |
| |
| DMEG: 伴有皮质发育不良的局灶性脑过度生长; 皮质发育不良可能是双侧的。 |
| |
| 面部浸润性脂肪瘤 |
| |
| 四肢 | 偏侧发育不全 |
|
| 大指 |
| |
| 纤维脂肪血管异常 |
| |
| 大指或或营养不良巨脂肪瘤 |
| |
| 淋巴管 7 | 孤立的淋巴管畸形:由淋巴管内皮细胞排列的扩张的血管通道 | 充满液体的囊肿通常与受累者的生长成比例地生长; 如果它们是浸润性的,则可能 → 疼痛和/或发病。 |
| 血管 7 | 血管畸形 | 包括毛细血管、静脉或混合畸形 |
| 皮肤 |
| 皮肤损伤通常是良性的。 |
过度生长会影响身体的任何部位,这取决于 PIK3CA 致病性变异在各种组织和器官中的分布。
DMEG = 发育不良性巨脑畸形; HMEG = 半巨脑畸形
- 1.
通常在组织学上以具有发育不良的神经元、球囊细胞和分层解体为特征
- 2.
- 3.
癫痫发作通常是部分性的,可能包括婴儿痉挛、强直性癫痫发作或 Ohtahara 综合征的电临床特征。
- 4.
可能与牙齿发育早熟、巨牙症、半巨舌症、舌头和颊黏膜上的突起以及黏膜神经瘤有关
- 5.
- 6.
由于静脉通道过度肌肉化,导致罕见的高流量变异。可能存在组织性血栓。
- 7.
毛细血管、淋巴和静脉通道的过度生长有时被称为 CLVM(毛细淋巴静脉畸形),其中扩张的淋巴通道与静脉和毛细血管成分结合。
Table 3
部分PIK3CA相关过度生长综合征表型
| 表型 1 | 过度生长类型 | 畸形/异常 | |||
|---|---|---|---|---|---|
| 皮肤和血管 | 肌肉骨骼 | 内脏 | 神经病学 | ||
| CLOVES (See Figure 1.) |
|
|
|
|
|
| CLAPO | 部分/全身软组织和骨骼 |
| |||
| FH or FAO |
|
|
|
| |
| HHML |
| 多发性脂肪瘤 | |||
| KTS | 单侧肢体骨骼和/或软组织过度生长 |
| 数字放大 | ||
| MCAP or M-CM (See Figures 2, 3.) |
| 皮肤血管畸形,尤其是面部皮肤毛细血管畸形 | 皮肤并指和轴后多指或多指 皮下脂肪瘤 | 肾母细胞瘤(少) |
|
| MPPH 3 | 大脑过度生长 4 |
|
| ||
CLAPO = 下唇毛细血管畸形、面部和颈部淋巴畸形、不对称和部分/全身过度生长; CLOVES = 先天的脂肪瘤过度生长、血管畸形、表皮痣、脊柱侧弯/骨骼和脊柱; DD = 发育迟缓; EN = 表皮痣; FH 或 FAO = 纤维脂肪增生或过度生长; HHML = 偏侧增生多发性脂肪瘤; HMEG = 半巨脑畸形; ID =智力障碍; KTS = Klippel-Trenaunay 综合征; MCAP 或 M-CM = 巨脑-毛细血管畸形; MPPH = 巨脑-多小脑回-多指-脑积水综合征
- 1.
最常见的发现;有关更多信息,请参见text。
- 2.
还可能包括皮质发育不良、多小脑回、Arnold-Chiari 畸形和脑室扩大
- 3.
- 4.
可能包括巨脑畸形、脑积水和多小脑回
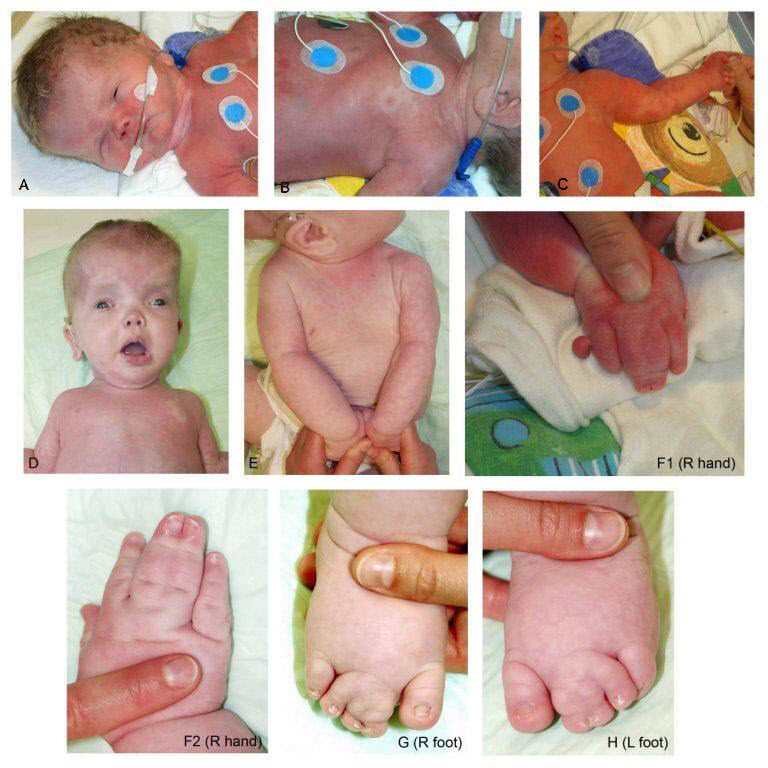
MCAP 综合征的特征。 MCAP 综合征患者的照片显示前额明显的大头畸形(D); 广泛的毛细血管畸形(A-F1); 双侧 2-3-4 脚趾并指(G,H); 3-4 指并指(F1,F2); 和右手轴后多指(F1)
来自Mirzaa et al [2012]。 经许可使用。
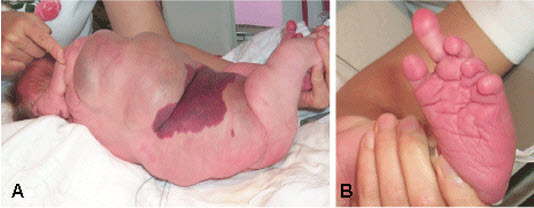
儿童 CLOVES 综合征的特征:(A) 大的脂肪瘤躯干肿块延伸到周围组织和上覆毛细血管畸形和 (B) 左脚大指

MCAP 综合征的特征。一个患有 MCAP 综合征的 40 个月大的男孩(左)和他未受影响的双胞胎妹妹(右)。注意左侧半肥大、典型的面部特征、双侧 2-3 趾并指和结缔组织发育不良伴松弛的多余皮肤。
来自Conway et al [2007b]。经许可使用
过度生长
PIK3CA 致病性功能获得性变异可导致几乎任何类型的组织过度生长。过度生长的开始通常是先天的或出生后早期,与婴儿后期或儿童时期的发病相反。
主要受累区域包括大脑、四肢(包括手指和脚趾)、躯干(包括腹部和胸部)和面部,所有这些区域通常呈不对称分布。
过度生长的组织可能具有“气球状”外观——即,受累的身体部位(通常是手指、脚趾和/或手或脚背)类似于膨胀的气球。
单方面受累比双边受累更常见。
过度生长可能包括以下部分或全部组织类型:
- 纤维性,包括纤维脂肪血管异常个体中环绕神经的致密纤维组织(见 Table 2)
- 神经(见 Brain Growth。)
- 血管(见Vascular Malformations。)
- 淋巴(见Lymphatic Malformations。)
- 骨骼(见Skeletal Findings。)
- 脂肪瘤(见 Lipomatous Overgrowth with or without Regional Reduction of Adipose Tissue。)
大脑发育
全脑过度生长可能伴有特定脑结构的继发性过度生长,导致脑室扩大、明显增厚的胼胝体和小脑扁桃体异位伴后颅窝拥挤[Conway et al 2007b, Mirzaa et al 2012]。成人 OFC 的范围从 +2 到高于平均值 +10 SD。
巨脑病。尽管大多数受影响的个体在出生时由于巨脑畸形 (MEG) 而患有大头畸形,但少数受影响的个体在出生时头部大小正常,但在出生后第一年内由于进行性 MEG 而发展为进行性大头畸形[Moore et al 1997, Conway et al 2007a, Conway et al 2007b, Mirzaa et al 2012].
在因阻塞性脑室扩大或脑积水而接受神经外科分流术的儿童中,头部生长明显以加速的速度继续,这表明 MEG 在将 MEG 作为其 PROS 发现的一部分的个体中的主要性质。
MCAP 综合征。 在对 21 名儿童的回顾中,出生后枕额周长 (OFC) 通常比胎龄平均值高出 +2 至 +7 个标准差 [Mirzaa et al 2012; 作者,未发表的数据]。
在大多数儿童中,OFC SD 在生命的第一年增加。 尽管头部生长在儿童早期可能会趋于平稳,但通常保持在平均值之上 +3 SD 或更多。
血管畸形
血管畸形可能包括毛细血管畸形、静脉畸形和不太常见的动脉或混合(毛细血管-淋巴-静脉或动静脉)畸形。 在躯干或四肢过度生长上可能发现低流量血管畸形(淋巴、静脉)。 血管畸形可能是浅表的或深部的(内脏的)。 其中许多病变只能通过 MRA/MRV 成像识别(见ISSVA Classification for Vascular Anomalies - 2018)。
- 皮肤毛细血管畸形可能位于面部中线(持续性单纯性痣)或遍及全身,呈网状外观,类似于橘皮。
- 包括动脉瘤在内的静脉畸形 (VM) 的特征是血管通道扩大和扭曲,随着时间的推移可能会增长并导致严重的发病率,包括出血、疼痛和毁容。
- 受影响的个体发生深静脉血栓形成和肺栓塞的风险可能增加,尤其是那些合并毛细血管-淋巴-静脉畸形的个体 [Douzgou et al 2022].
- 手术或硬化疗法后风险增加。
- 血栓形成风险也可能因慢性淤滞的其他原因而增加,包括行动不便(例如脱水、手术)、抗凝蛋白减少以及特定致病性 PIK3CA 变异对血管内皮的影响 [Keppler-Noreuil et al 2019].
- 也可能发生高流量血管畸形(动静脉),尤其是涉及脊髓-椎旁区域。
- 患有 Klippel-Trenaunay表型的人可能患有血管瘤和静脉和/或淋巴管畸形,并且有患 Kasabach-Merritt 综合征的风险,其特征是血小板减少和凝血障碍。
淋巴畸形
淋巴畸形可能出现在不同的位置(内部和/或外部),并可能导致各种临床问题,包括肿胀、疼痛和偶尔继发于外伤的局部出血。有些人可能有复杂的淋巴异常,尤其是全身性淋巴异常[Rodriguez-Laguna et al 2019]。复杂的淋巴异常可能具有侵袭性病程,难以治疗且预后不良,尤其是在靠近头颈部前部重要结构的情况下。
骨骼发现
手部的特征性发现包括宽阔的铲状手,手指张开或尺侧偏斜,一根或多根手指过度生长。
足部的特征性发现包括过度生长,大脚趾和第二脚趾之间有大的“凉鞋”间隙,大的球状脚趾,背侧和足底表面都有脂肪瘤,或者前掌宽阔,跖骨头之间有宽阔的间隙。
其他缺陷可能包括轴后、轴前或中央多指和皮肤并指,这通常涉及脚趾,但可能包括手指。皮肤并指以 2-3 趾、2-4 趾和 2-5 趾与凉鞋趾的模式出现。
可能会发生膝盖脱臼、腿长差异和模式软骨软化症。
一些受影响的个体可能有脊柱侧弯、脊椎异常、脊柱裂和/或胸肌异常,特别是在 CLOVES表型中。
FH 或FAO 表型患者有进行性骨骼过度生长。
具有 MCAP 表型的个体可能由于结缔组织发育不良而出现关节过度活动。
伴有或不伴有脂肪组织局部减少的脂肪瘤过度生长
脂肪瘤过度生长可能发生在血管畸形的同侧或对侧(如果存在)。特征性的躯干脂肪瘤浸润周围组织,通常需要手术切除。严重的脊柱侧弯、大的躯干肿块、伴有脊髓缺血的椎旁高流量病变、淋巴管畸形、皮肤囊泡、足部和手部的骨科问题以及中枢性静脉扩张症/血栓栓塞是需要积极或预防性医疗干预的重要疾病的例子(见Management)。
- 椎旁和椎管内延伸,更常见于 CLOVES 表型个体,对脊髓、鞘囊和神经根的压迫存在显著风险,导致包括脊髓病在内的主要神经功能缺损,需要及时诊断和多学科护理 [Alomari 2009].
- 脂肪瘤病可以是侵入性的,侵入髋关节和椎内空间,这会变得非常痛苦。
- 还描述了脂肪组织通过替换或压缩肌肉浸润到肌肉中,以及进入内脏(肝脏、脾脏、胰腺)、肠、纵隔和脊柱。
- 由于脂肪瘤组织的血管分布和血栓形成的风险,手术可能很困难。
- PIK3CA 致病性 功能获得性变异可导致脂肪组织的局部减少,这通常伴随着身体另一部分的显著过度生长。例如,在下半身或下肢/足部明显过度生长的患者中,已观察到上肢、胸部或上腹部的脂肪组织减少。
发育迟缓和智力障碍
智力障碍 (ID) 的程度似乎主要与癫痫发作、皮质发育不良(例如多小脑回)和脑积水(见Neuroimaging)的存在和严重程度有关。粗大运动延迟可能归因于受影响个体的多种因素,包括 MEG 的存在、皮质脑畸形、张力减退、肢体不对称或过度生长以及结缔组织发育不良。
MCAP 综合征
- 大多数患有 MCAP 综合征的人都有一些智力障碍;程度是可变的,从轻度学习障碍到严重残疾不等。
- 大多数有轻度中度延迟,但稳定进展,尽管速度较慢。
- 少数(<10%)有严重的障碍。 MCAP 综合征患者的预期里程碑获得范围尚未明确。
其他神经发育特征
张力减退。患有多小脑回(特别是额叶区域)的年幼儿童有张力减退,没有痉挛,并且可能有假性延髓问题;年龄较大的儿童通常有痉挛和假性延髓问题 [Mirzaa et al 2012].
婴儿喂养困难。尽管很少需要使用饲管,但许多儿童可能存在本质上通常是多因素的,,喂养困难(例如,由于肌张力减退、GERD)。
癫痫。估计有 30%-40% 的 PIK3CA 致病性变异个体患有癫痫症。报告的癫痫发作类型包括局灶性和强直阵挛(除其他外),但癫痫发作类型、严重程度、发病年龄以及任何相关的脑电图或神经影像异常主要取决于致病性 PIK3CA 变异的组织分布以及相关皮质的存在畸形与否。
小脑扁桃体异位(Chiari 畸形)的症状。婴儿可能有易怒、过度流口水、吞咽困难或呼吸问题,尤其是中枢性呼吸暂停。儿童可能有颈部疼痛或头痛、运动无力、感觉变化、视力问题、吞咽困难或行为改变。
行为问题和自闭症特征
一部分患有 MCAP 综合征的儿童 (6/21) 具有自闭症特征或临床诊断为自闭症 [Mirzaa et al 2012],这表明自闭症可能是少数将 MEG 作为 PROS 一部分的个体的神经认知特征的一部分 [McBride et al 2010]。在一个或几个受影响的个体中看到的其他行为异常:
- 注意力缺陷/多动障碍
- 强迫倾向
- 焦虑相关问题
神经影像学
患有大头畸形的 PROS 个体通常在出生后不久或出生后第一年内进行脑成像,从而早期识别以下关键神经成像特征(见
Figure 4) [Vogels et al 1998, Nyberg et al 2005, Conway et al 2007a, Conway et al 2007b, Martínez-Lage et al 2010, Mirzaa et al 2012].
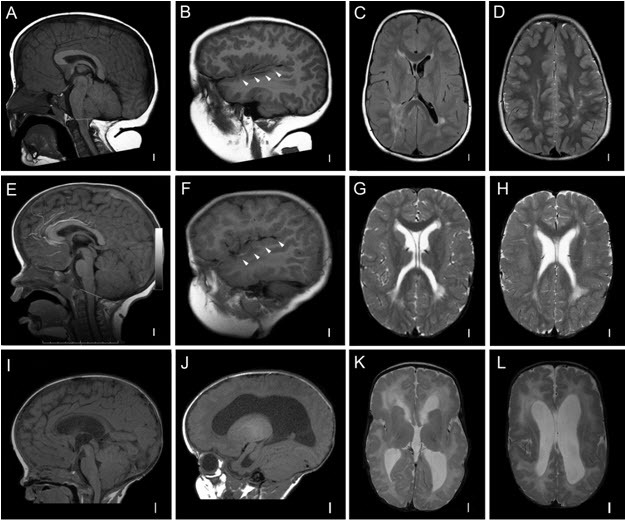
巨脑畸形 [Clayton-Smith et al 1997, Vogels et al 1998, Robertson et al 2000, Nyberg et al 2005, Coste et al 2012]。在某些情况下,产前超声检查可检测到 MEG(伴有或不伴有脑室扩大),并伴有胼胝体增厚。超过 90% 的受影响个体具有普遍进展的 先天的MEG。
脑室扩大和脑积水。大多数受影响的儿童在早期脑成像中有脑室扩张或脑室扩大的证据:
- 在对 MCAP 综合征患者的神经影像学检查结果的大型回顾中,65 名儿童中有 37 名(56%)患有脑室扩大,范围从轻度到明显的脑积水,伴有或不伴有小脑扁桃体异位[Conway et al 2007b]。
- 虽然尚不清楚所有这些个体的脑室扩大是否是阻塞性的,但超过一半的受影响儿童通常在出生后的第一年内接受心室分流术或第三脑室造口术。
小脑扁桃体异位(CBTE)。大的小脑合并小后颅窝导致小脑扁桃体异位(Chiari 畸形)和脊髓空洞症是常见的并发症,尤其是在 MCAP 综合征患者中:
- 在报告的 65 名 MCAP 患者中,有 15 名有 CBTE 伴或不伴疝的证据 [Conway et al 2007b]。
- 通过测量小脑扁桃体在枕骨大孔下方的距离,最好客观地评估异位的程度。
- 与脑室扩大不同,CBTE 在 MCAP 综合征患者中很少见于先天的。
- 在两个个体中,CBTE 在后续成像中的自发“消退”归因于不成比例地加速的颅骨过度生长[Mirzaa et al 2012]。
皮质脑畸形和多小脑回(PMG)。 PMG 可能存在于超过 50% 的受影响儿童中,最常见的是 MCAP 表型 [Conway et al 2007b; Gripp et al 2009; Mirzaa et al 2012;作者,未发表的数据]。
- MCAP 综合征患者最常见的 PMG 类型是双侧外侧裂 PMG,但也有其他类型,包括双侧额叶和局灶性 PMG。
- 广泛的 PMG,尤其是双侧外周 PMG,会增加以下风险:癫痫;口腔运动无力导致进食、吞咽和表达语言困难;发育迟缓;和音调异常。
肿瘤
良性肿瘤。最常见的是血管,被描述为(海绵状)血管瘤、血管瘤、血管平滑肌脂肪瘤和血管肿块[Clayton-Smith et al 1997, Moore et al 1997, Martínez-Glez et al 2010].
- 海绵状血管瘤已经发生在脑组织中,当它们扩大并引起疼痛时需要进行减瘤。
- 虽然最常见于皮肤或皮下组织,但血管瘤已在内脏和颅骨中被描述。
其他良性肿瘤包括两名患有脑膜瘤(不会扩大、扩散或转移)的 MCAP(年龄分别为 21 个月和 5 岁)患者;两名患有脊髓和主要神经纤维瘤的 PROS 患者;以及其他一些患有卵巢囊腺瘤、子宫肌瘤和脂肪瘤的患者 [Keppler-Noreuil et al 2014]。
恶性肿瘤。如果存在,大多数受影响的个体患有良性肿瘤,仅报告了少数恶性肿瘤 [Kurek et al 2012, Keppler-Noreuil et al 2014, Luks et al 2015, Gripp et al 2016, Hucthagowder et al 2017, Kuentz et al 2017, Peterman et al 2017, Postema et al 2017].。
肾母细胞瘤的估计频率范围为 1.4% 至 3.3%。在报告患有Wilms 瘤或肾母细胞瘤病的 12 人中,临床 PROS 诊断包括 CLOVES(8 人)、MCAP(2 人)和 KTS(2 人)。
- 诊断时的平均年龄为 27.4 个月(中位数 18 个月;范围 9-119 个月)。
- 肿瘤类型包括 7 名 (~60%) 的Wilms 瘤个体,4 名 (33%) 有肾母细胞瘤特征,以及 1 名 (8%) 的肾母细胞瘤病。
- 六个 (50%) 具有体细胞"热点" PIK3CA 变异(参见 Molecular Genetics)。
有几例 PROS 患者发生其他癌症的病例报告,包括以下 [Moore et al 1997, Schwartz et al 2002, Mills et al 2018]:
- 白血病(MCAP 表型)
- 前庭神经鞘瘤
- 视网膜母细胞瘤
目前系统数据不足以确定 PROS 与这些类型肿瘤的发展之间是否存在真正的关联,或者病例报告是否代表 PROS 与这些肿瘤的罕见共同发生。
其他
肾。肾脏畸形常见于患有 PROS 的个体,更具体地说是 CLOVES表型,包括盆腔扩张、输尿管扩张、肾积水、肾动脉重复、肾囊肿和肾脏增大。
皮肤。在 PROS 中观察到的异常情况:
- 皮肤黑色素细胞痣
- 咖啡牛奶斑
- 色素减退斑
- 橘子皮
- 色素痣
- 遵循 Blaschko 线条的片状色素沉着过度
- 线性角质形成细胞表皮痣,可能发生在身体的任何部位,并且可能遵循皮肤分布
- 脂溢性角化病和良性苔藓样皮肤病
- 由于结缔组织发育不良,MCAP 表型患者的皮肤弹性过度、松弛和皮下组织较厚
内分泌问题影响少数个体,最常见的包括低血糖症(主要是低胰岛素性低酮症低血糖症)、甲状腺功能减退和生长激素缺乏症 [Mirzaa et al 2016, Leiter et al 2017, Davis et al 2020].
- PROS 是先天的高胰岛素血症的临床表型(见Molecular Genetics),但低血糖时的血浆胰岛素浓度无法检测到。
- 在患有 MCAP 的个体和具有重叠形式的 PROS(包括大脑受累)的个体中都报告了这种情况。
心脏问题。在一些 MCAP 综合征患者中报告了结构性心脏缺陷(例如,心房和室间隔缺损)和/或大血管异常[Mirzaa et al 2016].
基因型-表型相关性
PIK3CA 中有几个突变热点(见Molecular Genetics和 Table 8),它们更常见与高度局灶性表型(丁香综合征、纤维脂肪增生、淋巴/血管畸形、半巨脑畸形和局灶性皮质发育不良)相关,即:p.Glu542Lys, p.Glu545Lys, p.His1047Arg, p.His1047Lys [Mirzaa et al 2016]。这些突变热点,当存在于大脑中时,也与更严重的癫痫表型相关[Pirozzi et al, in press]。在患有 PROS 的个体和患有 PIK3CA 相关癌症的未受影响个体中可能发现相同的 PIK3CA 错义变异(表 8 和 9)。
此外,对 PROS 个体的分析表明,特别是 MCAP 综合征主要与广泛分布在基因中的广泛 PIK3CA 变异相关,不太可能与 PIK3CA 突变热点相关 [Mirzaa et al 2016](参见Molecular Pathogenesis )。
命名法
由于体细胞 PIK3CA 致病性变异引起的疾病的表型变异性,2013 年 NIH 研讨会上的研究人员提出了 PIK3CA 相关过度生长谱的总称 [Keppler-Noreuil et al 2015]。 PIK3CA 相关的过度生长谱包括所有独特的、临床定义的实体,但突出了表型诊断之间的连续性和重叠。
患病率
由于确定的变化和广泛的表型谱,PIK3CA 相关过度生长谱 (PROS) 的患病率难以估计。已有超过 200 例 MCAP 综合征患者的报告。
遗传相关(等位基因)疾病
除本 GeneReview 中讨论的表型外,没有已知表型与 PIK3CA 中的胚系致病变异相关。
在没有任何其他 PROS 发现的情况下,作为单个肿瘤发生的散发性肿瘤(包括乳腺、结肠、子宫和其他肿瘤)通常在 PIK3CA 中含有 胚系中不存在的体细胞变异。 有关更多信息,请参阅Cancer and Benign Tumors.。
鉴别诊断
许多过度生长和巨脑畸形疾病与 PIK3CA 相关过度生长谱 (PROS) 重叠,总结在 Table 4.
Table 4
PIK3CA 相关过度生长谱 (PROS) 鉴别诊断中的相关基因
| 基因(s) | 鉴别诊断疾病 | MOI | 鉴别诊断的临床特征 | |
|---|---|---|---|---|
| 共同点 w/PROS | 与PROS鉴别点 | |||
| AKT1 | Proteus syndrome | NA (嵌合) | 局灶性体细胞过度生长、表皮痣、血管畸形、发育不良脂肪组织 | 脑结缔组织痣和出生后过度生长的发作(与 PROS 中的先天的发作相比)。 没有特征性的躯干脂肪血管肿块、脊柱椎旁快速血流病变、CLOVES综合征的肢端异常 |
| AKT3 CCND2 PIK3R2 | Megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndrome | AD (de novo) or 嵌合 | 脑过度生长 (MEG)、多小脑回、脑积水、多指、结缔组织或关节松弛 | 没有一致的血管/淋巴畸形或严重的局灶性体细胞过度生长 |
| HRAS KRAS NRAS | 线状皮脂腺痣综合征(LNSS) (OMIM 163200) | NA (嵌合) | 皮肤发现(包括表皮痣和血管畸形) | 没有明显的组织过度生长或更广泛的血管/淋巴畸形 |
| MTOR | Smith-Kingsmore 综合征 (SKS) (OMIM 616638) | AD (de novo) or 嵌合 | 脑过度生长 (MEG)、多小脑回、皮肤发现(包括色素沉着痣) | 没有持续的血管/淋巴畸形 |
| PTCH1 SUFU | Basal cell nevus syndrome | AD | 脑过度生长 (MEG)、多指、并指 | 煅烧钙化、BCC、颌囊肿、表皮囊肿、宽肋骨、许多其他骨骼和其他多系统特征 |
| PTEN | PTEN hamartoma tumor syndrome (PHTS) | AD | 脑过度生长 (MEG)、血管畸形(包括毛细血管畸形)、脂肪瘤 | 肠道错构瘤、阴茎色素斑、没有明显的局灶性躯体过度生长、肢端畸形、↑ 特定癌症类型的癌症风险 |
| SOLAMEN 综合征(phenotypic subtype of PHTS) | 见注解 1. | 节段性过度生长、脂肪瘤、动静脉畸形、表皮痣 | ↑ 癌症风险(卵巢囊腺瘤、多发性乳腺肿瘤、甲状腺腺瘤)、纤维囊性乳腺疾病、牙龈丘疹、多结节性甲状腺肿 | |
| TSC1 TSC2 | Tuberous sclerosis complex (TSC) | AD | 大脑过度生长 (MEG, HMEG, FCD) | 没有明显的局灶性过度生长和持续的血管/淋巴畸形; 存在白色痣、鱼鳞斑、↑ 多系统癌症风险 |
处理
已出版的PIK3CA相关过度生长谱的临床实践指南 [Douzgou et al 2022] (full text).
初步诊断后的评估
为了确定具有 PIK3CA 相关过度生长谱 (PROS) 的个体的疾病程度和需求,建议进行Table 5中总结的评估(如果未作为导致诊断的评估的一部分执行)。
注意:评估因患有这种疾病的个体的不同发现而变得复杂。 准确和彻底的病史评估对于评估血管畸形和其他临床特征是必要的。
Table 5
PIK3CA 相关过度生长谱个体初步诊断后的推荐评估
| 系统/器官 | 评估 | 备注 |
|---|---|---|
| 系统 (过度生长) | 测量生长参数,包括头围、全身长度、手臂、手、腿和脚的长度。 | 评估全身和节段性过度生长(包括腿长差异)和大头畸形 |
| 考虑全身MRI。 | 在那些 w/躯干过度生长 | |
| 考虑肢体 X 光片和随后的肢体 MRI。 | 在那些伴有肢体节段性或全身性过度生长的情况下 | |
| 考虑婴儿的脊柱超声和老年人的脊柱 MRI(w/MR 血管造影)。 | 在那些有脊柱受累证据的患者中(另请参见本表中的心血管/血管。) | |
| 疼痛和功能障碍的临床评估 | ||
| 系统 ( 生长迟缓或广义生长限制) | IGF1 和 IGFBP3 的测量 | 如果低,考虑转诊给内分泌科医生和可能的 GHD 评估。 |
| 神经系统 | 神经学评估 |
|
| 生长 | 发育评估 |
|
| 精神科/行为的 | 神经精神评估 | 年龄 >12 个月的人:筛查行为问题,包括睡眠障碍、ADHD、焦虑和/或暗示 ASD 的特征。 |
| 肌肉骨骼 | 骨科 / 物理医学与康复 / PT/OT 评估 | 包括评估:
|
| 胃肠道 | 腹部检查器官肿大和腹部肿块 | 考虑成像,包括腹部超声和/或 MRI。 |
| 心血管/血管 | 体格检查侧重于外部血管畸形,记录分布和可能的类型 | 如果血管畸形涉及四肢,考虑对手臂和/或腿部的主要血管进行多普勒超声检查; 如果发现,考虑进一步 MRI w/& w/o MRA。 |
| 脊柱的MRI & MRA | 评估可能的高位脊髓和椎旁血管病变 | |
| EKG & 超声心动图 | 用于评估心律失常、先天的心脏缺陷和/或临床指示的大血管异常以及具有 MCAP 表型的患者 | |
| 淋巴/脂肪瘤 | 不对称或非典型水肿或脂肪瘤的体格检查,尤其是臀部和脊柱周围 | 考虑对婴儿进行多普勒超声检查和/或对老年人进行 MRI/MRA/MRV 扫描。 |
| 泌尿生殖器 | 肾脏超声 | 评估肾母细胞瘤和/或肾畸形和/或肾积水 |
| 皮肤 | 全皮肤评估 | 用于血管畸形和色素异常的证据 |
| 内分泌 | 婴儿血糖水平的测量 | 评估低血糖的证据 |
| IGF1 & IGFBP3测量 | 对有/生长受限或生长不良的人进行 GHD 的评估 | |
| TSH & 游离T4 | 评估甲状腺功能减退 | |
| 血液 | 血液学咨询以获取基线评估的建议 |
|
| 感染 | 感染评估 | 可考虑对患有淋巴血管畸形的患者使用预防性抗生素; 每个血管外科医生和介入放射学血管专家 |
| 遗传咨询 | 通过遗传学专业人士 1 | 告知受影响的人及其家人有关 PROS 的性质、MOI 和影响,以促进医疗和个人决策 |
| 家庭支持 & 资源 | 评估:
|
ADHD =注意力缺陷/多动障碍; ASD = 自闭症谱系障碍; EEG = 脑电图; GHD = 生长激素缺乏症; IGF1 = 胰岛素样生长因子 1; IGFBP3 = 胰岛素样生长因子 BP3; MOI = 遗传模式; MRA = 磁共振血管造影; MRV = MR 静脉造影; OT = 职业治疗; PT = 物理治疗; T4 = 甲状腺素; TSH = 促甲状腺激素
- 1.
医学遗传学家、认证遗传咨询师、认证高级遗传护士
治疗表型
理想情况下,管理应通过包括外科医生、放射科医生、遗传学家、皮肤科医生、病理学家和血液科医生/肿瘤科医生在内的多学科团队的协调护理来完成,后者对于新兴的医疗管理和相关长期随访的协调至关重要
[Adams & Ricci 2019, Dekeuleneer et al 2020, Canaud et al 2021, Douzgou et al 2022].
Table 6
PIK3CA 相关过度生长谱个体表现的治疗
| 表型/关注 | 治疗 | 注意事项/其他 |
|---|---|---|
| 部分过度生长 | 可能需要减瘤手术 | 如果功能受限或疼痛为中度至重度 |
| 腿长差异 | 每个骨科医生的标准治疗 | 如果长度差异 >2 厘米,可能需要矫正鞋 |
| 巨脑畸形/脑室扩大 | 每位神经外科医生的标准治疗; 可能包括脑室腹腔分流术或第三脑室造口术 |
|
| 小脑扁桃体 异位或Chiari 畸形 | 每位神经外科医生的标准治疗; 可能包括后颅窝减压 |
|
| 癫痫 3 | 由经验丰富的神经科医生或癫痫科医生进行标准化的 ASM 治疗 |
|
| 考虑半球切除术或手术切除癫痫病灶 5 | 在那些通过神经影像学、脑电图或癫痫症的临床符号学发现的焦点或支持性发现中 | |
| DD/ID | 见 Developmental Delay / Intellectual Disability Management Issues. | |
| 精神科/行为的 | 每个精神科医生和/或发育儿科医生的标准治疗 | 见 Social/Behavioral Concerns. |
| 多指 | 考虑删除多余的数字。 | 每名骨科医生 |
| 足部畸形 /张开的脚趾 | 可以考虑手术干预; 每个骨科医生 | 考虑鞋子和改进功能 |
| 脊柱侧弯 | 每个骨科医生的标准治疗 | |
| 血管畸形 | 取决于血管畸形的类型:硬化疗法、激光疗法或口服药物(例如西罗莫司) |
|
| 心脏结构 缺陷/心律失常 | 每位心脏病专家的标准治疗 | |
| 淋巴管畸形 | 每个血管异常组的标准治疗 | 可能包括仔细的手术减瘤或口服药物(见 Therapies Under Investigation.) |
| 脂肪瘤 | 仔细手术切除浸润性肿块,通常需要多学科管理 6 | 椎旁和椎管内伸展对脊髓、硬膜囊和神经根的压迫有很大的风险。 |
| 肾脏异常 /肾积水 | 每个泌尿科医师和/或肾病科医师的标准治疗 | |
| Wilms 瘤 | 每个肿瘤学家的标准治疗 | |
| 凝血病或血栓形成 | 每个血液学家的标准治疗取决于凝血问题; 可能包括血栓形成的抗凝治疗或凝血病的新鲜冷冻血浆输注 | 那些带有 CLOVES表型的人特别有发生术后高凝状态→血栓形成的风险。 |
| 疼痛 | 评估疼痛来源并治疗根本原因,例如血管畸形、过度生长的继发影响(神经撞击、内脏器官受压)或功能障碍。 | |
| 甲状腺功能减退 | 每位内分泌专家的标准治疗 | 更有可能在那些 w/MCAP 或其他形式的包括大脑参与的 PROS 中7 |
| 低血糖 | 使用口服和/或静脉注射葡萄糖的标准治疗 | 通常只影响新生儿; 少数人在以后的生活中出现过低血糖。 |
| 如果低血糖是由于生长激素缺乏引起的,考虑 GH 治疗 | GH 治疗的数据有限,以及在该人群中是否禁忌。 | |
| 生长激素不足 | 考虑试用 GH 疗法. 7 |
|
| 家庭/社区 |
|
|
ASM = 抗癫痫药物; DD/ID = 发育迟缓/智力障碍; GH = 生长激素; OT = 职业治疗; PT = 物理治疗
- 1.
婴儿可能有易怒、过度流口水、吞咽困难或呼吸问题,尤其是中枢性呼吸暂停。
- 2.
儿童可能有颈部疼痛或头痛、运动无力、感觉变化、视力问题、吞咽困难或行为改变。
- 3.
- 4.
对父母/看护人进行有关常见癫痫发作的教育是适当的。 有关诊断为癫痫儿童的非医疗干预和应对策略的信息,请参阅 Epilepsy & My Child Toolkit.
- 5.
- 6.
- 7.
发育迟缓/智力残疾管理问题
以下信息代表美国发育迟缓/智力障碍人士的典型管理建议;标准建议可能因国家/地区而异。
年龄 0-3 岁。建议转诊至早期干预计划以获得职业、身体、言语和喂养治疗以及婴儿心理健康服务、特殊教育者和感觉障碍专家。在美国,早期干预是一项联邦政府资助的计划,在所有州都可以使用,提供家庭服务以满足个人治疗需求。
年龄3-5岁。在美国,建议通过当地公立学区进行发展学前教育。在安置之前,会进行评估以确定所需的服务和治疗,并为那些根据既定的运动、语言、社交或认知延迟而符合条件的人制定个性化教育计划 (IEP)。早期干预计划通常有助于这种过渡。发育学前班以中心为基础;对于因病情太不稳定而无法就诊的儿童,提供居家服务。
老少皆宜。建议咨询发育儿科医生,以确保适当的社区、州和教育机构(美国)的参与,并支持父母最大限度地提高生活质量。需要考虑的一些问题:
- 个性化教育计划 (IEP) 服务:
- IEP 为符合条件的儿童提供专门设计的指导和相关服务。
- IEP 服务将每年进行审查,以确定是否需要进行任何更改。
- 特殊教育法要求参与 IEP 的儿童在学校可能的限制在最小的环境中,并在适当的时间和地点尽可能多地纳入普通教育。
- 视力和听力顾问应该是孩子 IEP 团队的一部分,以支持获取学术材料。
- IEP 中将提供 PT、OT 和语音服务,前提是需求会影响孩子获得学术材料的机会。除此之外,可以考虑根据受影响个体的需求进行私人支持疗法。关于治疗类型的具体建议可以由发育儿科医生提出。
- 当孩子进入青少年时期时,应讨论过渡计划并将其纳入 IEP。对于接受 IEP 服务的人,公立学区必须提供服务直到 21 岁。
- 504 计划(第 504 条:禁止基于残疾的歧视的美国联邦法规)可以考虑为那些需要帮助的人考虑,例如前排座位、辅助技术设备、教室抄写员、课间额外时间、修改作业和放大的文本。
- 建议注册发育障碍管理局 (DDA)。 DDA 是一家美国公共机构,为合格的个人提供服务和支持。 资格因州而异,但通常由诊断和/或相关的认知/适应性障碍决定。
- 收入和资源有限的家庭也可能有资格为其残疾子女获得补充保障收入 (SSI)。
运动功能障碍
粗大运动功能障碍
- 建议进行物理治疗以最大限度地提高活动能力并降低后期发生的骨科并发症(例如挛缩、脊柱侧弯、髋关节脱位)的风险。
- 根据需要考虑使用耐用的医疗设备和定位装置(例如,轮椅、助行器、浴椅、矫形器、自适应婴儿车)。
- 对于肌张力异常,包括张力亢进或肌张力障碍,考虑让适当的专家协助管理使用巴氯芬、替扎尼定、肉毒杆菌毒素®、抗帕金森病药物或骨科手术。
精细运动功能障碍。对于影响适应功能的精细运动技能困难,如喂养、梳洗、穿衣和写作,建议进行职业治疗。
沟通问题。考虑为有表达语言困难的个人评估替代交流方式(例如,augmentative and alternative communication)。 AAC 评估可由具有该领域专业知识的语言病理学家完成。评估将考虑认知能力和感觉障碍,以确定最合适的交流方式。 AAC 设备的范围从低技术(如图片交换通信)到高科技(如语音生成设备)。与流行的看法相反,AAC 设备不会阻碍语音的发展,而是支持最佳的语音和语言发展。
社会/行为问题
应用行为 儿童可能有资格获得用于治疗自闭症谱系障碍的干预措施并从中受益,包括应用行为分析 (ABA)。 ABA 疗法针对个别孩子的行为、社交和适应性强项和弱点,通常与经过董事会认证的行为分析师一对一地进行。
咨询发育儿科医生可能有助于指导父母通过适当的行为管理策略或在必要时提供处方药,例如用于治疗注意力缺陷/多动障碍的药物。
儿科精神科医生可以解决对严重攻击性或破坏性行为的担忧。
Table 7
对具有 PIK3CA 相关过度生长谱的个体的推荐监测
| 系统/关注点 | 评估 | 频率 |
|---|---|---|
| 内容 (过度生长和 广义的 生长 限制) | 测量生长参数,包括头围、臂长、手、腿1 & 脚 2 | 每次随访 |
| 根据临床指示 | |
| 神经系统 | 系列头部 MRI 成像 | 取决于初步评估结果的严重程度和大脑成熟程度 3 |
| 每次随访 | |
| 发育 | 监控发展进度和教育需求。 | |
| 精神科/行为的 | 焦虑、注意力、攻击性或自残行为的行为评估 | 在儿童、青少年和成人的每次访问中 |
| 肌肉骨骼 |
| 每次随访 |
| 胃肠道 | 腹部触诊器官肿大和腹部肿块 | |
| 血管&淋巴畸形 | 临床评估和监测,最好由血管团队进行 6 | 根据临床指示 |
| 泌尿生殖器 | 考虑肾脏超声 | 每 3 个月一次,直到 8 岁 7 |
| 血液 | 血液学咨询/建议评估血栓形成和凝血病风险 | 任何手术干预后,尤其是那些带有丁香表型和/或血管畸形的患者。 |
| 家庭/社区 | 评估家庭对社会工作支持(例如姑息/暂托护理、家庭护理、其他当地资源)和护理协调的需求。 | 每次随访 |
OT = 职业治疗; PT = 物理治疗
- 1.
包括腿长差异
- 2.
如果发现特定身体区域快速增长,请考虑有针对性的随访,其中可能包括其他类型的监测技术,例如体积研究和血管造影 [Douzgou et al 2022].
- 3.
对于中枢神经系统过度生长或发育不良的患者:每 6 个月进行一次脑部 MRI,直到 2 岁,然后每年进行一次,直到 8 岁,以专门监测进行性脑积水和 Chiari 畸形 [Douzgou et al 2022]
- 4.
婴儿可能有易怒、过度流口水、吞咽困难或呼吸问题,尤其是中枢性呼吸暂停。
- 5.
儿童可能有颈部疼痛或头痛、运动无力、感觉变化、视力问题、吞咽困难或行为改变。
- 6.
团队可能包括皮肤病学、介入放射学和血液学/肿瘤学专家。
- 7.
鉴于研究表明 Wilms 肿瘤的发生率在 1.4% 和 3.3% 之间,因此对 Wilms 肿瘤进行肿瘤筛查是有争议的。在美国,如果肿瘤风险为 3% 或更高,通常会进行肿瘤筛查。需要进一步的纵向研究来评估对 PROS 患者进行Wilms 肿瘤筛查的必要性。
评估有风险的亲属
有关为遗传咨询目的而对高危亲属进行测试的相关问题,请参阅Genetic Counseling。
正在研究的疗法
一些研究已经证明哺乳动物雷帕霉素靶蛋白 (mTOR) 抑制剂西罗莫司对淋巴疾病的疗效 [Padilla et al 2019, Zenner et al 2019]。对 PI3K/AKT/mTOR 信号通路和 PROS 的自然史的持续了解将为开发新的靶向治疗策略提供基础。正在研究的 PROS 全身性药物靶向 PI3K 信号通路的不同成分。这些包括 mTOR、AKT 和 PI3K 基因的抑制剂:
- Sirolimus已在多项临床试验中对患有复杂淋巴异常、血管畸形和过度生长障碍的个体进行了研究 [Adams et al 2016, Hammer et al 2018, Adams & Ricci 2019, Parker et al 2019, Van Damme et al 2020](见ClinicalTrials.gov),其中一些人有 PROS。西罗莫司目前用于治疗这些疾病[Seront et al 2019, Dekeuleneer et al 2020]。西罗莫司已在多项 II 期研究中显示出对患有复杂血管异常的个体以及患有 PROS 和进行性过度生长的个体的疗效[Adams et al 2016, Erickson et al 2017, Hammer et al 2018, Parker et al 2019, Ricci et al 2019]。
- Miransertib 是一种 AKT1 抑制剂,已在扩大使用范围的临床试验和 I/II 期临床试验中进行了研究[Wassef et al 2015, Zampino et al 2019].
- Alpelisib 是一种 PI3K 抑制剂,在一项未经注册的试验中对 PROS 患者进行了调查,该试验以同情使用理由进行了治疗,并证明了疗效[Venot et al 2018]。未来的 II 期随机临床试验将可用于评估 PROS 患者的疗效、安全性和药代动力学。
在美国和欧洲的欧盟临床试验注册中搜索ClinicalTrials.gov,以获取有关各种疾病和病症的临床研究信息。注意:可能没有针对这种疾病的临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传方式和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况; 它并非旨在解决可能出现的所有个人、文化或道德问题,或替代与遗传学专业人士的咨询。 —编者。
遗传方式
PIK3CA 相关的过度生长障碍 (PROS) 不是遗传性的,因为大多数已确定的致病变异是体细胞性的(嵌合)。迄今为止,尚未报告确认的垂直传播或同胞发生。
家庭成员的风险
先证者的父母
- 体细胞嵌合 的PIK3CA 致病性变异儿童的父母尚未报告有任何显著的独特表现,即使考虑到这些遗传改变的体细胞嵌合性质,也不会出现此类发现。
- 理论上,由de novo 胚系PIK3CA致病性变异引起的 PROS 患儿的父母可能具有 胚系嵌合PIK3CA 致病性变异。父母胚系嵌合的理论风险估计小于 1%。
先证者的同胞
先证者的后代
- PROS 成人的生殖结果数据有限;没有这些疾病垂直传播的例子。虽然已经报道了患有 PROS 的成年人,但受影响个体的发育结果尚不清楚。具有明显神经系统受累的个体(例如 DMEG、HMEG)预后不良。
- 除了少数患有 PROS 的受累个体外,所有受累个体都有 PIK3CA 致病性变异的体细胞嵌合,这表明在多细胞胚胎的一个细胞中受精后发生了突变。因此,传播给后代的风险预计低于 50%。
- 几个 PROS 患者在 PIK3CA 中出现了 de novo 胚系 致病性变异 [Rivière et al 2012, Mirzaa et al 2016]。具有胚系 PIK3CA 致病性变异的个体的后代有 50% 的风险遗传致病性变异。
其他家庭成员。其他家庭成员的风险与普通人群相同。
相关遗传咨询问题
家庭计划
资源
GeneReviews 工作人员选择了以下特定疾病和/或支持组织和/或登记处,以造福患有这种疾病的个人及其家人。 GeneReviews 不对其他组织提供的信息负责。 有关选择标准的信息,请单击here.
- CLOVES Syndrome CommunityPO BOX 406West Kennebunk 04094Phone: 833-425-6837Email: info@clovessyndrome.org
- M-CM NetworkPO Box 97Chatham NY 12037Phone: 518-392-2150Email: hello@m-cm.net
- M-CM Network Contact RegistryThe M-CM Network contact registry will be used to inform individuals with M-CM and their guardians about: opportunities to participate in research, opportunities to contribute data, and discoveries about M-CM that may impact care decisions.
分子遗传
Molecular Genetics 和 OMIM 表格中的信息可能与 GeneReview 中其他地方的信息不同:表格可能包含更新的信息。 —编者。
Table A
PIK3CA 相关过度生长谱:基因和数据库
Table B
PIK3CA 相关过度生长谱的 OMIM 条目 (View All in OMIM)
| 155500 | MACRODACTYLY |
| 162900 | NEVUS, EPIDERMAL |
| 171834 | PHOSPHATIDYLINOSITOL 3-KINASE, CATALYTIC, ALPHA; PIK3CA |
| 602501 | MEGALENCEPHALY-CAPILLARY MALFORMATION-POLYMICROGYRIA SYNDROME; MCAP |
| 612918 | CONGENITAL LIPOMATOUS OVERGROWTH, VASCULAR MALFORMATIONS, AND EPIDERMAL NEVI |
| 613089 | CAPILLARY MALFORMATION OF THE LOWER LIP, LYMPHATIC MALFORMATION OF FACE AND NECK, ASYMMETRY OF FACE AND LIMBS, AND PARTIAL/GENERALIZED OVERGROWTH |
| 615108 | COWDEN SYNDROME 5; CWS5 |
分子发病机制
PIK3CA 相关过度生长谱系障碍通常由编码磷脂酰肌醇-3-激酶 (PI3K) 催化亚基 α (p110alpha) 的基因中的受精后体细胞变异引起 [Engelman et al 2006]。这些变异与 PI3K 信号通路的过度激活有关,其中包括多种下游效应物,如 AKT 和 mTOR,导致各种组织的异常生长和血管畸形。
PIK3CA 蛋白对于胰岛素降低血糖的作用和胰岛素样生长因子 1 (IGF1) 的作用至关重要,IGF1 通过与胰岛素受体非常相似的受体促进组织生长,并介导许多生长激素。病理激活的 PIK3CA 可以模拟细胞中胰岛素和/或 IGF1 的作用。为了使这种机制转化为临床上重要的内分泌病,靶组织需要承受高变异负荷。这种机制可以解释为什么一些受影响的婴儿具有类似于高胰岛素血症的临床特征。
发病机制。功能增益
PIK3CA 特定的实验室技术注意事项。由于大多数受影响的个体具有嵌合 PIK3CA 致病性变异,因此可能需要对适当的样本使用定制限制性片段长度多态性 (RFLP) 测定或数字PCR 。标准深度外显子组测序 可能会错过嵌合PIK3CA变异,特别是如果执行血液标本的时候。
- 为了检测新的或非常罕见的变异,通常需要对整个外显子进行测序。
- 只有当致病性变异 等位基因分数相对较高(~20%)时,才能使用 Sanger sequencing。
- 有针对性地捕获整个 PIK3CA 编码区,然后在非常深的覆盖范围内进行next-generation sequencing ,可能更适合体细胞变异检测,因为它允许检测整个基因中非常低水平的嵌合。
- 在 MCAP 综合征中,唾液或皮肤成纤维细胞(无论是否明显受影响)的 DNA 序列分析比外周血来源的 DNA 具有更高的检出率[Mirzaa et al 2016].
- 在局灶性脑过度生长障碍(HMEG、FCD、DMEG)中,来自受影响脑组织(例如,在癫痫手术时去除)的 DNA 分析比外周组织(血液或唾液)具有更高的检出率[Jansen et al 2015].
Table 8
常见PIK3CA 致病性变异
| 测序 | DNA 核酸改变 | 可能的 蛋白质变化 | 备注 | Reference |
|---|---|---|---|---|
| c.1624G>A | p.Glu542Lys | 突变热点与高度局灶性表型和大脑中更严重的癫痫相关 1 | D'Gama et al [2015] | |
| c.1633G>A | p.Glu545Lys | Rivière et al [2012] | ||
| c.3140A>G | p.His1047Arg | D'Gama et al [2015] | ||
| c.3141T>A | p.His1047Lys | Kuentz et al [2017] |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
CLOVES 综合征、纤维脂肪增生、淋巴管/血管畸形、半巨脑畸形和局灶性皮质发育不良(见 Genotype-Phenotype Correlations.)
癌症和良性肿瘤
PIK3CA 在许多癌症中发生体细胞突变,包括结肠直肠癌、卵巢癌、乳腺癌、肝细胞癌和胶质母细胞瘤。这些 PIK3CA 致病变异主要位于激酶结构域内的热点(由外显子20 编码),并导致与致癌性有关的功能获得 [Samuels et al 2004, Ikenoue et al 2005, Kang et al 2005].
大多数(>80%)在癌症(Table 9)和 PROS(Table 8)中激活 PIK3CA 致病变异聚集在三个热点:密码子 542 和 545 处的两个谷氨酸(Glu)残基和密码子1047的组氨酸(His)残基. PIK3CA 致病性变异在癌症中的分布来自癌症体细胞突变目录(COSMIC,v85,2018 年 5 月)。 PROS 中的 PIK3CA 致病变异包括来自较大队列研究的已发表病例。肿瘤发生和恶性肿瘤发展的风险是一个理论上的问题,因为 PIK3CA 在许多癌症中发生体细胞突变或过度表达。 PIK3CA 的致病性变异在癌症和大部分 PROS 个体中(尤其是那些具有 CLOVES、纤维脂肪增生和孤立的大指畸形的表型)最常见于螺旋结构域和激酶结构域内的热点。
在癌症体细胞突变目录(COSMIC)中列出的大约 160 个 PIK3CA 致病变异中,三个氨基酸的常见变异(外显子9 中的 p.Glu542Lys、p.Glu545Lys 和外显子20中的 p.His1047Arg 和 p.His1047Leu 20) 占肿瘤相关 PIK3CA 变体的 80%,并显示出最高的致癌活性[Samuels et al 2004, Samuels & Ericson 2006, Janku et al 2012]。见Table 9。
章节备注
作者备注
Ghayda Mirzaa 博士是华盛顿大学医学院医学遗传学和儿科副教授。她的研究专注于发育性脑部疾病,包括巨脑畸形,并发表了多篇与 PIK3CA 相关过度生长谱 (PROS) 相关的出版物,包括自然史、分子诊断和潜在疗法。
John M Graham Jr 博士是加州大学洛杉矶分校 David Geffen 医学院儿科名誉教授,也是 Cedars-Sinai 医学中心和 Harbour UCLA 医学中心的临床遗传学顾问。他对 PROS 有着长期的临床兴趣,并发表了许多关于该主题的文章。
Kim Keppler-Noreuil 博士是威斯康星大学医学与公共卫生学院的临床遗传学家和遗传学与代谢系主任;她拥有多篇与 PROS 相关的出版物,包括特邀评论文章和原创研究,特别关注临床和分子诊断,以及 PROS 的潜在疗法。
致谢
我们要感谢患者、他们的家人和我们的合作者,感谢他们对我们了解这些疾病的宝贵贡献。
作者历史
医学博士罗伯特·康威;韦恩州立大学 (2013-2021)
威廉·B·多宾斯,医学博士;西雅图儿童医院 (2013-2021)
John H Graham Jr,医学博士,科学博士(2013 年至今)
Kim Keppler-Noreuil,医学博士,FAAP,FACMG(2021 年至今)
Ghayda Mirzaa,医学博士,FAAP,FACMG(2013 年至今)
修订记录
- 2021 年 12 月 23 日(ma)实时发布全面更新
- 2013 年 8 月 15 日(我)评论发布现场
- 2013 年 3 月 11 日 (gm) 初稿提交
References
Literature Cited
- Adams DM, Ricci KW. Vascular anomalies: diagnosis of complicated anomalies and new medical treatment options. Hematol Oncol Clin North Am. 2019;33:455–70. [PubMed: 31030813]
- Adams DM, Trenor CC 3rd, Hammill AM, Vinks AA, Patel MN, Chaudry G, Wentzel MS, Mobberley-Schuman PS, Campbell LM, Brookbank C, Gupta A, Chute C, Eile J, McKenna J, Merrow AC, Fei L, Hornung L, Seid M, Dasgupta AR, Dickie BH, Elluru RG, Lucky AW, Weiss B, Azizkhan RG. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137:e20153257. [PMC free article: PMC4732362] [PubMed: 26783326]
- Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009;18:1–7. [PubMed: 19011570]
- Alomari AI. Comments on the diagnosis and management of CLOVES syndrome. Pediatr Dermatol. 2011;28:215–6. [PubMed: 21504461]
- Canaud G, Hammill AM, Adams D, Vikkula M, Keppler-Noreuil KM. A review of mechanisms of disease across PIK3CA-related disorder with vascular malformations. Orphanet J Rare Dis. 2021;16:306. [PMC free article: PMC8268514] [PubMed: 34238334]
- Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, Hennekam RC, Mueller RF, Brueton L, Super M, Steen-Johnsen J, Donnai D. Macrocephaly with cutis marmorata, haemangioma and syndactyly--a distinctive overgrowth syndrome. Clin Dysmorphol. 1997;6:291–302. [PubMed: 9354837]
- Conway RL, Danielpour M, Graham JM Jr. Surgical management of cerebellar tonsillar herniation in three patients with macrocephaly-cutis marmorata telangiectatica congenita. Report of three cases. J Neurosurg. 2007a;106:296–301. [PubMed: 17465364]
- Conway RL, Pressman BD, Dobyns WB, Danielpour M, Lee J, Sanchez-Lara PA, Butler MG, Zackai E, Campbell L, Saitta SC, Clericuzio CL, Milunsky JM, Hoyme HE, Shieh J, Moeschler JB, Crandall B, Lauzon JL, Viskochil DH, Harding B, Graham JM Jr. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A. 2007b;143A:2981–3008. [PMC free article: PMC6816457] [PubMed: 18000912]
- Coste K, Sarret C, Cisse A, Delabaere A, Francannet C, Vanlieferinghen P. Macrocephaly-capillary malformation. A neonatal case. Arch Pediatr. 2012;19:917–20. [PubMed: 22884750]
- Couto JA, Konczyk DJ, Vivero MP, Kozakewich HPW, Upton J, Fu X, Padwa BL, Mulliken JB, Warman ML, Greene AK. Somatic PIK3CA mutations are present in multiple tissues of facial infiltrating lipomatosis. Pediatr Res. 2017;82:850–4. [PMC free article: PMC5645230] [PubMed: 28665924]
- Davis S, Ware MA, Zeiger J, Deardorff MA, Grand K, Grimberg A, Hsu S, Kelsey M, Majidi S, Matthew RP, Napier M, Nokoff N, Prasad C, Riggs AC, McKinnon ML, Mirzaa G. Growth hormone deficiency in megalencephaly-capillary malformation syndrome: An association with activating mutations in PIK3CA. Am J Med Genet A. 2020;182:162–8. [PMC free article: PMC7262792] [PubMed: 31729162]
- Dekeuleneer V, Seront E, Van Damme A, Boon LM, Vikkula M. Theranostic advances in vascular malformations. J Invest Dermatol. 2020;140:756–63. [PubMed: 32200879]
- D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, Hossain A, Hatem NE, Barry BJ, Kwiatkowski DJ, Vinters HV, Barkovich AJ, Shendure J, Mathern GW, Walsh CA, Poduri A. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–5. [PMC free article: PMC4471336] [PubMed: 25599672]
- Di Rocco C, Battaglia D, Pietrini D, Piastra M, Massimi L. Hemimegalencephaly: clinical implications and surgical treatment. Childs Nerv Syst. 2006;22:852–66. [PubMed: 16821075]
- Douzgou S, Rawson M, Baselga E, Danielpour M, Faivre L, Kashanian A, Keppler-Noreuil KM, Kuentz P, Mancini GMS, Maniere MC, Martinez-Glez V, Parker VE, Semple RK, Srivastava S, Vabres P, De Wit MY, Graham JM Jr, Clayton-Smith J, Mirzaa GM, Biesecker LG. A standard of care for individuals with PIK3CA-related disorders: An international expert consensus statement. Clin Genet. 2022;101:32–47. [PMC free article: PMC8664971] [PubMed: 34240408]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. [PubMed: 16847462]
- Erickson J, McAuliffe W, Blennerhassett L, Halbert A. Fibroadipose vascular anomaly treated with sirolimus: successful outcome in two patients. Pediatr Dermatol. 2017;34:e317–e320. [PubMed: 29144050]
- Gripp KW, Baker L, Kandula V, Conard K, Scavina M, Napoli JA, Griffin GC, Thacker M, Knox RG, Clark GR, Parker VE, Semple R, Mirzaa G, Keppler-Noreuil KM. Nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. 2016;170:2559–69. [PMC free article: PMC5514817] [PubMed: 27191687]
- Gripp KW, Hopkins E, Vinkler C, Lev D, Malinger G, Lerman-Sagie T, Dobyns WB. Significant overlap and possible identity of macrocephaly capillary malformation and megalencephaly polymicrogyria-polydactyly hydrocephalus syndromes. Am J Med Genet A. 2009;149A:868–76. [PubMed: 19353582]
- Hammer J, Seront E, Duez S, Dupont S, Van Damme A, Schmitz S, Hoyoux C, Chopinet C, Clapuyt P, Hammer F, Vikkula M, Boon LM. Sirolimus is efficacious in treatment for extensive and/or complex slow-flow vascular malformations: a monocentric prospective phase II study. Orphanet J Rare Dis. 2018;13:191. [PMC free article: PMC6206885] [PubMed: 30373605]
- Hucthagowder V, Shenoy A, Corliss M, Vigh-Conrad KA, Storer C, Grange DK, Cottrell CE. Utility of clinical high-depth next generation sequencing for somatic variant detection in the PIK3CA-related overgrowth spectrum. Clin Genet. 2017;91:79–85. [PubMed: 27307077]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–7. [PubMed: 15930273]
- Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS, Kurzrock R. PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012;3:1566–75. [PMC free article: PMC3681495] [PubMed: 23248156]
- Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Rivière JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, Dobyns WB. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–28. [PMC free article: PMC4614119] [PubMed: 25722288]
- Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA. 2005;102:802–7. [PMC free article: PMC545580] [PubMed: 15647370]
- Keppler-Noreuil KM, Lozier J, Oden N, Taneja A, Burton-Akright J, Sapp JC, Biesecker LG. Thrombosis risk factors in PIK3CA-related overgrowth spectrum and Proteus syndrome. Am J Med Genet C Semin Med Genet. 2019;181:571–81. [PMC free article: PMC8513083] [PubMed: 31490637]
- Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, Alomari A, Ezaki M, Dobyns W, Biesecker LG. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A:287–95. [PMC free article: PMC4480633] [PubMed: 25557259]
- Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, Tosi LL, Huson SM, Whitehouse RW, Jakkula E, Grant I, Balasubramanian M, Chandler KE, Fraser JL, Gucev Z, Crow YJ, Brennan LM, Clark R, Sellars EA, Pena LD, Krishnamurty V, Shuen A, Braverman N, Cunningham ML, Sutton VR, Tasic V, Graham JM Jr, Geer J Jr, Henderson A, Semple RK, Biesecker LG. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. 2014;164A:1713–33. [PMC free article: PMC4320693] [PubMed: 24782230]
- Kuentz P, St-Onge J, Duffourd Y, Courcet JB, Carmignac V, Jouan T, Sorlin A, Abasq-Thomas C, Albuisson J, Amiel J, Amram D, Arpin S, Attie-Bitach T, Bahi-Buisson N, Barbarot S, Baujat G, Bessis D, Boccara O, Bonnière M, Boute O, Bursztejn AC, Chiaverini C, Cormier-Daire V, Coubes C, Delobel B, Edery P, Chehadeh SE, Francannet C, Geneviève D, Goldenberg A, Haye D, Isidor B, Jacquemont ML, Khau Van Kien P, Lacombe D, Martin L, Martinovic J, Maruani A, Mathieu-Dramard M, Mazereeuw-Hautier J, Michot C, Mignot C, Miquel J, Morice-Picard F, Petit F, Phan A, Rossi M, Touraine R, Verloes A, Vincent M, Vincent-Delorme C, Whalen S, Willems M, Marle N, Lehalle D, Thevenon J, Thauvin-Robinet C, Hadj-Rabia S, Faivre L, Vabres P, Rivière JB. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet Med. 2017;19:989–97. [PubMed: 28151489]
- Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, Warman ML. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–15. [PMC free article: PMC3370283] [PubMed: 22658544]
- Kwan SY, Shyu HY, Lin JH, Wong TT, Chang KP, Yiu CH. Corpus callosotomy in a patient of hemimegalencephaly and Lennox-Gastaut syndrome. Brain Dev. 2008;30:643–6. [PubMed: 18439776]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–5. [PMC free article: PMC4417942] [PubMed: 22729223]
- Leiter SM, Parker VER, Welters A, Knox R, Rocha N, Clark G, Payne F, Lotta L, Harris J, Guerrero-Fernández J, González-Casado I, García-Miñaur S, Gordo G, Wareham N, Martínez-Glez V, Allison M, O'Rahilly S, Barroso I, Meissner T, Davies S, Hussain K, Temple K, Barreda-Bonis AC, Kummer S, Semple RK. Hypoinsulinaemic, hypoketotic hypoglycaemia due to mosaic genetic activation of PI3-kinase. Eur J Endocrinol. 2017;177:175–86. [PMC free article: PMC5488397] [PubMed: 28566443]
- Lindhurst MJ, Parker VE, Payne F, Sapp JC, Rudge S, Harris J, Witkowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, Cunningham ML, Darling TN, Geer J, Gucev Z, Sutton VR, Tziotzios C, Dixon AK, Helliwell T, O'Rahilly S, Savage DB, Wakelam MJ, Barroso I, Biesecker LG, Semple RK. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928–33. [PMC free article: PMC3461408] [PubMed: 22729222]
- Luks VL, Kamitaki N, Vivero MP, Uller W, Rab R, Bovée JV, Rialon KL, Guevara CJ, Alomari AI, Greene AK, Fishman SJ, Kozakewich HP, Maclellan RA, Mulliken JB, Rahbar R, Spencer SA, Trenor CC 3rd, Upton J, Zurakowski D, Perkins JA, Kirsh A, Bennett JT, Dobyns WB, Kurek KC, Warman ML, McCarroll SA, Murillo R. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048–54. [PMC free article: PMC4498659] [PubMed: 25681199]
- Martínez-Glez V, Romanelli V, Mori MA, Gracia R, Segovia M, González-Meneses A, López-Gutierrez JC, Gean E, Martorell L, Lapunzina P. Macrocephaly-capillary malformation: Analysis of 13 patients and review of the diagnostic criteria. Am J Med Genet A. 2010;152A:3101–6. [PubMed: 21077203]
- Martínez-Lage JF, Guillén-Navarro E, Almagro MJ, Felipe-Murcia M, López López-Guerrero A, Galarza M. Hydrocephalus and Chiari type 1 malformation in macrocephaly-cutis marmorata telangiectatica congenita: a case-based update. Childs Nerv Syst. 2010;26:13–8. [PubMed: 19763591]
- McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137–41. [PubMed: 20533527]
- Mills JR, Moyer AM, Kipp BR, Poplawski AB, Messiaen LM, Babovic-Vuksanovic D. Unilateral vestibular schwannoma and meningiomas in a patient with PIK3CA-related segmental overgrowth: Co-occurrence of mosaicism for 2 rare disorders. Clin Genet. 2018;93:187–90. [PubMed: 28737257]
- Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries LS, Lev D, Kramer N, Hopkins E, Graham JM Jr, Dobyns WB. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A:269–91. [PubMed: 22228622]
- Mirzaa G, Timms AE, Conti V, Boyle EA, Girisha KM, Martin B, Kircher M, Olds C, Juusola J, Collins S, Park K, Carter M, Glass I, Krägeloh-Mann I, Chitayat D, Parikh AS, Bradshaw R, Torti E, Braddock S, Burke L, Ghedia S, Stephan M, Stewart F, Prasad C, Napier M, Saitta S, Straussberg R, Gabbett M, O'Connor BC, Keegan CE, Yin LJ, Lai AHM, Martin N, McKinnon M, Addor MC, Boccuto L, Schwartz CE, Lanoel A, Conway RL, Devriendt K, Tatton-Brown K, Pierpont ME, Painter M, Worgan L, Reggin J, Hennekam R, Tsuchiya K, Pritchard CC, Aracena M, Gripp KW, Cordisco M, Van Esch H, Garavelli L, Curry C, Goriely A, Kayserilli H, Shendure J, Graham J Jr, Guerrini R, Dobyns WB. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight. 2016;1:e87623. [PMC free article: PMC5019182] [PubMed: 27631024]
- Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, Higgins JV, Stevens CA, Twersky S, Weksberg R, Dobyns WB. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. 1997;70:67–73. [PubMed: 9129744]
- Nyberg RH, Uotila J, Kirkinen P, Rosendahl H. Macrocephaly-cutis marmorata telangiectatica congenita syndrome--prenatal signs in ultrasonography. Prenat Diagn. 2005;25:129–32. [PubMed: 15712320]
- Padilla CA, Bárcena JA, López-Grueso MJ, Requejo-Aguilar R. The regulation of TORC1 pathway by the yeast chaperones Hsp31 is mediated by SFP1 and affects proteasomal activity. Biochim Biophys Acta Gen Subj. 2019;1863:534–46. [PubMed: 30578832]
- Parker VE, Keppler-Noreuil KM, Faivre L, Luu M, Oden NL, De Silva L, Sapp JC, Andrews K, Bardou M, Chen KY, Darling TN, Gautier E, Goldspiel BR, Hadj-Rabia S, Harris J, Kounidas G, Kumar P, Lindhurst MJ, Loffroy R, Martin L, Phan A, Rother KI, Widemann BC, Wolters PL, Coubes C, Pinson L, Willems M, Vincent-Delorme C, Vabres P, Semple RK, Biesecker LG, et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genet Med. 2019;21:1189–98. [PMC free article: PMC6752269] [PubMed: 30270358]
- Peterman CM, Fevurly RD, Alomari AI, Trenor CC 3rd, Adams DM, Vadeboncoeur S, Liang MG, Greene AK, Mulliken JB, Fishman SJ. Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer. 2017;64(12) [PubMed: 28627003]
- Pirozzi F, Berkseth M, Shear R, Gonzalez L, Timms AE, Sulc J, Pao E, Oyama N, Forzano F, Conti V, Guerrini R, Doherty ES, Saitta SC, Lockwood CM, Pritchard CC, Dobyns WB, Novotny E, Wright J, Saneto RP, Friedman S, Hauptman J, Ojemann J, Kapur RP, Mirzaa GMM. Profiling PI3K-AKT-MTOR variants in focal brain malformations reveals new insights for diagnostic care. Brain. In press.
- Postema FAM, Hopman SMJ, Aalfs CM, Berger LPV, Bleeker FE, Dommering CJ, Jongmans MCJ, Letteboer TGW, Olderode-Berends MJW, Wagner A, Hennekam RC, Merks JHM. Childhood tumours with a high probability of being part of a tumour predisposition syndrome; reason for referral for genetic consultation. Eur J Cancer. 2017;80:48–54. [PubMed: 28544908]
- Ricci KW, Hammill AM, Mobberley-Schuman P, Nelson SC, Blatt J, Bender JLG, McCuaig CC, Synakiewicz A, Frieden IJ, Adams DM. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham-Stout disease. Pediatr Blood Cancer. 2019;66:e27614. [PMC free article: PMC6428616] [PubMed: 30672136]
- Rivière JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL., Finding of Rare Disease Genes (FORGE) Canada Consortium. Majewski J, Bulman DE, O'Driscoll M, Shendure J, Graham JM Jr, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40. [PMC free article: PMC3408813] [PubMed: 22729224]
- Robertson SP, Gattas M, Rogers M, Adès LC. Macrocephaly--cutis marmorata telangiectatica congenita: report of five patients and a review of the literature. Clin Dysmorphol. 2000;9:1–9. [PubMed: 10649789]
- Rodriguez-Laguna L, Agra N, Ibañez K, Oliva-Molina G, Gordo G, Khurana N, Hominick D, Beato M, Colmenero I, Herranz G, Torres Canizalez JM, Rodríguez Pena R, Vallespín E, Martín-Arenas R, Del Pozo Á, Villaverde C, Bustamante A, Ayuso C, Lapunzina P, Lopez-Gutierrez JC, Dellinger MT, Martinez-Glez V. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J Exp Med. 2019;216:407–18. [PMC free article: PMC6363432] [PubMed: 30591517]
- Sadick M, Müller-Wille R, Wildgruber M, Wohlgemuth WA. Vascular anomalies (part i): classification and diagnostics of vascular anomalies. Rofo. 2018;190:825–35. [PubMed: 29874693]
- Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. [PubMed: 16357568]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [PubMed: 15016963]
- Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007;143A:2944–58. [PubMed: 17963221]
- Schwartz IVD, Felix TM, Riegel M, Schüler-Faccini L. Atypical macrocephaly-cutis marmorata telangiectatica congenita with retinoblastoma. Clin Dysmorphol. 2002;11:199–202. [PubMed: 12072801]
- Seront E, Van Damme A, Boon LM, Vikkula M. Rapamycin and treatment of venous malformations. Curr Opin Hematol. 2019;26:185–92. [PubMed: 30855337]
- Van Damme A, Seront E, Dekeuleneer V, Boon LM, Vikkula M. New and emerging targeted therapies for vascular malformations. Am J Clin Dermatol. 2020;21:657–68. [PubMed: 32557381]
- Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, Blanc E, Johnson SC, Hoguin C, Boccara O, Sarnacki S, Boddaert N, Pannier S, Martinez F, Magassa S, Yamaguchi J, Knebelmann B, Merville P, Grenier N, Joly D, Cormier-Daire V, Michot C, Bole-Feysot C, Picard A, Soupre V, Lyonnet S, Sadoine J, Slimani L, Chaussain C, Laroche-Raynaud C, Guibaud L, Broissand C, Amiel J, Legendre C, Terzi F, Canaud G. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. 2018;558:540–6. [PMC free article: PMC7610773] [PubMed: 29899452]
- Vogels A, Devriendt K, Legius E, Decock P, Marien J, Hendrickx G, Fryns JP. The macrocephaly-cutis marmorata telangiectatica congenita syndrome. Long-term follow-up data in 4 children and adolescents. Genet Couns. 1998;9:245–53. [PubMed: 9894160]
- Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M, et al. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:e203–14. [PubMed: 26055853]
- Zampino G, Leoni C, Buonuomo PS, Rana I, Onesimo R, Macchiaiolo M, et al. An open-label, Phase I/II study of miransertib (ARQ 092), an oral pan-AKT inhibitor, in patients (pts) with PIK3CA-related overgrowth spectrum (PROS) and Proteus syndrome (PS): study design and preliminary results (NCT03094832). Gothenburg, Sweden: European Society of Human Genetics Conference; June 15-18, 2019; Abstract C 18.6.
- Zenner K, Cheng CV, Jensen DM, Timms AE, Shivaram G, Bly R, Ganti S, Whitlock KB, Dobyns WB, Perkins J, Bennett JT. Genotype correlates with clinical severity in PIK3CA-associated lymphatic malformations. JCI Insight. 2019;4:e129884. [PMC free article: PMC6948764] [PubMed: 31536475]