
小结
临床特点.
进行性假性类风湿发育不良 (PPD)是以关节软骨受累为特征的一种骨骼发育不良,在无炎症情况下伴有进行性关节僵硬和增大。发病通常在3-6岁之间,开始于指间关节。随着时间的推移,大关节和脊柱的累及会导致明显的关节挛缩、步态紊乱、脊柱侧弯和/或脊柱后凸,导致异常的姿势和显著的发病率。尽管有严重的关节病,但疼痛不是此病的主要表现特征。
最初的身高是正常的;然而,随着骨骼变化的进展,身材矮小(<3厘米)在青春期变得明显。
诊断/检测.
虽然影像学检查对PPD的诊断具有较高的准确性,但一个 先证者 最终诊断的明确是依赖于影像学发现特征性的表现和 分子遗传学检测在CCN6 (原WISP3) 基因上发现 双等位基因的 致病性变异.非甾体抗炎药可对继发性骨关节炎引起的疼痛起作用
管理.
对症治疗:支持治疗。非甾体抗炎药可对继发性骨关节炎引起的疼痛起作用。其他抗炎药物可能具有有限的治疗效果,但考虑到其显著的副作用,最好避免使用。物理治疗可以帮助保持关节的灵活性。相关手术干预,包括下肢复位、关节置换术和/或椎管狭窄的治疗。
监测: 监测包括骨畸形在内的骨科并发症,继发性关节病,脊柱畸形,和疼痛。
应避免的因素/情况:长时间活动受限(例如,固定一个姿势)。
妊娠管理: 骨盆畸形可能需要剖宫产分娩。
遗传咨询.
PPD以 常染色体隐性遗传 模式遗传。理论上, 受累的 个体,其每一个同胞都有 25% 的机会成为患者,50%的机会成为无临床症状的 携带者, 25% 的机会既不患病也不携带。如果在受累的家庭成员中发现了CCN6 双位点致病变异,则对高危亲属进行携带者检测和对高危妊娠胎儿进行产前检测是恰当的。
诊断
进行性假性类风湿发育不良(PPD)是一种脊柱骨骺发育不良的特殊形式,伴明显关节疾病。
目前缺乏正式的诊断标准。
推测性结论
有下列临床和放射学表现的患者应怀疑为进行性假性类风湿发育不良。
临床特征
- 出生时健康
- 关节病发病于儿童早期,通常在三至六岁之间
- 手部指间关节扩大 (Figure 1)
- 所有关节活动进行性受限
- 步态异常
- 膝外翻/膝内翻
- 进行性髋关节疾病(晚期通常为髋内翻)
- 关节疼痛
- 运动减弱易疲劳
- 儿童期晚期和青春期伴胸腰椎后凸畸形的脊柱受累导致躯干短
- 成人身高低于第三百分位数
- 无炎症迹象
- 正常的红细胞沉降率(ESR)和c反应蛋白(CRP)水平
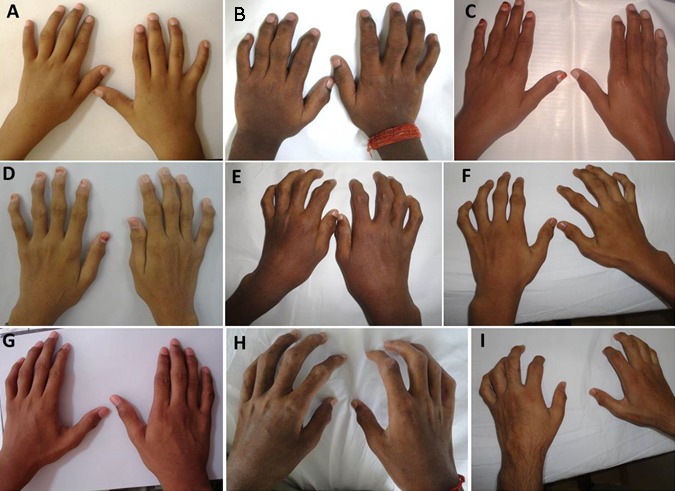
Figure 1.
患者的手呈进行性肿胀,指间关节活动范围受限。A. 5岁 B.7岁 C.11岁 D.12岁 E.13岁 F.15岁 G.16岁 H.17岁 I.23岁
影像学特征 包括脊柱骨骺发育不良、全身关节病、手部特殊关节畸形(表面类似于幼年特发性关节炎),晚期表现为弥漫性骨质疏松。
- 手. 掌指关节和指间关节的骨骺增大,干骺增宽,关节间隙缩小或消失,尤其是近端指间关节 (Figure 2) [Dalal et al 2012]。屈曲指常出现在成年期。
- 骨盆. 髋部显示大而扁平的股骨骨骺和短而宽的股骨颈 (Figure 3) [Dalal et al 2012, Garcia Segarra et al 2012]。 髂嵴增宽,髋臼顶不规则 (Figure 3)。
- 脊柱. 所有患者均可观察到椎终板骨化和椎板平滑肌的进行性不规则性,并伴有椎间盘间隙的丢失或狭窄(Figure 4)。在青春期前可见椎体前喙状突起 [Garcia Segarra et al 2012].
- 肩膀和膝盖. 可见骨赘形成和关节周围钙化。
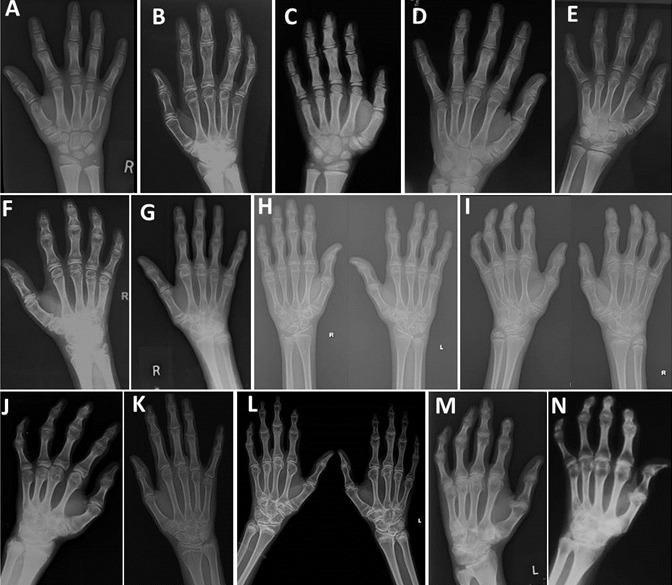
Figure 2.
手部x光片显示掌骨和指骨的大骨骺和宽形骺。关节空间也减小了。 A. 5 岁 B.6岁 C.7岁 D.8岁 E.9岁 F.10岁 G.11岁 H.12岁 I.13岁 J.15岁 K.16岁 L.17岁 M.30岁 N.50岁
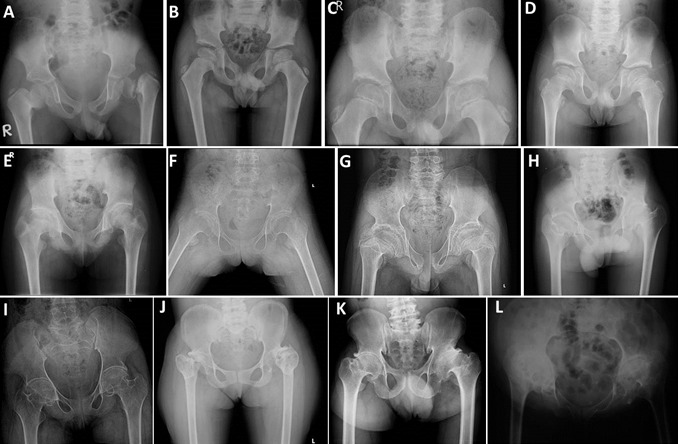
Figure 3.
骨盆x光片显示髋关节间隙缩小,大股骨骺,股骨颈短而宽,髋臼顶不规则。在青春期,髂嵴也是不规则的。A.5岁 B.7岁 C.8岁 D.9岁 E.11岁 F.12岁 G.13岁 H.15岁 I.16岁 J.19岁 K.30岁 L.50岁
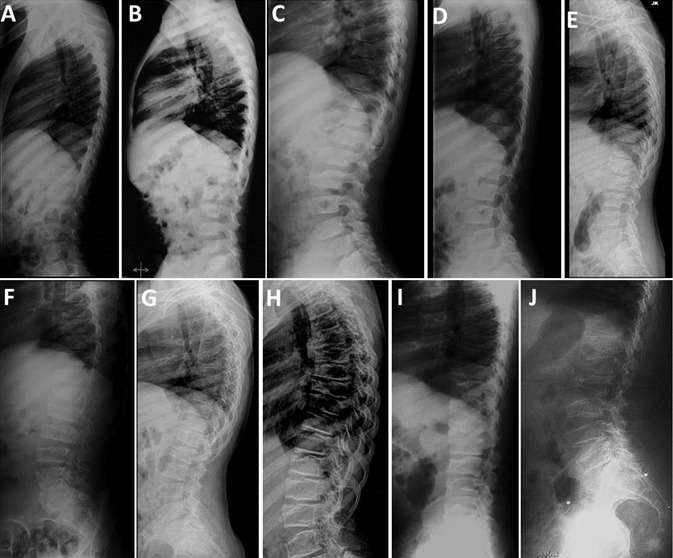
Figure 4.
由于上、下关节软骨的侵蚀导致进行性椎板病 A. 5 岁 B.7岁 C.9岁 D.11岁 E.13岁 F.15岁G.16岁 H.17岁 I.30岁 J.50岁
建立诊断
一般来说,影像学检查对PPD的诊断具有较高的准确性。但进行性假性类风湿发育不良症诊断的确立依赖于用 分子遗传学检测 (见 Table 1)在 先证者 中发现CCN6 (之前的 WISP3) 双等位基因的 致病性变异。
分子检测方法包括 单-基因 检测, 多基因检测包 的使用和 基因组的 检测.
单-基因 检测. 首先对CCN6 进行序列分析。如果只发现一个致病性变异或没有 致病性变异 发现,可能需要通过培养的皮肤成纤维细胞进行 cDNA 的 序列分析 来检测 内含子的 致病性变异导致的 剪接 畸变 [Garcia Segarra et al 2012] (见 分子遗传学).
致病性变异的靶向分析可包括以下内容:
- c.156C>A (p.Cys52Ter) 是土耳其患者中最常见的 致病性变异 ,在全球致病性变异中占大约 28% [Author, review of the literature]。
- c.1010G>A (p.Cys337Tyr) 和 c.233G>A (p.Cys78Tyr) 是印度人群中最常见的致病性变异。
包括CCN6 和其他感兴趣基因(见鉴别诊断)的 一个 多基因检测包 可以考虑应用。注意点: (1) 基因包里包含的基因以及每个 基因 使用的检测方法的诊断 敏感性 因实验室而异也可能随时间而改变。 (2) 一些多基因检测包可能包括与本 GeneReview 中讨论的疾病无关的基因;因此,临床医生需要决定哪一个多基因检测包可能更经济合理地鉴定出引起该疾病的真证遗传学病因,而对 意义不确定 的变异和和无法解释 表型 的致病性变异检出有限。 (3)在一些实验室,检测包的选择包括实验室自定义设计的检测包和/或关注表型的 外显子组 分析包括由临床医生指出的基因。(4) 检测包使用的方法可能包括 序列分析, 缺失/重复分析, 和/或其他非基于序列的检测。
有关多基因检测包的介绍,请点击 这里。 临床医生订购基因检测的更多详细信息可以在 这里 找到。
当一系列的单-基因 试验(和/或使用包含CCN6 的 多基因检测包 )未能确认具有PPD特征的个体的诊断时,更多全面的 基因组的 检测 (有条件使用的) 包括 外显子组测序 和 基因组测序 可以被考虑应用。这种检测可以提供或提示以前没有考虑到的诊断 (例如,不同基因的突变导致相似的临床表现)。
关于全面 基因组的 检测的介绍请点击 这里 。 临床医生订购基因检测的更多详细信息可以点击 这里.
Table 1.
Table1 进行性假性类风湿发育不良的分子遗传学检测
| 基因 1 | 检测方法 | 应用此方法在具有特征性影像学表现的先证者中检出致病性变异2 的比例 |
|---|---|---|
| CCN6 | 测序分析3 | 近 100% 4 |
临床特征
临床描述
进行性假性类风湿发育不良(PPD)是一种骨骼发育不良,其特征为关节软骨受损伤,进行性关节僵硬和增大,且无炎症征象 [Dalal et al 2012]。疾病的进展严重影响步态和姿势,并导致显著的发病率。
PPD无任何骨骼外临床表现,如颅面特征或认知受累。
临床发病特征.
患有PPD的儿童在出生时和婴儿期是正常的。发病年龄通常在3至6岁之间[Wynne-Davies et al 1982, Garcia Segarra et al 2012];发病年龄范围为1岁至16岁 [Delague et al 2005, Dalal et al 2012]。
大多数患者最初表现为指间关节肿胀、疼痛和步态异常。随着时间的推移,关节畸形变得明显。目前发现的症状关节疼痛很少,且关节疼痛的轻微程度与关节病变的严重程度不成比例。
关节. 手部的增大、僵硬和活动范围受限开始于近端指间关节,并向远端指间关节发展 (Figure 1) [Garcia Segarra et al 2012]。 这些关节发生进行性挛缩。
关节增大、僵硬、活动范围受限逐渐累及所有大关节 (也就是膝盖、臀部、手腕和肘部) [Dalal et al 2012, Garcia Segarra et al 2012]. 髋关节受累通常在病程后期表现为髋内翻。膝盖表现为内翻或外翻。通常情况下,肩关节并不严重 受累的 [Dalal et al 2012].
脊柱. 大多数 受累的 个体在青春期被注意到脊柱侧凸和/或脊柱后凸。 前凸也可见。颈部只是偶尔涉及 [Dalal et al 2012]。
身高. 身高最初是正常的;然而,随着骨骼变化的进展,矮小的身材变得明显。成年人身高一般在第三百分位以下 [Garcia Segarra et al 2012]. 臀部和膝盖的屈曲畸形以及脊柱的变化是造成身材矮小的部分原因。
基因型-表型相关性
未观察到 基因型-表型的相关性 。
不同家庭中出现的发病年龄、严重程度和进展的轻微差异不能用致病变异的类型或位置来解释。
已观察到家庭内部的差异 [Bhavani et al 2015].
命名法
其名称“进行性假性类风湿发育不良”反映了其与幼年型类风湿性关节炎的相似之处。
PPD以前被称为脊柱骨骺发育不良伴进行性关节病。
患病率
据估计,在英国PPD的患病率为百万分之一 (流行率为<1-9:1,000,000) [Wynne-Davies et al 1982]。然而,该疾病可能由于与青少年 特发性 关节炎的临床特征重叠而诊断不足。到目前为止,已经报道了超过160个分子确诊的PPD家系。
PPD在近亲婚配的群体更加常见 [Delague et al 2005, Dalal et al 2012]. 最大的家系已经在印度被报道 [Dalal et al 2012, Bhavani et al 2015].
PPD 已经在 近亲婚配 率高的科威特、黎巴嫩、伊朗、约旦、沙特阿拉伯、叙利亚、巴勒斯坦和摩洛哥人群中被发现。
该基因涉及的疾病(等位基因)
除GeneReview中讨论的表型外,没有已知的表型与CCN6(以前的WISP3)的致病变异相关。
鉴别诊断
青少年特发性关节炎和所有累及骨骺和脊椎骨的骨骼发育不良均应在与进行性假性类风湿发育不良(PPD)的鉴别诊断中予以考虑。
青少年 特发性 关节炎 (JIA) 是最常与 PPD 相混淆的疾病.。最主要的区别点:
- JIA有关节炎症(压痛和温热)。
- JIA有ESR和CRP升高,而PPD没有。然而,在某些情况下,这些急性反应物在JIA中的升高也可能很小。
- 在JIA中,x光片上可见关节破坏。在PPD中, x光片上显示发育不良伴骨骺增大和颈阔肌。
X连锁型脊柱骨骺发育不良 (X连锁 SEDT) 发生于青春期或成年期,其特征是不成比例的身材矮小、椎体后隆起的颈阔肌症和不累及周围关节的骨关节炎。 X-连锁 SEDT 是由 TRAPPC2 (SEDL) 基因突变引起的。
在粘多糖病(MPSs) 中,椎体畸形(钩状椎体下喙状或平直椎体中央喙状)和进行性关节限制与PPD相似。MPSs中观察到的骨骼外表现(如粗糙相、角膜混浊、肝肿大、智力残疾)在PPD中不存在。(见 MPS I, MPS II, MPS IV A, MPS IVB, 和 粘多糖病:OMIM 表型系列 来查阅OMIM中与该 表型 相关的基因。)
非典型性 COL2A1 相关脊柱骨骺发育不良可能与PPD有共同特征:
脊柱干骺端发育不良,角部骨折型,也被称为Sutcliffe型,可能和PPD的一些表型变异类似。
疾病管理
初步诊断后的评估
为了确定诊断为进行性假性类风湿发育不良(PPD)患者的疾病程度和需求,建议进行以下评估:
- 完整的骨骼检查,如果还没有完成则作为诊断评估的一部分
- 转诊到儿科骨科医生或治疗骨发育不良的专家
- 咨询临床遗传学家和/或遗传咨询或多学科骨骼发育不良专家团队
对症治疗
支持治疗。目前还没有针对PPD的特定疗法。
非甾体抗炎药可对继发性骨关节炎引起的疼痛起作用。其他抗炎药物包括类固醇和免疫抑制药物(环孢素和甲氨蝶呤)在治疗中作用有限,考虑到其显著的副作用,最好避免使用。
物理治疗可以帮助保持关节的灵活性。应避免长时间活动受限(例如,固定一个姿势)。
大多数关节病变需要骨科医生和/或物理治疗师的干预。
- 下肢角形畸形. 手术矫正的适应症是恢复下肢的正常排列,并减轻步态紊乱、不稳定和/或疼痛。
- 进行性关节僵硬
- 大关节僵硬可以通过物理治疗、活动调整和辅助行走来管理。
- 小型关节病变由专业治疗师处理,他可能会建议使用适应性设备、活动调整和/或专业培训。
- 关节疼痛. 晚期骨关节炎引起的严重关节疼痛可通过关节成形术(即髋关节和膝关节置换术)治疗。早期髋关节置换术(生命的第二个十年)可以成功缓解疼痛和恢复行走能力。
- 脊柱进行性僵硬和畸形
- 脊柱侧凸和轻度后凸可用支具治疗。
- 椎管狭窄可通过减压、融合和内固定治疗。
监测
没有发表具体的监测指南。
建议包括以下内容:
- 监测骨科并发症,包括骨畸形、继发性关节疾病、脊柱畸形和疼痛
- 每年由骨骼发育不良专家或多学科骨骼发育不良诊所作定期评估
应避免的因素/环境
避免长时间活动受限(例如,固定一个姿势) 。
孕期管理
患有PPD的孕妇有骨盆畸形可能需要剖宫产。
还在探索中的治疗方法
搜索美国的 ClinicalTrials.gov 和欧洲的 EU Clinical Trials Register 网站,可获得各种疾病的临床研究信息。注意:这种疾病可能没有临床试验。
遗传咨询
遗传咨询的内容是向个人和家庭提供有关遗传性疾病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们作出基于足够医疗背景知识,以及符合个人情况的决定。下面章节主要讨论遗传风险评估,根据家族史和基因检测来确定家庭成员的遗传状态。这部分的目的并不是为了解决所有患者可能面临的所有个人、文化和伦理问题,也不能替代遗传学专业人员的咨询 。 —ED
遗传模式
进行性假性类风湿发育不良(PPD) 是以 常染色体隐性遗传 模式遗传。.
家庭成员的风险
先证者的父母
- 杂合子是无临床症状的,没有患病风险。
先证者的同胞
- 理论上,每个受累个体的同胞有25%的的机会患病,50%的机会成为一个无症状携带者,25%的机会不患病不携带。
- 杂合子是无症状的,没有患病风险。
携带者 (杂合子) 检测
对高危亲属进行携带者检测需要事先确认检出该家族中 CCN6 致病变异位点。
相关遗传咨询问题
生育规划
- 确定遗传风险,鉴定携带者,讨论产前诊断如何进行的最佳时间是在怀孕之前。
DNA样本库 是用来存储以备将来可能使用的DNA(通常从白细胞中提取)的。因为检测方法和我们对于基因、等位基因变异以及疾病的理解在未来都可能被更新,所以应该考虑储存 受累的 个体的DNA。
产前检测和植入前遗传检测
一旦在一个受累的 家系成员中确认了CCN6 致病变异,可选择对高风险妊娠的胎儿行产前诊断和 植入前遗传学检测 。
医学专业人员之间和家庭成员之间,关于是否采用产前检测可能存在分歧,特别是考虑进行产前检测的目的是终止妊娠而不是早期诊断的情况下。 虽然大多数中心会认为使用产前诊断的决定是父母的选择,但应深入讨论这些问题。
资源
GeneReviews工作人员选择了下列特定疾病组织和支持机构,使患者和其家庭受益。GeneReviews 对其他组织提供的信息不负责。 有关选择标准的信息,请点击这里。
- Arthritis Foundation1330 W. Peachtree StreetSuite 100Atlanta GA 30309Phone: 800-283-7800 (toll-free); 404-872-7100
- National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)1 AMS CircleBethesda MD 20892-3675Phone: 877-226-4267 (toll-free); 301-565-2966 (TTY)Fax: 301-718-6366Email: niamsinfo@mail.nih.gov
- International Skeletal Dysplasia RegistryUCLA615 Charles E. Young DriveSouth Room 410Los Angeles CA 90095-7358Phone: 310-825-8998Fax: 310-206-5266Email: Salon@mednet.ucla.edu
- Skeletal Dysplasia Network, European (ESDN)Institute of Genetic MedicineNewcastle University, International Centre for LifeCentral ParkwayNewcastle upon Tyne NE1 3BZUnited KingdomEmail: info@esdn.org
分子遗传学
下面分子遗传学和 OMIM 列表中的信息可能与 GeneReview 里其他部分的信息不一致:表中可能包含更近期的信息。 — ED
表Table A.
Table A. 进行性假性类风湿发育不良:基因和数据库
| 基因 | 染色体位置 | 蛋白质 | 位置特异性数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| CCN6 | 6q21 | Cellular communication network factor 6 | WISP3 database | CCN6 | CCN6 |
Table B.
Table B. 进行性假性类风湿发育不良的OMIM入口 (在OMIM中查看所有)
基因结构.CCN6 (原 WISP3) 由5个外显子组成。 Ensembl 列出了 由各类 剪接 (GRCh38) 产生的11 种转录本。最常使用和临床相关的参考序列为 NM_003880.3 ,长度为1,235 bp。 基因 和蛋白质信息的详细汇总,见 Table A, 基因。
致病性变异. 据报道,CCN6 有60多种致病性变异。已被描述的有错义变异,缺失,无义 变异,重复 ,和 剪接位点 变异以及像插入缺失等复杂变异。此外,有研究从成纤维细胞 cDNA 中鉴定出两个深度内含子的 错义 变异导致可变 剪接 [Garcia Segarra et al 2012]。
所有种族中最常见的 致病性变异 是 p.Cys52Ter (~28%)。
其他两种致病性变异是只在印度人群中发现的是位于5号 外显子 的 p.Cys337Tyr 和位于2号外显子的 p.Cys78Tyr。
Table 2.
Table 2. 此 GeneReview 中讨论的 CCN6 致病性变异
| DNA 核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
| c.156C>A | p.Cys52Ter | NM_003880 NP_003871 |
| c.233G>A | p.Cys78Tyr | |
| c.1010G>A | p.Cys337Tyr |
表中所列的变异由作者提供。 GeneReviews 工作人员未对变异进行独立分级。
GeneReviews 遵循人类基因组变异协会 (varnomen
- .hgvs.org ) 的标准命名惯例。 命名的解释见 Quick Reference 。
正常 基因产物. CCN6 编码354个氨基酸的Wnt1诱导信号通路蛋白3 (WISP-3),分子量约为40 kd。WISP-3包括五个功能域:信号肽序列,胰岛素样生长因子结合蛋白(IGF-BP)类 结构域,血管性血友病因子C型(VWC)重复域,血小板反应蛋白I型结构域,和半胱氨酸结构域 [Hurvitz et al 1999]。
WISP-3 是生长因子CCN(结缔组织生长因子/富含半胱氨酸/肾母细胞瘤过表达)家族的一员 [Pennica et al 1998]。 WISP-3 通过激活SOX9转录因子调控II型胶原和蛋白聚糖的表达 [Sen et al 2004] ,它通过抑制软骨细胞增殖和促进软骨细胞前体细胞分化,在软骨稳态中发挥重要作用 [Wang et al 2013]。
异常基因产物. CCN6 变异导致PPD的病理生理异常尚未完全了解。然而,研究表明,在PPD患者的软骨细胞中,WISP-3的表达显著降低 [Zhou et al 2007]。极低水平WISP-3表达的突变关节软骨细胞增殖率增加,细胞活力增加,凋亡减少,提示它们处于不成熟和过度增殖状态,这可能解释了PPD患者观察到的干骺增大。突变的WISP-3在软骨细胞的细胞质和细胞膜中显示异常聚集 [Wang et al 2013]。由于突变的WISP-3延迟了细胞内胶原合成并抑制细胞外胶原分泌,PPD患者的软骨柔韧性减弱。
癌症和良性肿瘤. WISP-3的缺失或下调与乳腺癌、结直肠癌和肝细胞癌相关,因为WISP-3的减少会刺激肿瘤的发生。COSMIC(癌症中体细胞突变目录)数据库显示了与癌症相关的组织中45个特异的 CCN6 变异。
参考文献
引用文献
- Bhavani GS, Shah H, Dalal AB, Shukla A, Danda S, Aggarwal S, Phadke SR, Gupta N, Kabra M, Gowrishankar K, Gupta A, Bhat M, Puri RD, Bijarnia-Mahay S, Nampoothiri S, Mohanasundaram KM, Rajeswari S, Kulkarni AM, Kulkarni ML, Ranganath P, Ramadevi AR, Hariharan SV, Girisha KM. Novel and recurrent mutations in WISP3 and an atypical phenotype. Am J Med Genet A. 2015;167A:2481 - 4. [PubMed: 25988854]
- Dalal A, Bhavani G SL, Togarrati PP, Bierhals T, Nandineni MR, Danda S, Danda D, Shah H, Vijayan S, Gowrishankar K, Phadke SR, Bidchol AM, Rao AP, Nampoothiri S, Kutsche K, Girisha KM. Analysis of the WISP3 gene in Indian families with progressive pseudorheumatoid dysplasia. Am J Med Genet A. 2012;158A:2820 - 8. [PubMed: 22987568]
- Delague V, Chouery E, Corbani S, Ghanem I, Aamar S, Fischer J, Levy-Lahad E, Urtizberea JA, Mégarbané A. Molecular study of WISP3 in nine families originating from the Middle-East and presenting with progressive pseudorheumatoid dysplasia: identification of two novel mutations, and description of a founder effect. Am J Med Genet A. 2005;138A:118 - 26. [PubMed: 16152649]
- Garcia Segarra N, Mittaz L, Campos-Xavier AB, Bartels CF, Tuysuz B, Alanay Y, Cimaz R, Cormier-Daire V, Di Rocco M, Duba HC, Elcioglu NH, Forzano F, Hospach T, Kilic E, Kuemmerle-Deschner JB, Mortier G, Mrusek S, Nampoothiri S, Obersztyn E, Pauli RM, Selicorni A, Tenconi R, Unger S, Utine GE, Wright M, Zabel B, Warman ML, Superti-Furga A, Bonafé L. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet C Semin Med Genet. 2012;160C:217 - 29. [PubMed: 22791401]
- Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J, Holderbaum D, Pauli RM, Herd JK, Van Hul EV, Rezai-Delui H, Legius E, Le Merrer M, Al-Alami J, Bahabri SA, Warman ML. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet. 1999;23:94 - 8. [PubMed: 10471507]
- Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, Brush J, Taneyhill LA, Deuel B, Lew M, Watanabe C, Cohen RL, Melhem MF, Finley GG, Quirke P, Goddard AD, Hillan KJ, Gurney AL, Botstein D, Levine AJ. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci U S A. 1998;95:14717 - 22. [PMC free article: PMC24515] [PubMed: 9843955]
- Sen M, Cheng YH, Goldring MB, Lotz MK, Carson DA. WISP3-dependent regulation of type II collagen and aggrecan production in chondrocytes. Arthritis Rheum. 2004;50:488 - 97. [PubMed: 14872491]
- Wang M, Man XF, Liu YQ, Liao EY, Shen ZF, Luo XH, Guo LJ, Wu XP, Zhou HD. Dysfunction of collagen synthesis and secretion in chondrocytes induced by wisp3 mutation. Int J Endocrinol. 2013;2013:679763 [PMC free article: PMC3614060] [PubMed: 23573089]
- Wynne-Davies R, Hall C, Ansell BM. Spondylo-epiphysial dysplasia tarda with progressive arthropathy. A "new" disorder of autosomal recessive inheritance. J Bone Joint Surg Br. 1982;64:442 - 5. [PubMed: 6807993]
- Zhou HD, Bu YH, Peng YQ, Xie H, Wang M, Yuan LQ, Jiang Y, Li D, Wei QY, He YL, Xiao T, Ni JD, Liao EY. Cellular and molecular responses in progressive pseudorheumatoid dysplasia articular cartilage associated with compound heterozygous WISP3 gene mutation. J Mol Med (Berl). 2007;85:985 - 96. [PubMed: 17483925]
章节注
致谢
- 印度医学研究委员会-遗传性关节病和椎体多节段缺损的临床和分子评估 (BMS 54/2/2013)
- 科技部 – 应用自合子定位和 外显子组测序 鉴定骨骼发育障碍的遗传基础 (SB/SO/HS/005/2014)
修订历史
- 2015年11月25日(me)综述发布
- 2015年6月30日(kmg) 初始提交