
简介
临床特征。交界型大疱性表皮松解症(JEB)的特征是皮肤和粘膜易碎,起泡时几乎没有创伤。水疱可能很严重,并且口腔和鼻腔,手指和脚趾周围以及上呼吸道内部可能会在皮肤上形成肉芽组织。水泡一般都能愈合,没有明显的疤痕。 JEB的广泛分类包括Herlitz JEB(又称致命)和非Herlitz JEB(又称非致命)。在经典的重度JEB形式的Herlitz JEB中,水疱在出生时就已出现,或在新生儿期变得明显。先天性尿道和膀胱畸形也可能发生。在非Herlitz JEB中,表型可能是轻度的,水疱出现在手,脚,膝盖和肘部,可有或无肾脏或输尿管受累。有些人在新生儿期后没有水疱。 JEB和大疱性表皮松解(EB)的共有的其他特征包括先天的皮肤局部缺乏(先天性皮肤发育不良),粟丘疹,指甲营养不良,瘢痕性脱发,毛发不全,假性挛缩。
诊断/检测。由于所有类型EB的临床特征均存在明显重叠,因此通常需要通过透射电子显微镜(TEM)和/或免疫荧光抗体/抗原谱检查皮肤活检,以建立JEB的诊断,尤其是在婴儿中。已知导致JEB致病性变异的四个基因是LAMB3(占所有JEB的70%),COL17A1(占12%),LAMC2(占9%)和LAMA3(占9%)。
管理。表型的处理:穿刺并干燥水泡和穿三层衣服(第一层:不粘连;第二层:用于稳定和保护;第三层:弹性以确保完整性)。保护皮肤免受剪力;教养看护者正确看护婴幼儿;用高效局部类固醇,硝酸银,电灼或自体皮肤移植物治疗肉芽组织;扩张食管狭窄;胃食管疾病的标准治疗;气管切开术(如果合适);定期牙科护理;适当的鞋类和物理疗法,以促进/保持步行;社会心理支持,包括社会服务和心理咨询;适当管理慢性疼痛;使用标准疗法治疗泌尿科和肾脏疾病。
预防继发并发症:用于治疗伤口感染的杀菌剂;注意严重受累的婴儿的体液和电解质平衡;额外的营养支持,包括在必要时喂养胃造口术;钙,维生素D,锌和铁的补充剂。
监视:常规筛查缺铁性贫血,锌缺乏,骨质减少和/或骨质疏松症;定期超声心动图以评估扩张型心肌病;在生命的第二个十年中,对鳞状细胞癌进行监测。
避免接触的药物/情况:普通医用胶带或Band-Aids®,穿着不良或质地较粗糙的衣服和鞋类,通常会伤害皮肤的活动(例如远足,骑山地自行车,接触运动)。
其他:考虑剖腹产以减少对受累的胎儿的皮肤伤害。
遗传咨询。JEB以常染色体隐性遗传的方式遗传。受累的孩子父母通常是肯定杂合子(即携带者)。由于已经报道了胚系嵌合和单亲二倍体等,需要通过分子遗传学检测来确认父母的携带者身份。 父母都是携带者的受影响个体的每个同胞都有25%的机会患病,成为无症状携带者的机会为50%, 25%不受影响。具有常染色体隐性遗传性JEB的个体的后代是致病性变异的肯定杂合子(携带者)。如果在家庭中同时发现了两种致病变异,则可以对风险较高的家庭成员进行携带者检测,对风险较高的妊娠进行产前诊断。
诊断
提示性发现
对于皮肤脆弱的个体,应怀疑交界型大疱性表皮松解症(JEB):
- 水泡很少或没有创伤。 水泡可能是轻度或严重的; 但是,水泡通常可以治愈,并且没有明显的疤痕。
- 大量口腔和粘膜受累
注意:水泡可能很严重,并且在口腔和鼻腔,手指和脚趾周围以及上呼吸道和气管的内部和周围可能会在皮肤上形成肉芽组织(请参见
Figure 1, Figure 2).
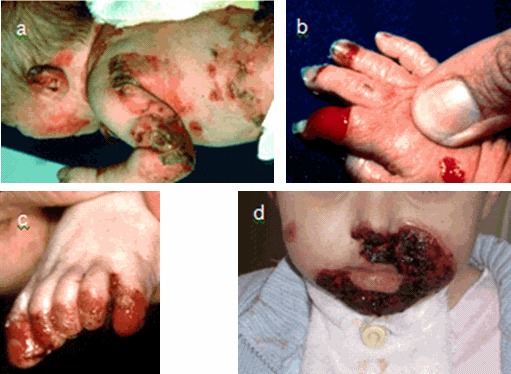
Figure 1.
JEB全身严重型 a 耳朵上广泛分布的水泡和肉芽组织
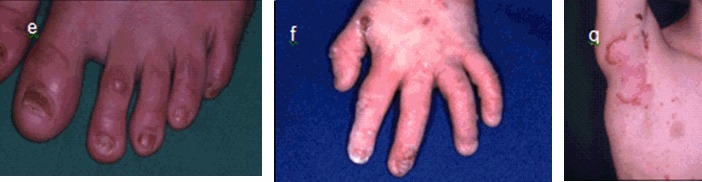
Figure 2.
JEB泛发中间型 e 大龄儿童的轻度指甲营养不良
建立诊断
JEB的诊断由具有以下一项或两项的先证者确定:
- 通过 分子遗传学检测鉴定Table 1所列基因之一中双等位基因的致病变异
- 使用透射电子显微镜(TEM)和/或免疫荧光抗体/抗原谱进行皮肤活检(请参阅Skin Biopsy)
注意:基因检测是首选的诊断方法。除非没有确定的分子遗传学检测,否则不再常规进行用于诊断的皮肤活检。
分子遗传学测试 分子遗传学检测方法可以包括针对靶向基因的检测(multigene panel,靶向分子遗传学检测)和基于表型综合基因组的检测(外显子组测序, 基因组测序, exome array)组合。
以基因为目标的测试要求临床医生确定可能涉及哪些基因,而 基因组的测试则不需要。由于交界型大疱性表皮松解症的表型较宽, 可使用“Suggestive Findings”中所述基因靶向检测方法进行诊断(参见Option 1),而表型与许多其他遗传性皮肤脆弱性疾病没有区别或在明显的后遗症(如旺盛的肉芽肿组织)之前如新生儿期有表现,使用基因组测试更可能诊断(请参阅 Option 2)。
选项1
当表型和实验室发现提示结缔性表皮松解性大疱的诊断时, 分子遗传学检测方法可包括使用 multigene panel 或靶向分子检测[Lucky et al 2018].
包含COL17A1,ITGB4,LAMB3,LAMA3,LAMC2和其他(见Differential Diagnosis)与交界型大疱性表皮松解症有关基因multigene panel 最有可能以最合理的成本鉴定出该病的遗传原因,同时限制了对这些变异的鉴定。无法解释潜在表型的基因的意义不确定和致病性变异。注意:(1)基因套餐中包含的基因和每个基因所用测试的诊断敏感性因实验室而异,并可能随时间变化。 (2)一些基因套餐可能包含与本基因综述中讨论的病症无关的基因。 (3)在某些实验室中,套餐选项可能包括定制的实验室设计面板和/或定制的针对表型的 外显子组分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括序列分析, deletion/duplication analysis和/或其他非基于序列的测试。对于这种疾病,建议同时进行多基因分析(包括删除/重复分析)(见Table 1)。
有关多基因套餐的介绍,请单击 here。有关订购基因检测的临床医生的更多详细信息,请参见here.。
选项2
当该表型与其他许多以皮肤脆弱和起泡为特征的遗传性疾病没有区别时,综合基因组的测试(不需要临床医生确定可能涉及哪些基因)是最佳选择。 外显子组测序是最常用的方法。 基因组测序也是可能的。
如果外显子组测序不能诊断,可以考虑外显子组阵列(临床上可用时)。
有关全面的 基因组的测试的介绍,请单击 here。 临床医生的更多详细信息可在here找到。
Table 1.
交界型大疱性表皮松解症的分子遗传学检测
| 基因 1, 2 | 致病性基因占比 | 致病性变异 3 所用方法 | |
|---|---|---|---|
| 测序 4 | 基因靶向 deletion/duplication analysis 5 | ||
| COL17A1 | 12% | >98% | <2% 6 |
| ITGB4 | <1% 7 | ~100% | <1% |
| LAMA3 | 9% | >98% 8 | <1% 6 |
| LAMB3 | 70% | >98% | <2% 9 |
| LAMC2 | 9% | >98% | <2% 6 |
- 1.
- 2.
- 3.
有关该基因中检测到的等位基因变异的信息见Molecular Genetics .
- 4.
- 5.
通过序列分析检测的致病变异可能包括小的基因内缺失/插入和 错义, nonsense和剪接位点变异; 通常,不会检测到外显子或全基因缺失/重复。 有关在解释序列分析结果时要考虑的问题,请单击here。
- 6.
鉴定基因组的DNA的编码和侧翼 内含子的区域的序列分析不容易检测到缺失/重复; 缺失/重复分析各种可用方法包括: quantitative PCR,长程PCR,依赖多重连接的探针扩增(MLPA)和包括该 基因/染色体片段的 染色体芯片(CMA)。
- 7.
Pulkkinen et al [1997], Takizawa et al [2000b], Fassihi et al [2005], Varki et al [2006]
- 8.
针对特定种族人群的致病性变异的目标分析应该是第一步(请参阅Testing Strategy,目标分子遗传学检测)。
- 9.
必须注意对LAMA3最长转录本的基因组的区域进行测序,而不是对较短转录本变异之一进行测序。
皮肤活检 有时通过皮肤活检进行检查(1)透射电子显微镜(TEM)和/或(2)免疫荧光抗体/抗原谱可以诊断JEB。
包括整个基底膜区的打孔活检是优选的。 活检应从新鲜(小于12小时龄)水泡的前缘或机械性水泡进行,并应包括一些正常的邻近皮肤。 (年龄较大的水泡发生变化,可能会掩盖诊断形态,并可能产生误导。)
注意:
- 透射电子显微镜
- 标本必须放置在进行测试的实验室指定的固定介质(例如戊二醛)中。
- 甲醛固定的样品不能用于电子显微镜。
- 免疫荧光抗体/抗原谱
- 样品应按照执行测试的实验室规定,在无菌携带介质(例如Michel's或Zeus's)中发送。
- 一些实验室更喜欢速冻组织。
- 在某些实验室中,作图仅通过使用基底膜不同层的各种标记抗体来指定切割水平。 优选具有EB中感兴趣蛋白质抗原的实验室,因为可以评估裂解水平以及EB中突变的特定基因产物的存在与否。
- 样品应按照执行测试的实验室规定,在无菌携带介质(例如Michel's或Zeus's)中发送。
- 光学显微镜不足以准确诊断任何亚型的EB。
透射电子显微镜(TEM)用于检查基底膜区域结构的数量和形态-特别是:锚定原纤维的数量和形态;血丝小体,锚定丝和角蛋白中间丝的存在和形态;以及微泡显示组织分裂平面。
JEB中关于TEM的发现包括以下内容 [Shinkuma et al 2011]:
- 各种形式的JEB。在表皮基底膜的透明层中或在角质形成细胞层最低水平的半桥粒体水平的基底膜正上方,可见分裂。
- JEB泛发严重型。血球小体发育不全,数量减少。锚定细丝明显减少或不存在。
- JEB泛发中间型。锚丝可以减少;血小体可能减少或发育不良。
免疫荧光抗体/抗原谱。 发现包括:
- JEB泛发严重型或JEB泛发中间型LAMA3,LAMB3或LAMC2的致病性变异导致的层粘连蛋白332(aka LAM5)抗体异常染色或缺乏染色[Aumailley et al 2005]
- 由COL17A1中的致病性变异引起的JEB中XVII胶原蛋白抗体异常染色或缺失
其他抗原(例如胶原蛋白VII,角蛋白5和14)的正常染色可确诊JEB。
注意:尤其是在较轻型的EB形式中,间接免疫荧光研究通常不足以做出诊断,因为检测到的抗原水平接近正常且未观察到切割平面。在这些情况下,必须进行皮肤活检的电子显微镜检查。
临床特征
临床表现
交界型大疱性表皮松解症(JEB)的特征是皮肤和粘膜易损,起泡时几乎没有或没有创伤。 JEB的广泛分类基于生命的最初几年的严重程度和生存率包括JEB泛发严重型和JEB泛发中间型[Yuen et al 2013, Kelly-Mancuso et al 2014].
JEB泛发严重型(以前称为JEB Herlitz)。出生时会出现严重的水疱,或者在新生儿期间会出现严重的水疱,并可能导致受累皮肤的大部分区域出现明显的肉芽组织。肉芽组织典型地出现在鼻子,嘴,耳朵,手指和脚趾的尖端周围,以及容易受摩擦的区域,例如臀部和头后部。脸上的永久性斑块很难治疗。肉芽组织表现为大的侵蚀斑块和斑块,通常具有锯齿状或环形边界,易碎且容易出血。可能会大量流失血液,液体和蛋白质。这种侵蚀通常会危及生命,因为它们使这些婴儿容易出现电解质失衡和感染,包括败血症和猝死。如果婴儿幸存下来,起泡可能会持续一生,除非有严重的继发感染, 通常不会留下疤痕。在一些幸存的JEB泛发严重型患者中,已经看到手脚出现假性的指状融合,使手指融合到“手套状”的手和脚中,导致功能严重丧失[Fine et al 1999]。在一项队列研究中,五年内出生的71名儿童中有73%的平均年龄为5个月 [Kelly-Mancuso et al 2014].
除皮肤受累外,还可看到口腔,上呼吸道,食道,膀胱,尿道和角膜的粘膜受累。牙釉质点蚀引起的成釉不全很常见。气道周围的肉芽组织的积聚通常是声门下的,第一个表现是微弱,嘶哑的哭声。最终,气道的压缩和阻塞导致喘鸣和呼吸窘迫。除非进行气管切开术,否则许多儿童会因呼吸系统并发症而夭折。然而,在皮肤如此脆弱的孩子中进行气管切开术是困难的 [Ida et al 2012].
膀胱和尿道上皮受累会导致排尿困难,尿储留,尿路感染以及最终的肾功能不全。肾脏和输尿管异常包括增生/多囊肾,肾积水/输尿管积水,急性肾小管坏死,阻塞性尿路病,输尿管膨出,多重肾脏收集系统和膀胱缺失 [Puvabanditsin et al 1997, Kambham et al 2000, Nakano et al 2000, Wallerstein et al 2000, Fine et al 2004, Varki et al 2006, Pfendner et al 2007].
食管变窄已有报道,但比 常染色体隐性遗传性营养不良性EB患儿少。
JEB全身严重型继发并发症包括营养不良和生长迟缓,贫血,脱发,皮肤感染,败血症,电解质失衡,骨质疏松[Fewtrell et al 2006],扩张型心肌病,鳞状细胞癌 [Yuen & Jonkman 2011], 和牙釉质发育不良点蚀[Krämer 2010, Stellingsma et al 2011].
大多数患有JEB泛发重症的儿童无法生存到第一年。
JEB泛发中间型(以前称为JEB non-Herlitz)包含比JEB泛发重症较轻的临床表型。该表型可能是轻度的,起泡于手,脚,膝盖和肘部,有或没有肾脏,输尿管或食道受累。或相对更广泛,包括弯曲区域和躯干。刚出生后,有些孩子几乎不会起泡。在JEB泛发严重型的个体中看到的严重的肉芽组织和呼吸系统损害很少见。
各种类型的脱发和甲状肌营养不良以及牙釉质点蚀仍然是这类JEB的标志。
JEB泛发严重型和JEB泛发中间型的其他表现包括:
- 先天性局限性皮肤缺失(先天性角膜缺乏症)
- 丰富的肉芽组织
- 指甲营养不良
- 疤痕性脱发
- JEB泛发中间型患者的鳞状细胞癌 [Montaudié et al 2016]
- 假性和其他挛缩。 假性指的是指手或脚的任何手指之间的部分或全部间隔丢失(少有)。
- 粟丘疹(少有)
- 疤痕(少有)
基因型-表型相关
JEB泛发严重型是两个等位基因上致病性变异失活的结果,导致功能蛋白很少或没有[Varki et al 2006]。 对于移码变异,其严重性可能与终止密码子的位置以及是否形成了任何功能性(尽管被截短的)蛋白质有关。 一些功能性蛋白质的存在似乎是减轻疾病严重程度的最重要因素[Kiritsi et al 2013].
JEB泛发中间型通常由氨基酸取代和剪接连接变异产生,尽管由于已确定的等位变异的广泛表型变异性和范围而难以归纳[Varki et al 2006]。 另外,通过非移码跳过含有 nonsense 或移码变异的外显子,预期会出现严重的表型 [McGrath et al 1999, Kowalewski et al 2016].
命名法
Table 2:
交界型大疱性表皮松解症命名
| 当前术语 | 2008 命名法 | 2013 "Onion Skin" 命名法 | 其他过去使用特定的JEB名称 | ||
|---|---|---|---|---|---|
| JEB亚型 | 简称 | ||||
| 泛发 | JEB, 泛发重型 | JEB-gen sev | Herlitz JEB (H-JEB) | JEB 泛发重型 , 层粘连蛋白332缺失, LAMA3, LAMB3, or LAMC2 致病性变异 (特异型) |
|
| JEB, 泛发中间型 | JEB-gen intermed | Non-Herlitz JEB (NH-JEB) | JEB泛发中间型, 层粘连蛋白332或胶原蛋白XVII 染色减少型,LAMA3, LAMB3, LAMC2, or COL17A1致病型 (特异型) |
| |
| JEB 幽门闭锁型 | JEB-PA | ||||
| JEB-晚发型 | JEB-LO | ||||
| JEB 呼吸和肾脏受累型 | JEB-RR | ||||
| 局部 | JEB, 局限型 | JEB-loc | |||
| JEB, inversa | JEB-inv; JEB-I | ||||
| JEB-LOC 综合征 | |||||
Adapted from Fine et al [2014]
JEB =交界型大疱性表皮松解症
患病率
根据国家EB注册机构的统计,在美国人口中,所有类型的JEB的患病率为每百万人0.44[Fine et al 1999]。 最近的数据估计,每年JEB的发病率在每百万3.59至6.7之间,死亡率为73%[Kelly-Mancuso et al 2014, Hammersen et al 2016].
- JEB全身重症的患病率估计为每百万例0.4,但可能不足。 JEB全身性严重发病率也很低(百万分之0.41),但可能被低估了:许多JEB全身性严重病例未报告,因为婴儿在新生儿期死于该病(据报道婴儿死亡率为73%)[Kelly-Mancuso et al 2014].
- JEB全身中间型发病率为每百万人2.0。
- 在美国人口中,各种形式的JEB的携带者风险经计算为1:270 [作者,个人通讯]。
- JEB全身重度的携带者风险经计算为1:781[Nakano et al 2000, Pfendner et al 2001].
遗传相关(等位基因)疾病
与JEB泛发中间型相似的表型特征的等位基因疾病:
- LAMA3.Punjabi印第安人和具有伊朗血统的个体中描述了一种相关的疾病,喉气管皮肤综合症(LOCS或Shabbir综合征; OMIM 245660)。 LOCS具有许多表型特征,类似于JEB泛发重度。皮肤脆弱表现为轻度的水疱和手和面部的糜烂,并扩散到身体的其他部位,并伴有结硬皮缺损。新生儿可能会嘶哑,然后出现喉咙异常和生长,结膜疾病,指甲异常和牙釉质增生。最终,结膜疾病可能导致失明,而喉疾病可能导致需要气管切开术的威胁生命的气道阻塞[Cohn & Murrell 2010].两个LAMA3致病变异c.151dupG和Gln57Ter位于三个LAMA3异型体之一的外显子39上,已被确定为致病性[McLean et al 2003, Barzegar et al 2013]。遗传是常染色体隐性遗传.LOCS的诊断可能因TEM上没有确定的切割平面而变得复杂,并且通过免疫荧光对基底膜蛋白进行的层粘连蛋白5染色减少但不缺少。编码所有三个异型体的LAMA3的所有76个外显子的序列分析对于提供明确的诊断是必要的,尤其是在新生儿中。
- ITGB4. ITGB4中的双等位基因致病变异与幽门闭锁性大疱性表皮松解症(EB-PA)更常见。 EB-PA中已经描述了跨越所有ITGB4的100多种致病变异。 在两个等位基因上导致过早终止密码子的致病变异会导致最严重的表型,在新生儿期常常致命。 EB-PA的特征是皮肤和粘膜易碎,起泡时几乎没有或没有创伤。 先天的幽门闭锁; 输尿管和肾脏异常(增生/多囊肾,肾积水/输尿管,输尿管膨出,重复的肾脏收集系统,无膀胱)。 ITGB4中的双等位基因致病变体很少引起epidermolysis bullosa simplex (EBS)或幽门闭锁,伴有脱皮性肠病且无皮肤病[Salvestrini et al 2008]。
其他等位基因疾病(不在JEB的鉴别诊断中):
- COL17A1. COL17A1中的致病变异也可能与通常与epidermolysis bullosa simplex有关的表皮角质形成细胞裂解平面有关。
- LAMB3和LAMA3致病变异与发育不良性釉质生成不全有关 [Yuen et al 2012, Kim et al 2013, Poulter et al 2014, Lee et al 2015, Wang et al 2015, Gostyńska et al 2016, Du et al 2018].
LAMC2. 除JEB外,没有其他表型与LAMC2中的致病变异有关。
鉴别诊断
由14种不同基因的致病变异引起的EB的四种主要类型是epidermolysis bullosa simplex (EBS),交界型大疱性表皮松解症(JEB),dystrophic epidermolysis bullosa(DEB)和Kindler syndrome。EB的主要类型和罕见亚型的诊断标准最近已扩展到包括Kindler综合征,喉-眼皮肤综合征和许多罕见形式的EBS [Fine et al 2009, Intong & Murrell 2012].诊断标准旨在涵盖其他新发现的水疱性疾病。优秀的临床评价包括Principles and Practice of Medical Genetics中有关EB的一章[Anton-Lamprecht & Gedde-Dahl 2002]和 Fine's Revised Classification System[Fine et al 1999, Fine et al 2000, Fine et al 2008].
EB的四种主要类型具有皮肤脆弱性,表现为几乎没有创伤的水疱。所有类型的EB都有一个阳性的Nikolsky症(摩擦后未受累的皮肤起水泡)。没有特定类型的临床发现;因此,确定EB的类型需要从新发的水疱进行新鲜的皮肤活检,然后通过间接免疫荧光对关键的基底膜蛋白成分进行染色或对诊断进行分子遗传学确认。使用皮肤活检时,可通过免疫荧光抗体/抗原作图及其分布确定TEM上的切割平面以及这些蛋白质成分的存在/不存在来建立诊断。电子显微镜也是诊断性的,通常在较温和的EB模式中更有用。
临床检查可用于确定水泡程度,口腔和其他粘膜损伤的存在以及疤痕的存在和程度。
仅临床发现而确定EB类型的存在以下局限性:
- 在幼儿和新生儿中,水泡和瘢痕形成的程度和严重程度不足以识别EB类型。
- 粘膜和指甲受累以及存在或不存在粟粒可能不是有助于区分的因素(请参阅 Clinical Characteristics)。
- 炎症后的变化(例如在EBS,Dowling-Meara型(EBS-DM)中看到的变化)通常被误认为是瘢痕形成或斑驳的色素沉着。
- 由于腐蚀或刮擦的感染,EBS和JEB中可能出现疤痕,这进一步损坏了裸露的表面。
- 在任何类型的EB中都可以看到先天性皮肤缺失,这并不是一种可鉴别的诊断特征(请参阅Clinical Characteristics)。
特定类型EB的典型临床发现包括:
- 角膜糜烂,食道狭窄和指甲受累可能表明DEB。
- 在较轻的病例中,仅限于手和脚的水疱和疤痕表明常染色体显性遗传DEB(DDEB)。
- 年龄较大的儿童和成人中的假性挛缩(假畸形)和挛缩通常提示 常染色体隐性遗传DEB(RDEB)。
- 声音嘶哑和呼吸窘迫提示H-JEB。
- 肉芽组织提示JEB。
- 手掌和脚底过度角化提示EBS,尤其是DM型。
Epidermolysis bullosa simplex(EBS)的特征是皮肤(在某些情况下包括粘膜上皮)的脆弱性,可导致很少或没有创伤的无疤痕水疱。当前的大疱性表皮松解症(EB)的分类包括两种主要的EBS类型和12种次要的EBS类型。所有这些在超微结构水平上均具有真皮-表皮交界处水泡的共同特征。 EBS的四种最常见的子类型是:
- EBS,局限型(EBS-loc;以前称为Weber-Cockayne型)
- EBS ,Dowling-Meara型(EBS-DM)
- EBS,其他泛发型(EBS,gen-nonDM;以前称为Koebner型)
- 斑驳色素沉着的EBS(EBS-MP)
EBS的这四个亚型的表型范围从相对轻度的手脚水疱到更普遍的水疱和广泛的过度角化,这可能是致命的。
- 在EBS-loc中,出生时很少出现水疱或水疱很少,大约18个月大时,可能会出现膝盖和小腿爬行或脚上的水疱; 有些人在青春期或成年早期就表现出这种疾病。 水疱通常局限于手和脚,但如果创伤严重,可能会发生在任何地方。
- 在EBS(非糖尿病DM)中,水疱可能在出生时出现或在生命的最初几个月内发展。 程度比EBS-loc广泛,但通常比EBS-DM轻。
- 在EBS-MP中,皮肤脆弱性在出生时就很明显,并且在临床上与EBS-DM没有区别。 随着时间的流逝,在躯干和四肢逐渐出现渐进的褐色色素沉着,并有色素沉着的斑点,成人时期色素沉着逐渐消失。 可能发生局灶性掌和足底角化过度。
- 在EBS-DM中,发病通常在出生时开始。 家庭内部和家庭之间的严重程度差异很大。 典型的广泛性和严重性水疱和/或多团小水疱,出血性水疱是常见的。 改善发生在儿童中期至晚期。 EBS-DM在某些人体内似乎随着温暖而改善。 手掌和脚掌进行性过度角化症始于童年,可能是成年后受累的主要不适。 指甲营养不良很常见。 色素沉着过度和色素减退均可发生。 EBS-DM的粘膜受累可能会干扰进食。 水泡可能严重到导致新生儿或婴儿死亡。
单纯型大疱性表皮松解症(EBS)的其他类型。当前的分类系统根据表皮中水泡的位置将EBS分为两个亚型。在EBS的上基底形式中,水泡发生在基底角质形成细胞上方。 EBS的上基底形式极为罕见,包括:EBS浅表肌; EBS,Plakophilin-1缺乏症(也称为外胚层发育不良/皮肤脆性综合征);和EBS,致死性棘皮病。
- EBS,缺乏Plakophilin-1的特征是皮肤轻度脆弱,伴有口周龟裂和唇炎,皮下垂或脱发,疼痛和裂开的掌角化病。它是由PKP1中的功能丧失性变异引起的(有关综述,请参见McGrath & Mellerio [2010]).
- EBS致死性棘皮分解症是由DSP尾部的致病变异引起的,该变异编码去氨白蛋白 [Jonkman et al 2005, Bolling et al 2010, Hobbs et al 2010]。患病的新生儿进行性糜烂而无水泡,脱发和指甲脱落。出生后头几天内因严重的体液和电解质失衡而死亡是常见的。
Hemmidesmosomal EB.Pulkkinen & Uitto [1999]提出,具有肌营养不良症的EB(EB-MD)和患有幽门闭锁的EB(EB-PA)被认为是“ 半桥粒JEB”,因为所涉及的蛋白质位于半桥粒。在基底角质形成细胞内,血凝素定位于半桥粒的内部斑块,它们是发育不良的,并且与角蛋白丝的结合性较差。皮肤活检的电子显微镜检查显示在角质小体上方的基底角质形成细胞底层内有一个切割平面(分离水平)。 (请参阅EB-PA。)
但是,“半桥粒大疱性表皮松解性”并不是公认的名称,也不包含在2008年的EB分类中。 EBS(由PLEC1,ITGA6或ITGB4中的双等位基因的致病性变异引起)或JEB(由ITGA6或ITGB4的双等位基因致病性变异引起)通常包括以下三种类型:
- EB-MD(OMIM)。 全世界已经报告了大约50例EB-MD病例。 一些因PLEC1突变而患有EB的人会发展为肌肉营养不良[Smith et al 1996, Charlesworth et al 2003, Koss-Harnes et al 2004, Schara et al 2004, Pfendner et al 2005]。 水泡较早发生,通常是轻度的。 肌营养不良症可能要等到儿童期,青春期或成年后才出现,并可能导致行动不便,并最终在生命的后期死亡。 整个PLEC1致病性变异都被报道了,但似乎聚集在两个长开放阅读框中,该阅读框包含 基因3'末端的外显子。 已经报道了无义,错义,插入/缺失和剪接连接变异。 最温和的表型通常与 非移码插入或删除相关,不会改变microRNA (mRNA) 的阅读框架[Pfendner et al 2005]. 遗传是 常染色体隐性遗传。
由于PLEC1致病性变异导致的常染色体隐性EBS致死病例报道[Charlesworth et al 2003]. Kunz et al [2000] 报道了由于PLEC1中的致病性变异而导致严重粘膜受累的EBS病例。
由PLEC1中的变异引起的EBS可能从相对温和的(以前称为Ogna形式 (OMIM) [Koss-Harnes et al 2002],到更严重甚至致命的 [Kunz et al 2000, Charlesworth et al 2003]. 最近的数据表明,多达8%的EBS病例可能是由PLEC1突变引起的[Bolling et al 2014]. - EB-PA. 在一些美国和日本家庭中,患有幽门闭锁的EB与PLEC1中的过早终止变异有关[Nakamura et al 2005, Pfendner & Uitto 2005],更常见的相关基因是编码β4整联蛋白(ITGB4)的基因。 EB-PA罕见病例与α6整合素基因(ITGA6)中的致病变异有关。尽管疾病过程很严重,在新生儿期通常是致命的,但有非致命形式报道。在编码α6或β4整联蛋白的基因中具有致病变异的个体也可能显示肾脏和输尿管异常,包括增生/多囊肾,积水性肾病/输尿管,急性肾小管坏死,阻塞性尿毒症,输尿管膨出,多重肾收集系统和膀胱缺如 [Puvabanditsin et al 1997, Kambham et al 2000, Nakano et al 2000, Wallerstein et al 2000, Varki et al 2006, Pfendner et al 2007].。有时,由于羊水过少,有或无α-胎蛋白和乙酰胆碱酯酶水平升高以及羊水中的回声物质,可能会在妊娠期间怀疑幽门闭锁[Dolan et al 1993, Azarian et al 2006].
Dystrophic EB(DEB).水泡在基底膜下方形成,并且基底膜附着在水泡顶上,导致水泡愈合时形成疤痕。 编码VII型胶原的 基因COL7A1的致病变异已在所有形式的DEB中呈现,包括显性和隐性 [Varki et al 2007].
管理
初步诊断后的评估
为了确定诊断为交界型大疱性表皮松解症(JEB)的个体的疾病程度和需求,建议进行以下评估:
- 对儿童进行性嘶哑或喘鸣的水疱形成部位的评估,包括口腔,食道和气道
- 由经验丰富的耳鼻喉科医生直接对气道进行检查,并使用适当的小型且润滑的仪器,以确定气道受损的程度,以便与家人讨论有关气管切开术的决定
- 胃食管反流疾病的评估,可能会导致上呼吸道的额外创伤[Ida et al 2012]
- 通过骨骼射线照相或DEXA扫描评估现有的骨质减少
- 通过临床评估和/或超声心动图评估心脏[Fine et all 2008]
- 测量血红蛋白和电解质以评估贫血和电解质失衡
- 临床患病婴儿的皮肤细菌培养和血液培养决定适当的抗生素治疗
- 临床遗传学咨询
表型治疗
皮肤. 需要保护皮肤免受剪切力的影响,看护者需要学习如何处理EB患儿 [Denyer 2010, Pope et al 2012].
应将新的水泡刺穿并干燥,以防止流体压力进一步扩散。 在大多数情况下,水泡敷料包括三层:
- 一种主要的非粘附性敷料,不会剥离表皮的顶层。 对不同的主要层的耐受是不同的。 主要层包括以下内容:
- 普通Band-Aids®
- 浸有凡士林或外用防腐剂等润肤剂的敷料(e.g., Vaseline® gauze, Adaptic®, Xeroform®)
- 不粘产品 (e.g., Telfa®, N-terface®)
- 不含粘合剂的有机硅产品(e.g., Mepitel®, Mepilex®)
- 第二层为第一层提供稳定性,并添加填充以允许更多活动。 通常使用纱布卷(例如Kerlix®)。
- 通常具有一定弹性的第三层,可确保敷料的完整性(例如Coban®或直径不同的弹性管纱网,例如BandNet®)
其他。最常见的继发性并发症是感染。除了伤口护理之外,治疗伤口的慢性感染也是一个挑战。许多受累的人感染了耐药菌,最常见的是耐甲氧西林的金黄色葡萄球菌(MRSA)和铜绿假单胞菌。抗生素和防腐剂都需要使用。
食道狭窄和网状组织可反复扩张以改善吞咽 [Azizkhan et al 2007].
婴儿嘶哑的哭声应警惕肉芽组织阻塞气道的可能性。关于气管切开术的决定应涉及家庭,并考虑婴儿的医疗状况。由于这些婴儿的预后差,剧烈疼痛和不适,因此与家庭和医院伦理委员会的讨论通常有助于确定要提供的干预和舒适护理的类型[Yan et al 2007, Ida et al 2012].
如果出现胃食管反流病,应象普通人群一样对待。
一些孩子由于水泡和角化过度而延迟或行走困难。适当的鞋类和理疗对于保持下肢活动至关重要。
可以尝试使用高效的局部类固醇,硝酸银,电灼或自体皮肤移植物来治疗肉芽组织。
社会心理支持,包括社会服务和心理咨询,是必不可少的[Lucky et al 2007].
疼痛管理已成为日常护理的重要组成部分[Goldschneider & Lucky 2010]。对于那些难以控制的疼痛,可以考虑转诊给疼痛管理专家。
由于固有的釉质异常,必须进行牙科护理[Kirkham et al 2000, Krämer et al 2012].
在这个人群中,泌尿科和肾脏疾病可能很严重 [Kajbafzadeh et al 2010]. 对于那些受累的幸存者,可以考虑转诊给泌尿科医师。
预防继发并发症
液体和电解质问题在新生儿期和患广泛疾病的婴儿中可能非常严重,甚至危及生命,需要谨慎处理。
对于受累程度较重的JEB婴儿和儿童,成长可能是一个问题,需要额外的营养支持,包括在必要时进行肠胃造口术以确保摄入足够的热量[Haynes 2006].
在新生儿中幸存的儿童,在发现以下营养缺陷时也必须解决:
- 钙和维生素D替代治疗骨质疏松症和骨质疏松症
- 补锌促进伤口愈合[Mellerio et al 2007]
- 口服或静脉输注铁血和红细胞输血治疗缺铁性贫血(慢性问题)
伤口感染应使用适当的抗微生物剂或抗菌药物治疗。
监视
应常规筛查缺铁性贫血,并进行全血细胞计数和血清铁浓度测量,以在必要时补充铁。
通常应通过测量血清锌浓度来筛查锌缺乏症,以在必要时补充锌以增强伤口愈合。
通过骨矿物质密度扫描进行筛查可以发现早期骨质减少和/或骨质疏松症。尚未建立有关应开始年龄的指导原则。
扩张型心肌病的筛查可通过常规超声心动图检查来完成[Lara-Corrales et al 2010].
由于存在鳞状细胞癌的风险,因此至关重要的是,在生命的第二个十年中对不愈合,疤痕组织旺盛或外观异常的伤口进行监测。可能需要对可疑病变进行频繁的活检,然后局部切除。
避免的药物/情况
大多数患有JEB的人不能使用普通医用胶带或Band-Aids®。有机硅产品可以很好地替代胶带。
应避免穿着不合适或质地较粗的衣服和鞋类,因为它们可能导致外伤。
通常应避免伤害皮肤的活动(例如远足,骑山地自行车,接触运动);应鼓励有志参加此类活动的 受累的寻找保护皮肤的创新方法。
评估亲戚处于危险中
有关与遗传咨询目的有关的高危亲属测试的问题,请参见Genetic Counseling。
怀孕管理
建议剖腹产以减少分娩过程中 受累的 胎儿的皮肤创伤。
正在调查的疗法
已经提出了几种针对JEB的基因治疗的方法,其专注于体外表皮细胞的逆转录病毒修饰[Robbins et al 2001, Ortiz-Urda et al 2003]。一项成功的临床试验是使用遗传修饰的表皮干细胞在具有 双等位基因的LAMB3致病变异的 受累的个体中移植而进行的[Mavilio et al 2006, Di Nunzio et al 2008]。动物模型包括在小鼠中进行羊膜内产羊胎蛋白332递送 [Mühle et al 2006]和在狗中自发形成JEB [Capt et al 2005, Spirito et al 2006].
正在使用从非水疱性皮肤的斑块中培养的自体回复细胞来研究自然l基因治疗[Pasmooij et al 2012].
所有JEB相关基因的基因敲除小鼠模型应促进这些治疗方法的发展[Jiang & Uitto 2005]. Natsuga et al [2010].综述了用于研究EB的动物模型。
诱导多能干细胞(IPS)正在全球多个实验室进行研究,以解决其他类型EB的治疗[Tolar et al 2013, Osborn et al 2013].
已经对LAMB3的蛋白质替代疗法进行了体外研究[Igoucheva et al 2008],并获得了可喜的结果。
搜索ClinicalTrials.gov,以获取有关各种疾病和状况的临床研究信息。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 下一节讨论遗传风险评估以及家族史和遗传测试的使用,以阐明家庭成员的遗传状况。 本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不是代替与遗传学专业人员进行咨询。 —编者。
遗传方式
交界型大疱性表皮松解症(JEB)以 常染色体隐性遗传的方式遗传。
迄今为止,尚无证据表明LAMA3,LAMB3,LAMC2或COL17A1中存在单个(即 杂合的) 致病性变异导致JEB。然而,已经报道了具有单个LAMB3或COL17A1变异的个体中的牙齿异常[McGrath et al 1996, Almaani et al 2009, Poulter et al 2014].
家庭成员的风险
先证者的父母
- 受累的孩子的父母通常是肯定杂合子,因此每个父母都携带一个突变的等位基因。因为已经报道了胚系嵌合和单亲二倍体[Pulkkinen et al 1997, Takizawa et al 2000b, Cserhalmi-Friedman et al 2002, Fassihi et al 2005],所以父母的携带者身份需要通过 分子遗传学检测来确认。
- 杂合子(携带者)是无症状的,除了在少数罕见情况下,据报道,COL17A1或LAMB3 致病性变异的携带者患有牙釉质发育不全和/或产生龋齿[Nakamura et al 2006, Murrell et al 2007, Kim et al 2013, Poulter et al 2014].
先证者的同胞
- 父母都是携带者的受累个人的同胞有25%的几率患病,有50%的几率为无症状的携带者, 不受到影响的几率为25% 。
- 一旦知道一个高风险同胞不受影响,则他/她成为携带者的风险为2/3。
- 杂合子(携带者)是无症状的,除了COL17A1或LAMB3致病变异,其中携带者可能表现出牙釉质发育不全和/或斑点和龋齿 [Nakamura et al 2006, Murrell et al 2007, Kim et al 2013, Poulter et al 2014].
先证者的后代。具有常染色体隐性遗传JEB的个体的后代是致病性变异的肯定杂合子(携带者)。
先证者的其他家庭成员。先证者的父母每个携带者的同胞都有50%风险成为携带者。
携带检测
如果已经确定了该家族中的致病变异,则可以对高危家庭成员进行携带者测试。
相关的遗传咨询问题
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑受累的银行DNA。
产前检查和植入前遗传学诊断
分子遗传学测试。一旦在 受累的家庭成员中确定了LAMA3,LAMB3,LAMC2或COL17A1的致病变异,就可以选择产前诊断和植入前遗传诊断来减少JEB风险。
子宫镜检查。通过胎儿镜检查获得的胎儿皮肤活检的电子显微镜评估在JEB中也可诊断。胎儿镜检查比CVS或羊膜穿刺术对妊娠的风险更大,并且在妊娠后期(18-20周)进行。目前尚未在美国使用胎儿镜检查对JEB进行产前诊断,但在欧洲可能会进行。
资源
GeneReviews工作人员选择了以下特定疾病和/或雨伞支持组织和/或注册表,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- DEBRA InternationalAm Heumarkt 27/3Vienna 1030AustriaPhone: +43 1 876 40 30-0Fax: +43 1 876 40 30-30Email: office@debra-international.org
- DebRA of America, Inc. (Dystrophic Epidermolysis Bullosa Research Association)16 East 41st Street3rd FloorNew York NY 10017Phone: 866-332-7276 (toll-free); 212-868-1573Email: staff@debra.org
- DebRA UKDebRA House13 Wellington Business ParkCrowthorne Berkshire RG45 6LSUnited KingdomPhone: +44 01344 771961Fax: +44 01344 762661Email: debra@debra.org.uk
- My46 Trait Profile
- Medline Plus
- EBCare RegistryThe EBCare Registry is a resource for individuals and families affected by all forms of epidermolysis bullosa (EB) and qualified researchers working on approved EB research projects.Phone: 866-332-7276Fax: 888-363-0790Email: coordinator@EBCare.org
分子遗传学
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。 —编者。
Table A.
交界型大疱性表皮松解症
Table B.
交界型大疱性表皮松解症OMIM 条目(View All in OMIM)
| 113811 | COLLAGEN, TYPE XVII, ALPHA-1; COL17A1 |
| 147557 | INTEGRIN, BETA-4; ITGB4 |
| 150292 | LAMININ, GAMMA-2; LAMC2 |
| 150310 | LAMININ, BETA-3; LAMB3 |
| 226650 | EPIDERMOLYSIS BULLOSA, JUNCTIONAL, NON-HERLITZ TYPE |
| 226700 | EPIDERMOLYSIS BULLOSA, JUNCTIONAL, HERLITZ TYPE |
| 226730 | EPIDERMOLYSIS BULLOSA JUNCTIONALIS WITH PYLORIC ATRESIA |
| 600805 | LAMININ, ALPHA-3; LAMA3 |
分子遗传机制
由LAMA3,LAMB3和LAMC2编码的蛋白质组装成层粘连蛋白332异源三聚体(aka LAM5 [Aumailley et al 2005])。这些基因中的 致病性变异可能导致对轻度创伤的抵抗力降低,并导致粘膜皮肤起水泡,这是交界型大疱性表皮松解症(JEB)的标志( Kiritsi et al [2013] 综述)。变异的类型,取代氨基酸的生化特性(如果存在)及其位置决定了水泡表型的严重性(请参阅Genotype-Phenotype Correlations)。致病的nonsense 变异在JEB的严重形式中占主导地位,并导致组装到层粘连蛋白332中的三种蛋白质之一不存在。蛋白质亚基关键位置的致病性错义变异影响层粘连蛋白α3,β3和γ2的能力。多肽组装成三聚体分子,其二级结构,以及其形成椎板的细胞内锚定原纤维的能力。
XVII胶原蛋白是半桥粒的组成部分,具有细胞内和细胞外成分。有证据表明它与半桥粒体内的α-6整联蛋白相互作用。血小体-由几种蛋白质成分组成的结构,包括COLXVII,α-6beta-4整联蛋白,BPAG1和Plectin-将表皮细胞锚定在底层真皮上。 COL17A1中变异的类型和位置决定了是否制造了某些部分功能的蛋白质,并且还影响皮肤的切割平面水平。在某些情况下,影响细胞内 结构域的变异会在通常与交界型大疱性表皮松解症(EBS)相关的基底角质形成细胞最低水平内产生一个切割平面[Charlesworth et al 2003].
整联蛋白成对结合,包含一对α链和一个β链。 α6β4整联蛋白包含来自蛋白质整联蛋白家族的一种α6和一种β4整联蛋白,并且是表皮的半桥粒的组分。已知整联蛋白参与细胞粘附以及细胞表面介导的信号传导。已经描述了α6和β4整联蛋白中的插入/缺失,剪接连接和氨基酸取代变异,这些变异将导致EB-PA [Ruzzi et al 1997, Gache et al 1998, Lépinard et al 2000, Varki et al 2006, Masunaga et al 2015, Mencía et al 2016, Masunaga et al 2017].
COL17A1 基因结构。 cDNA NM_000494.3在56个外显子中编码1,497个氨基酸。有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
良性变异。有一种剪接的 mRNA变异[Ruzzi et al 2001].
致病变异。 COL17A1中的致病性变异编码胶原XVII蛋白(半桥粒的组成部分),通常会导致JEB泛发中间型 [Gatalica et al 1997, Pulkkinen et al 1999, Takizawa et al 2000a, van Leusden et al 2001, Pasmooij et al 2004],尽管已经描述了一些由COL17A1致病性变异导致的致命性JEB的个体 [Varki et al 2006, Murrell et al 2007]。已经描述了遍及整个基因分布的所有类型的变异(包括提前终止密码子,废话,插入/缺失,剪接点和错义变异)。变异的类型和位置以及细胞对变异的反应决定了表型,其范围从轻度到严重,在某些情况下是致命的。已经描述了恢复正常表型的方法[Pasmooij et al 2005, Pasmooij et al 2012].
正常基因产物。 XVII胶原蛋白NP_000485.3(也称为BP180)具有1,497个氨基酸,由被跨膜 结构域分隔的胞内和胞外域组成,该跨膜结构域将XVII胶原与其他胶原家族成员区分开。细胞内结构域位于基底角质形成细胞内;胞外域位于细胞外部,并与基底膜区域的其他组件关联。胶原蛋白XVII的羧基末端一半为916个氨基酸,由15个可变长度的胶原蛋白结构域(15至242个氨基酸)组成,这些胶原蛋白结构域由短距离的非胶原蛋白序列隔开。胶原结构域缔合以形成所有胶原家族成员特征性分子的同三聚体三螺旋节段。
异常基因产物。导致 无效 等位基因 的过早终止密码子致病性变异会导致皮肤脆弱,牙齿异常和脱发,通常在具有JEB泛发中间型的个体中发现。其他致病性变异可能导致不同的表型严重性。尽管COL17A1变异通常不会导致致命性,但已描述了几例新生儿致命表型的案例[Varki et al 2006, Murrell et al 2007]。影响细胞内结构域的致病变异可能导致切割平面与EBS更一致,并且在基于电子显微镜活检结果的诊断方面会产生误导。影响跨膜结构域的致病变异可能导致胶原XVII蛋白在细胞内积聚。尽管已经描述了COL17A1中的甘氨酸取代,但尚未鉴定出导致皮肤脆弱的常染色体显性遗传变异。甘氨酸取代[Nakamura et al 2006]或其他COL17A1变体[Murrell et al 2007] 的杂合子携带者可能表现出牙釉质点蚀,并且该特征可以诊断患有受累的患儿的家庭的COL17A1致病性变异[Murrell et al 2007].
ITGB4 基因结构。正常的全长cDNA被编码在41个外显子中,跨度为36 kb基因组的。 cDNA包含6,033 bp,具有5,469个核苷酸的开放阅读框架,编码1,822个氨基酸。两个剪接变异表达蛋白质的不同 异型体[Pulkkinen et al 1997c]。最常见的表皮变异不表达 外显子33。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病变异。在患有EB-PA的个体中已经描述了超过100种ITGB4的致病变异[Pulkkinen et al 1997b, Pulkkinen et al 1997c, Pulkkinen et al 1998a, Pulkkinen et al 1998b, Ashton et al 2001, Nakano et al 2001, Iacovacci et al 2003, Varki et al 2006, Masunaga et al 2015]。在两个等位基因上导致过早终止密码子的致病变异会导致最严重的表型,在新生儿期常常致命。其他类型的变异,包括氨基酸取代和剪接变异,可能导致表型不那么严重 [Mellerio et al 1998, Pulkkinen et al 1998b, Chavanas et al 1999, Varki et al 2006]。在一个个体中,从ITGB4中的 纯合性 错义变异描述了没有幽门闭锁的严重水疱 [Inoue et al 2000],在另一个人中,有错义和PTC ITGB4变异的纯合子[Inoue et al 2000, Jonkman et al 2000]。很少有复发性致病变异的描述。然而,p.Cys61Tyr变异在美国具有JEB-PA的西班牙裔个体中很常见 [Varki et al 2006],参见 Table 3.Table 3.
本GeneReview中讨论的ITGB4致病变异
| DNA 核苷酸改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.182G>A | p.Cys61Tyr | NM_000213 NP_000204.3 |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen- .hgvs.org ). 的标准命名约定。 有关命名法的说明,请参见Quick Reference。
正常基因产物。整联蛋白成对结合,包含一对α链和一个β链。 α6β4整联蛋白包含来自整联蛋白家族的一种α6和一种β4整联蛋白,并且是表皮半桥粒的组分。在半桥粒体内,α6β4整联蛋白与XVII胶原形成附着,以履行其在蛋白质网络中的作用,从而赋予表皮强度和完整性,并通过半桥粒与基底膜的粘附将表皮细胞锚定在下面的真皮上。还已经证明α6β4整联蛋白参与细胞信号转导并且可能在癌变中起作用[Chung et al 2004, Guo et al 2006, Yoon et al 2006, Folgiero et al 2007].
异常基因产物。无效的等位基因可能导致抗α6β4整联蛋白抗体染色时几乎看不到蛋白质或没有蛋白质。在某些较温和的情况下,由于氨基酸取代或剪接连接变异而导致的染色减少。
LAMA3 基因结构。所有LAMA3都以76个外显子编码,位于染色体18q11.2,长度为318 kb。转录变异是通过交替剪接产生的(见Table 4和McLean et al [2003])。有关 基因信息以及其他转录本和蛋白质异型体的详细摘要,请参见Table A,基因。
致病变异。无义, 错义, 剪接和插入/缺失变异已有报道[Nakano et al 2002a, Varki et al 2006]。两个等位基因上的过早终止密码子致病性变异在大多数情况下都会导致JEB泛发严重。已经报道了一些具有早期终止密码子变异的JEB轻度 受累的个体 [Nakano et al 2002a]。氨基酸取代和剪接变异可能导致较温和的 表型 [Posteraro et al 1998, Nakano et al 2002a]。可能存在重叠表型,其中LAMA3中的致病变异会导致眼睛和喉部受累而导致皮肤脆弱[Varki et al 2006, Figueira et al 2007].
Table 4.
GeneReview讨论的LAMA3致病变异
| DNA 核苷酸改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.151dupG 1 | p.Val51GlyfsTer4 | NM_000227 NP_000218 |
| c.1981C>T 2 | p.Arg661Ter |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen- .hgvs.org ). 的标准命名约定。 有关命名法的说明,请参见Quick Reference。- 1.
- 2.
McGrath et al [1996]引用了c.1948C> T(p.Arg650Ter),没有参考序列。 此处的坐标来自OMIM600805(等位基因变异0.0002),具有rs17852757的dbSNP标识,给出了此表中参考序列的HGVS坐标。
Table 5.
该GeneReview中讨论的LAMA3转录变异和蛋白质异型体
| Transcript Variants | Protein Isoforms | ||||||
|---|---|---|---|---|---|---|---|
| Name(s) | Accession # | Length (nt) | Total # Exons | Name(s) | Accession # | Amino Acids (#) | Encoded by Exon #s 1 |
| 2, 3a | NM_000227 | 5,623 | 38 | α3a, 2, LAMA3a | NP_000218 | 1,724 | 39-76 |
| 1, 3b1 | NM_198129 | 10,666 | 75 | α3b1, 1, LAM3b1 | NP_937762 | 3,333 | 1-38 & 40-76 |
| 3, 3b2 | NM_001127717 | 10,498 | 74 | α3b2, 3, LAM3b2 | NP_001121189 | 3,277 | 1-9, 11-38, & 40-76 |
See LAMA3, 基因组的 sequence NC_000018
- .10 - 1.
基于McLean et al [2003]外显子数目。
- 2.
正常基因产物。交替剪接产生了三种异型体(α3a,α3b1,α3b2)(Table 4)。注意,较短的1,724个氨基酸的α3b2亚型在38个外显子(LAMA3的外显子39-76)中编码,并且独特之处在于表达了 外显子39。
层粘连蛋白A3蛋白与层粘连蛋白B3和C2蛋白缔合以形成层粘连蛋白332异三聚体,其包含表皮中的锚定纤丝。锚定的原纤维将基底层的各层保持在一起,并与真皮侧的胶原蛋白VII和表皮侧的半脂质体的凝集素和α6β4整联蛋白形成结合。这种相互作用使得表皮的蛋白质网络得以形成,从而形成了抵抗创伤的灵活而有弹性的屏障。
异常基因产物。参见Molecular Genetic Pathogenesis。在所有三个基因(LAMB3,LAMC2和LAMA3)中,氨基酸取代,剪接变异以及非移码缺失和插入都可能导致某些部分功能性蛋白的形成,从而导致表型更温和。特定的氨基酸取代(例如半胱氨酸残基的取代)抑制二硫键的形成,导致层粘连蛋白332的分子内和分子间缔合改变,并可能导致更严重的表型。在用免疫荧光法研究的皮肤活检中,如果其中一种蛋白质的合成被破坏,其他两种蛋白质的染色通常也会受到影响。
LAMB3 基因结构。正常的LAMB3 cDNA在23个外显子中有3,516个核苷酸的 开放阅读框架,全长29 kb。有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病变异。无义,错义, 剪接和 插入/缺失变异已有报道[Nakano et al 2002b, Varki et al 2006]。已经报道了一些具有早期终止密码子变异的轻度受累的 [Pulkkinen et al 1998a, Nakano et al 2002a]。氨基酸取代和剪接变异可以产生较温和的表型 [Mellerio et al 1998, Posteraro et al 1998, Nakano et al 2002a]。在美国约有45%的H-JEB患者中存在以下LAMB3致病性变体,即p.Arg635Ter, p.Gln243Ter, c.957ins77, p.Arg42Ter [Varki et al 2006]。这些致病变异总是导致过早终止密码子,并且当在两个等位基因上均发现时,会导致JEB泛发严重型。
Table 6.
GeneReview讨论的LAMB3致病变异
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|---|---|
| c.124C>T | p.Arg42Ter | NM_000228 NP_000219 |
| c.727C>T | p.Gln243Ter | |
| c.957ins77 | p.Glu320Ter | |
| c.1903C>T | p.Arg635Ter |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen- .hgvs.org ). 的标准命名约定。 有关命名法的说明,请参见Quick Reference。
正常基因产物。层粘连蛋白B3蛋白具有1,172个氨基酸。它与层粘连蛋白A3和C2蛋白结合形成层粘连蛋白332异源三聚体,该异源三聚体包含表皮中的锚定纤维。
异常基因产物。参见Molecular Genetic Pathogenesis。在所有三个基因(LAMB3,LAMC2和LAMA3)中,氨基酸取代, 剪接变异以及非移码缺失和插入都可能导致某些部分功能性蛋白的形成,从而导致表型更温和。特定的氨基酸取代(例如半胱氨酸残基的取代)抑制二硫键的形成,导致层粘连蛋白332的分子内和分子间缔合改变,并可能导致更严重的表型。在用免疫荧光法研究的皮肤活检中,如果其中一种蛋白质的合成被破坏,其他两种蛋白质的染色通常也会受到影响。已经描述了通过LAMB3嵌合将其恢复为正常表型,并且对治疗有影响[Pasmooij et al 2007].
LAMC2 基因结构。两个LAMC2转录本变异是通过剪接产生的。较长的LAMC2 cDNA转录本 NM_005562.2在表皮中表达,并在23个外显子中编码1,193个氨基酸,跨越55 kb。较短的转录物变异 NM_018891.2在肾脏的大脑皮层,肺和远端小管中表达。有关基因,转录本和蛋白质信息的详细摘要,请参见Table A,基因。
致病变异。无义,错义, 剪接和插入/缺失变异已有报道[Castiglia et al 2001, Nakano et al 2002b, Varki et al 2006]。氨基酸取代和剪接变异可以产生较温和的表型[Posteraro et al 1998, Castiglia et al 2001, Nakano et al 2002a].
Table 7.
GeneReview讨论的LAMC2致病变异
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|---|---|
| c.283C>T | p.Arg95Ter | NM_005562 NP_005553 |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen- .hgvs.org ). 的标准命名约定。 有关命名法的说明,请参见Quick Reference。
正常基因产物。 层粘连蛋白C2蛋白与层粘连蛋白A3和C2结合形成层粘连蛋白332异源三聚体,后者构成表皮中的锚定纤维。
基因基因产物。 参见 Molecular Genetic Pathogenesis。 在所有三个基因(LAMB3,LAMC2和LAMA3)中,氨基酸取代,剪接变异以及 非移码缺失和插入都可能导致某些部分功能性蛋白的形成,从而导致表型更温和。 特定的氨基酸取代(例如半胱氨酸残基的取代)抑制二硫键的形成,导致层粘连蛋白332的分子内和分子间缔合改变,并可能导致更严重的表型。 在用免疫荧光法研究的皮肤活检中,如果其中一种蛋白质的合成被破坏,其他两种蛋白质的染色通常也会受到影响。
参考文献
Literature Cited
- Almaani N, Liu L, Dopping-Hepenstal PJ, Lovell PA, Lai-Cheong JE, Graham RM, Mellerio JE, McGrath JA. Autosomal dominant junctional epidermolysis bullosa. Br J Dermatol. 2009;160:1094 - 7. [PubMed: 19120338]
- Anton-Lamprecht I, Gedde-Dahl T. Epidermolysis bullosa. In: Rimoin DL, Connor MJ, Pyeritz RE, Korf BR, Emery AEH, eds. Emery and Rimoin’s Principles and Practice of Medical Genetics. 4 ed. London, UK: Churchill Livingstone Publishers; 2002:3810-97.
- Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol. 2005;24:326 - 32. [PubMed: 15979864]
- Azarian M, Dreux S, Vuillard E, Meneguzzi G, Haber S, Guimiot F, Muller F. Prenatal diagnosis of inherited epidermolysis bullosa in a patient with no family history: a case report and literature review. Prenat Diagn. 2006;26:57 - 9. [PubMed: 16378325]
- Azizkhan RG, Denyer JE, Mellerio JE, González R, Bacigalupo M, Kantor A, Passalacqua G, Palisson F, Lucky AW. Surgical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile. Int J Dermatol. 2007;46:801 - 8. [PubMed: 17651160]
- Bolling MC, Veenstra MJ, Jonkman MF, Diercks GF, Curry CJ, Fisher J, Pas HH, Bruckner AL. Lethal acantholytic epidermolysis bullosa due to a novel homozygous deletion in DSP: expanding the phenotype and implications for desmoplakin function in skin and heart. Br J Dermatol. 2010;162:1388 - 94. [PubMed: 20302578]
- Bolling MC, Jongbloed JD, Boven LG, Diercks GF, Smith FJ, McLean WH, Jonkman MF. Plectin mutations underlie epidermolysis bullosa simplex in 8% of patients. J Invest Dermatol. 2014;134:273 - 6. [PubMed: 23774525]
- Capt A, Spirito F, Guaguere E, Spadafora A, Ortonne JP, Meneguzzi G. Inherited junctional epidermolysis bullosa in the German Pointer: establishment of a large animal model. J Invest Dermatol. 2005;124:530 - 5. [PubMed: 15737193]
- Castiglia D, Posteraro P, Spirito F, Pinola M, Angelo C, Puddu P, Meneguzzi G, Zambruno G. Novel mutations in the LAMC2 gene in non-Herlitz junctional epidermolysis bullosa: effects on laminin-5 assembly, secretion, and deposition. J Invest Dermatol. 2001;117:731 - 9. [PubMed: 11564184]
- Charlesworth A, Gagnoux-Palacios L, Bonduelle M, Ortonne JP, De Raeve L, Meneguzzi G. Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J Invest Dermatol. 2003;121:1344 - 8. [PubMed: 14675180]
- Cohn HI, Murrell DF. Laryngo-onycho-cutaneous syndrome. Dermatol Clin. 2010;28:89 - 92. [PubMed: 19945620]
- Cserhalmi-Friedman PB, Anyane-Yeboa K, Christiano AM. Paternal germline mosaicism in Herlitz junctional epidermolysis bullosa. Exp Dermatol. 2002;11:468 - 70. [PubMed: 12366701]
- Cserhalmi-Friedman PB, Baden H, Burgeson RE, Christiano AM. Molecular basis of non-lethal junctional epidermolysis bullosa: identification of a 38 basepair insertion and a splice site mutation in exon 14 of the LAMB3 gene. Exp Dermatol. 1998;7:105 - 11. [PubMed: 9583749]
- Denyer JE. Wound management for children with epidermolysis bullosa. Dermatol Clin. 2010;28:257 - 64. [PubMed: 20447488]
- Di Nunzio F, Maruggi G, Ferrari S, Di Iorio E, Poletti V, Garcia M, Del Rio M, De Luca M, Larcher F, Pellegrini G, Mavilio F. Correction of laminin-5 deficiency in human epidermal stem cells by transcriptionally targeted lentiviral vectors. Mol Ther. 2008;16:1977 - 85. [PubMed: 18813277]
- Dolan CR, Smith LT, Sybert VP. Prenatal detection of epidermolysis bullosa letalis with pyloric atresia in a fetus by abnormal ultrasound and elevated alpha-fetoprotein. Am J Med Genet. 1993;47:395 - 400. [PubMed: 7510931]
- Fassihi H, Wessagowit V, Ashton GH, Moss C, Ward R, Denyer J, Mellerio JE, McGrath JA. Complete paternal uniparental isodisomy of chromosome 1 resulting in Herlitz junctional epidermolysis bullosa. Clin Exp Dermatol. 2005;30:71 - 4. [PubMed: 15663509]
- Fewtrell MS, Allgrove J, Gordon I, Brain C, Atherton D, Harper J, Mellerio JE, Martinez AE. Bone mineralization in children with epidermolysis bullosa. Br J Dermatol. 2006;154:959 - 62. [PubMed: 16634901]
- Figueira EC, Crotty A, Challinor CJ, Coroneo MT, Murrell DF. Granulation tissue in the eyelid margin and conjunctiva in junctional epidermolysis bullosa with features of laryngo-onycho-cutaneous syndrome. Clin Experiment Ophthalmol. 2007;35:163 - 6. [PubMed: 17362460]
- Fine JD, Bauer EA, McGuire J Moshell A, eds. Epidermolysis Bullosa; Clinical Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. Baltimore, MD: Johns Hopkins University Press; 1999.
- Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, Heagerty A, Hintner H, Jonkman MF, McGrath J, McGuire J, Moshell A, Shimizu H, Tadini G, Uitto J. Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051 - 66. [PubMed: 10827412]
- Fine JD, Hall M, Weiner M, Li KP, Suchindran C. The risk of cardiomyopathy in inherited epidermolysis bullosa. Br J Dermatol. 2008;159:677 - 82. [PMC free article: PMC2592258] [PubMed: 18616785]
- Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009;60:203 - 11. [PubMed: 19026465]
- Fine JD, Johnson LB, Weiner M, Stein A, Cash S, DeLeoz J, Devries DT, Suchindran C., National Epidermolysis Bullosa Registry. Inherited epidermolysis bullosa and the risk of death from renal disease: experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis. 2004;44:651 - 60. [PubMed: 15384016]
- Gatalica B, Pulkkinen L, Li K, Kuokkanen K, Ryynänen M, McGrath JA, Uitto J. Cloning of the human type XVII collagen gene (COL17A1), and detection of novel mutations in generalized atrophic benign epidermolysis bullosa. Am J Hum Genet. 1997;60:352 - 65. [PMC free article: PMC1712405] [PubMed: 9012408]
- Goldschneider KR, Lucky AW. Pain management in epidermolysis bullosa. Dermatol Clin. 2010;28:273 - 82. ix. [PubMed: 20447492]
- Haynes L. Nutritional support for children with epidermolysis bullosa. Br J Nurs. 2006;15:1097 - 101. [PubMed: 17170656]
- Hobbs RP, Han SY, van der Zwaag PA, Bolling MC, Jongbloed JD, Jonkman MF, Getsios S, Paller AS, Green KJ. Insights from a desmoplakin mutation identified in lethal acantholytic epidermolysis bullosa. J Invest Dermatol. 2010;130:2680 - 3. [PMC free article: PMC3061313] [PubMed: 20613772]
- Huber M, Floeth M, Borradori L, Schäcke H, Rugg EL, Lane EB, Frenk E, Hohl D, Bruckner-Tuderman L. Deletion of the cytoplasmatic domain of BP180/collagen XVII causes a phenotype with predominant features of epidermolysis bullosa simplex. J Invest Dermatol. 2002;118:185 - 92. [PubMed: 11851893]
- Ida JB, Livshitz I, Azizkhan RG, Lucky AW, Elluru RG. Upper airway complications of junctional epidermolysis bullosa. J Pediatr. 2012;160:657 - 61.e1. [PubMed: 22050875]
- Igoucheva O, Kelly A, Uitto J, Alexeev V. Protein therapeutics for junctional epidermolysis bullosa: incorporation of recombinant beta3 chain into laminin 332 in beta3-/- keratinocytes in vitro. J Invest Dermatol. 2008;128:1476 - 86. [PMC free article: PMC3357058] [PubMed: 18079746]
- Inoue M, Tamai K, Shimizu H, Owaribe K, Nakama T, Hashimoto T, McGrath JA. A homozygous missense mutation in the cytoplasmic tail of beta4 integrin, G931D, that disrupts hemidesmosome assembly and underlies Non-Herlitz junctional epidermolysis bullosa without pyloric atresia? J Invest Dermatol. 2000;114:1061 - 4. [PubMed: 10792571]
- Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30:70 - 7. [PubMed: 22137229]
- Irvine AD, McLean WH. The molecular genetics of the genodermatoses: progress to date and future directions. Br J Dermatol. 2003;148:1 - 13. [PubMed: 12534588]
- Jiang QJ, Uitto J. Animal models of epidermolysis bullosa--targets for gene therapy. J Invest Dermatol. 2005;124:xi鈥搙iii. [PubMed: 15812910]
- Jonkman MF, Pas HH, Nijenhuis M, Kloosterhuis G, Steege G. Deletion of a cytoplasmic domain of integrin beta4 causes epidermolysis bullosa simplex. J Invest Dermatol. 2002;119:1275 - 81. [PubMed: 12485428]
- Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, Pas HH. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 2005;77:653 - 60. [PMC free article: PMC1275614] [PubMed: 16175511]
- Kajbafzadeh AM, Elmi A, Mazaheri P, Talab SS, Jan D. Genitourinary involvement in epidermolysis bullosa: clinical presentations and therapeutic challenges. BJU Int. 2010;106:1763 - 6. [PubMed: 20477826]
- Kambham N, Tanji N, Seigle RL, Markowitz GS, Pulkkinen L, Uitto J, D'Agati VD. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis. 2000;36:190 - 6. [PubMed: 10873890]
- Kelly-Mancuso G, Kopelan B, Azizkhan RG, Lucky AW. Junctional epidermolysis bullosa incidence and survival: 5-year experience of the Dystrophic Epidermolysis Bullosa Research Association of America (DebRA) Nurse Educator. Pediatr Dermatol. 2014;31:159 - 62. [PubMed: 23721227]
- Kim JW, Seymen F, Lee KE, Ko J, Yildirim M, Tuna EB, Gencay K, Shin TJ, Kyun HK, Simmer JP, Hu JC. LAMB3 mutations causing autosomal dominant amelogenesis imperfecta. J Dent Res. 2013;92:899 - 904. [PMC free article: PMC3775375] [PubMed: 23958762]
- Kirkham J, Robinson C, Strafford SM, Shore RC, Bonass WA, Brookes SJ, Wright JT. The chemical composition of tooth enamel in junctional epidermolysis bullosa. Arch Oral Biol. 2000;45:377 - 86. [PubMed: 10739859]
- Koss-Harnes D, Høyheim B, Anton-Lamprecht I, Gjesti A, Jørgensen RS, Jahnsen FL, Olaisen B, Wiche G, Gedde-Dahl T Jr. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol. 2002;118:87 - 93. [PubMed: 11851880]
- Koss-Harnes D, Høyheim B, Jonkman MF, de Groot WP, de Weerdt CJ, Nikolic B, Wiche G, Gedde-Dahl T Jr. Life-long course and molecular characterization of the original Dutch family with epidermolysis bullosa simplex with muscular dystrophy due to a homozygous novel plectin point mutation. Acta Derm Venereol. 2004;84:124 - 31. [PubMed: 15206692]
- Krämer SM, Serrano MC, Zillmann G, Gálvez P, Araya I, Yanine N, Carrasco-Labra A, Oliva P, Brignardello-Petersen R, Villanueva J. DEBRA International. Oral health care for patients with epidermolysis bullosa--best clinical practice guidelines. Int J Paediatr Dent. 2012;22 Suppl 1:1 - 35. [PubMed: 22937908]
- Krämer SM. Oral care and dental management for patients with epidermolysis bullosa. Dermatol Clin. 2010;28:303 - 9. [PubMed: 20447495]
- Kunz M, Rouan F, Pulkkinen L, Hamm H, Jeschke R, Bruckner-Tuderman L, Bröcker EB, Wiche G, Uitto J, Zillikens D. Mutation reports: epidermolysis bullosa simplex associated with severe mucous membrane involvement and novel mutations in the plectin gene. J Invest Dermatol. 2000;114:376 - 80. [PubMed: 10652001]
- Laimer M, Lanschuetzer CM, Diem A, Bauer JW. Herlitz junctional epidermolysis bullosa. Dermatol Clin. 2010;28:55 - 60. [PubMed: 19945616]
- Lara-Corrales I, Mellerio JE, Martinez AE, Green A, Lucky AW, Azizkhan RG, Murrell DF, Agero AL, Kantor PF, Pope E. Dilated cardiomyopathy in epidermolysis bullosa: a retrospective, multicenter study. Pediatr Dermatol. 2010;27:238 - 43. [PubMed: 20609141]
- Lucky AW, Pfendner E, Pillay E, Paskel J, Weiner M, Palisson F. Psychosocial aspects of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile. Int J Dermatol. 2007;46:809 - 14. [PubMed: 17651161]
- Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, Maruggi G, Ferrari G, Provasi E, Bonini C, Capurro S, Conti A, Magnoni C, Giannetti A, De Luca M. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397 - 402. [PubMed: 17115047]
- McGrath JA, Ashton GH, Mellerio JE, Salas-Alanis JC, Swensson O, McMillan JR, Eady RA. Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. J Invest Dermatol. 1999;113:314 - 21. [PubMed: 10469327]
- McGrath JA, Mellerio JE. Ectodermal dysplasia-skin fragility syndrome. Dermatol Clin. 2010;28:125 - 9. [PubMed: 19945625]
- McGrath JA, Kivirikko S, Ciatti S, Moss C, Christiano AM, Uitto J. A recurrent homozygous nonsense mutation within the LAMA3 gene as a cause of Herlitz junctional epidermolysis bullosa in patients of Pakistani ancestry: evidence for a founder effect. J Invest Dermatol. 1996;106:781 - 4. [PubMed: 8618022]
- McLean WH, Irvine AD, Hamill KJ, Whittock NV, Coleman-Campbell CM, Mellerio JE, Ashton GS, Dopping-Hepenstal PJ, Eady RA, Jamil T, Phillips R, Shabbir SG, Haroon TS, Khurshid K, Moore JE, Page B, Darling J, Atherton DJ, Van Steensel MA, Munro CS, Smith FJ, McGrath JA. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum Mol Genet. 2003;12:2395 - 409. [PubMed: 12915477]
- Mellerio JE, Eady RA, Atherton DJ, Lake BD, McGrath JA. E210K mutation in the gene encoding the beta3 chain of laminin-5 (LAMB3) is predictive of a phenotype of generalized atrophic benign epidermolysis bullosa. Br J Dermatol. 1998;139:325 - 31. [PubMed: 9767254]
- Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, Palisson F. Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile. Int J Dermatol. 2007;46:795 - 800. [PubMed: 17651159]
- Micheloni A, De Luca N, Tadini G, Zambruno G, D'Alessio M. Intracellular degradation of beta4 integrin in lethal junctional epidermolysis bullosa with pyloric atresia. Br J Dermatol. 2004;151:796 - 802. [PubMed: 15491419]
- Mühle C, Neuner A, Park J, Pacho F, Jiang Q, Waddington SN, Schneider H. Evaluation of prenatal intra-amniotic LAMB3 gene delivery in a mouse model of Herlitz disease. Gene Ther. 2006;13:1665 - 76. [PubMed: 16871230]
- Murrell DF, Pasmooij AM, Pas HH, Marr P, Klingberg S, Pfendner E, Uitto J, Sadowski S, Collins F, Widmer R, Jonkman MF. Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J Invest Dermatol. 2007;127:1772 - 5. [PubMed: 17344927]
- Nakamura H, Sawamura D, Goto M, Nakamura H, Kida M, Ariga T, Sakiyama Y, Tomizawa K, Mitsui H, Tamaki K, Shimizu H. Analysis of the COL17A1 in non-Herlitz junctional epidermolysis bullosa and amelogenesis imperfecta. Int J Mol Med. 2006;18:333 - 7. [PubMed: 16820943]
- Nakamura H, Sawamura D, Goto M, Nakamura H, McMillan JR, Park S, Kono S, Hasegawa S, Paku S, Nakamura T, Ogiso Y, Shimizu H. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn. 2005;7:28 - 35. [PMC free article: PMC1867514] [PubMed: 15681471]
- Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, Uitto J. Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet. 2002a;110:41 - 51. [PubMed: 11810295]
- Nakano A, Lestringant GG, Paperna T, Bergman R, Gershoni R, Frossard P, Kanaan M, Meneguzzi G, Richard G, Pfendner E, Uitto J, Pulkkinen L, Sprecher E. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. J Am Acad Dermatol. 2002b;46:510 - 6. [PubMed: 11907499]
- Nakano A, Pfendner E, Hashimoto I, Uitto J. Herlitz junctional epidermolysis bullosa: novel and recurrent mutations in the LAMB3 gene and the population carrier frequency. J Invest Dermatol. 2000;115:493 - 8. [PubMed: 11023379]
- Natsuga K, Shinkuma S, Nishie W, Shimizu H. Animal models of epidermolysis bullosa. Dermatol Clin. 2010;28:137 - 42. [PubMed: 19945627]
- Ortiz-Urda S, Lin Q, Yant SR, Keene D, Kay MA, Khavari PA. Sustainable correction of junctional epidermolysis bullosa via transposon-mediated nonviral gene transfer. Gene Ther. 2003;10:1099 - 104. [PubMed: 12808440]
- Osborn MJ, Starker CG, McElroy AN, Webber BR, Riddle MJ, Xia L, DeFeo AP, Gabriel R, Schmidt M, von Kalle C, Carlson DF, Maeder ML, Joung JK, Wagner JE, Voytas DF, Blazar BR, Tolar J. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21:1151 - 9. [PMC free article: PMC3677309] [PubMed: 23546300]
- Pasmooij AM, Nijenhuis M, Brander R, Jonkman MF. Natural gene therapy may occur in all patients with generalized non-Herlitz junctional epidermolysis bullosa with COL17A1 mutations. J Invest Dermatol. 2012;132:1374 - 83. [PubMed: 22318390]
- Pasmooij AM, Pas HH, Bolling MC, Jonkman MF. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J Clin Invest. 2007;117:1240 - 8. [PMC free article: PMC1857245] [PubMed: 17476356]
- Pasmooij AM, Pas HH, Deviaene FC, Nijenhuis M, Jonkman MF. Multiple correcting COL17A1 mutations in patients with revertant mosaicism of epidermolysis bullosa. Am J Hum Genet. 2005;77:727 - 40. [PMC free article: PMC1271383] [PubMed: 16252234]
- Pasmooij AM, van der Steege G, Pas HH, Smitt JH, Nijenhuis AM, Zuiderveen J, Jonkman MF. Features of epidermolysis bullosa simplex due to mutations in the ectodomain of type XVII collagen. Br J Dermatol. 2004a;151:669 - 74. [PubMed: 15377356]
- Pasmooij AM, van Zalen S, Nijenhuis AM, Kloosterhuis AJ, Zuiderveen J, Jonkman MF, Pas HH. A very mild form of non-Herlitz junctional epidermolysis bullosa: BP180 rescue by outsplicing of mutated exon 30 coding for the COL15 domain. Exp Dermatol. 2004b;13:125 - 8. [PubMed: 15009107]
- Pfendner E, Rouan F, Uitto J. Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations. Exp Dermatol. 2005;14:241 - 9. [PubMed: 15810881]
- Pfendner E, Uitto J, Fine JD. Epidermolysis bullosa carrier frequencies in the US population. J Invest Dermatol. 2001;116:483 - 4. [PubMed: 11231335]
- Pfendner E, Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol. 2005;124:111 - 5. [PubMed: 15654962]
- Pfendner EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Basic science of epidermolysis bullosa and diagnostic and molecular characterization: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile. Int J Dermatol. 2007;46:781 - 94. [PubMed: 17651158]
- Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, Goodman L, Coutts P, Wagner J, Allen U, Sibbald G. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67:904 - 17. [PMC free article: PMC3655403] [PubMed: 22387035]
- Posteraro P, De Luca N, Meneguzzi G, El Hachem M, Angelo C, Gobello T, Tadini G, Zambruno G, Castiglia D. Laminin-5 mutational analysis in an Italian cohort of patients with junctional epidermolysis bullosa. J Invest Dermatol. 2004;123:639 - 48. [PubMed: 15373767]
- Posteraro P, Sorvillo S, Gagnoux-Palacios L, Angelo C, Paradisi M, Meneguzzi G, Castiglia D, Zambruno G. Compound heterozygosity for an out-of-frame deletion and a splice site mutation in the LAMB3 gene causes nonlethal junctional epidermolysis bullosa. Biochem Biophys Res Commun. 1998;243:758 - 64. [PubMed: 9501007]
- Poulter JA, El-Sayed W, Shore RC, Kirkham J, Inglehearn CF, Mighell AJ. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 2014;22:132 - 5. [PMC free article: PMC3865405] [PubMed: 23632796]
- Pulkkinen L, Bruckner-Tuderman L, August C, Uitto J. Compound heterozygosity for missense (L156P) and nonsense (R554X) mutations in the beta4 integrin gene (ITGB4) underlies mild, nonlethal phenotype of epidermolysis bullosa with pyloric atresia. Am J Pathol. 1998;152:935 - 41. [PMC free article: PMC1858243] [PubMed: 9546354]
- Pulkkinen L, Bullrich F, Czarnecki P, Weiss L, Uitto J. Maternal uniparental disomy of chromosome 1 with reduction to homozygosity of the LAMB3 locus in a patient with Herlitz junctional epidermolysis bullosa. Am J Hum Genet. 1997;61:611 - 9. [PMC free article: PMC1715967] [PubMed: 9326326]
- Pulkkinen L, Marinkovich MP, Tran HT, Lin L, Herron GS, Uitto J. Compound heterozygosity for novel splice site mutations in the BPAG2/COL17A1 gene underlies generalized atrophic benign epidermolysis bullosa. J Invest Dermatol. 1999;113:1114 - 8. [PubMed: 10636730]
- Pulkkinen L, McGrath JA, Christiano AM, Uitto J. Detection of sequence variants in the gene encoding the beta 3 chain of laminin 5 (LAMB3). Hum Mutat. 1995;6:77 - 84. [PubMed: 7550237]
- Pulkkinen L, Uitto J. Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999;18:29 - 42. [PubMed: 10367729]
- Puvabanditsin S, Garrow E, Samransamraujkit R, Lopez LA, Lambert WC. Epidermolysis bullosa associated with congenital localized absence of skin, fetal abdominal mass, and pyloric atresia. Pediatr Dermatol. 1997;14:359 - 62. [PubMed: 9336805]
- Robbins PB, Lin Q, Goodnough JB, Tian H, Chen X, Khavari PA. In vivo restoration of laminin 5 beta 3 expression and function in junctional epidermolysis bullosa. Proc Natl Acad Sci U S A. 2001;98:5193 - 8. [PMC free article: PMC33186] [PubMed: 11296269]
- Ruzzi L, Pas H, Posteraro P, Mazzanti C, Didona B, Owaribe K, Meneguzzi G, Zambruno G, Castiglia D, D'Alessio M. A homozygous nonsense mutation in type XVII collagen gene (COL17A1) uncovers an alternatively spliced mRNA accounting for an unusually mild form of non-Herlitz junctional epidermolysis bullosa. J Invest Dermatol. 2001;116:182 - 7. [PubMed: 11168815]
- Schara U, Tücke J, Mortier W, Nüsslein T, Rouan F, Pfendner E, Zillikens D, Bruckner-Tuderman L, Uitto J, Wiche G, Schröder R. Severe mucous membrane involvement in epidermolysis bullosa simplex with muscular dystrophy due to a novel plectin gene mutation. Eur J Pediatr. 2004;163:218 - 22. [PubMed: 14963703]
- Shinkuma S, McMillan JR, Shimizu H. Ultrastructure and molecular pathogenesis of epidermolysis bullosa. Clin Dermatol. 2011;29:412 - 9. [PubMed: 21679868]
- Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH, Lane EB. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450 - 7. [PubMed: 8696340]
- Spirito F, Capt A, Del Rio M, Larcher F, Guaguere E, Danos O, Meneguzzi G. Sustained phenotypic reversion of junctional epidermolysis bullosa dog keratinocytes: Establishment of an immunocompetent animal model for cutaneous gene therapy. Biochem Biophys Res Commun. 2006;339:769 - 78. [PubMed: 16316622]
- Stellingsma C, Dijkstra PU, Dijkstra J, Duipmans JC, Jonkman MF, Dekker R. Restrictions in oral functions caused by oral manifestations of epidermolysis bullosa. Eur J Dermatol. 2011;21:405 - 9. [PubMed: 21609900]
- Takizawa Y, Hiraoka Y, Takahashi H, Ishiko A, Yasuraoka I, Hashimoto I, Aiso S, Nishikawa T, Shimizu H. Compound heterozygosity for a point mutation and a deletion located at splice acceptor sites in the LAMB3 gene leads to generalized atrophic benign epidermolysis bullosa. J Invest Dermatol. 2000a;115:312 - 6. [PubMed: 10951252]
- Takizawa Y, Pulkkinen L, Chao SC, Nakajima H, Nakano Y, Shimizu H, Uitto J. Mutation report: complete paternal uniparental isodisomy of chromosome 1: a novel mechanism for Herlitz junctional epidermolysis bullosa. J Invest Dermatol. 2000b;115:307 - 11. [PubMed: 10951251]
- Tolar J, Xia L, Lees CJ, Riddle M, McElroy A, Keene DR, Lund TC, Osborn MJ, Marinkovich MP, Blazar BR, Wagner JE. Keratinocytes from induced pluripotent stem cells in junctional epidermolysis bullosa. J Invest Dermatol. 2013;133:562 - 5. [PMC free article: PMC3514565] [PubMed: 22931927]
- Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005;23:33 - 40. [PubMed: 15708287]
- van Leusden MR, Pas HH, Gedde-Dahl T Jr, Sonnenberg A, Jonkman MF. Truncated typeXVII collagen expression in a patient with non-herlitz junctional epidermolysis bullosa caused by a homozygous splice-site mutation. Lab Invest. 2001;81:887 - 94. [PubMed: 11406649]
- Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006;43:641 - 52. [PMC free article: PMC2564586] [PubMed: 16473856]
- Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44:181 - 92. [PMC free article: PMC2598021] [PubMed: 16971478]
- Wallerstein R, Klein ML, Genieser N, Pulkkinen L, Uitto J. Epidermolysis bullosa, pyloric atresia, and obstructive uropathy: a report of two case reports with molecular correlation and clinical management. Pediatr Dermatol. 2000;17:286 - 9. [PubMed: 10990577]
- Yan EG, Paris JJ, Ahluwalia J, Lane AT, Bruckner AL. Treatment decision-making for patients with the Herlitz subtype of junctional epidermolysis bullosa. J Perinatol. 2007;27:307 - 11. [PubMed: 17363907]
- Yuen WY, Huizinga J, Jonkman MF. Punch grafting of chronic ulcers in patients with laminin-332-deficient, non-Herlitz junctional epidermolysis bullosa. J Am Acad Dermatol. 2013;68:93 - 7. [PubMed: 22633040]
- Yuen WY, Jonkman MF. Risk of squamous cell carcinoma in junctional epidermolysis bullosa, non-Herlitz type: report of 7 cases and a review of the literature. J Am Acad Dermatol. 2011;65:780 - 9. [PubMed: 21624701]
Suggested Reading
- Van Coster R, Pulkkinen L. Epidermolysis bullosa: the disease of the cutaneous basement membrane zone. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). New York, NY: McGraw-Hill. Chap 222. Available online.
本章节的注解
更新历史
- 2 January 2014 (me) 系统性更新发布到公开网页上
- 22 February 2008 (me) 内容发布到公开网页上
- 10 May 2007 (egp)最早稿件