
简介
临床特征。DFNA2非综合征听力损失的特征是对称的、主要是高频的感音神经性听力损失(SNHL),其在所有频率上都逐步进展。 年轻时在低频时听力损失较轻,高频时则中等听力损失 。在老年人中,低频的听力中等损失,而高频时则严重损失。尽管在对学龄儿童进行例行听力评估时通常会发现听力障碍,但从出生起听力就有可能受损,尤其是在高频率时。大多数 受累的人在10至40岁之间最初需要助听器来帮助。到70岁时,所有患有DFNA2非综合征性听力损失的人都有严重广泛的听力障碍。
诊断/测试。DFNA2非综合征性听力损失的诊断是建立在具有特征性听觉特征,与常染色体显性遗传一致的家族史以及在KCNQ4中鉴定 杂合的 致病性变异的个体中。
管理。表型治疗:轻度至中度听力损失者使用助听器;当听力损失严重广泛时,应考虑人工耳蜗;在学校为听力受损的儿童和青少年提供特别援助。
监测:至少每年一次听力图,以追踪听力下降的情况。
应避免的药物/情况:避免暴露于嘈杂的噪音下可能会降低高频SNHL的发展速度。
对有风险的亲属的评估:确定婴儿或幼儿期先证者的家庭成员是否已遗传了KCNQ4中致病性变异,从而可以对孩子和家庭进行早期支持和管理。
遗传咨询。DFNA2非综合征性听力损失以 常染色体显性遗传方式遗传。患有DFNA2非综合征性听力损失的大多数人的父母都有听力损失。 de novoKCNQ4 致病性变异的个体比例尚未知。患有DFNA2非综合征性听力损失的个体的每个孩子都有50%的几率遗传KCNQ4致病变异。一旦在家庭成员中发现了KCNQ4致病变异,就可以对风险增加的妊娠进行产前检查,并针对DFNA2非综合征性听力损失进行植入前遗传诊断。
诊断
提示性发现
具有以下临床,影像学和家族史发现的个体应考虑DFNA2非综合征听力损失:
临床发现
- 对称的,主要是高频的感音神经性听力损失(SNHL),在所有频率上都是渐进的:
- 年轻时在低频时倾向于轻度听力损失,高频时则中等听力损失。
- 在老年人中,低频时中等听力损失,而高频时严重听力损失。
- 体检正常
影像学发现。 颞骨成像(即内耳CT)是正常的。 具体而言,不应出现诸如前庭导水管扩张(也称为扩大的前庭导水管)和Mondini发育不良(即内耳发育不全)等异常情况。
家族史。有听力损失的家族史,并且呈常染色体显性遗传 。
建立诊断 DFNA2非综合征性听力损失的诊断建立在具有上述特征音频特征的先证者中,并通过分子遗传学检测在KCNQ4中鉴定 杂合的 致病性变异(参见Table 1)。
由于DFNA2非综合征性听力损失的 表型与许多其他遗传性听力障碍疾病没有区别,因此推荐的分子遗传学检测方法包括使用 multigene panel 或全基因组的测试。
注意:单基因检测(KCNQ4的序列分析,然后进行基因靶向的 deletion/duplication analysis)很少有用,通常不建议这样做。
多基因听力损失和耳聋套餐 由KCNQ4和其他相关基因组成的多基因听力损失和耳聋套餐(见Hereditary Hearing Loss and Deafness Overview)最有可能以最合理的成本鉴定出该病的遗传原因,同时减少了意义不确定和不能解释潜在表型的基因中的致病变异出现。注意:(1)套餐中包含的基因和每个基因所用测试的诊断敏感性因实验室而异,并可能随时间而变化。 (2)一些多基因套餐可能包含与本《基因综述》中讨论的病症无关的基因。 (3)在某些实验室中,套餐选项可能包括定制的实验室设计套餐和/或定制的以表型为重点的外显子组分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括 序列分析, deletion/duplication analysis和/或其他非基于序列的测试。对于这种疾病,建议同时进行包含缺失/重复的multigene panel (见 Table 1)。
有关多基因套餐的介绍,请单击here。有关临床医生订购基因检测的更多详细信息,请参见 here。
全基因组的测试(不需要临床医生确定可能涉及哪些基因)是另一个不错的选择。
外显子组测序是最常用的方法。基因组测序也是可用的。如果外显子组测序不能诊断,则可以考虑外显子组阵列(如果在临床上可用),尤其是当证据支持 常染色体显性遗传时。
有关全基因组的测试的介绍,请单击here。有关订购基因组测试的临床医生的更多详细信息,请参见 here。
Table1
用于DFNA2非综合征听力损失的分子遗传学测试
| 基因 1 | 方法 | 此方可检测的具有致病变异 2的先证者所占的比例 |
|---|---|---|
| KCNQ4 | 测序 3 | 99% 4 |
| 靶向基因 deletion/duplication analysis 5 | 1% 6 |
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics
- 3.
- 4.
- 5.
基因靶向deletion/duplication analysis检测基因内的缺失或重复。 所使用的方法可能包括quantitative PCR,远程PCR,多重连接依赖探针扩增(MLPA),以及旨在检测单外显子缺失或重复的基因靶向微阵列。
- 6.
几乎可以在针对性耳聋的目标测序套餐中包括的任何基因中检测到拷贝数变异(CNV)。 在使用OtoSCOPE对2,506名听力损失者进行顺序测试时,确定了一个KCNQ4的CNV [作者,个人观察]。
临床特征
临床表现
患有DFNA2非综合征听力损失的所有个体都有对称的,主要是高频的听力障碍,在所有频率上都是渐进进展的。最初, 受累的是高频; 后期在所有频率上听力损失都会变得严重。 De Leenheer et al [2002a]对被诊断为DFNA2非综合征听力损失的个体的临床表现和预后进行了全面回顾。
一般在儿童早期或青春期就有听力丧失的报道。但是,听力也可能会出生即受到损害,尤其是在高频时。听力损失通常在学龄儿童的标准听力评估过程中被发现, 有时在儿童语言延迟发展评估过程中被发现。
在所有受累的人中,高频时听力损失更为严重,导致特征性的音频曲线向下倾斜,到50岁时,其听力阈值在500 Hz时在50到90 dB之间,在2-4 kHz时在90到120 dB之间。 Figure 1.显示了具有DFNA2非综合征性听力损失的青少年的典型听力图。
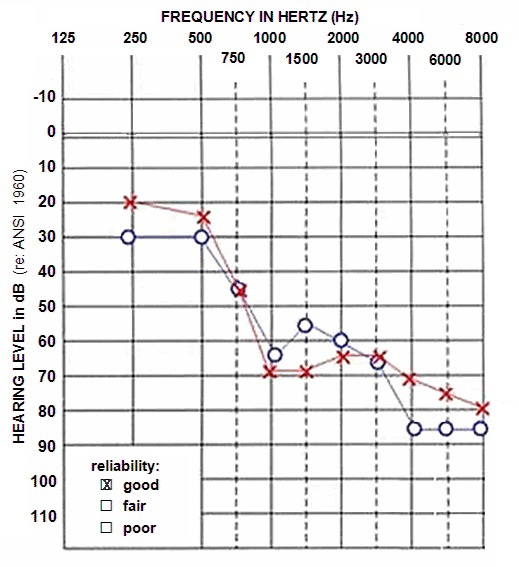
Figure 1.
年龄为12岁且患有DFNA2听力损失的个人的听力图。 请注意,在这个年龄段,高频下的损失更大; 随着时间的流逝,所有频率的听力都会逐渐下降。
尽管家庭中成员的发病年龄各不相同,但DFNA2非综合征听力损失年阈值却以相对均匀的〜1 dB /年计算 [Coucke et al 1999, Talebizadeh et al 1999, Ensink et al 2000, Van Hauwe et al 2000, Akita et al 2001, De Leenheer et al 2002a, De Leenheer et al 2002b, Van Camp et al 2002]。多数患有DFNA2非综合征性听力损失的人首先都配备了助听器,以辅助10至40岁之间的声音放大 [De Leenheer et al 2002a]。到70岁时,所有归因于KCNQ4的致病性变异的听力损失者都患有严重的听力损失。
其他发现
- 前庭功能。在两个患有DFNA2非综合征听力损失的家庭(荷兰家庭1和4;Table 3)中,有30%的人前庭眼反射活动增强[Marres et al 1997, De Leenheer et al 2002b]。在其他任何患有DFNA2非综合征性听力损失的家庭中均未观察到前庭问题。
- 语音识别分数。在几个荷兰家庭中进行测量时,考虑到纯音阈值,语音识别得分相对较高 [De Leenheer et al 2002b, Van Camp et al 2002].
基因型-表型的相关性
杂合的KCNQ4致病性错义变异相关的表型在所有家族中都相似:主要是在儿童中可检测到的高频感觉神经性听力损失(SNHL),并在所有频率上均呈进展性。在较年轻的年龄段,在低频时倾向于轻度听力损失,而高频时则中等听力损失。在老年人中,低频时中等听力损失,而高频时听力损失则严重。
尽管在一个带有 p.Trp276Ser 变异的荷兰家庭中已经报道了DFNA2非综合征听力丧失的 先天的发作 [De Leenheer et al 2002b, Van Camp et al 2002],但在其他具有该变异的家庭中尚未见报道。在这个家庭中,高频听力损失是进行性的,在生命的最初几十年中并没有实质性的语音识别能力丧失 [De Leenheer et al 2002b].
杂合的KCNQ4截短变异相关的 表型不同于与KCNQ4致病性 错义变异相关的表型。在两个家族中,KCNQ4的小移码缺失(c.211_223del和 c.211delC) 导致的截断蛋白质要么与正常等位基因翻译的正常蛋白质不相互作用,要么可能不保留在细胞中而介导 nonsense变异。与这种剂量效应相关的听力损失在低频和中频情况下较轻,在高频情况下则严重,并且发作年龄比致病性错义变异所致的听力下降晚[Coucke et al 1999, Akita et al 2001].
外显率
完全外显率。 具有杂合的KCNQ4 致病性变异的所有个体均表现出听力损失表型。 发病年龄和严重程度是各异的。
患病率
在2012年至2014年之间进行的一项研究中,耳鼻喉科和肾脏分子研究实验室使用多基因OtoSCOPE套餐测试对总共1,119名听力下降的个体进行了临床诊断测试。 已确定,在440位有遗传性听力损失的个体中,KCNQ4致病变异占常染色体显性遗传神经感觉性听力损失(ADNSHL)的9.5% [Sloan-Heggen et al 2016].
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,没有其他表型与KCNQ4中的胚系致病变异相关。鉴别诊断
完整的鉴别诊断见Hereditary Hearing Loss and Deafness Overview 。 注意: Table 4 完整列出了 常染色体显性遗传非综合征听力障碍的基因和临床表现。
处理
初步诊断后的评估
为了确定诊断为DFNA2非综合征性听力损失的个体的听力损失程度和需求,建议如下评估(如果未作为诊断的评估的一部分进行):
- 听力检查,包括骨传导测试
- 咨询临床遗传学家和/或遗传咨询师
表型治疗
有关治疗的完整讨论,请参见Hereditary Hearing Loss and Deafness Overview
当听力损失为轻度至中度时,应确保安装助听器以提供更好的放大效果。
当听力损失变得全面严重时,可以考虑采用人工耳蜗(CIs)。对于低频听力保持或相对良好,高频损失全面严重个体,可以考虑使用混合(短)人工耳蜗。混合植入物结合了两种成熟的技术-声音放大和植入物技术-提供电声听力。耦合到人工耳蜗声音处理器的声学组件会放大残留的低频听力;使用电气组件耳蜗植入技术为高频听力提供电刺激。
对于学龄儿童或青少年,可能需要为听力受损的人提供特殊帮助,并应在可能的情况下提供适当的帮助。
监测
应当每年获取听力图,以追踪听力下降的情况。
避免的药物/环境
通过鼓励患有DFNA2非综合征听力损失的人避免在工作场所和娱乐期间暴露于嘈杂的噪音,可以降低高频听力损失的进展速度。
评估处于危险中亲戚
确定患有DFNA2非综合征性听力损失的婴儿或幼儿亲属是否继承了KCNQ4致病性变异,可以为孩子和家庭提供早期支持和管理。
有关与遗传咨询目的相关的高危亲属测试的问题,请参见Genetic Counseling。
正在调查的疗法
在美国搜索ClinicalTrials.gov,在EU Clinical Trials Register,以获取有关各种疾病和状况的临床研究信息。 注意:可能没有针对该疾病的临床试验。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分介绍了遗传风险评估以及家族史和基因检测的使用,以阐明家族成员的遗传状况。 本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —编者。
遗传方式
DFNA2非综合征性听力损失以常染色体显性遗传方式遗传。
家庭成员的风险
先证者的父母
- 大多数被诊断患有DFNA2非综合征性听力损失的人都有聋哑父母。
- 患有DFNA2非综合征性听力损失的先证者可能会因 de novoKCNQ4变异而出现耳聋。尚不知新发CNCN4 致病性变异的个体比例。 Sloan-Heggen et al [2016]在序列研究中报告的六名临床诊断的个体中,有五名被证实具有 常染色体显性遗传,而其中一名代表单发的病例(即,该家族中单发) 。因此,似乎在大多数情况下,KCNQ4相关听力损失是从受累的父母那里继承的 [Sloan-Heggen et al 2016].
- 对具有明显de novo 致病性变异的先证者父母进行评估的建议包括听力测定法和分子遗传学检测.
- 如果在任一亲本的白细胞DNA中都无法检测到 先证者中发现的致病性变异,则可能的解释包括先证者de novo变异或父母胚系嵌合的致病性变异。尽管从理论上讲是可能的,但尚未报道胚系嵌合的情况。
- 某些患有DFNA2非综合征性听力损失的人的家族史似乎是阴性的,父母在听力损失发作之前过早死亡或父母的听力丧失发作较晚,未能诊断出表现较轻的表型表现的父母,因此,在对先证者的父母进行适当的临床评估和分子遗传学检测之前,无法确定阴性家族史。
- 注意:如果父母是首次发生 致病性变异的个体,则他/她可能具有体细胞嵌合致病性变异,并伴有轻度听力损失。
先证者的同胞。先证者同胞有无耳聋的可能性取决于先证者父母的遗传状况:
- 如果先证者的父母失聪,则每个同胞都有50%的几率失聪。
- 如果在任一亲本的白细胞DNA中均未检测到先证者中发现的KCNQ4致病性变异,则由于胚系嵌合的理论可能性,同胞将患有DFNA2非综合征性听力损失的可能性估计为1%[Rahbari et al 2016].
- 如果父母没有经过KNCQ4 致病性变异的测试,但都听力正常 ,则先证者同胞耳聋的可能性很低。 (注:由于父母的胚系嵌合理论上的可能性,推测患有临床上未受影响的父母的先证者的同胞具有增加的患DFNA2非综合征性听力损失的可能性。)
先证者的后代。患有DFNA2非综合征听力损失的个体的每个孩子都有50%的几率遗传致病性变异。
其他家庭成员。其他家庭成员失聪的可能性取决于先证者'父母的状态:如果父母失聪,则他或她的家庭成员也可能失聪或发展为失聪。
相关的遗传咨询问题
有关出于早期诊断和管理目的而评估先证者亲属的信息,请参阅《管理》,Evaluation of Relatives at Risk。
以下几点值得注意:
- 与聋人社区成员和标志的沟通需要熟练的口译员的服务。
- 聋人社区的成员可能将耳聋视为一种区别特征,而不是需要“治疗”或“治愈”或“预防”的障碍,损害或医疗状况。
- 许多聋人对获取有关其自身失聪原因的信息感兴趣,包括有关医疗,教育和社会服务的信息,而不是有关预防,生殖或计划生育的信息。重要的是要确定并解决家庭/个人的问题。
- 最好使用某些术语:概率或机会与风险的关系;耳聋和听力障碍与听力损失。应避免使用诸如“异常”之类的术语。
具有明显de novo变异的家庭的注意事项。当患有DFNA2非综合征听力损失的先证者的父母都没有 致病性变异或耳聋的临床证据时,先证者很可能具有新发变异。但是,要考虑可能的非医学解释,包括 非生物学父亲或产妇(即辅助生殖)或未公开收养。
家庭计划
- 确定遗传状态和讨论是否可以进行产前检查的最佳时间是在怀孕之前。
- 向患有DFNA2非综合征听力损失的年轻人提供遗传咨询(包括讨论后代将成为耳聋和生殖选择的可能性)是适当的。
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。因为将来测试方法和我们对基因,等位基因变异和耳聋的理解可能会改善,所以应考虑患有DFNA2非综合征听力损失的人的DNA储备。
产前检测和植入前遗传学诊断
一旦在家庭成员中发现了KCNQ4致病性变异,就可以对风险增加的妊娠进行产前检查,并可以对DFNA2非综合征听力损失进行 植入前遗传诊断。
在医疗专业人员之间以及在家庭内部,关于使用产前检查的观点可能存在差异,尤其是如果考虑将检查用于终止妊娠而不是早期诊断的目的时,尤其如此。尽管大多数中心将有关产前检查的决定视为父母的选择,但对这些问题的讨论是适当的。
相关组织机构
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或登记册,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- National Library of Medicine Genetics Home Reference
- Alexander Graham Bell Association for the Deaf and Hard of Hearing3417 Volta Place NorthwestWashington DC 20007Phone: 866-337-5220 (toll-free); 202-337-5220; 202-337-5221 (TTY)Fax: 202-337-8314Email: info@agbell.org
- American Society for Deaf Children (ASDC)800 Florida Avenue NortheastSuite 2047Washington DC 20002-3695Phone: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)Fax: 410-795-0965Email: info@deafchildren.org; asdc@deafchildren.org
- BabyHearing.orgThis site, developed with support from the National Institute on Deafness and Other Communication Disorders, provides information about newborn hearing screening and hearing loss.
- My46 Trait Profile
- National Association of the Deaf (NAD)8630 Fenton StreetSuite 820Silver Spring MD 20910Phone: 301-587-1788; 301-587-1789 (TTY)Fax: 301-587-1791Email: nad.info@nad.org
- NCBI Genes and Disease
分子遗传
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。 —编者.
Table A
DFNA2非综合征性听力损失:基因和数据库
| 基因座 | 基因 | 染色体定位 | 蛋白 | 基因座数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|---|
| DFNA2 | KCNQ4 | 1p34 | Potassium voltage-gated channel subfamily KQT member 4 | KCNQ4 database Deafness Variation Database - KCNQ4 | KCNQ4 | KCNQ4 |
Table B
DFNA2非综合征性听力损失OMIM条目 (View All in OMIM)
基因结构。KCNQ4的转录本长度为2,335个碱基对。 转录由14个外显子组成。 有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
临床意义不确定的变异。 导致同义氨基酸变化的两个变异具有不确定的临床意义(参见Table 2)。 这些核苷酸变异是在具有非综合征性听力损失的185位个体的筛查中检测到的。 据报告,这些人患有非综合征性听力损失。 没有提供有关家族史的信息。 迄今为止,这些变异临床意义仍不确定,因为据作者所知,基因表达尚未得到验证(目前假设这些变异会破坏外显子剪接增强子并干扰正常基因剪接)。
Table 2
该GeneReview中讨论临床意义不确定的KCNQ4变异
| DNA 核酸改变1 | 蛋白改变 1 | 蛋白质区域 | 人群 | 发病年龄 2 | 参考文献 |
|---|---|---|---|---|---|
| c.648C>T | p.Arg216= 3 | S4 transmembrane 结构域 | 台湾人 | 儿童 | Su et al [2007] |
| c.1503C>T | p.Thr501= 3 | Distal to S6 transmembrane 结构域 |
表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
GeneReviews遵循人类基因组变异学会 (varnomen
- .hgvs.org )的标准命名约定。 有关命名法的说明,请参见Quick Reference。- 1.
KCNQ4的参考序列: NM_004700
- .2 , NP_004691- .2 - 2.
对于表中所述的所有致病变异,病理是高频听力障碍,并且在耳蜗外毛细胞和大脑中组织特异性表达。
- 3.
预计不会对蛋白质水平产生影响。
致病变异。 大多数KCNQ4致病变异聚集在外显子5、6和7中,它们编码形成通道孔的高度保守的氨基酸序列。 主要的致病变异是引起显性负效效应的错义变异。 p.Trp276Ser变异似乎是最常见的,已在四个无关家庭中鉴定出,包括五个患有DFNA2非综合征性听力损失的荷兰家庭中的三个和一个日本家庭(参见 Table 3)。
Table 3
该GeneReview中讨论的KCNQ4致病变异
| DNA 核酸改变 (Alias 1) | 可能的蛋白质改变 (Alias 1) | 蛋白质区域 | 人群 | 发病年龄 2 | 参考文献 |
|---|---|---|---|---|---|
| c.211_223del13 (211del13) | p.Gln71ProfsTer64 (Q71fsTer134) | N-terminal cytoplasmic | 比利时人 | Adolescence | Coucke et al [1999] |
| c.211delC | p.Gln71SerfsTer68 (FS71) | N-terminal cytoplasmic | 日本人 | Adolescence | Kamada et al [2006], Ishikawa et al [2014] |
| c.827G>C | p.Trp276Ser | P-loop | 荷兰人,日本人 | Childhood | Coucke et al [1999], Van Camp et al [2002], Topsakal et al [2005] |
| c.853G>T | p.Gly285Cys | P-loop | 北美洲人 | Childhood | Coucke et al [1999] |
| c.853G>A | p.Gly285Ser | P-loop | 北欧人,汉族人 | Childhood | Kubisch et al [1999], Wang et al [2014] |
表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
GeneReviews遵循人类基因组变异学会 (varnomen
- .hgvs.org )的标准命名约定。 有关命名法的说明,请参见Quick Reference。KCNQ4参考序列: NM_004700
- .2 , NP_004691- .2 - 1.
不符合当前命名约定的变异名称
- 2.
对于表中所述的所有致病变异,病理是高频听力障碍,并且在耳蜗外毛细胞和大脑中组织特异性表达。
正常基因产物。 KCNQ4编码的蛋白质长度为695个氨基酸,并形成具有六个跨膜结构域和一个P环区的钾通道,形成通道孔。 P-环内高度保守的甘氨酸-酪氨酸-甘氨酸(GYG)序列包含选择性过滤器,该过滤器可区分钾离子以进行选择性转运[Kubisch et al 1999]。致病性变异聚集在通道孔区域中,并且某些变异(即p.Gly285Ser和p.Gly285Cys)直接影响选择性过滤器。
异常基因产物。大多数KCNQ4致病变异是错义改变,通过 显性负效效应导致听力丧失。该表型反映了内耳中KCNQ4蛋白缺陷的后果。该蛋白质组装成四聚体以形成由四个亚基组成的钾通道。在具有一种致病性错义变异的人中,编码蛋白总量的一半是有缺陷的,因此,每16个通道中只有一个正常的包含四个蛋白亚基通道[Kubisch et al 1999]。随着时间的流逝,结果是认为内耳钾循环的逐渐丧失。由于钾离子对于毛细胞的转导至关重要,因此无法回收这些离子会导致听力损失。大多数致病变异影响位于通道孔内或附近的氨基酸。异常蛋白质亚基的存在会干扰内耳中四聚体通道蛋白的组装和/或功能。
一些KCNQ4致病变异导致 单倍剂量不足的缺失。结果,内耳细胞产生的功能性KCNQ4蛋白不足,并且随着时间的流逝,听觉功能受到损害。
关于位点异质性的证据。基于在两个中国小家庭中鉴定出的两个不同的GJB3基因序列变异,GJB3被认为是DFNA2基因座的耳聋相关基因。来自两个家庭的个体都有双侧感觉神经性听力损失(SNHL),其特征在于听力图从正常的听力阈值1,000毫秒以下缓慢下降到高频的中等程度的听力损失。
但GJB3与听力损失相关联的证据既不充分也不令人信服:
- 在两个家庭中,其他听力正常的个体均报告了GJB3的致病变异,这一发现与完全 外显率不一致,几乎在所有类型的常染色体显性遗传SNHL中都观察到了这一发现。
- 怀疑在这两个家族中排除了KCNQ4致病性变异,这是在1998年报道的,因为KCNQ4致病性变异直到1999年才确定与常染色体显性遗传SNHL有关。
- 尚无其他具有常染色体显性遗传SNHL的家族分离出GJB3致病变异的报道。
- GJB3中的特定致病变异会导致变异性红斑角质病。
参考文献
引用文献
- Akita J, Abe S, Shinkawa H, Kimberling WJ, Usami S. Clinical and genetic features of nonsyndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a gene responsible in Japanese. J Hum Genet. 2001;46:355 - 61. [PubMed: 11450843]
- Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJ, Cremers CW, Van de Heyning P, Willems PJ, Smith SD, Van Camp G. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet. 1999;8:1321 - 8. [PubMed: 10369879]
- De Leenheer EM, Ensink RJ, Kunst HP, Marres HA, Talebizadeh Z, Declau F, Smith SD, Usami S, Van de Heyning PH, Van Camp G, Huygen PL, Cremers CW. DFNA2/KCNQ4 and its manifestations. Adv Otorhinolaryngol. 2002a;61:41 - 6. [PubMed: 12408061]
- De Leenheer EM, Huygen PL, Coucke PJ, Admiraal RJ, van Camp G, Cremers CW. Longitudinal and cross-sectional phenotype analysis in a new, large Dutch DFNA2/KCNQ4 family. Ann Otol Rhinol Laryngol. 2002b;111:267 - 74. [PubMed: 11915881]
- Ensink RJ, Huygen PL, Van Hauwe P, Coucke P, Cremers CW, Van Camp G. A Dutch family with progressive sensorineural hearing impairment linked to the DFNA2 region. Eur Arch Otorhinolaryngol. 2000;257:62 - 7. [PubMed: 10784363]
- Ishikawa K, Naito T, Nishio SY, Iwasa Y, Nakamura K, Usami S, Ichimura K. A Japanese family showing high-frequency hearing loss with KCNQ4 and TECTA mutations. Acta Otolaryngol. 2014;134:557 - 63. [PubMed: 24655070]
- Kamada F, Kure S, Kudo T, Suzuki Y, Oshima T, Ichinohe A, Kojima K, Niihori T, Kanno J, Narumi Y, Narisawa A, Kato K, Aoki Y, Ikeda K, Kobayashi T, Matsubara Y. A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet. 2006;51:455 - 60. [PubMed: 16596322]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437 - 46. [PubMed: 10025409]
- Marres H, van Ewijk M, Huygen P, Kunst H, van Camp G, Coucke P, Willems P, Cremers C. Inherited nonsyndromic hearing loss. An audiovestibular study in a large family with autosomal dominant progressive hearing loss related to DFNA2. Arch Otolaryngol Head Neck Surg. 1997;123:573 - 7. [PubMed: 9193215]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126 - 33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441 - 50. [PMC free article: PMC4796320] [PubMed: 26969326]
- Su CC, Yang JJ, Shieh JC, Su MC, Li SY. Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol Neurootol. 2007;12:20 - 6. [PubMed: 17033161]
- Talebizadeh Z, Kelley PM, Askew JW, Beisel KW, Smith SD. Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat. 1999;14:493 - 501. [PubMed: 10571947]
- Topsakal V, Pennings RJ, te Brinke H, Hamel B, Huygen PL, Kremer H, Cremers CW. Phenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S). Otol Neurotol. 2005;26:52 - 8. [PubMed: 15699719]
- Van Camp G, Coucke PJ, Akita J, Fransen E, Abe S, De Leenheer EM, Huygen PL, Cremers CW, Usami S. A mutational hot spot in the KCNQ4 gene responsible for autosomal dominant hearing impairment. Hum Mutat. 2002;20:15 - 9. [PubMed: 12112653]
- Van Hauwe P, Coucke PJ, Ensink RJ, Huygen P, Cremers CW, Van Camp G. Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet. 2000;93:184 - 7. [PubMed: 10925378]
- Wang H, Zhao Y, Yi Y, Gao Y, Liu Q, Wang D, Li Q, Lan L, Li N, Guan J, Yin Z, Han B, Zhao F, Zong L, Xiong W, Yu L, Song L, Yi X, Yang L, Petit C, Wang Q. Targeted high-throughput sequencing identifies pathogenic mutations in KCNQ4 in two large Chinese families with autosomal dominant hearing loss. PLoS One. 2014;9:e103133. [PMC free article: PMC4130520] [PubMed: 25116015]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet. 1998;20:370 - 3. [PubMed: 9843210]
本章节备注
作者备注
Molecular Otolaryngology Research Laboratories home page
Hereditary Hearing Loss home page
Deafness Variation Database. 耳聋变异数据库(DVD)整理来自主要公共数据库的数据,以基于收集的证据为每个变异提供单一分类; 它由遗传性听力损失专家策划,目的是为侧重于耳聋的变异解释提供单一来源指南。
更新历史
- 10 May 2018 (bp)系统性更新发布到公开网页上
- 20 August 2015 (me) 系统性更新发布到公开网页上20 June 2013 (me) 系统性更新发布到公开网页上
- 17 February 2011 (me) 系统性更新发布到公开网页上
- 4 April 2008 (me) 内容发布到公开网页上
- 19 December 2007 (rjhs) 初稿提交