
概述
临床特征.
常染色体隐性多囊肾病(ARPKD)属于 先天的 肝肾纤维囊性综合征的一种,是导致儿童肝肾相关疾病及死亡的重要原因。在新生儿期,大多数ARPKD患者的肾脏出现明显扩大且回声增强。肾脏疾病的特征是肾肥大、高血压和不同程度的肾功能不全。超过50%的ARPKD 受累的 患者在出生后10年内进展为终末期肾病(ESRD);ESRD患者可能需要肾移植。
部分 受累的 新生儿因妊娠期羊水过少引发肺发育不全。约有30%在新生儿期或出生后一年内死于呼吸功能不全或者肺部感染并发症。通过新生儿呼吸支持和肾替代治疗,这些婴儿的长期生存率提高到80%以上。
随着肾替代治疗和肾移植治疗的发展,ARPKD患者的长期生存率有所提高,临床上肝胆疾病可能成为ARPKD自然历史的主要特征。此外,小部分患有此类疾病的人被诊断为肝脾肿大;肾脏疾病症状通常较轻微,可能在腹部影像学检查中被偶然发现。
尽管婴儿在出生时总是会出现组织学上的肝脏纤维化,但约有50%的婴儿在诊断时有肝脏损伤的临床证据。这可导致进行性门静脉高压,并导致食管/胃底静脉曲张、痔疮增大、脾肿大、脾功能亢进、蛋白丢失性肠病和胃肠道出血。其他肝脏检查还发现患者体内存在肝内胆管囊状扩张(Caroli综合征)和胆总管扩张,由于胆管扩张和胆汁流动停滞,这将可能导致复发性或持续性细菌性上行胆管炎。越来越多存活下来的 受累的 新生儿最终需进行门体静脉分流或肝移植,来治疗门静脉高压症或胆管炎的并发症。
尽管ARPKD是典型的常发生于新生儿的疾病,但患者年龄和呈现的临床症状有显著的变异性,这与肾脏和胆汁异常的相对程度有关。
诊断/检测.
ARPKD的诊断怀疑是基于先证者的临床表现及先证者的亲生父母没有肾脏疾病。最近的一项研究表明,DZIP1L可能是第二个与ARPKD相关的基因;ARPKD的诊断怀疑是基于 先证者 的临床表现及先证者的亲生父母没有肾脏疾病。通过对 受累的 患者的PKHD1 双等位基因的 致病变异进行鉴定,确定诊断结果。最近的一项研究表明,DZIP1L可能是第二个与ARPKD相关的 基因 ;还需要更多的证据来证明这一点。
管理.
临床症状的治疗: 如果肾脏严重扩大限制了橫膈运动,则需要通过机械通气和(很少)单侧或双侧肾切除术来恢复受累的 新生儿的呼吸功能。少尿或无尿的新生儿在出生后的最初几天可能需要进行腹膜透析,因此对脱水和高血压的早期识别和治疗至关重要。患有严重慢性肾脏疾病的患儿应采用现代儿科ESRD治疗的所有模式进行治疗。胆道功能障碍的治疗重点在于(1)营养素和脂溶性维生素吸收不良,以及(2)上行胆管炎的风险,包括使用合成胆汁酸以及对上行胆管炎的早期识别和治疗。对进行性门静脉高压症的患者,可能需要进行内窥镜联合硬化治疗或静脉曲张套扎,可能需要进行门体静脉分流和/或考虑肝移植。ESRD和严重门静脉高压症的患者可能需要双肾/肝移植。
预防继发性并发症: 熊去氧胆酸治疗可能会增加胆汁酸的含量和/或减少胆结石的形成。建议对于患有严重门静脉高压和脾功能障碍的患者进行免疫接种,以对抗包裹菌。对于患有慢性肺病和/或早产的24个月以下的儿童,推荐使用帕利珠单抗 (Synagis®)。
监测: 定期监测血压、肾功能、血清电解质浓度、水合状况、营养状况和生长情况。通过体格检查评估肝脾大小以监测肝胆功能不全导致的门静脉脉高压。除血清白蛋白水平、PT / PTT、25-OH维生素D、维生素E水平和脂溶性维生素水平外,还应定期进行血小板计数。定期进行超声检查,如果出现肝肿大和/或脾肿大,请转诊给肝病专家;定期行食管胃十二指肠镜检查(EGD)排查食管静脉曲张。推荐在开始时进行MR胆管造影,这是一种敏感的胆道扩张测量方式。
要避免的情况和药物:用于高血压的拟交感神经药物;肾毒性药物(非甾体抗炎药和氨基糖苷)。潜在的肝毒性药物(例如,剂量>30 mg/kg/天的对乙酰氨基酚,中草药和酒精)应尽量减少。临床数据表明,除非临床需要,应避免使用咖啡因、茶碱类药物和钙通道阻滞剂。
亲属风险评估: 如果该家族中的致病变异未知,可通过高分辨率肾脏、肝脏超声检查和系统血压监测排查先证者同胞是否患病.
遗传咨询.
ARPKD以 常染色体隐性遗传 方式遗传。先证者 的每个同胞都有25%的机率会遗传致病性变异从而受累的,有50%的机率遗传致病性变异 并成为 携带者,有25%的机率既没有遗传致病性变异也不成为携带者。如果在家庭中发现了两种致病变异,则可以对高危亲属进行携带者检测,推荐对高危妊娠者进行产前检测。在25%ARPKD确诊风险的孕妇中,尚无系统的数据可用于确定产前超声检查的 敏感性和特异性。
诊断
提示性发现
双侧肾脏增大且回声增强的患者可怀疑是常染色体隐性遗传多囊肾病(ARPKD)。其诊断通常是基于临床表现和影像学检查 [Sweeney & Avner 2011, Telega et al 2013, Hartung & Guay-Woodford 2014, Sweeney & Avner 2014, Hoyer 2015, Sweeney et al 2016]。ARPKD的特异性诊断标准 (参考 Zerres et al [1996]):
- 肾脏影像学的经典发现 :包括
- 以下一项或多项::
- 影像学检查显示胆管扩张 (见 超声检查)
- 肝胆管病理学表现为特征性的发育性胆道管板异常并导致CHF(见 儿童期和青年期)
- 高分辨率 (HRUS)超声检查显示父母双侧无肾增大和/或特征性影像学表现
影像学经典发现
超声检查(US) i是评估胎儿和儿童ARPKD的首选诊断方法,因为它经济有效、无痛、运用广泛,而且不需要射线或镇静。主要用于诊断肾脏是否异常,但对于ARPKD患者,腹部超声也可能显示胆管受累或脾肿大。然而,只凭肾脏US是无法确诊的。(见 Polycystic Kidney Disease, Autosomal Dominant).
超声检查发现ARPKD的肾脏诊断标准为:
- 回声增强;
- 皮髓质分界不清。
产前:
- 在严重的ARPKD病例中,超声检查可能显示肾脏明显增大伴强回声,羊水过少或胎儿膀胱内无尿。
- 产前超声检查发现大肾型回声肾伴皮质髓质分化不良及羊水过少,提示ARPKD,但也可能是其他疾病的诊断。
新生儿:
- 双侧可触及的包块但无特征明显慢性肺疾病的新生儿,有羊水过少史或新生儿自发性气胸、高血压高度提示ARPKD,但并无诊断意义。.
- 如上所述的胆道检查结果,以及门脉高压的表征,例如肝脾肿大,使得诊断ARPKD的可能性更高。
儿童期和青年期:
- 肾影像学的发现如上所述,随着纤维化的进展,肾的大小实际上可能会随着年龄的增长而减小。
- 进行性门静脉高压常以肝胆异常为主要表现。
微小的囊性肾脏病变可能在早期出现,随后发展为肉眼可见的囊肿。研究表明,HRUS可以显著提高对轻微疾病的诊断,并提供无创的、详细的肾脏表现定义,而无需大量使用电离辐射或造影剂 [Turkbey et al 2009, Gunay-Aygun et al 2010b]。在ARPKD的诊断中,磁共振成像与HRUS或基因检测相比并无优势。
磁共振胰胆管造影(MRCP). MRCP的胆总管扩张影像学检查可以清楚地描述胆总管系统。 MRCP是胆道解剖学的一个敏感指标。结合肾脏的影像学表现,胆道异常可以作为ARPKD的诊断标准,并在很大程度上取代了通过肝脏活检取得导管解剖这种更具侵入性的分析方法 [Turkbey et al 2009, Gunay-Aygun et al 2010a, Gunay-Aygun et al 2013]。
注意: 肾活检不用于诊断ARPKD。
病理 (活检或尸检)
发育性导管板异常的组织学表现,包括胆管增生、胆道扩张和门脉周围纤维化,存在于所有ARPKD患者中 [Kamath & Piccoli 2003]。
- ARPKD的肝胆疾病是发育缺陷的结果,其中导管板重塑失败导致胚胎胆管结构的持久性异常最终将导致胆管扩张。
- 扩张的胆管可能演变成肉眼可见的囊肿,这些囊肿与肝内胆管相连,可通过影像学检查,特别是MRCP检查发现。
- 相关的门静脉通常异常,表现为扩张,门静脉分支较小。
- 甚至在出生时,在门脉中也可以看到大量的纤维化,并且随着受累的孩子的年龄增长,门静脉周围纤维化的数量增加,导致肝肿大和进行性门脉高压。
有趣的是,由于不清楚的原因,ARPKD受累的肝脏的左叶往往比右叶 [Gunay-Aygun et al 2013]。
建立诊断
ARPKD的诊断是通过先证者出现先天性肾囊肿、先天肝纤维化和分子遗传学检测中发现的PKHD1中 双等位基因的的致病变异来确定的(见 表1)。
最近,由于在已知的PKD相关基因中没有可识别的致病性变异,在4个不相关的具有ARPKD特征的 近亲婚配的家系中发现7个受累的个体的DZIP1L中具有双等位基因的致病突变,这使得作者认为DZIP1L是ARPKD的第二个基因位点[Lu et al 2017]。但是,DZIP1L突变小鼠的罕见发生和异常表型表明,需要更多证据证明DZIP1L是ARPKD的第二个基因位点。
分子遗传学检测方法可以包括单基因 检测,多基因组panel以及更全面的基因组检测。
- 也可以考虑包括PKHD1,DZIP1L和其他感兴趣的基因的多基因组 (参考鉴别诊断)。注意:(1) 每组中包含的基因以及每个基因检测的诊断敏感性因实验室而异,很可能会随着时间而改变。(2) 一些多基因组可能包含与本GeneReview中讨论的病症无关的基因;因此,临床医生需要确定哪个多基因组能够以最合理的成本来最好的鉴定疾病的遗传方式。(3)专家组中使用的方法可能包括序列分析,缺失/重复分析和/或其他非基于序列的测试。
- 如果序列单基因检测(和/或使用包括PKHD1和DZIP1L在内的多基因组 panel)未能确认ARPKD特征个体的诊断,可以考虑进行更全面的基因组检测(如果有的话),包括外显子组测序和基因组测序。这种检测可以提供或提示以前没有考虑到的诊断(例如,不同基因的突变或导致相似临床表现的基因)。
- 有关全面基因组测试的介绍,请单击这里。有关临床医生订购基因组检测的更多详细信息,请点击这里。
Table 1.
ARPKD的分子遗传学检测 没有有关基因靶向缺失/重复分析检测率的数据
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
在四个家庭七个受累的 个体中鉴定出纯合的致病变异[Lu et al 2017]。
- 8.
临床特征
临床概述
常染色体隐性多囊肾病(ARPKD)属于一组先天的肝、肾纤维囊性综合征,是儿童肾、肝相关疾病和死亡率较高的原因之一。主要受累的两个器官系统是肾和肝;在其他一些器官系统中也可以看到次级效应。
表现. 大多数受累的个体表现在新生儿期。借助现代产科超声检查,若在产前超声检查发现异常,可能会怀疑诊断。在产前超声检查中发现的受严重影响的胎儿表现出“Potter”羊水过少 表征,并伴有致命的肺发育不全和大量回声的肾脏时可危及正常分娩。
由于广泛的临床可变性,ARPKD的诊断可以在儿童的任何阶段进行,在极少数情况下可直到青春期或成年期才出现相关临床症状[Gunay-Aygun et al 2010b]。少数受累的的个体以在大龄的儿童或年轻人中出现,ARPKD明显的表现特征是肝功能障碍是(见 肝脏)。
肾脏. 体格检查时,常出现双侧大肿块(肾肥大症)。
- 排尿量通常不会减少;多尿和多饮的临床表现与肾脏浓缩缺陷一致。但是,在出生的第一周可能会出现尿少和明显的急性肾衰竭。
- 低钠血症通常发生在新生儿期,若不存在肾功能衰竭,通常可以治愈。
- 肾功能(如血肌酐和血尿素氮[BUN]的浓度所反映)经常受损。随着生命的最初三年正常肾脏的发育进展,肾脏功能随时间的推移出现明显改善。
- 高血压通常在生命的最初几周内被注意到,但随着年龄的增长可能会改善。
终末期肾脏疾病 (ESRD). M通常在生命的头十年,超过50%的(见 管理)个体会发展为ESRD [Hoyer 2015, Sweeney et al 2016]。ESRD可能需要肾脏移植(见 管理)。
- 高分辨率超声检查显示的围产期表现和皮质髓质受累与肾脏疾病进展更快有关 [Gunay-Aygun et al 2010b].
肝脏. 随着肾脏替代疗法和肾脏移植技术的进步提高了患者的长期生存率,临床肝胆疾病可能会成为ARPKD自然病史的主要特征 [Sweeney & Avner 2011, Sweeney & Avner 2014, Sweeney et al 2016].
先天性肝纤维化 (CHF). PKHD1致病变异(又称CHF)引起的ARPKD的非变异肝损害是一种胆道导管板重构的发育异常。由致病性DZIP1L变异引起的ARPKD患者中CHF的患病率尚未充分确定;到目前为止,在发现的7个DZIP1L致病变异个体中,只有一个个体进行了这一发现的检测,并发现轻度 [Lu et al 2017]。
- 高达70%受累的 个体(包括具有典型症状的长期幸存者和肝胆疾病为主的患者)由于进行性门静脉周围纤维化而发展为门脉高压;食管静脉曲张的出血是导致该疾病死亡的重要因素[Gunay-Aygun et al 2013, Sweeney & Avner 2014, Sweeney et al 2016]。
- Although his尽管出生时总是存在组织学上的肝纤维化,但新生儿可能没有肝病的临床表现、影像学或实验室证据[Shneider & Magid 2005]。Zerres et al [1996]发现在115位平均年龄为29天的ARPKD患儿中,仅45%的患者有确切临床证据显示他们肝脏受累。
Caroli 综合征. 除CHF之外,超过60%的ARPKD患者出现肝内胆管无阻塞性扩张(Caroli综合征)和胆总管扩张。
- 肝胆引流异常是复发性或持续细菌性升胆管炎合并脓毒症的重要危险因素。
- 胆汁淤积还可能导致脂溶性维生素(A,D,E和K)的吸收障碍。
肝脾肿大. 一组ARPKD患者被诊断为肝脾肿大[Roy et al 1997];肾脏疾病通常是轻症的,可能偶然发现于腹部影像学检查中。
- 在美国国立卫生研究院自然史研究的78名受累的 个体中证实ARPKD的确诊年龄的存在巨大差异,其中受影响个体的年龄范围为1至56岁[Gunay-Aygun et al 2010a, Gunay-Aygun et al 2010b],表明ARPKD / CHF的临床范围比以前假设的要广得多。i
肺. 羊水过少引起的肺不同程度的发育不全在许多受累的婴儿中出现,这是新生儿期ARPKD发病和死亡的主要原因。由于膈肌偏移受限,肾脏大量肿大,也可能导致换气不足和呼吸窘迫。
- 与其他疾病合并羊水过少的新生儿相比,在妊娠晚期少量羊水过少或低羊水过多的新生儿可能出现相对较轻的肺部疾病[Sweeney & Avner 2011]。其原因尚不清楚,作者推测宫内肾脏分泌过多的对肺发育至关重要的生长因子(包括表皮生长因子的成员),可能对肺发育有着尚未清楚的积极影响。
生理缺陷.可能存在与羊水过少相关的面部特征,包括耳朵位置低、小颌、鼻子扁平、肢体定位缺陷和生长缺陷。.
其他
- 最近的数据表明,在积极的营养支持下,相当一部分儿童的生长是正常的[Sweeney & Avner 2011, Sweeney et al 2016]。对于即使有明显肾功能损害和门脉高压的儿童,在生命的头两年积极的营养支持也显著提高了其生长速度[Telega et al 2013]。
- 进食困难可由增大的肾脏、肝脏或脾脏对胃的机械压迫引起,后者是门脉高压的并发症。另外,严重的肾脏损害可能导致进食困难、食欲不振和/或胃动力受损。
- 据报道,在两名成人和一名儿童ARPKD中,脑动脉瘤是常染色体显性多囊疾病(ADPKD)潜在的严重并发症[Chalhoub et al 2013]。尽管ADPKD中动脉瘤的发病率和ADPKD和ARPKD蛋白之间的相互作用增加,但迄今没有证据表明动脉瘤是ARPKD的肾外表现[Sweeney & Gunay-Aygun et al 2016]。
死亡率. 尽管ARPKD的短期和长期死亡率非常高,但通过现代新生儿呼吸支持和肾脏替代疗法,ARPKD患儿的生存率显著提高。
- 在存活超过1年的患者中(使用透析、肾脏和/或肝移植),1年生存率约为85%-87% [Guay-Woodford & Desmond 2003, Bergmann et al 2005],10年生存率为82% [Hoyer 2015].
- 尽管生存率有所提高,但由于以下原因,这种双器官疾病的发病率非常高:
- 肾集合管扩张囊肿和明显的肾增大,导致全身高血压和进行性肾功能衰竭
肾脏和肝脏移植. For对于接受肾移植的ARPKD患者,其异体移植物存活率与未接受ARPKD的患者相当[Telega et al 2013]。据估计,大约10%受累的儿童在新生儿期存活下来,需要进行肝移植[Wen 2011]。
在肾移植后死亡的患者中,64%-80%的死亡直接归因于肝胆疾病引起的胆管炎/脓毒症 [Telega et al 2013]。
基因型-表型相关性
迄今为止,PKHD1和DZIP1L的基因型-表型 相关性 尚未建立。大多数PKHD1致病变异是 "私有的" 或 只存在于单个家庭 [Bergmann et al 2004].
在一项对73名不同年龄的PKHD1致病变异引起的ARPKD患者的研究中,其结果表明PKHD1变异类型与肾脏大小和功能无关[Gunay-Aygun et al 2010a]. Frank et al [2014] 确定了4名受累的的患者,尽管他们各自携带纯合性 PKHD1 致病性变异 ,但都存活了下来,这些变异预计会导致翻译提前终止(通常与生命不相容)。
基因中其他非编码区域的修饰基因、 表观遗传 变化和变异被认为是导致家族间广泛的临床变异的原因 [Sweeney & Avner 2014, Sweeney et al 2016]。
外显率
对于PKHD1致病变异体,ARPKD的外显率完全的;疾病严重程度存在显著的家族内变异 [Sweeney & Avner 2011, Sweeney & Avner 2014, Sweeney et al 2016]。到目前为止,与ARPKD相关的肾脏异常的外显率在DZIP1L致病变异的报告中是完整的(增大回声性肾脏,皮质髓质分化不良,系统性高血压,和不同程度的肾功能不全)。先天性肝纤维化是在与PKHD1相关的ARPKD中发现的恒定存在的,在患有DZIP1L病原体变异的单个受累的 个体中较轻 [Lu et al 2017]。
命名
ARPKD也被称为“婴儿多囊肾病” 。在 Blyth & Ockenden [1971] 对儿童多囊性肾脏疾病的最初描述中,依据肾脏和肝脏的临床和组织学发现将儿童PKD分为围产期、新生儿期、婴儿期和青少年期,提示了四种不同的疾病或“疾病分期” 。随后,依据多个 受累的患者同胞的家庭(见, 例如,Kaplan et al [1988], Guay-Woodford & Desmond [2003])提供的证据表明,这些区别是没有意义的。
最近的趋势是将这种情况称为ARPKD / CHF。至少一个患者倡导组织,ARPKD / CHF联盟,采用了此术语 (见 Resources)。
患病率
ARPKD的发生率约为1:10,000至1:40,000。由于未能正确诊断从新生儿到年轻人的所有年龄段的人,ARPKD的实际发生率可能被低估了 [Adeva et al 2006, Gunay-Aygun et al 2010a]。由DZIP1L致病性变异引起的ARPKD病例数非常少,以致ARPKD的患病率不会受到与DZIP1L相关的任何其他病例的影响。
在一般人群中,PKHD1 致病性变异的携带者携带频率大约为1:70 [Zerres et al 1998]。
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,目前还未知其他表型与PKHD1或DZIP1L的致病变异相关。
鉴别诊断
肾脏表现
囊性肾病的疾病包括:
- 常染色体显性遗传性多囊肾病(ADPKD)的特征是进行性囊肿发育和双侧多囊肾增大。ADPKD是一种全身性疾病,伴有其他器官(例如肝脏,精囊,胰腺和蛛网膜)囊肿和非囊性异常(例如,颅内动脉瘤和结节扩张,主动脉根部扩张和胸主动脉夹层,二尖瓣脱垂,结肠憩室和腹壁疝)。约85%ADPKD患者的病因是PKD1的致病性变异;约15%ADPKD患者的病因为PKD2。尽管ADPKD大多发生在成年期,但1%-2% 受累的 的患者发生在新生儿期,其症状和体征通常与ARPKD没有区别[Guay-Woodford et al 1998, Sweeney & Avner 2011, Sweeney & Avner 2014, Sweeney et al 2016]。肾脏超声检查可以区分两者:双侧大囊肿是ADPKD的典型特征。在ADPKD的早期,尤其是年幼儿童,可能是侵犯单侧肾脏。随着ADPKD的进展,累及成双侧;囊肿会变得巨大。由于ADPKD可能要到生命的第三或第四十年才出现,因此无症状的父母可能直到受累的儿童出生后才被确定为[Fick et al 1993]。需要对任何非典型ARPKD或怀疑ADPKD的患者的父母进行肾脏超声检查,以评估是否可能存在先前未诊断的ADPKD。值得注意的是:
- Pei et al [2009]观察到,在一小部分人(即有PKD2致病变异的人)中,40岁之前可能无法排除ADPKD的诊断;
- 肾小球囊性肾病(GCKD)是一种通常在新生儿期出现并伴有腹部肿块的疾病,临床上可能与ARPKD难以区分。肾脏超声检查的结果与ARPKD相似:弥漫性增大回声的肾脏及偶有大囊肿。组织学检查示鲍曼氏囊扩张和不典型增生,髓质分化异常。10%的患者累及肝内胆管,类似于ARPKD的胆道管板异常。GCKD可以是ADPKD的一个亚型;然而,在至少一个大的亲缘中,两个ADPKD位点的连锁被排除在外[Sharp et al 1997]。GCKD也作为遗传疾病的一部分发生,包括tuberous sclerosis complex, orofacial digital syndrome type 1,13型三体,近中肾综合征和短肋-多指综合征。
- 肾囊肿与糖尿病综合征 (RCAD) (OMIM 137920)。 基因里编码转录因子肝细胞核因子1(HNF1β) 的致病变异是发展性肾脏疾病最常见的已知单遗传病因 [Clissold et al 2015, Verhave et al 2016]。这种疾病是导致胎儿肾脏回声增大或正常大小的主要原因,常导致ARPKD的误诊。尽管其他肾脏表型(单肾、肾发育不全/发育异常)也可能发生,肾囊肿是HNF1β肾组织相关肾病最常见的特征表型。肾外表型很常见,HNF1β高表达基因首次被确定为糖尿病的疾病基因(MODY5)。报道的其他表型(用于区分该遗传疾病与ARPKD)包括生殖器畸形、自闭症、癫痫、痛风、低镁血症、甲状腺功能亢进、肝脏和肠道异常,以及一种罕见的肾癌[Bockenhauer & Jaureguiberry 2016, Limwongse 2016]。
- 弥漫性囊性发育异常的特征是超声表现为大回声的肾脏,组织学表现为组织紊乱、分化差的伴有软骨等原始成分的肾元节段 [Watkins et al 1999]。弥散性囊性发育异常可偶尔出现,或作为众多综合征的组成部分常出现 (如, Joubert syndrome, Meckel-Gruber 综合征, Jeune 窒息性胸营养不良) [Limwongse et al 1999, Limwongse 2016]。在这些综合征中,临床上以肾外或肝外异常为主;弥漫性囊性发育异常仍是较次要的特征。
- 肾脏囊肿是许多疾病的病理表现。与ARPKD不同的是,肾脏囊肿只是该综合征发育异常的其中一个表现 [Limwongse 2016]。新生儿糖尿病, 先天的甲状腺功能减退,肝纤维化,PKD,先天性青光眼的综合征在两例中有提及。肝活检证实了CHF的典型发现;未对肾脏进行组织学评估(OMIM 601331)。
其他累及肾脏的疾病 在新生儿期,与ARPKD相似的疾病包括恶性肿瘤,如白血病或Wilms肿瘤 (见 Wilms Tumor Overview),双侧肾静脉血栓形成和造影剂肾病 [Guay-Woodford et al 1998, Sweeney & Avner 2011, Sweeney et al 2016]。
肝脏表现Liver Manifestations
其他以肾囊性变和肝纤维化为特征的肝肾疾病包括已经提到的一些疾病:青少年肾炎和多系统疾病如Meckel-Gruber综合征, Bardet-Biedl syndrome, Joubert syndrome, 和Jeune窒息性胸营养不良 [Johnson et al 2003]。与ARPKD的肿大肾脏相比,这些常染色体隐性遗传 疾病的肾脏通常较小或正常大小。
管理
初步诊断后评估Evaluations Following Initial Diagnosis
为了确定诊断为常染色体隐性遗传 多囊肾病(ARPKD)的患者的疾病程度和需求,建议进行以下评估:
- 呼吸状况,包括体格检查、脉搏血氧饱和度和胸片(如有需要)
- 检测肾功能,包括血清尿素氮(BUN)、肌酐和胱抑素C的浓度,从而更准确地估计肾小球滤过率(GFR)[Gunay-Aygun et al 2010a]
- 检测电解质异常(如低钠血症、高钾血症)的血清电解质浓度
- 尿液分析,以评估尿浓度和蛋白尿。临床评估血管内容量状态,检查可能的容量不足或超载。注:小儿ARPKD尿液中常见白细胞,不代表感染。如果临床怀疑有尿路感染,应在开始治疗前进行尿培养。
- 肾脏超声检查(可用时考虑高分辨率技术)。
- 测量血压。如果血压升高,家庭血压监测有助于区分固定高血压和“白大褂”高血压(即在体检中出现的高血压)。
- 评估喂养、体重增加和线性增长,并提供适当的正式营养咨询
- 测量肝转氨酶、血清胆汁酸、肝脏合成功能(例如,通过评估血清白蛋白浓度、25-OH维生素D的水平,维生素E水平和凝血功能), 脂溶性维生素水平,完成全血细胞计数、肝肿大/脾肿大的体格检查,腹部超声评估肝脏受累的程度
- 咨询临床遗传学家和/或遗传顾问
对症治疗
(详细的管理策略见近期综述,包括Sweeney & Avner [2011], Telega et al [2013], Guay-Woodford et al [2014], Sweeney & Avner [2014], 和 Sweeney et al [2016] 。)
对 受累的 婴儿的初步治疗以稳定呼吸功能为中心。
呼吸
- 机械通气对于治疗肺发育不全(以喷气或振荡式100%氧气通气仍不能充氧为特征)或肾体积增大导致的低通气(以氧合充足但二氧化碳分压升高为特征)可能是必要的。也可能需要在产后48-72小时内确定是否存在肺发育不全或可逆性肺部疾病。
- 当增大的肾脏阻止了膈肌偏移和/或引起严重的进食不耐受时,一些人主张单侧或双侧肾切除术 [Shukla et al 2004]。
- 经验表明,单侧肾切除术的价值可能有限,因为在单侧肾切除术后,对侧肾常显示明显的增大[作者,未发表的观察]。
- 双侧肾切除术放置腹膜透析导管,然后进行短时间的血液透析,通常可使腹膜愈合,为慢性腹膜透析做准备[Sweeney & Avner 2011, Sweeney et al 2016]。这些程序的时机,以及与先行活体供体移植的潜在协调将取决于患者的年龄、大小、临床状况以及活体供体的可用性等因素。
肾脏
- 有少尿或无尿的新生儿在出生后的头几天可能需要腹膜透析。
- 低钠血症很常见,应根据个体的血容量状况进行治疗。
- 脱水的早期识别和治疗至关重要。可能需要通过鼻胃管或胃造瘘管进行补充喂养或液体治疗。
- 高血压通常对血管紧张素转换酶(ACE)抑制剂或血管紧张素受体抑制剂(ARBs)反应良好,这是治疗的首选。在许多情况下,高血压可能严重到需要多种抗高血压药物联合治疗。
- 患有严重慢性肾病的儿童应采用现代儿科终末期肾病(ESRD)治疗的所有模式,包括透析和肾移植。
- 慢性肾病III期或更高期儿童的贫血可能需要补充铁和促红细胞生成素(ESAs)的治疗。
肝脏。胆管功能障碍的治疗重点是:(1)营养物质和脂溶性维生素的吸收不良;(2)上行性胆管炎的风险。
- 使用合成胆汁酸:
血清脂溶性维生素(K, D, E, A)水平低或体重增加不佳,尽管有足够的热量补充,可能表明需要补充胆汁酸。
- 临床怀疑胆汁酸缺乏可通过测定血清胆汁酸来验证。
如果有明显的肝内导管扩张(Caroli综合征)的证据,可以通过磁共振胰胆管造影(MRCP)鉴别,则表明可以使用合成胆汁酸。
- 细菌性胆管炎通常是肝脏受累者的未被诊断的并发症,可表现为反复出现的菌血症和肠道病原体,没有胆管炎的典型临床特征。持续性发热,特别是右上腹疼痛,应进行评估和积极治疗。
- 食管静脉曲张应根据需要采用内窥镜扎带或硬化治疗。
双器官移植。已有小病例系列报道了对ARPKD患者成功进行肝肾移植[Harps et al 2011, Chapal et al 2012, Brinkert et al 2013, Telega et al 2013].
在此之前,只有一小部分ARPKD患者,特别是那些后来确诊的患者需要肝移植。然而,随着生存率的提高和肾替代治疗的进展,需要肝移植的ARPKD患者的数量可能会增加。
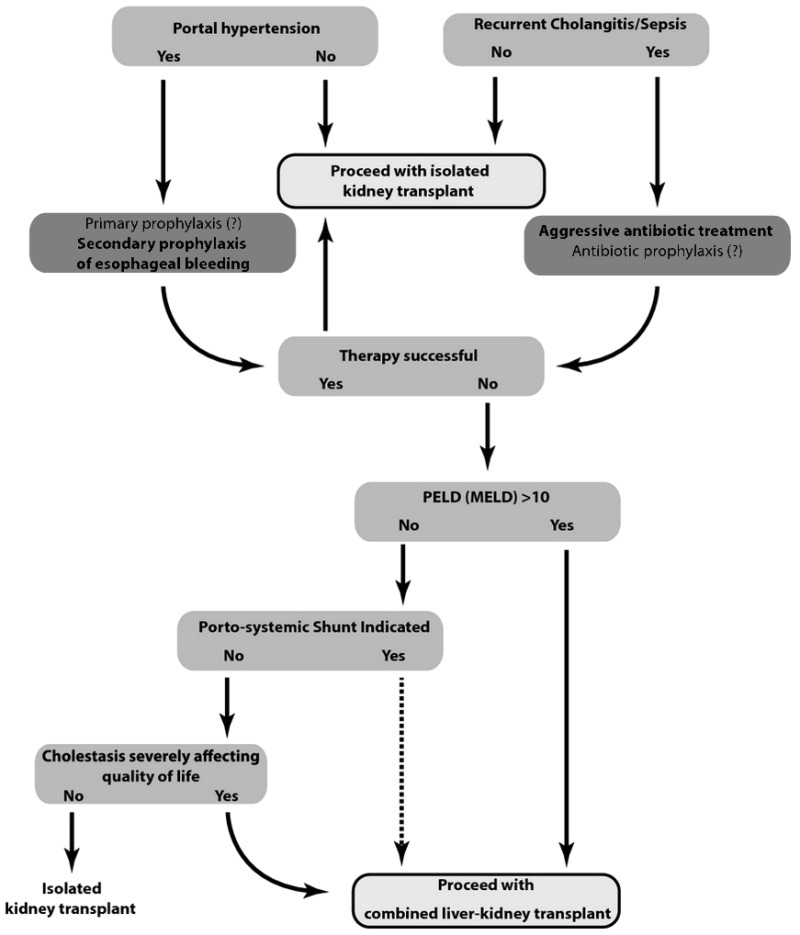
已经提出了一种评估患有严重肝肾疾病的ARPKD患者行双器官移植风险收益的算法,以帮助临床医生进行决策 [Telega et al 2013]。见 Table 2 (pdf) 和 Figure 1.
喂养和成长
- 即使在没有肾功能不全的情况下,喂养不耐受和生长衰竭也可能是很严重的,尤其是在年幼的婴儿中。为了优化体重的增加成长,通常需要积极的营养支持,包括通过鼻胃管或胃造瘘管进行补充喂食 [Dell et al 2009, Sweeney & Avner 2011, Hartung & Guay-Woodford 2014, Sweeney & Avner 2014].
- 生长衰竭的儿童可能会受益于生长激素的治疗[Lilova et al 2003]。开始生长激素治疗的最佳年龄取决于儿童的生长速度;研究表明,治疗两岁或两岁以下的慢性肾病儿童是有益的[Seikaly et al 2007, Schaeffer 2016]。
预防继发性并发症
熊去氧胆酸(熊二醇或Actigall®)是一种胆汁酸,可用于增加胆汁的数量和流量,以及/或减少患有严重肝胆疾病的ARPKD患者胆结石的发生。
严重门静脉高压及脾功能障碍患者,需要免疫接种包裹菌(肺炎球菌;乙型H型流感;脑膜炎球菌)。
最新指南建议,年龄小于24个月、患有慢性肺病和/或有早产史的高危儿童应服用帕利珠单抗(Synagis®) [Committee on Infectious Diseases 2009].
尽管慢性抗生素预防对所有ARPKD患儿的作用仍存在争议,但建议对有上行性胆管炎高风险的ARPKD患者,包括那些过去有过上行性胆管炎发作的患者,使用抗生素预防。
监测
根据病程和并发症定期监测以下情况:
- 在医生定期来访时监测血压,如有必要,也需进行家庭血压监测 (见Evaluations Following Initial Diagnosis)
- 电解质平衡,通过获得血清中钠、钾和氯的浓度来监测。矿物质平衡,通过评估钙和磷来监测。如果钙水平较低,则应评估镁浓度 [Dell et al 2009, Sweeney & Avner 2011, Sweeney & Avner 2014].
- 水合状态
- 营养状况,生长情况显示在标准生长图表和营养咨询中
- 肝门静脉管受累,除血清白蛋白水平、PT/PTT、25-OH维生素D、维生素E水平和脂溶性维生素水平外,还需进行体格检查和血小板计数
- 如果存在肝肿大和/或脾肿大,则应进行额外的监测,包括定期的超声检查或MRI。对于肝脾肿大,建议转诊,请儿科肝病专家进行评估,并通过食管胃十二指肠镜(EGD)进行定期监测以发现食管静脉曲张 [Telega et al 2013].
- 考虑MR胆管造影术,这是一种对胆管扩张更为敏感的测量方法,在基线时,按照指示进行测量 [Shneider & Magid 2005, Telega et al 2013, Sweeney et al 2016]
避免的事宜
应避免下列情况:
- 对高血压 受累的 患者,拟交感神经药物
一般来说,除非临床情况允许使用,已知的肾毒性药物包括非甾体抗炎药(NSAIDs)和氨基糖苷类药物
潜在的肝毒性药物(例如醋氨酚30毫克/公斤/天、草药补充剂和酒精)应尽量减少使用。
细胞和动物模型的研究表明,咖啡因、茶碱类药物和钙通道阻滞剂可能会加剧肾囊肿的形成和生长。然而,在ARPKD或ADPKD患者中,这还没有得到严格的研究。
亲属风险评估
为了尽早确定哪些人能从及时采取治疗和预防措施中获益,对明显无症状先证者 的老少同胞进行评估是合理的。
评估可以包括:
- 如果已知家族中的致病变异,则进行分子遗传检测;
- 如果家族中致病性变异体尚不清楚,进行高分辨率肾脏及肝脏超声波检查及全身血压监测。
关于 遗传咨询 的目的,请参阅 Genetic Counseling,以了解与高危亲属检测相关的问题。
治疗在研究中
最近在ARPKD的两个动物模型上的研究表明,tesevatinib (TSV) ,一种独特的多激酶抑制剂,能显著减缓肾囊性疾病和肝胆疾病的进展[Sweeney et al 2017]. 这些数据以及ADPKD中TSV的I-II期多中心临床试验(也称为KD-019)生成的安全性数据(ClinicalTrials.gov标识符:NCT01559363)导致了TSV的I-II期多中心临床试验的初步开展计划于2016年下半年或2017年初对ARPKD婴幼儿进行治疗。
在美国的ClinicalTrials.gov 和欧盟的 EU Clinical Trials Register 搜索以获取广泛疾病情况的临床研究信息。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质、遗传和影响的信息,从而帮助他们做出知情的医疗选择和个人决定的过程。下面部分涉及遗传风险评估和使用家族史和基因测试来阐明家族成员的遗传状况。这部分不打算解决所有个人,文化或者个人可能面临的伦理问题,或者代替遗传学专家的咨询。
遗传模式
A常染色体隐性遗传多囊性肾病(ARPKD)是由PKHD1双等位基因致病变异引起的。
注:DZIP1L中的双等位基因致病变异也可能与ARPKD有关。在明确证明DZIP1L是第二个DZIP1L相关 基因之前,还需要更多的证据。
对家庭成员的风险
先证者的父母
- PKHD1致病变异的杂合子无症状,不存在发生ARPKD的风险。
- 对于临床诊断为ARPKD的患儿父母进行肾脏超声检查是很重要的,(即未经PKHD1分子遗传学检测确定的诊断)以排除ADPKD的可能性(见鉴别诊断)。
先证者的同胞
- PKHD1中致病性变异的杂合子是无症状的,不存在发生ARPKD的风险。
先证者的后代
- ARPKD个体的后代是PKHD1致病性变异的专性杂合子(携带者) 。
携带者(杂合子)检测
高危亲属的携带者测试需要事先鉴定家族中PKHD1的致病变异。
有关评估高危亲属以进行早期诊断和治疗的信息,请参阅管理、风险亲属评估。
计划生育
- 确定遗传风险、澄清携带者状态和讨论产前检测可用性的最佳时间是在怀孕前。
DNA库是为将来可能使用而储存的DNA(通常从白细胞中提取)。由于测试方法和我们对基因、等位基因变异和疾病的理解很可能在未来得到改善,因此应该考虑储存受影响个体的DNA。
产前检测和植入前基因检测
高危妊娠(即根据家族史风险为25%的孕妇)。一旦在患病患病家庭成员中发现PKHD1致病变异,最优处理方式对高危妊娠进行产前检测和植入前基因检测。
注:已制定并成功实施了一项针对既携带PKHD1 致病性变异 并希望怀上未受ARPKD影响的孩子的夫妇的PGD协议[Lau et al 2010]。使用多重置换扩增(MDA)对单个卵裂球进行基因组扩增,并使用来自PKHD1和侧翼序列的新型短串联重复序列(STR)标记进行单倍型分析[Lau et al 2010].。(注:本PGD方案和基因内STR标记可从威斯康星州儿童医院免费获得 [联系信息]。)
没有系统的数据可用于诊断产前超声检查在25%风险的孕妇中ARPKD的敏感性和特异性。
低风险妊娠(即产前常规超声检查发现囊性肾增大,但是不知道风险增加的孕妇)
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或登记册,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。有关选择标准的信息,请单击此处。
- ARPKD/CHF 联盟柯克伍德PA 17536 邮政信箱70电话: 800-708-8892(免费);717-529-5555传真: 800-807-9110(免费)电子邮件: info@arpkdchf.org
- 国家医学遗传学图书馆首页
- PKD基金会堪萨斯城MO 64114-2000 ,Suite 510,8330 Ward Parkway电话: 800-753-2873 (免费); 816-931-2600传真: 816-931-8655电子邮件: pkdcure@pkdcure.org
- Ciliopathy 联盟英国电话: 44 20 7387 0543
- 欧洲罕见肾病参考网络(ERKNet)德国海德堡电话: 49-6221-56-2349传真: 49-6221-56-5166电子邮件: contact@erknet.org
- 加拿大肾脏基金会加拿大安大略省蒙特利尔H3X 2H9,310-5160 DecarieBlvd电话: 800-361-7494 (toll-free); 514-369-4806传真: 514-369-2472电子邮件: info@kidney.ca
- 国家肾脏基金会(NKF)纽约州纽约市东33街30号,纽约10016电话: 800-622-9010 (toll-free); 212-889-2210电子邮件: info@kidney.org
分子遗传学
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。
Table A.
多囊肾病,常染色体隐性遗传:基因和数据库
| 基因 | 染色体位点 | 蛋白 | 特定地点的数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| PKHD1 | 6p12 | 纤维囊蛋白 | 常染色体隐性多囊肾疾病突变数据库PKHD1数据库 | PKHD1 | PKHD1 |
Table B.
分子发病机制
尽管鉴定了PKHD1及其蛋白产物纤维囊蛋白,但常染色体隐性遗传多囊性肾病(ARPKD)的发病机制仍不清楚[Sweeney & Avner 2011, Sweeney & Avner 2014, Sweeney et al 2016]。纤维囊蛋白功能的降低或缺乏被认为是疾病发病机理的基础[Hiesberger et al 2004, Zhang et al 2004]。最近的研究表明,许多与PKD相关的蛋白质与原发纤毛的功能有关,原发纤毛是位于包括肾小管和胆管细胞在内的大多数上皮细胞的顶表面的细胞器 [Lina & Satlinb 2004, Pazour 2004]。具有双等位基因的DZIP1L致病性变异的个体的人成纤维细胞在纤毛上的定位和PKD1和PKD2的表达异常[Lu et al 2017]。初级纤毛的结构和/或功能异常会导致其机械感觉特性发生变化,这可能会导致下游第二信使路径(尤其是循环AMP系统)的激活[Nauli et al 2003, Pazour 2004]。这些途径被认为可以激活已知的成囊过程,例如细胞增殖和体液分泌。所有增生性囊性上皮细胞的一个一致特征是EGFR轴的定性和定量异常表达(综述于Sweeney & Avner [2011]和Sweeney & Avner [2014])。基因缺陷、纤毛异常、蛋白复合物形成和EGFR异常之间的分子联系仍是推测性的。
除蛋白产物的直接相互作用外,还显示了纤维囊藻蛋白与多囊藻蛋白1和多囊藻蛋白2(涉及常染色体显性多囊肾疾病)在分子水平上相互作用。
这些纤溶蛋白除了纤毛以外,还以多聚体蛋白复合物的形式存在于多个部位。这些多囊藻毒素复合物位于顶细胞表面,与粘附连接相邻的侧细胞表面以及与粘着斑激酶相关的基底细胞膜[Avner & Sweeney 2006, Sweeney et al 2016]。多聚体蛋白复合物下游信号整合可能与ARPKD的分子和细胞病理生理学联系在一起。c-Src已被确认为纤维囊蛋白异常信号传导的关键中间体[Sweeney et al 2008, Sweeney et al 2017]。
据人们所了解ADPKD中的高血压是由肾素-血管紧张素系统(RAS)介导的。但是,ARPKD中的支持数据有限。过去十年的研究强调了RAS系统的复杂性和“局部”(例如,肾脏特异性的)RAS激活的重要性,而这在系统测量中可能没有反映。一项组织学研究支持了局部肾脏RAS在ARPKD高血压发病中的潜在作用,该研究表明ARPKD个体的两个肾脏中几种肾素-血管紧张素轴成分的表达增加[Loghman-Adham et al 2005]。 ARPKD动物模型中的最新数据表明,受影响动物的肾脏以及肝脏中都有RAS激活[Goto et al 2010a, Goto et al 2010b]。这就提出了一个问题,即RAS活化是否可能是ARPKD发病机制的更基本特征,而不是慢性肾脏疾病的非特异性表现。
基因结构.PKHD1是一个非常大的基因,包含86个编码外显子[Onuchic et al 2002, Ward et al 2002, Bergmann et al 2004]。最大的阅读框包含67个外显子(NM_138694.3),但已描述了多个交替剪接的转录本 [Bergmann et al 2004]。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病变体. 不同类型的变体分布在整个基因中。请参阅表 A中的LSDB和HGMD数据库 。
正常基因产物. PKHD1产物是具有受体样性质的大蛋白[Onuchic et al 2002, Ward et al 2002]。它局限于肾脏,胆管和胰腺。此外,已显示纤维囊蛋白定位于肾小管上皮细胞中的原发纤毛以及其他离散位置,提示在某些情况下多种途径可能与睫状功能障碍有关 [Ward et al 2003],或多聚体蛋白复合物信号传导。囊性上皮和内皮。睫状结构和功能异常可能参与许多不同类型的胆囊性肾脏疾病的发病机制[Ong & Wheatley 2003] (请参阅分子发病机制)。
异常基因产物. 纤维囊蛋白功能的降低或缺乏被认为是疾病发病机理的基础[Hiesberger et al 2004, Zhang et al 2004](具体参见分子发病机制)。
相关研究
引用文献
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85:1 - 21. [PubMed: 16523049]
- Avner ED, Sweeney WE. Renal cystic disease: new insights for the clinician. Pediatr Clin North Am. 2006;53:889 - 909. [PubMed: 17027616]
- Bergmann C, Senderek J, Küpper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Büttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004;23:453 - 63. [PubMed: 15108277]
- Bergmann C, Senderek J, Windelen E, Küpper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schöneborn S, Konrad M, Schmitt CP, Seeman T, Neuhaus TJ, Vester U, Kirfel J, Büttner R, Zerres K. APN (Arbeitsgemeinschaft für Pädiatrische Nephrologie). Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005;67:829 - 48. [PubMed: 15698423]
- Blyth H, Ockenden BG. Polycystic disease of kidney and liver presenting in childhood. J Med Genet. 1971;8:257 - 84. [PMC free article: PMC1469189] [PubMed: 5097134]
- Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2016;31:707 - 14. [PubMed: 26160100]
- Brinkert F, Lehnhardt A, Montoya C, Helmke K, Schaefer H, Fischer L, Nashan B, Bergmann C, Ganschow R, Kemper MJ. Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): indication and outcome. Transpl Int. 2013;26:640 - 50. [PubMed: 23582048]
- Chalhoub V, Abi-Rafeh L, Hachem K, Ayoub E, Yazbeck P. Intracranial aneurysm and recessive polycystic kidney disease: the third reported case. JAMA Neurol. 2013;70:114 - 6. [PubMed: 23318517]
- Chandar J, Garcia J, Jorge L, Tekin A. Transplantation in autosomal recessive polycystic kidney disease: liver and/or kidney? Pediatr Nephrol. 2015;30:1233 - 42. [PubMed: 25115876]
- Chapal M, Debout A, Dufay A, Salomon R, Roussey G, Burtey S, Launay EA, Vigneau C, Blancho G, Loirat C, Hourmant M, Fakhouri F. Kidney and liver transplantation in patients with autosomal recessive polycystic kidney disease: a multicentric study. Nephrol Dial Transplant. 2012;27:2083 - 8. [PubMed: 22076432]
- Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease -- an expanding clinical spectrum. Nat Rev Nephrol. 2015;11:102 - 12. [PubMed: 25536396]
- Committee on Infectious Diseases. From the American Academy of Pediatrics: Policy statements--Modified recommendations for use of palivizumab for prevention of respiratory syncytial virus infections. Pediatrics. 2009;124:1694 - 701. [PubMed: 19736258]
- Dell KM, Sweeney WE, Avner ED. Polycystic kidney disease. In: Avner ED, Harmon W, Niadet P, Yoshikawa N, eds. Pediatric Nephrology. 6 ed. Heidelberg, Germany: Springer-Verlag; 2009:849-88.
- Fick GM, Johnson AM, Strain JD, Kimberling WJ, Kumar S, Manco-Johnson ML, Duley IT, Gabow PA. Characteristics of very early onset autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3:1863 - 70. [PubMed: 8338917]
- Fonck C, Chauveau D, Gagnadoux MF, Pirson Y, Grünfeld JP. Autosomal recessive polycystic kidney disease in adulthood. Nephrol Dial Transplant. 2001;16:1648 - 52. [PubMed: 11477168]
- Frank V, Zerres K, Bergmann C. Transcriptional complexity in autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9:1729 - 36. [PMC free article: PMC4186505] [PubMed: 25104275]
- Goto M, Hoxha N, Osman R, Dell KM. The renin-angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2010a;25:2449 - 57. [PubMed: 20798958]
- Goto M, Hoxha N, Osman R, Wen J, Wells R, Dell KM. Renin-angiotensin system (RAS) activation in congenital hepatic fibrosis in the PCK rat model of autosomal recessive polycystic kidney disease (ARPKD). J Pediatr Gastroenterol Nutr. 2010b;50:639 - 44. [PMC free article: PMC4241057] [PubMed: 20400910]
- Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, Emre S, Heller T, Kamath BM, Murray KF, Moise K, Eichenwald EE, Evans J, Keller RL, Wilkins-Haug L, Bergmann C, Gunay-Aygun M, Hooper SR, Hardy KK, Hartung EA, Streisand R, Perrone R, Moxey-Mims M. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014;165:611 - 7. [PMC free article: PMC4723266] [PubMed: 25015577]
- Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111:1072 - 80. [PubMed: 12728091]
- Guay-Woodford LM, Galliani CA, Musulman-Mroczek E, Spear GS, Guillot AP, Bernstein J. Diffuse renal cystic disease in children: morphologic and genetic correlations. Pediatr Nephrol. 1998;12:173 - 82. [PubMed: 9630032]
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112 - 121.e2. [PMC free article: PMC4162098] [PubMed: 23041322]
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman M, Graf J, Bryant JC, Kleta R, Garcia A, Edwards H, Piwnica-Worms K, Adams D, Bernardini I, Fischer RE, Krasnewich D, Oden N, Ling A, Quezado Z, Zak C, Daryanani KT, Turkbey B, Choyke P, Guay-Woodford LM, Gahl WA. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin J Am Soc Nephrol. 2010b;5:972 - 84. [PMC free article: PMC2879301] [PubMed: 20413436]
- Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica-Worms K, Bryant J, Bernardini I, Fischer R, Huizing M, Guay-Woodford L, Gahl WA. PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab. 2010a;99:160 - 73. [PMC free article: PMC2818513] [PubMed: 19914852]
- Harps E, Brinkert F, Ganschow R, Briem-Richter A, van Husen M, Schmidtke S, Herden U, Nashan B, Fischer L, Kemper MJ. Immediate postoperative intensive care treatment of pediatric combined liver-kidney transplantation: outcome and prognostic factors. Transplantation. 2011;91:1127 - 31. [PubMed: 21544033]
- Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014;134:e833 - 45. [PMC free article: PMC4143997] [PubMed: 25113295]
- Hartung EA, Matheson M, Lande MB, Dell KM, Guay-Woodford LM, Gerson AC, Warady BA, Hooper SR, Furth SL. Neurocognition in children with autosomal recessive polycystic kidney disease in the CKiD cohort study. Pediatr Nephrol. 2014;29:1957 - 65. [PMC free article: PMC4167962] [PubMed: 24828609]
- Hiesberger T, Bai Y, Shao X, McNally BT, Sinclair AM, Tian X, Somlo S, Igarashi P. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest. 2004;113:814 - 25. [PMC free article: PMC362119] [PubMed: 15067314]
- Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr Opin Pediatr. 2015;27:186 - 92. [PubMed: 25689455]
- Jahnukainen T, Kirjavainen T, Luoto T, Ylinen E, Linkosalo L, Arikoski P, Pakarinen M, Jalanko H. Long-term pulmonary function in children with recessive polycystic kidney disease. Arch Dis Child. 2015;100:944 - 7. [PubMed: 26163120]
- Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J Med Genet. 2003;40:311 - 9. [PMC free article: PMC1735460] [PubMed: 12746391]
- Kamath BM, Piccoli DA. Heritable disorders of the bile ducts. Gastroenterol Clin North Am. 2003;32:857 - 75. [PubMed: 14562578]
- Kaplan BS, Kaplan P, de Chadarevian JP, Jequier S, O'Regan S, Russo P. Variable expression of autosomal recessive polycystic kidney disease and congenital hepatic fibrosis within a family. Am J Med Genet. 1988;29:639 - 47. [PubMed: 3377007]
- Lau EC, Janson MM, Roesler MR, Avner ED, Strawn EY, Bick DP. Birth of a healthy infant following preimplantation PKHD1 haplotyping for autosomal recessive polycystic kidney disease using multiple displacement amplification. J Assist Reprod Genet. 2010;27:397 - 407. [PMC free article: PMC2922704] [PubMed: 20490649]
- Lilova M, Kaplan BS, Meyers KE. Recombinant human growth hormone therapy in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2003;18:57 - 61. [PubMed: 12488992]
- Limwongse C. Developmental syndromes and malformations of the urinary tract. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Francesco E, Goldstein S, eds. Pediatric Nephrology. 7 ed. Springer. 2016:135-78.
- Limwongse C, Clarren SK, Cassidy SB. Syndromes and malformations of the urinary tract. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric Nephrology. 4 ed. Baltimore, MD: Lippincott Williams & Wilkins. 1999:427-52.
- Lina F, Satlinb LM. Polycystic kidney disease: the cilium as a common pathway in cystogenesis. Curr Opin Pediatr. 2004;16:171 - 6. [PubMed: 15021197]
- Loghman-Adham M, Soto CE, Inagami T, Sotelo-Avila C. Expression of components of the renin-angiotensin system in autosomal recessive polycystic kidney disease. J Histochem Cytochem. 2005;53:979 - 88. [PubMed: 15879580]
- Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya IP, Kramer C, Ortiz-Brüchle N, Hilger N, Metzis V, Hiersche M, Tay SY, Tunningley R, Vij S, Courtney AD, Whittle B, Wühl E, Vester U, Hartleben B, Steffen Neuber S, Frank V, Little MH, Daniel Epting D, Papathanasiou P, Perkins AC, Wright GD, Hunziker W, Gee HY, Otto EA, Zerres K, Hildebrandt F, Sudipto Roy S, Wicking C, Bergmann C. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017;49:1025 - 34. [PMC free article: PMC5687889] [PubMed: 28530676]
- Melchionda S, Palladino T, Castellana S, Giordano M, Benetti E, De Bonis P, Zelante L, Bisceglia L. Expanding the mutation spectrum in 130 probands with ARPKD: identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. J Hum Genet. 2016;61:811 - 21. [PubMed: 27225849]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129 - 37. [PubMed: 12514735]
- O'Brien K, Font-Montgomery E, Lukose L, Bryant J, Piwnica-Worms K, Edwards H, Riney L, Garcia A, Daryanani K, Choyke P, Mohan P, Heller T, Gahl WA, Gunay-Aygun M. Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease. J Pediatr Gastroenterol Nutr. 2012;54:83 - 9. [PubMed: 21694639]
- Ong AC, Wheatley DN. Polycystic kidney disease--the ciliary connection. Lancet. 2003;361:774 - 6. [PubMed: 12620752]
- Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin- transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305 - 17. [PMC free article: PMC447605] [PubMed: 11898128]
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol. 2004;15:2528 - 36. [PubMed: 15466257]
- Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205 - 12. [PMC free article: PMC2615723] [PubMed: 18945943]
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302 - 6. [PubMed: 9203177]
- Schaeffer F. Endocrine and growth abnormallities in chronic kidney disease. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Francesco E, Goldstein S, eds. Pediatric Nephrology. 7 ed. Springer. 2016:2295-348.
- Seikaly MG, Salhab N, Warady BA, Stablein D. Use of rhGH in children with chronic kidney disease: lessons from NAPRTCS. Pediatr Nephrol. 2007;22:1195 - 204. [PubMed: 17530299]
- Sharp CK, Bergman SM, Stockwin JM, Robbin ML, Galliani C, Guay-Woodford LM. Dominantly transmitted glomerulocystic kidney disease: a distinct genetic entity. J Am Soc Nephrol. 1997;8:77 - 84. [PubMed: 9013451]
- Shneider BL, Magid MS. Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transplant. 2005;9:634 - 9. [PubMed: 16176423]
- Shukla AR, Kiddoo DA, Canning DA. Unilateral nephrectomy as palliative therapy in an infant with autosomal recessive polycystic kidney disease. J Urol. 2004;172:2000 - 1. [PubMed: 15540776]
- Sweeney W Jr, Gunay-Aygun M, Patil A, Avner ED. Childhood polycystic kidney disease. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Francesco E, Goldstein S, eds. Pediatric Nephrology. 7 ed. Springer. 2016:1103-53.
- Sweeney WE Jr, Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol. 2011;26:675 - 92. [PubMed: 21046169]
- Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014;75:148 - 57. [PMC free article: PMC3953890] [PubMed: 24336431]
- Sweeney WE Jr, von Vigier RO, Frost P, Avner ED. Src Inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331 - 41. [PMC free article: PMC2440293] [PubMed: 18385429]
- Sweeney WE, Frost P, Avner ED. Tesevatinib ameliorates progression of ARPKD in rodent models of autosomal recessive polycystic kidney disease. World J Nephrol. 2017;6:188 - 200. [PMC free article: PMC5500456] [PubMed: 28729967]
- Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatr Transplant. 2013;17:328 - 35. [PMC free article: PMC3663883] [PubMed: 23593929]
- Tsimaratos M, Cloarec S, Roquelaure B, Retornaz K, Picon G, Chabrol B, Guys JM, Sarles J, Nivet H. Chronic renal failure and portal hypertension--is portosystemic shunt indicated? Pediatr Nephrol. 2000;14:856 - 8. [PubMed: 10955945]
- Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol. 2009;39:100 - 11. [PMC free article: PMC2918426] [PubMed: 19089418]
- Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. hepatocyte nuclear factor 1beta-associated kidney disease: more than renal cysts and diabetes. J Am Soc Nephrol. 2016;27:345 - 53. [PMC free article: PMC4731131] [PubMed: 26319241]
- Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259 - 69. [PubMed: 11919560]
- Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, Harris PC. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703 - 10. [PubMed: 12925574]
- Watkins SL, McDonald RA, Avner ED. Renal dysplasia, hypoplasia and miscellaneous cystic disorders. In: Barratt TM, Avner ED, Harmon WE, eds. Pediatric Nephrology. 4 ed. Baltimore, MD: Lippincott Williams & Wilkins. 1999:415-25.
- Wen J. Congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Clin Transl Sci. 2011;4:460 - 5. [PMC free article: PMC3252199] [PubMed: 22212229]
- Zerres K, Rudnik-Schöneborn S, Deget F, Holtkamp U, Brodehl J, Geisert J, Scharer K. Autosomal recessive polycystic kidney disease in 115 children: clinical presentation, course and influence of gender. Arbeitsgemeinschaft fü Pädiatrische, Nephrologie. Acta Paediatr. 1996;85:437 - 45. [PubMed: 8740301]
- Zerres K, Rudnik-Schöneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med. 1998;76:303 - 9. [PubMed: 9587064]
- Zhang MZ, Mai W, Li C, Cho SY, Hao C, Moeckel G, Zhao R, Kim I, Wang J, Xiong H, Wang H, Sato Y, Wu Y, Nakanuma Y, Lilova M, Pei Y, Harris RC, Li S, Coffey RJ, Sun L, Wu D, Chen XZ, Breyer MD, Zhao ZJ, McKanna JA, Wu G. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci U S A. 2004;101:2311 - 6. [PMC free article: PMC356947] [PubMed: 14983006]
- Zvereff V, Yao S, Ramsey J, Mikhail FM, Vijzelaar R, Messiaen L. Identification of PKHD1 multiexon deletions using multiplex ligation-dependent probe amplification and quantitative polymerase chain reaction. Genet Test Mol Biomarkers. 2010;14:505 - 10. [PubMed: 20575693]
章节注释
作者须知
威斯康星州儿童医院的联系信息(植入前遗传诊断 方案)
Estil Y Strawn,医学博士
威斯康星州,妇产科医学院院长,
弗罗德特医院
威斯康星大街900 W
密尔沃基WI 53226
414-805-3666
estrawn@mcw.edu
作者历史
医学博士Ellis D Avner(2001年—)
医学博士Katherine MacRae Dell ,凯斯西储大学医学院(2001-2014)
威廉•E•斯威尼(MS)(2014年—)