
摘要
临床表现 经典型MLC以早期起病的巨颅为特点,常伴有大运动的轻度发育迟缓及抽搐;逐渐出现共济失调、痉挛,有时可有椎体外系表现;病程晚期,常出现轻度的智力倒退。所有的患儿均有巨颅,它可于出生时即出现,但更多的于生后1年内出现。巨颅的轻重程度不一,部分患儿头围可比平均值高4-6个标准差。1岁以后,头围增长速度恢复正常,增长曲线常与第98百分位数增长曲线平行,但高数个厘米。几乎所有的患儿在幼年时即有癫痫。大多数患儿早期智力及运动发育正常。独走常不稳,随后出现躯干及四肢的共济失调、微小的锥体束受损症状及深部腱反射亢进。智力倒退出现较晚且程度较轻。独立行走能力严重程度轻重不一,部分仅可独走数年,但部分可保留独走能力致五十几岁。部分患者于十几岁或二十几岁去世,部分患者可存活致四十几岁。
非经典的改善型在疾病初期与经典型表现相似,但不伴智力或运动的倒退,相反有一个改善的临床病程:其运动及认知功能可改善或恢复正常;巨颅常持续存在,但也有部分患儿头围最终正常;患儿可遗留有肌张力减低和笨拙等或神经系统检查正常。部分患儿可有稳定的智力障碍,伴或不伴孤独症样表现。
诊断/检测 MLC的诊断建立在患儿典型的临床表现及头颅MRI表现上,包括大脑半球白质肿胀及异常信号,前颞叶和常见的额顶叶皮层下囊肿。MLC1双等位基因突变导致的经典型称为MLC1型;HEPACAM双等位基因突变导致MLC2A型。改善型与HEPACAM杂合突变相关,称为MLC2B型。约有75%MLC患者可检测到MLC1致病性变异,约20%患儿可检测到HEPACAM突变。
管理 对症处理:使用抗癫痫药物(Antiepileptic drugs,AEDs)控制癫痫发作;康复训练改善患儿运动功能;特殊教育。
遗传咨询 MLC1型及MLC2A型为常染色体隐性遗传模式。理论上,受累个体的每个同胞均有25%的风险成为患者,50%的可能性为无症状携带者,25%的可能性为不携带不患病的正常个体。如果家系中两个致病性等位基因突变均已确定,那么可对家系中风险亲属及高风险妊娠分别进行携带者检测及产前诊断。
MLC2B型为常染色体显性遗传模式。新生的致病性变异很常见。MLC2B型患者的每一个子女均有50%的可能性遗传到致病性变异。如果家系的致病突变得以确定,那么可对高危妊娠进行产前诊断。
诊断
临床诊断
如果患者具有MLC典型的临床表现及特征性的头颅MRI特点,则很有把握将该患者诊断为MLC [van der Knaap et al 1995, Singhal et al 1996, Topçu et al 1998]。
典型临床表现
经典型
- 所有患者均有出生时或者生后1年内(更常见)出现的巨颅。1岁以后,头围增长速度正常,头围增长曲线位于第98百分位增长曲线上方,并与其平行。
- 早期发育正常或者轻度落后。大多数(但不是所有)患儿可获得独立行走的能力。
- 儿童早期或晚期开始出现小脑共济失调及轻度痉挛所致缓慢运动功能倒退。大多数受累儿童在十几岁即需要轮椅。
- 逐渐出现构音障碍,也可出现吞咽困难。
- 一些患儿可出现椎体外系运动异常,包括肌张力障碍和手足徐动,且常于晚期出现。也可出现抽动。
- 智力倒退出现得较运动倒退晚,并且程度较轻。
- 一些患儿可出现行为问题。
- 大多数患儿有癫痫发作,一般易被药物控制住,但是也有一些患儿可出现癫痫持续状态。
- 轻微的头部外伤可致部分患儿出现短暂性倒退,大多数表现为抽搐、长时程意识障碍或者是可缓慢恢复的急性运动倒退。
MLC1双等位基因突变导致的经典型称为MLC1型;HEPACAM双等位基因突变导致MLC2A型。
改善型
- 所有患者均有出生时或者生后1年内(更常见)出现的巨颅。1岁以后,头围增长速度正常,头围增长曲线位于第98百分位增长曲线上方,并与其平行。少数患儿头围最终正常。
- 早期发育正常或者轻度落后。所有患儿均可获得独立行走能力。
- 1岁以后,运动功能逐渐好转,部分患儿遗留笨拙或肌张力减低。
- 部分患儿有癫痫发作,一般易被药物控制,但是也有一些患儿可出现癫痫持续状态。
- 部分患儿智力障碍程度稳定,伴或不伴孤独症。
- 不会有运动或智力的倒退。
改善型与HEPACAM杂合突变相关,称为MLC2B型。
MRI标准
头颅MRI具有诊断性意义。(见图 1)
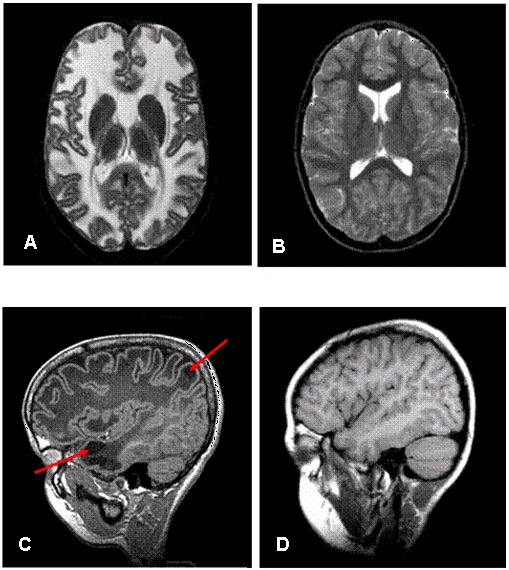
- 大脑半球白质弥漫性异常信号及轻度肿胀;见图 1A(MLC1型患者),图 1B(正常个体)
- 中央白质结构,包括胼胝体、内囊和脑干虽然会受累,但较其他结构保存较好。
- 小脑白质常有轻度异常信号,但不伴肿胀。
- 几乎所有患儿均有皮层下囊肿,可出现在前颞叶,但更多的是在额顶叶。见图1C(MLC1型患儿),图1D(正常个体)
- 随着病程进展,白质肿胀减轻,开始出现大脑萎缩。皮层下囊肿数量及体积均增加。在部分患儿中,囊肿体积增致巨大,占据大量额顶叶白质位置。而另外一些患儿,大脑白质异常逐渐消失,大脑白质异常信号强度较前减弱。
- 弥散加权像(Diffusion-weighted imaging,DWI)上异常白质弥散增强[Itoh et al 2006, van der Voorn et al 2006]。
- MLC2B型患儿1岁以内的MRI同上所述,但随后明显好转。部分患儿MRI可在数年内恢复正常,但部分患儿可遗留微小的额叶、颞叶皮层下白质异常以及前颞叶的囊肿。
检测
常规的实验室检测,包括脑脊液分析,均为正常。
神经病理学检查 脑活检主要的表现为髓鞘外层大量囊泡的形成,提示髓鞘板层沿着周期内线裂开或者是髓鞘板层不完整 [van der Knaap et al 1996]。此外,在星形胶质细胞终足处也可见小体积的囊泡 [Duarri et al 2011]。
分子遗传学检测
基因 MLC是由MLC1或HEPACAM致病性变异所致。
可能遗传异质性(此处不知如何翻译,根据下述内容翻译)当一个患儿具有MLC典型的临床表现及头颅MRI特征性表现,但未检测到MLC1或HEPACAM突变时,不能排除MLC的诊断,因为可能存在以下原因:(1)所采用的检测方法的局限性未检测到MLC1或HEPACAM突变;(2)患儿的MLC表型可能由其他未被确定的基因突变导致。
MLC分子遗传学检测小结
基因1 | 在MLC患者中所占比例 | 检测方法 | 可检测到的变异2 |
MLC1 | 75%3 | 序列分析4 | 序列变异 |
缺失/重复分析5 | (多个)外显子或全基因的缺失/重复6 | ||
HEPACAM | 20%7 | 序列分析 | 序列变异4 |
- 见表A. 染色体位点与蛋白的基因与数据库.
- 等位基因变异信息见分子遗传学。
- 对具有MLC临床及MRI表现的患者进行MLC1基因组DNA编码区序列分析可检测到65%-70%患儿有纯合或复合杂合变异。MLC1大片段缺失或深部内含子变异需要采用缺失/重复分析或cDNA测序。如果基因组DNA及cDNA分析均未能发现MLC1上的致病性变异,那么该患者很有可能有其他基因的突变,最常见的为HEPACAM。
由于并不是所有时候都可获得患儿的cDNA,所以当对MLC1基因组DNA的外显子及外显子-内含子交界区域进行测序后,仍会有一部分MLC1变异检测不到。
- 序列分析检测可检测到良性、可能良性、意义不确定、可能致病性或致病性变异。致病性变异包括小的基因内部的缺失/插入、错义、无义以及剪接位点变异; 但通常检测不到外显子或者全基因缺失/重复。有关序列分析结果解读的相关问题 点击这里.。
- 对基因组DNA编码区与侧翼内含子的区序列分析不能检测到缺失/重复变异,但定量PCR,长片段PCR,多重连接依赖的探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)以及靶向染色体芯片分析技术(基因特异性或片段特异性)可用于检测缺失/重复突变。用于检测基因组缺失/重复的染色体芯片分析也可能包含该基因/片段。
- Leegwater et al [2002], Ilja Boor et al [2006]
- 对未检测到MLC1致病性变异的MLC患者进行HEPACAM基因组DNA编码区进行测序分析,约有75%检测到HEPACAM突变:2/3为HEPACAM杂合突变(如MLC2B型),1/3具有HEPACAM双等位基因致病性变异(如MLC2A型)[López-Hernández et al 2011]。
检测结果说明
- 如果在MLC1或者HEPACAM上检测到双等位基因突变,则可分别诊断为MLC1型或MLC2B型,临床上表现为经典型。
- 如果检测到HEPACAM杂合突变,结合随访中患儿头颅MRI改善的临床特点,可诊断为常染色体显性遗传的MLC2B型,临床上表现为改善型。
- 当一个患儿具有MLC典型的临床表现及头颅MRI特征性表现,但未检测到MLC1或HEPACAM突变时,不能排除MLC的诊断,因为可能存在以下原因:(1)所采用的检测方法的局限性未检测到MLC1或HEPACAM突变;(2)患儿的MLC表型可能由其他未被确定的基因突变导致。
检测策略
先证者的确诊
- 当患儿符合MLC临床诊断标准,包括头颅MRI表现时,即可诊断为MLC。
- 如果先证者不符合MLC的诊断标准,比如,不具有典型的巨颅或头颅MRI表现不够典型,那么MLC的确诊依赖于分子遗传学检测结果。
- 如果先证者具有经典型的特点,那么建议首先对先证者行MLC1的分子遗传学检测,如果MLC1没有检测到突变,则进行HEPACAM的分子遗传学检测。
- 如果先证者具有改善型的特点,那么建议首先对先证者进行HEPACAM的分子遗传学检测。
亲属中携带者的检测 对常染色体隐性遗传的MLC1型和MLC2A型家系中高风险成员进行携带者检测必须建立在已经确定家系致病性突变的基础上。
携带有分别可导致MLC1型和MLC2A型表型的MLC1和HEPACAM杂合变异者,不会有发展为MLC的风险。
产前诊断和植入前遗传诊断对高风险妊娠进行胚胎植入前产前诊断需建立在确定了家系致病性突变的基础上。
临床特点
临床表现
MLC有经典型及非经典改善型两种表型。MLC1双等位基因突变导致的经典型称为MLC1型;HEPACAM双等位基因突变导致MLC2A型。改善型与HEPACAM杂合突变相关,称为MLC2B型。
在目前的所有报道中,所有MLC患儿均有巨颅的临床表现[van der Knaap et al 1995, Goutières et al 1996, Singhal et al 1996, Topçu et al 1998, Ben-Zeev et al 2001]。它可在出生时即有,但更多的是在生后1年内出现。巨颅的程度轻重不一:部分患儿的头围可比平均值高4-6个标准差。1岁以后,头围增长速度正常,增长曲线与第98百分位数增长曲线平行。非经典改善型患儿头围在起病时与经典型患儿一样大,但随后可恢复正常。
经典型MLC(MLC1型和MLC2A型) 大多数患儿最初的智力及运动发育正常,少数轻度落后。除了进展性的巨颅外,患儿第一个临床表现常为独立行走晚于正常婴幼儿。行走经常不稳,并且经常摔倒。随后,随着年龄增长,患儿可由于躯干及四肢共济失调而逐渐出现运动功能倒退。锥体束受损的症状出现得较晚且较轻,而且常常被小脑性共济失调的症状所覆盖。一些患者踝关节肌张力偏高,但大多数肌张力偏低。深部腱反射亢进,巴氏征阳性。随着病程进展,患儿逐渐丧失独立行走能力,许多患儿在十几岁或二十几岁时即完全需要依靠轮椅。
几乎所有患儿在疾病早期即有癫痫,但大多数易被药物所控制[Yalcinkaya et al 2003]。
智力倒退出现得较晚且较轻。小学的后几年级,患儿在校表现欠佳会变得比较明显。少数患儿在早期即有智力的轻度障碍。部分患儿可伴有行为问题[Sugiura et al 2006]。逐渐出现构音障碍,也可出现吞咽困难。一些患儿出现椎体外系运动异常,包括肌张力障碍和手足徐动。部分可有抽动[Sugiura et al 2006]。
轻微的头部外伤可致部分患儿出现短暂性倒退,大多数表现为抽搐或癫痫持续状态、长时程意识障碍或者是可缓慢恢复的急性运动倒退[Bugiani et al 2003]。
部分患儿病程更重,仅能独走几年,甚至终身不能获得独立行走能力。
部分患儿病程较为良性,直到青少年时期,也仅仅表现为巨颅。部分患者在其四十几岁仍能独立行走。
由于MLC认识的时间尚短,患者平均寿命的研究资料不足。部分患儿于十几岁或二十几岁去世,而部分可以存活至五十几岁。
改善型MLC(MLC2B型) MLC2B患儿在疾病早期与经典型患者表现相同:大多数患儿智力及运动发育正常,少数轻度落后。除了进展性的巨颅,第一个明显的临床表现为行走较晚。行走经常不稳,且易摔跤。
在2-3岁,大多数患儿运动功能不断改善甚至恢复正常。神经系统检查可正常,但也有部分患儿遗留有肌张力减低及笨拙。不会发生运动功能倒退。
认知功能可正常,但部分患儿也可有智力障碍,伴或不伴自闭症。不会出现智力功能倒退。
患儿可有癫痫或癫痫持续状态。
大多数患儿遗留有巨颅,但也有部分患儿头围值恢复正常。
由于疾病认识的时间尚短,几乎尚无患者平均寿命的研究。仅有1例患儿在4岁由于癫痫持续状态死亡 [作者,尚未发表]。
当一个家系中有两代或超过两代的个体受累时,先证者常为小孩,随后患病的父母才会得以确诊。具有相同致病性变异的父母常常有巨颅,但其余功能一般正常。部分可有认知/行为问题或运动笨拙。考虑到带有HEPACAM杂合突变的父母身体状况正常,所以推测杂合的HEPACAM突变可能不会影响患者的寿命,但目前尚无正式的研究强调这一点。
基因型-表现关系
经典型MLC 目前尚无研究表明由MLC1突变(MLC1型)和HEPACAM突变(MLC2A型)导致的经典型患者表型有差异。
- MLC1型和MLC2A型患者表型严重程度与突变无关。
- 同一家系中不同患者的疾病严重程度及病程差异很大。
改善型MLC 同一家系中不同患者的疾病严重程度及病程差异很大。同一家中与先证者具有相同的变异的父母或者同胞可完全正常或者仅有巨颅。
命名
既往MLC也被叫做
- 良性病程的肿胀性白质脑病 (Leukoencephalopathy with swelling and a discrepantly mild course)
- 伴肿胀及囊肿形成的白质脑病 (Leukoencephalopathy with swelling and cysts)
- 伴巨颅的婴儿白质脑病 (Infantile leukoencephalopathy and megalencephaly)
- 囊泡形成性白质脑病 (Vacuolating leukoencephalopathy)
流行病学
MLC是一种罕见病,在人群中携带者频率很低。因此,本病在低近亲婚配的群体中发生率更低,但在高近亲婚配群体中发生率更高。许多经典型MLC的父母为近亲婚配。
MLC1致病性变异导致经典型MLC(MLC1型)
- MLC1型在近亲婚配常见的群体中,如土耳其,发生率更高[Topçu et al 1998]。
- 几乎所有东印度群岛MLC1型患者均为阿加渥人种,患病的个体均具有相同的致病性变异(c.135insC, p.Cys46LeufsTer34),说明此突变具有建立者效应[Leegwater et al 2002, Singhal et al 2003, Gorospe et al 2004]。
- MLC1型在利比亚犹太人中也相对常见[Ben-Zeev et al 2001]。在5个无生物学关联的利比亚犹太家系中检测到一个相同的致病性变异(c.176G>A, p.Gly59Glu) [Ben-Zeev et al 2002]。在一个具有相同祖先的犹太人种土耳其家系中的数个受累个体里也检测到相同变异。该变异在利比亚犹太人中估计的携带者频率为1/81,但通过对200例正常的利比亚犹太人进行该位点的检测,发现该位点携带者频率为1/40。尚未在非犹太人种的土耳其患病个体中检测到相同变异。
- MLC1的致病性变异c.278C>T, p.Ser93Leu虽然在日本患者中常见[Saijo et al 2003, Tsujino et al 2003],但在芬兰、土耳其、意大利患儿中也有报道[Leegwater et al 2001;Montagna et al 2006]。
基因相关性疾病(等位基因病)
MLC1突变除了导致在GeneReview中所描述的表型外,可能还与紧张型精神分裂症(catatonic schizophrenia)相关。
- Meyer[2001]等认为一个假定的显性作用的错义变异,即p.Leu309Met,在某一周期性紧张症(periodic catatonia)大家系中符合共分离。
- Rubie[2003]等对140例周期性紧张症的个体以及5个MLC患者进行MLC1分析,其结果与Devaney[2002]等及Kaganovich[2004]等的研究结果可能可排除MLC1为精神分裂症易感基因。
- Verma[2005]等认为MLC1的罕见及常见变异参与精神分裂症及双相情感障碍(bipolar disorder)。
- Selch[2007]等对Verma[2005]的数据重新分析后将MLC1变异缩窄到仅与周期性紧张相关,并且认为“有一种更大可能性:MLC1上相关的单核苷酸多态性(single nucleotide polymorphism,SNP)与一个潜在的附近的“真实”致病基因之间存在连锁不平衡(linkage disequilibrium)。“
阐明MLC1的功能以及与精神分裂症某一亚群体相关的MLC1变异的影响,对于解决目前这个争论十分必要。
鉴别诊断
主要与为数不多的几种以巨颅及弥漫性白质脑病为特点的疾病相鉴别,包括Canavan病(Canavan disease)、亚历山大病(Alexander disease)和GM2神经节苷脂沉积症婴儿型(infantile-onset GM2 gangliosidosis),有时还需要与GM1神经节苷脂沉积症婴儿型(infantile-onset GM1 gangliosidosis)、L-2羟基戊二酸尿症(L-2-hydroxyglutaric aciduria)相鉴别。部分层粘连蛋白α2链缺陷导致的先天性肌营养不良(muscular dystrophy caused by laminin alpha-2 (merosin) deficiency,MDC1A)患儿也有巨颅的临床表现。这些疾病的临床特点与病程与常MLC不同。如果患者的头围在1岁以内都是正常的,那么这个患儿不是MLC的可能性非常大。这些病的头颅MRI特点也与MLC不同。
层粘连蛋白α2链缺陷 虽然层粘连蛋白α2链缺陷的白质异常改变与MLC非常相似,但是前者没有典型的皮层下囊肿[van der Knaap et al 1997]。此外,层粘连蛋白α2链缺乏病患儿有非常显著的无力和肌张力减低,这些都不是MLC的特点。分子遗传学检测有助于确诊(见先天性肌营养不良)。
Canavan病 典型的头颅MRI表现为丘脑及苍白球受累,受累相对较轻的壳核及尾状核形成双侧新月形区域。而苍白球及丘脑在MLC中不受累。Canavan病白质也可囊性变,但是不是MLC那种特征性的皮层下囊肿。尿液中N-乙酰基天冬氨酸(N-acetylaspartic acid ,NAA)升高和/或ARSA分子遗传学分析有助于新生儿/婴儿Canavan病的诊断。
亚历山大病 巨颅及头颅MRI表现为额叶主要受累的白质脑脑病见于亚历山大病[van der Knaap et al 2001]。MLC与亚历山大病不同,没有前头部主要受累的特点。亚历山大病中一些特殊的脑结构始终有对比增强,这一点也与MLC不同。亚历山大病也可有囊样退行性变,但发生的位置与MLC不同:前者主要出现在额叶深部白质。GFAP的分子遗传学检测有助于确诊亚历山大病。
GM2神经节苷脂沉积症婴儿型 除了白质异常外,基底节、丘脑受累为其典型的头颅MRI特征。血清、白细胞或培养的皮肤成纤维细胞中氨基己糖苷酶A(hexosaminidase A)及氨基己糖苷酶B(hexosaminidase B)的活性分析可确诊。
GM1神经节苷脂沉积症婴儿型 头颅MRI表现与GM2神经节苷脂沉积症非常相似[Chen et al 1998]。白细胞或者培养的皮肤成纤维细胞中β-半乳糖苷酶(beta-galactosidase)活性缺陷可确诊本病。
管理
初诊后评估/初诊流程/初诊评估
为确定MLC患者的疾病程度,建议进行如下的评估:
- 神经系统检测
- 头颅MRI
- 物理治疗/作业疗法认知评定
- 认知障碍评估(神经心理检查)
对症治疗
主要包括以下支持疗法
- 如果有抽搐,则使用抗癫痫药物
- 物理治疗改善运动功能
- 特殊教育
- 如有必要,行语言训练
预防继发性并发症
轻度的头部外伤可导致暂时性运动倒退或(极少见)昏迷。所以当患儿具有较高风险发生头部外伤时应佩戴头盔。
避免以下事物/环境
轻度的头部外伤可导致暂时性运动倒退或(极少见)昏迷。因此,患儿应避免接触性运动以及其他易发生头部外伤的活动。
高风险亲属的评估
以遗传咨询为目的的高风险亲属评估相关问题见遗传咨询。
正在研发的治疗手段
可于“ClinicalTrials.gov” 上查询大量疾病相关临床研究信息。注意:可能目前暂无关于此病的研究。
其他
利尿剂、乙酰唑胺以及肌酸已被证实无效。
遗传咨询
遗传咨询是为个体及家系提供遗传性疾病的本质、遗传方式及并发症等信息来帮助他们做出知情的医学及个体决定的过程。以下几个方面涉及到遗传风险评估以及家族史和遗传检测的使用,用以阐明家系成员遗传状态。这一部分并不能解决个体可能面临的个人、文化及伦理问题,也不能代替遗传学专家的咨询。-ED
遗传模式
MLC1或HEPACAM双等位基因突变所致(前者为MLC1型,后者为MLC2A型)的经典型MLC为常染色体隐性遗传。
HEPACAM杂合突变(MLC2B型)导致的改善型MLC为常染色体显性遗传。
家系成员风险-常染色体隐性遗传(MLC1型及MLC2A型)
先证者之父母
- 其父母必定是杂合子,因此各携带一个突变的等位基因。
- 杂合子(携带者)无症状。目前在杂合子(携带者)中尚未发现临床表现或头颅MRI异常。
先证者之同胞
- 理论上,患者的任何一个同胞均有25%的可能性成为患者,50%可能性成为无症状携带者以及25%可能性成为既不受累也不携带的正常个体。
- 一旦一个同胞已经确定不是患者,那么他是携带者的可能性为2/3。
- 杂合子(携带者)无症状。目前在杂合子(携带者)中尚未发现临床表现或头颅MRI异常。
先证者之子代 其子代必定是携带一个致病性变异的杂合子。
先证者其他家系成员 先证者父母的任何的同胞均有50%可能性为携带者。
携带者检测
一旦家系的致病性变异得以确定,则对先证者亲属进行携带者检测则成为可能。
家系成员风险-常染色体显性遗传(MLC2B型)
先证者之父母
- 许多MLC2B型患儿的父亲或母亲有巨颅。考虑到此种表型的神经功能可改善,因此其父或母不会出现神经功能倒退。虽然目前尚无研究说明HEPACAM杂合突变是否会在疾病晚期导致其他症状的出现,但目前尚无证据表明患者会在晚期出现病情进展。
- MLC2B型的先证者的突变可为新生突变。
- MLC2B型患者中新生突变的比例目前尚不清楚,但研究者们认为新生突变的发生相当频繁[López-Hernández et al 2011]。
- 如果先证者父母白细胞DNA未能检测到相应致病性变异,有两种可能性可解释这一现象:父或母为胚系嵌合、先证者发生新生突变。虽然目前尚无生殖细胞嵌合的报道,但这仍是可能的现象。
- 对带有新生致病性变异的先证者的父母进行的评估包括分子遗传学检测。先证者的父母一方可能为受累个体,但由于既往未能识别此种综合征或其表型太轻而致漏诊。因此,当先证者强调阴性家族史时,也需得等到相应的评估均完成后才能确定。
先证者之同胞
- 先证者同胞受累的风险依赖于先证者父母的遗传状态。
- 如果先证者的父或母具有HEPACAM杂合致病性变异,那么先证者同胞获得变异的风险为50%。
- 即使先证者的父母没有临床表型,先证者的同胞仍有一定风险,因为先证者的父或母的外显率可能较低。
- 如果先证者父母白细胞DNA未能检测到相应致病性变异,那么先证者同胞的患病风险较低但仍高于普通人群,因为先证者父母有可能存在胚系嵌合。
先证者之子代 MLC2B型患者的子代均有50%的可能性获得致病性变异。
先证者其他家系成员 先证者其他家系成员患病的风险依赖于先证者父母的遗传状态。如果先证者之父或母受累,那么父亲或者母亲的家系成员有一定风险患病。
遗传咨询相关问题
生育计划
- 评估遗传风险、明确携带者状态以及讨论产前检查可行性的最佳时间均都在妊娠前。
- 应对所有受累个体、携带者以及有风险为携带者的有生育需求的年轻成年人提供遗传咨询(包括子代潜在受累风险及生育选择)。
DNA银行是指以备将来所需而进行的DNA储备(通常是从白细胞中提取)。因为我们对检测手段以及对基因、等位基因变异以及疾病的认识在将来都很有可能会有提高,所以现在应该对受累的个体建立DNA银行。
产前检查以及植入前遗传学诊断
一旦受累家系的致病性变异得以确定,那么对高危妊娠进行产前诊断或者植入前遗传诊断则成为可能。
资源
GeneReviews的成员为MLC患者挑选出以下疾病特异性机构和/或伞支持机构和/或登记处。但GeneReviews不为这些机构所提供的信息负责。进一步获取入选标准,请点击此处。
- 澳大利亚脑白质营养不良支持小组
神经中心
澳大利亚,布莱克本维多利亚 3130,铁路道 54号
联系电话:1800-141-400 (受话方付费电话); +61 3 98452831
传真:+61 3 95834379
邮箱:mail@alds.org.au
www.alds.org.au
- 欧洲脑白质营养不良协会
法国,Laxou Cedex 54521,B.P. 61024米洛斯-维恩斯街2号
联系电话:03833093 34
传真:03833000 68
邮箱:ela@ela-asso.com
- 脑白质营养不良联合基金会
DeKalb IL 60115,Suite 2,北二街224号
联系电话:800-728-5483 (受话方付费电话); 815-748-3211
传真:815-748-0844
邮箱:office@ulf.org
www.ulf.org
- 髓鞘疾病生物反应器项目
分子遗传学
Molecular Genetics和OMIM表格信息可能与GeneReview中的信息不同:表格中可能包含有更多最新的认识。-ED
伴皮层下囊肿的巨脑性白质脑病:基因及数据库
基因 | 染色体位置 | 蛋白质 | 基因座特异性数据库 | 人类基因组突变数据库(HGMD) | Clin Var |
HEPACAM | 11q24-.2 | 肝细胞黏附分子 | HEPACAM | HEPACAM | |
MLC1 | 22q13-.33 | 膜蛋白MLC1 | MLC1数据库 | MLC1 | MLC1 |
数据来自于下列标准参考资料:基因信息来自于HGNC,染色体基因座、基因座名称、关键区域、互补群来自于OMIM;蛋白质来自于UniProt。点击此处获取更多有关数据库(基因座特异性数据库, HGMD, ClinVar) 链接。
MLC的OMIM入口
604004 | 伴皮层下囊肿的巨脑性白质脑病1型;MLC1型 |
605908 | MLC1基因;MLC1 |
611642 | 肝细胞黏附分子;HEPACAM |
613925 | 伴皮层下囊肿的巨脑性白质脑病2A型;MLC2A型 |
613926 | 伴皮层下囊肿的巨脑性白质脑病2B型;MLC2B型 |
MLC1
基因结构 MLC1包含有26,214个碱基和13个外显子;其cDNA包含有3435个碱基对。MLC1主要表达在脑中,但在白细胞中也有表达。脑内尾状核、丘脑和海马中的表达量最高。关于此基因及蛋白的详细信息见表A,基因。
致病性等位基因变异 根据最近的MLC1变异更新[Ilja Boor et al 2006],大约50%致病性变异为错义,28%为缺失或插入所致框移,22%为剪接位点的致病性变异,1个(1/50)为无义变异.(详情见表A)
Montagna et al [2006] 报道了9个新的变异。
如果可以获得患者的cDNA,那么cDNA分析是检测深部内含子区域的单核苷酸突变的最佳方法,可直接阐明异常剪接导致了什么的转录本。比如:导致MLC1的第4和第5外显子及二者之间的内含子区域缺失的c.298_423del126+108del [Ilja Boor et al 2006]和发生在第一内含子区域的c.178-10T>A。理论上,后者不影响剪接,但是cDNA测序发现该突变可导致编码第60至89氨基酸的第二外显子的剪接跳跃[MS van der Knaap & GC Scheper, 未发表]。
部分MLC1等位基因变异
变异分类 | DNA核苷酸改变 | 预测的蛋白质改变 | 参考序列 |
良性 | c.512G>T 1 | p.Cys171Phe | NM_015166-.3 NP_055981-.1 |
c.654C>A 1 | p.Asn218Lys | ||
c.925C>A 1 | p.Leu309Met | ||
c.1031A>G 1 | p.Asn344Ser | ||
致病性 | c.135insC | p.Cys46LeufsTer34 | |
c.176G>A | p.Gly59Glu | ||
c.178-10T>A | -- | ||
c.298_423del126+108del 2 | p.Thr99fsTer | ||
c.278C>T | p.Ser93Leu |
变异分类备注:表格中所列变异均由相应作者提供。GeneReviews工作人员没有对变异进行独立分类。
命名备注:GeneReviews采用人类基因组变异学会(Human Genome Variation Society ,varnomen-.hgvs.org)的命名标准进行命名。命名详细解释请点击此处。
1.正常变异频率:c.512G>T (9%); c.654C>A (1.5%); c.925C>A (0.7%); c.1031A>G (11%)
2.描述外显子126个核苷酸缺失以及供位剪接位点突变导致423位核苷酸后108个核苷酸缺失。外显子内含子一共缺失234个核苷酸。
正常基因产物 MLC1蛋白有377个氨基酸,大小预计约为41千道尔顿。MLC1是功能尚不清楚的完整膜蛋白[HUGE Protein Database]。蛋白主要位于血管旁、室管膜下和软脑膜下的星形胶质细胞终足。其定位提示MLC1可能在血脑屏障及脑脑脊液屏障上参与转运[Boor et al 2005, Teijido et al 2007]。Teijido et al [2004]报道在成年小鼠脑组织中,MLC1主要位于部分神经元的纤维及轴突束中。在人组织中MLC1蛋白与抗营养糖蛋白-相关复合物(dystrophin glycoprotein-associated complex,DGC)共定位[Boor et al 2007]。
异常基因产物 Teijido et al [2004]和Montagna et al [2006]通过分析蛋白表达及在胞膜上的分布量来检测MLC1变异的致病性。所有检测的MLC1致病性变异均导致MLC1表达量降低及后续的在胞膜表面的分布减少。
在大鼠原代星形胶质细胞中,MLC1表达减少后出现胞内囊泡[Duarri et al 2011]。
HEPACAM
基因结构 HEPACAM包含有17,076个碱基及7个外显子。cDNA包含有3563个碱基对。基因及蛋白的详细信息见表A,基因。
致病性等位基因变异 HEPACAM致病性变异中导致经典型表型(即MLC2A型)的大多数为错义突变,但也可为无义突变及框移突变。在改善型患者中(即MLC2B型),仅可见HEPACAM错义突变[López-Hernández et al 2011].。
部分HEPACAM等位基因变异
变异分类 | DNA核苷酸改变 | 预测的蛋白质改变 | 参考序列 |
致病性(MLC2A型) | c.275G>A 1 | p.Arg92Gln | NM_152722-.4 NP_689935-.2 |
致病性(MLC2B型) | c.265G>A2 | p.Gly89Ser | |
c.274C>T | p.Arg92Trp |
变异分类备注:表格中所列变异均由相应作者提供。GeneReviews工作人员没有对变异进行独立分类。
命名备注:GeneReviews采用人类基因组变异学会(Human Genome Variation Society ,varnomen-.hgvs.org)的命名标准进行命名。命名详细解释请点击此处。
1.致病性错义变异c.274C>T, p.Arg92Trp,影响这一相同氨基酸的突变,是MLC2B型中最常见致病性变异之一。
2.是MLC2B型最常见的致病性变异(López-Hernández et al [2011]中16例患儿有6例检测到此突变)。
正常基因产物 HEPACAM编码的肝细胞黏附分子(也叫HepaCAM,或GlialCAM)蛋白有417个氨基酸,预计其分子大小约为46千道尔顿[Chung Moh et al 2005, Favre-Kontula et al 2008]。
HEPACAM主要表达在脑,在中枢神经系统白质束、脑室旁室管膜细胞以及脊髓中央通道中表达量最高[Favre-Kontula et al 2008]。GlialCAM是一个单次跨膜蛋白,有两个胞外免疫球蛋白功能域。虽然该蛋白最先在肝脏细胞肿瘤细胞系中发现(因此命名为HepaCAM),但是其最主要表达于脑中,并且在星形胶质细胞、少突胶质细胞及神经元中均有表达。
GlialCAM在生后小鼠脑组织发育过程中表达上调,并与髓鞘碱性蛋白(myelin basic protein,MBP)同步升高,提示其可能参与髓鞘化过程。此外,在体外环境中,GlialCAM在少突胶质细胞及星形胶质细胞发育的各个阶段以及细胞连接处中均有表达[Favre-Kontula et al 2008]。
异常基因产物 突变后的GlialCAM蛋白不再主要分布于胞膜或细胞-细胞连接处。
在培养的大鼠元代星形胶质细胞中,常染色体隐性或显性致病性变异可致MLC1定位异常。常染色体隐性遗传的GlialCAM突变共表达野生型GlialCAM后可纠正其对MLC1的定位的有害影响,但是共表达野生型GlialCAM不能纠正常染色体显性遗传的GlialCAM突变对MLC1的定位的有害影响[López-Hernández et al 2011]。
参考文献
引用文献
-
Ben-Zeev B, Gross V, Kushnir T, Shalev R, Hoffman C, Shinar Y, Pras E, Brand N. Vacuolating megalencephalic leukoencephalopathy in 12 Israeli patients. J Child Neurol. 2001;16:93-9. [PubMed: 11292232]
-
Ben-Zeev B, Levy-Nissenbaum E, Lahat H, Anikster Y, Shinar Y, Brand N, Gross-Tzur V, MacGregor D, Sidi R, Kleta R, Frydman M, Pras E. Megalencephalic leukoencephalopathy with subcortical cysts; a founder effect in Israeli patients and a higher than expected carrier rate among Libyan Jews. Hum Genet. 2002;111:214-8. [PubMed: 12189496]
-
Boor I, Nagtegaal M, Kamphorst W, van der Valk P, Pronk JC, van Horssen J, Dinopoulos A, Bove KE, Pascual-Castroviejo I, Muntoni F, Estévez R, Scheper GC, van der Knaap MS. MLC1 is associated with the dystrophin-glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 2007;114:403-10. [PMC free article: PMC2039857] [PubMed: 17628813]
-
Boor PK, de Groot K, Waisfisz Q, Kamphorst W, Oudejans CB, Powers JM, Pronk JC, Scheper GC, van der Knaap MS. MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol. 2005;64:412-9. [PubMed: 15892299]
-
Bugiani M, Moroni I, Bizzi A, Nardocci N, Bettecken T, Gärtner J, Uziel G. Consciousness disturbances in megalencephalic leukoencephalopathy with subcortical cysts. Neuropediatrics. 2003;34:211-14. [PubMed: 12973663]
-
Chen CY, Zimmerman RA, Lee CC, Chen FH, Yuh YS, Hsiao HS. Neuroimaging findings in late infantile GM1 gangliosidosis. AJNR Am J Neuroradiol. 1998;19:1628-30. [PubMed: 9802482]
-
Chung Moh M, Hoon Lee L, Shen S. Cloning and characterization of hepaCAM, a novel Ig-like cell adhesion molecule suppressed in human hepatocellular carcinoma. J Hepatol. 2005;42:833-41. [PubMed: 15885354]
-
Devaney JM, Donarum EA, Brown KM, Meyer J, Stöber G, Lesch KP, Nestadt G, Stephan DA, Pulver AE. No missense mutation of WKL1 in a subgroup of probands with schizophrenia. Mol Psychiatry. 2002;7:419-23. [PubMed: 11986987]
-
Duarri A, Lopez de Heredia M, Capdevila-Nortes X, Ridder MC, Montolio M, López-Hernández T, Boor I, Lien CF, Hagemann T, Messing A, Gorecki DC, Scheper GC, Martínez A, Nunes V, van der Knaap MS, Estévez R. Knockdown of MLC1 in primary astrocytes causes cell vacuolation: A MLC disease cell model. Neurobiol Dis. 2011;43:228-38. [PMC free article: PMC3885813] [PubMed: 21440627]
-
Favre-Kontula L, Rolland A, Bernasconi L, Karmirantzou M, Power C, Antonsson B, Boschert U. GlialCAM, an immunoglobulin-like cell adhesion molecule is expressed in glial cells of the central nervous system. Glia. 2008;56:633-45. [PubMed: 18293412]
-
Gorospe JR, Singhal BS, Kainu T, Wu F, Stephan D, Trent J, Hoffman EP, Naidu S. Indian Agarwal megalencephalic leukodystrophy with cysts is caused by a common MLC1 mutation. Neurology. 2004;62:878-82. [PubMed: 15037685]
-
Goutières F, Boulloche J, Bourgeois M, Aicardi J. Leukoencephalopathy, megalencephaly, and mild clinical course. A recently individualized familial leukodystrophy. Report on five new cases. J Child Neurol. 1996;11:439-44. [PubMed: 9120220]
-
Ilja Boor PK, de Groot K, Mejaski-Bosnjak V, Brenner C, van der Knaap MS, Scheper GC, Pronk JC. Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum Mutat. 2006;27:505-12. [PubMed: 16652334]
-
Itoh N, Maeda M, Naito Y, Narita Y, Kuzuhara S. An adult case of megalencephalic leukoencephalopathy with subcortical cysts with S93L mutation in MLC1 gene: a case report and diffusion MRI. Eur Neurol. 2006;56:243-5. [PubMed: 17077634]
-
Kaganovich M, Peretz A, Ritsner M, Bening Abu-Shach U, Attali B, Navon R. Is the WKL1 gene associated with schizophrenia? Am J Med Genet B Neuropsychiatr Genet. 2004;125B:31-7. [PubMed: 14755440]
-
Leegwater PA, Boor PK, Yuan BQ, van der Steen J, Visser A, Konst AA, Oudejans CB, Schutgens RB, Pronk JC, van der Knaap MS. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum Genet. 2002;110:279-83. [PubMed: 11935341]
-
Leegwater PA, Yuan BQ, van der Steen J, Mulders J, Konst AA, Boor PK, Mejaski-Bosnjak V, van der Maarel SM, Frants RR, Oudejans CB, Schutgens RB, Pronk JC, van der Knaap MS. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831-8. [PMC free article: PMC1275636] [PubMed: 11254442]
-
López-Hernández T, Ridder MC, Montolio M, Capdevila-Nortes X, Polder E, Sirisi S, Duarri A, Schulte U, Fakler B, Nunes V, Scheper GC, Martínez A, Estévez R, van der Knaap MS. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422-32. [PMC free article: PMC3071909] [PubMed: 21419380]
-
Meyer J, Huberth A, Ortega G, Syagailo YV, Jatzke S, Mossner R, Strom TM, Ulzheimer-Teuber I, Stober G, Schmitt A, Lesch KP. A missense mutation in a novel gene encoding a putative cation channel is associated with catatonic schizophrenia in a large pedigree. Mol Psychiatry. 2001;6:302-6. [PubMed: 11326298]
-
Montagna G, Teijido O, Eymard-Pierre E, Muraki K, Cohen B, Loizzo A, Grosso P, Tedeschi G, Palacín M, Boespflug-Tanguy O, Bertini E, Santorelli FM, Estévez R. Vacuolating megalencephalic leukoencephalopathy with subcortical cysts: functional studies of novel variants in MLC1. Hum Mutat. 2006;27:292. [PubMed: 16470554]
-
Rubie C, Lichtner P, Gartner J, Siekiera M, Uziel G, Kohlmann B, Kohlschutter A, Meitinger T, Stober G, Bettecken T. Sequence diversity of KIAA0027/MLC1: are megalencephalic leukoencephalopathy and schizophrenia allelic disorders? Hum Mutat. 2003;21:45-52. [PubMed: 12497630]
-
Saijo H, Nakayama H, Ezoe T, Araki K, Sone S, Hamaguchi H, Suzuki H, Shiroma N, Kanazawa N, Tsujino S, Hirayama Y, Arima M. A case of megalencephalic leukoencephalopathy with subcortical cysts (van der Knaap disease): molecular genetic study. Brain Dev. 2003;25:362-6. [PubMed: 12850517]
-
Selch S, Strobel A, Haderlein J, Meyer J, Jacob CP, Schmitt A, Lesch KP, Reif A. MLC1 polymorphisms are specifically associated with periodic catatonia, a subgroup of chronic schizophrenia. Biol Psychiatry. 2007;61:1211-4. [PubMed: 17210142]
-
Singhal BS, Gorospe JR, Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts. J Child Neurol. 2003;18:646-52. [PubMed: 14572144]
-
Singhal BS, Gursahani RD, Udani VP, Biniwale AA. Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr Neurol. 1996;14:291-6. [PubMed: 8805171]
-
Sugiura C, Shiota M, Maegaki Y, Yoshida K, Koeda T, Kitahara T, Ohno K. Late-onset neuropsychological symptoms in a Japanese patient with megalencephalic leukoencephalopathy with subcortical cysts. Neuropediatrics. 2006;37:286-90. [PubMed: 17236107]
-
Teijido O, Casaroli-Marano R, Kharkovets T, Aguado F, Zorzano A, Palacín M, Soriano E, Martínez A, Estévez R. Expression patterns of MLC1 protein in the central and peripheral nervous systems. Neurobiol Dis. 2007;26:532-45. [PubMed: 17434314]
-
Teijido O, Martínez A, Pusch M, Zorzano A, Soriano E, Del Río JA, Palacín M, Estévez R. Localization and functional analyses of the MLC1 protein involved in megalencephalic leukoencephalopathy with subcortical cysts. Hum Mol Genet. 2004;13:2581-94. [PubMed: 15367490]
-
Topçu M, Saatci I, Topcuoglu MA, Kose G, Kunak B. Megalencephaly and leukodystrophy with mild clinical course: a report on 12 new cases. Brain Dev. 1998;20:142-53. [PubMed: 9628190]
-
Tsujino S, Kanazawa N, Yoneyama H, Shimono M, Kawakami A, Hatanaka Y, Shimizu T, Oba H. A common mutation and a novel mutation in Japanese patients with van der Knaap disease. J Hum Genet. 2003;48:605-8. [PubMed: 14615938]
-
van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WF, Hoogenraad F, Valk J. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37:324-34. [PubMed: 7695231]
-
van der Knaap MS, Barth PG, Vrensen GF, Valk J. Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol (Berl) 1996;92:206-12. [PubMed: 8841668]
-
van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begeer JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM. Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol. 2001;22:541-52. [PubMed: 11237983]
-
van der Knaap MS, Smit LM, Barth PG, Catsman-Berrevoets CE, Brouwer OF, Begeer JH, de Coo IF, Valk J. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol. 1997;42:50-9. [PubMed: 9225685]
-
van der Voorn JP, Pouwels PJW, Hart AA, Serrarens J, Willemsen MA, Kremer HP, Barkhof F, van der Knaap MS. Childhood white matter disorders: quantitative MR imaging and spectroscopy. Radiology. 2006;241:510-17. [PubMed: 17057071]
-
Verma R, Mukerji M, Grover D. B-Rao C, Das SK, Kubendran S, Jain S, Brahmachari SK. MLC1 gene is associated with schizophrenia and bipolar disorder in Southern India. Biol Psychiatry. 2005 Jul 1;58(1):16-22. [PubMed: 15992519]
-
Yalcinkaya C, Yuksel A, Comu S, Kilic G, Cokar O, Dervent A. Epilepsy in vacuolating megalencephalic leukoencephalopathy with subcortical cysts. Seizure. 2003;12:388-96. [PubMed: 12915085]
建议阅读
Suggested Reading
-
Kazusa Human cDNA Project. Database of human unidentified gene-encoded (HUGE) large proteins analyzed.
-
van der Knaap MS, Lai V, Köhler W, Salih MA, Fonseca MJ, Benke TA, Wilson C, Jayakar P, Aine MR, Dom L, Lynch B, Kálmánchey R, Pietsch P, Errami A, Scheper GC. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann Neurol. 2010;67:834-7. [PubMed: 20517947]
Chapter Notes
Author History
JC Pronk, PhD; Vrije Universiteit Medical Center, Amsterdam (2003-2008)
Gert C Scheper, PhD (2008-present)
Marjo S van der Knaap, MD, PhD (2003-present)
Revision History
-
3 November 2011 (me) Comprehensive update posted live
-
29 July 2008 (me) Comprehensive update posted live
-
29 November 2005 (me) Comprehensive update posted live
-
11 August 2003 (me) Review posted to live Web site
-
12 June 2003 (mvdk) Original submission