
简介
临床特征
孤立的甲基丙二酸血症/尿症是本GeneReview 的重点,是由甲基丙二酰辅酶 A 变位酶(分别为 Mut0型和Mut-型)的完全或部分缺陷引起的,或其辅因子腺苷钴胺素(cblA、cblB 或 cblD-MMA,即VitB12)的转运或合成缺陷,或缺乏甲基丙二酰辅酶 A异构酶。 孤立的甲基丙二酸血症/酸尿症的发病范围从新生儿期到成年期。所有表型都以相对健康和间歇性代谢失代偿期为特征,通常与并发感染和应急有关。
- 在新生儿期,该疾病可表现为嗜睡、呕吐、肌张力减退、体温过低、呼吸窘迫、严重的酮症酸中毒、高氨血症、中性粒细胞减少和血小板减少,并可能导致出生后前 4 周内死亡。
- 在婴儿型/非VitB12有效型表型中,婴儿在出生时是正常的,但在几周到几个月内会出现嗜睡、呕吐、脱水、发育迟缓、肝肿大、肌张力减退和脑病。
- 在新生儿中偶尔可以观察到中间型VitB12有效型表型,但通常在生命的最初几个月或几年观察到; 受累的儿童表现出厌食、生长落后、肌张力减退和发育迟缓,有时在摄入蛋白质后会出现蛋白质厌恶和/或呕吐和嗜睡。
- 不典型和“良性”/成人甲基丙二酸血症表型与甲基丙二酸的尿排泄增加(尽管轻微)相关。
甲基丙二酸血症的主要继发性并发症包括:智力障碍(多种);肾小管间质性肾炎伴进行性肾功能衰竭; “代谢性中风”(急性和慢性基底节损伤)导致残疾运动障碍伴舞蹈手足徐动症、肌张力障碍和下肢/四肢轻瘫;胰腺炎;生长落后;功能性免疫损伤和视神经萎缩。
诊断/测试
孤立的甲基丙二酸血症的诊断依赖于通过气液色谱和质谱分析血浆和/或尿液中的有机酸。建立甲基丙二酸血症的特定亚型需要进行细胞生化研究(包括 14C丙酸和B12有效型互补分析,以及钴胺素VitB12分布测定)和分子遗传学检测。在与孤立性甲基丙二酸血症相关的五种基因(MMUT、MMAA、MMAB、MCEE 和 MMADHC)之一中发现双等位基因的致病变异——加上父母携带者的状态——可以建立诊断。
管理
对症处理:通过恢复容量状态和酸碱平衡来稳定危重患者;减少或限制蛋白质摄入;通过含高葡萄糖的液体和胰岛素提供更多的卡路里来阻止分解代谢;监测血清电解质和氨、静脉或动脉血气和尿量。管理包括低脂肪氨基酸的高热量饮食;羟钴胺素VitB12肌内注射;肉碱补充剂;抗生素,如新霉素或甲硝唑,以减少肠道菌群产生的丙酸盐;根据需要放置胃造口管;并积极治疗感染。在少数患者中使用的其他疗法包括用于治疗急性高氨血症发作的 N-氨基甲酰谷氨酸;肝、肾或肝肾联合移植;和抗氧化剂用于治疗视神经萎缩。
预防主要表现:在某些情况下,新生儿筛查可以在症状前发现受累的新生儿并进行早期治疗。
要避免的药物/情况:禁食和增加膳食蛋白质。
其他:Medic Alert® 手镯和最新、易于访问、详细的紧急治疗方案。
遗传咨询
孤立的甲基丙二酸血症以常染色体隐性遗传方式遗传。受累的个体的每个同胞有25%的机会患病,有50%的机会成为无症状携带者,以及 25% 的机会不患病也不是携带者。如果家族中的致病变异已知,则可以使用分子遗传学技术对高危家庭成员进行携带者检测和对高危妊娠进行产前检测。在某些情况下,通过对培养的胎儿细胞进行酶分析和代谢物测量(通过绒毛膜绒毛取样或羊膜穿刺术获得),可以对风险增加的妊娠进行产前诊断。
基因谱
| 孤立的甲基丙二酸血症/尿症:包括的表型 |
|---|
|
诊断
在这篇综述中,术语“孤立的甲基丙二酸血症”是指一组与血液和尿液中甲基丙二酸 (MMA) 浓度升高相关的先天性代谢障碍,这些障碍是由于未能将线粒体基质中丙酰辅酶 A 代谢中的甲基丙二酰辅酶 A 转化为琥珀酰辅酶 A而导致的,不伴有高同型半胱氨酸血症或同型胱氨酸尿症、低蛋氨酸血症或其他代谢物(如丙二酸)的变化(Figure 1)。
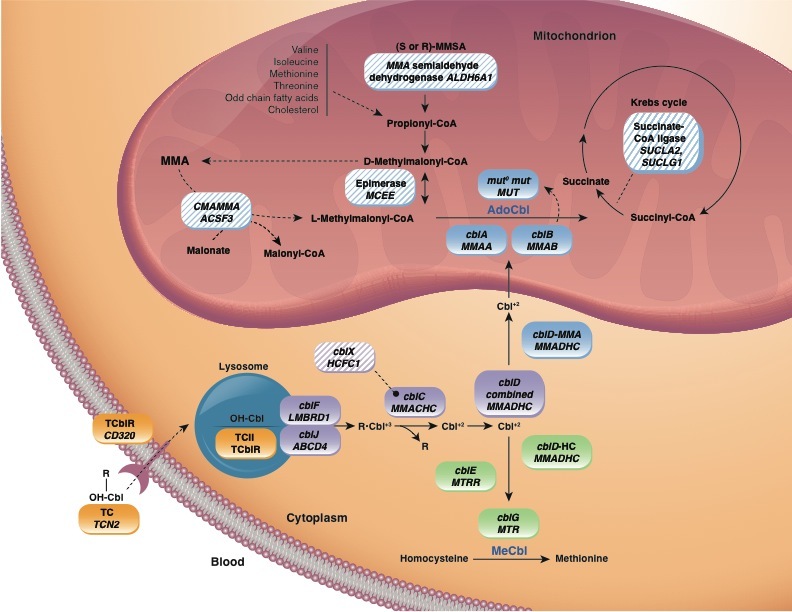
孤立的甲基丙二酸血症由以下任何一种引起:
- MMUT编码的甲基丙二酰辅酶A变位酶完全(mut0型)缺乏或部分(mut–型)缺乏
- 其辅因子 5'-脱氧腺苷钴胺素VitB12合成减少,与分别由 MMAA、MMAB 或 MMADHC 中双等位基因的致病变异引起的 cblA、cblB 或 cblD-MMA 互补组相关
- MCEE编码的甲基丙二酰辅酶A差向异构酶活性不足
请注意,以下疾病不包括在本 GeneReview 的范围内(见Differential Diagnosis):
- 由 SUCLA2 或 SUCLG1 突变引起的与琥珀酰辅酶 A 连接酶缺乏相关的甲基丙二酸血症分别在 SUCLA2-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria和SUCLG1-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria讨论.
- 由细胞内钴胺素VitB12代谢的其他步骤中的缺陷引起的与高同型半胱氨酸血症或同型半胱氨酸尿症相关的甲基丙二酸血症在Disorders of Intracellular Cobalamin Metabolism讨论。
- 罕见的缺陷,如合并丙二酸和甲基丙二酸血症、甲基丙二酸半醛脱氢酶缺乏症、转钴胺素受体缺乏症以及合并甲基丙二酸血症和同型半胱氨酸血症,cblX 型,在Differential Diagnosis中讨论。
提示性发现
由于 孤立的甲基丙二酸血症的体征和症状是非特异性的,因此提示性发现可能包括以下内容:
- 新生儿:嗜睡、呕吐、肌张力减退、体温过低、呼吸窘迫、严重酮症酸中毒、高氨血症、中性粒细胞减少症和血小板减少症
- 注意:在有扩大新生儿筛查潜在项目的州,孤立的甲基丙二酸血症可在出生正常的新生儿急性失代偿发作之前被诊断。
- 在较大的婴儿和儿童中:发育迟缓、肾综合征和肌张力减退、智力障碍或其他急性(基底节中风)和慢性神经系统症状。
在部分 mut缺乏、cblA 或 cblB 的患者中,不同年龄的提示性发现可能包括以下内容:
- 减弱MMA 表型 [Lerner-Ellis et al 2004, Lerner-Ellis et al 2006, Hörster et al 2007]
- 孤立性肾小管酸中毒或慢性肾功能衰竭 [Dudley et al 1998, Coman et al 2006]
- 基底节代谢性卒中[Korf et al 1986, Heidenreich et al 1988]
- 并发疾病后的灾难性/致命性酮症酸中毒 [Ciani et al 2000]
建立诊断
Figure 1描绘了intracellular propionate and cobalamin metabolism的概述。 Figure 2提供了对尿液和/或血浆中甲基丙二酸升高的人进行检查的流程图,这是一种改进的流程图,包括考虑 甲基丙二酰辅酶A差向异构酶缺乏症、琥珀酰辅酶A连接酶缺乏症和该途径中的其他罕见缺陷,以及体内VitB12反应性检查在任何年龄患有甲基丙二酸血症的个体中的使用。
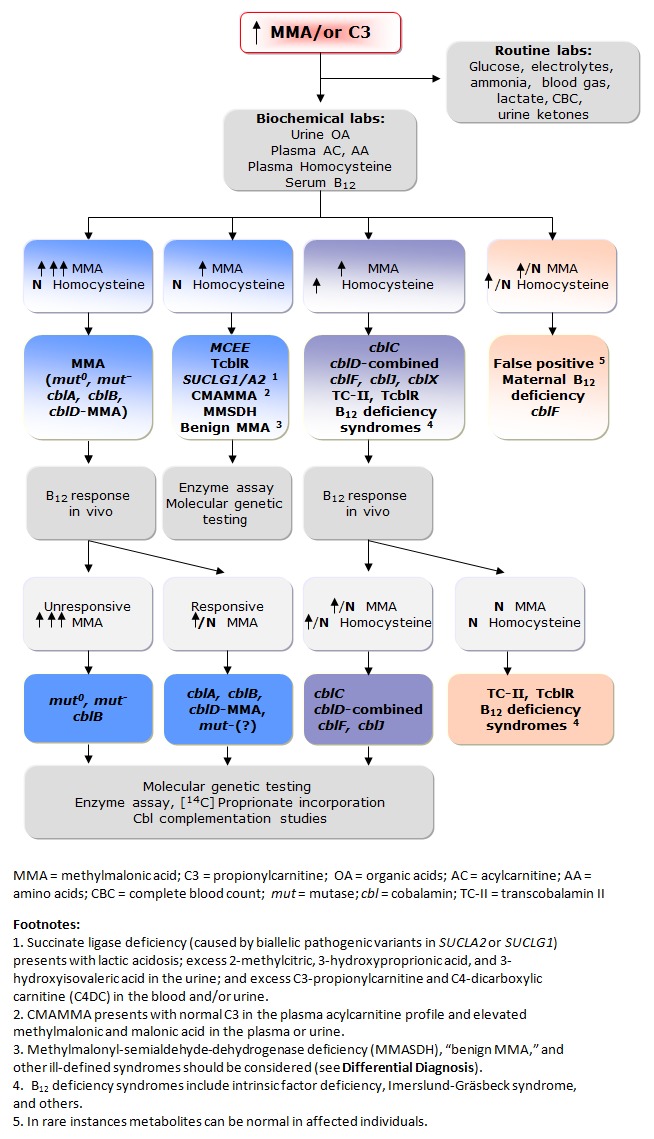
步骤 1. 在临床表现可疑且 MMA 尿有机酸筛查阳性的先证者中,有助于确定诊断的实验室检测包括:葡萄糖、电解质、氨、血气、乳酸、CBC 和尿酮体,血浆 MMA、tHcy 和VitB12水平、血浆氨基酸和酰基肉碱谱。相关发现:
- 高血浆和尿液 MMA,VitB12、tHcy 和蛋氨酸水平正常
- 升高的丙酰肉碱 (C3)
- 动脉或静脉血气检测中的高阴离子间隙代谢性酸中毒和尿液中大量酮体和乳酸
- 高氨血症
- 高甘氨酸血症
- 乳酸性酸中毒
- CBC显示中性粒细胞减少、血小板减少、贫血
第 2 步:在通过扩大 新生儿筛查遗传风险(遗传风险增加)发现丙酰肉碱升高的新生儿和患有该疾病的高风险个体(例如,先证者的同胞)中,首先要确定是否存在丙酰肉碱甲基丙二酸显著升高,最好通过尿液有机酸分析(通过 GC/MS)和血浆酰基肉碱谱(通过 TMS)来完成。注意:同时,获得血浆 MMA、氨基酸、血浆同型半胱氨酸和血清VitB12的水平(新生儿和母亲)有助于进一步区分甲基丙二酸血症的原因(见步骤 3)。
除了升高的甲基丙二酸外,还可以看到以下生化结果:
- 在尿液的 GC/MS 分析中检测到存在 3-羟基丙酸、2-甲基柠檬酸和甘氨酰甘氨酸
- 血浆氨基酸分析中甘氨酸血浆浓度升高
- 通过 TMS 测量的丙酰肉碱 (C3) 血浆浓度升高和 C4-二羧酸或甲基丙二酸/琥珀酰肉碱 (C4DC) 的不同程度升高
步骤 3. 一旦确定了甲基丙二酸血症和酸尿症的升高,正常的血浆同型半胱氨酸和VitB12水平可以帮助区分孤立的MMA 与其他疾病(见 Figure 2,左两列)。 注意:虽然可以精确定量血浆和/或尿液甲基丙二酸浓度(Table 1),但通常不需要立即用于诊断目的。
Table 1
甲基丙二酸血症表型及其亚型
| 甲基丙二酸血症表型/亚型 1 | 甲基丙二酸浓度 | |
|---|---|---|
| 尿 2 | 血 | |
| 婴儿/ VitB12无反应型 3 mut0, mut–, cblB | 1,000-10,000 mmol/mol Cr | 100-1,000 µmol/L |
| VitB12-反应型 3cblA, cblD-MMA cblB, mut– (rare) | Tens - hundreds mmol/mol Cr | 5-100 µmol/L |
| “良性”/成人甲基丙二酸血症4 | 10-100 mmol/mol Cr | 100 µmol/L |
| MCEE 缺陷 5 | 50-1,500 mmol/mol Cr | 7 µmol/L |
| 正常6 | <4 mmol/mol Cr 7 | <0.27 µmol/L 7 |
MCEE = 甲基丙二酰辅酶A差向异构酶; ND = 未确定
- 1.
生化参数和临床表型并不总是一致的,部分原因是肾功能会影响血浆 MMA 浓度[Kruszka et al 2013, Manoli et al 2013]。肾衰竭患者的血浆 MMA 大幅升高,可超过 5,000 µmol/L。
- 2.
在一些中心,通过 1H-NMR光谱分析尿液也可用于证明甲基丙二酸浓度增加[Iles et al 1986].
- 3.
作者有约超过 80 名具有VitB12反应型和VitB12无反应型患者的经验
- 4.
来自Giorgio et al [1976] 并转换为 µmol/L 的血浆浓度
- 5.
- 6.
- 7.
正常值并非完全来自儿童或新生儿。一些实验室以 mg/g/Cr(正常:<3 mg/g/Cr)报告尿 MMA 浓度,以 nmol/L(正常:<271 nmol/L)报告血清浓度。 MMA的分子量为118 g/mol。
步骤 4 应确定所有受累的个体对VitB12的体内反应。没有标准的方案被记录在案。当病情稳定时,受影响的个体可以每天肌肉或静脉注射 1.0 mg 羟钴胺素 VitB12(OH-Cbl)(见备注),持续一到两周,然后通过连续尿液有机酸分析和/或测量 MMA、丙酰肉碱和同型半胱氨酸的血浆浓度,以评估 MMA 和相关代谢物(3-OH-丙酸、2-柠檬酸甲酯)。代谢物产生和血浆浓度显著降低 (>50%) 被认为反应性[Fowler et al 2008, Kruszka et al 2013]。在所有 cblA 和少数 cblB个体中报告了体内反应 [Hörster et al 2007]。
注意:羟钴胺素(不是氰钴胺素)是治疗甲基丙二酸血症的首选制剂;因此,如果体内对肌注羟钴胺素的反应存在疑问或处于临界状态,则应继续给予VitB12,并进行皮肤活检以分离成纤维细胞,通过体外14C丙酸盐试验以评估对VitB12的反应性。
步骤 5. 分子遗传学检测(Table 2)可用于通过识别五种基因(MMUT、MMAA、MMAB、MCEE 和 MMADHC)之一的双等位基因的致病变异并确认来确定孤立的MMA 的诊断及确定父母的携带者身份。此外,由于细胞生化检测的可及性、成本、侵入性等原因,分离甲基丙二酸血症的亚型主要由 分子遗传学检测确定。
分子测试方法可以包括以下:分层单基因测试。 由于孤立的甲基丙二酸血症的表型可以是相同的,而与基因无关,因此分子遗传学检测可以按照以下顺序进行:
1.VitB12无反应型个体的 UT 和 MMAB
2.对VitB12反应型的个体中的 MMAA
3. 如果前三个基因(MMUT、MMAB 和 MMAA)的检测结果无异常,则进行 MCEE 和 MMADHC 检测注意:对于所有基因,首先进行序列分析,如果仅检测到一个 致病性变异,则进行deletion/duplication analysis。
使用包括这五个基因和代谢途径中的其他基因的multigene panel(见Differential Diagnosis)。 注意:每个基因组和诊断基因组中包含的基因和用于基因组测试的敏感性可能因实验室而异。
有关多基因套餐的介绍,请单击 here。 有关订购基因检测的临床医生的更多详细信息,请参见 here。
Table 2
用于孤立性甲基丙二酸血症的分子遗传学检测
| 基因 1 | 归因于该基因突变的孤立型MMA 的比例 2 | 该方法检测到的变异比例 | |
|---|---|---|---|
| 测序 3 | 缺失/重复 a分析 4 | ||
| MMUT | 60% (78% mut0 型, 22% mut– 型) | 96% 5, 6 | 不详, 无报道 |
| MMAA | 25% | 97% 7 | 不详, 无报道 |
| MMAB | 12% | 98% 8 | 不详, 无报道 |
| MCEE | 不详 | 4 probands/families 9 | 不详, 无报道 |
| MMADHC | 不详 | 6 probands/families 10 | 不详, 无报道 |
- 1.
Table A. Genes and Databases 可查染色体 位点 和蛋白。见Molecular Genetics 可查 基因的等位基因变异鉴定。
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
步骤 6. 皮肤成纤维细胞的细胞生化检测是体外确定 MMA 亚型和VitB12反应型的金标准,当上述检测方法无法提供明确的诊断以指导管理时,它是有用的。有关生化测试的详细信息,请单击here(pdf)。
新生儿筛查
在过去的十年中,美国和世界上许多国家实施了 新生儿筛查质谱法(Mass Spectrometry),通过检测血液中丙酰肉碱(C3)的浓度升高,后者为甲基丙二酸血症和相关丙酸血症患者血液中的代谢物,确定了新生儿甲基丙二酸血症[Chace et al 2001, Therrell et al 2014].
注意:由于丙酰肉碱是最常导致假阳性结果的分析物之一,因此建议将 C3/C2、C3/C0、C3/C16 和新的生物标志物(如 C16:1OH)与高血浓度 C3 结合使用,作为新生儿筛查MS/MS 对甲基丙二酸血症和丙酸血症的分析标准[Lindner et al 2008]。
3-羟基丙酸、甲基丙二酸和/或 2-甲基柠檬酸的二级检测可用于降低与假阳性结果相关的成本和焦虑[Matern et al 2007, la Marca et al 2008].
- 如果 C3 和 C5OH 升高,则需要考虑全羧化酶缺乏和/或biotinidase deficiency的诊断。
- 升高的 C4-二羧酸酰基肉碱 (C4DC) 是甲基丙二酰肉碱和琥珀酰肉碱的标志物,提示酰辅酶 A 连接酶缺乏相关的甲基丙二酸尿症 [Fowler et al 2008, Morava et al 2009].
推荐行动 (ACT) 表和确认算法描述了对筛查呈阳性的婴儿进行随访所涉及的基本必要步骤;参见美国医学遗传学会 (ACMG) Newborn Screening ACT Sheet和 National Academy of Clinical Biochemistry Guidelines (pdf)[Dietzen et al 2009].
临床特征
临床表现
下文描述的与mut0型、 mut– 型、cblA、cblB 和 cblD-MMA 相关的 孤立的甲基丙二酸血症的表型具有共同的临床表现,以相对健康期和间歇性代谢失代偿期为特征的自然病程,通常与并发感染和应急有关[Zwickler et al 2012]。每一次这样的失代偿都可能危及生命。值得注意的是,孤立性甲基丙二酸血症的自然病程需要进一步研究,特别是关于包括肾脏疾病在内的医学并发症、实体器官移植的影响和分子病理学。
婴儿/VitB12无反应型表型(mut0型,cblB)。婴儿期最常见的 孤立的甲基丙二酸血症表型。婴儿在出生时是正常的,但在开始喂食含蛋白质时会迅速出现嗜睡、呕吐和脱水。就诊时,它们表现出肝肿大、肌张力减退,并且在许多情况下表现为高氨血症性脑病。实验室检查结果通常显示严重的高阴离子间隙代谢性酸中毒、酮症和酮尿(新生儿高度异常强烈提示有机酸尿)、高氨血症和高甘氨酸血症[Matsui et al 1983, Kölker et al 2015a]。尤其是当高氨血症严重且持续时,可能需要透析。
可见血小板减少和中性粒细胞减少,提示新生儿败血症。
尽管积极干预, 孤立的甲基丙二酸血症的新生儿表现可导致灾难性死亡。具有VitB12反应性mut–型或 cblA 的婴儿也可能出现急性新生儿危象。
部分缺陷或VitB12反应型表型(mut–型、cblA、cblB [罕见]、cblD-MMA)。这种孤立的甲基丙二酸血症的中间 表型可发生在生命的最初几个月或几年。受影响的婴儿可能会出现喂养问题(通常是厌食和呕吐)、生长迟缓、张力减退和发育迟缓。有些人在摄入蛋白质后会出现蛋白质厌恶和/或呕吐和嗜睡的临床症状。
在确诊和开始治疗之前,这些婴儿有发生灾难性失代偿的风险(就像新生儿一样)[Shapira et al 1991, Lerner-Ellis et al 2004, Lerner-Ellis et al 2006, Hörster et al 2007].
在这种严重失代偿发作期间,如果没有针对 MMA 进行及时治疗并且症状被误诊为例如糖尿病酮症酸中毒,尽管进行了强化干预,儿童仍可能死亡[Ciani et al 2000].
在 新生儿筛查之前,cblA 或 mut–型的婴儿会出现破坏性神经节损伤(更具体地说是苍白球腔隙性梗死),导致衰弱性运动障碍 [Korf et al 1986, Heidenreich et al 1988].
部分mut 缺乏、cblA 或 cblB 的患者也可出现孤立的肾小管酸中毒或慢性肾功能衰竭[Dudley et al 1998, Coman et al 2006].
甲基丙二酰辅酶A差向异构酶缺乏症。 MCEE 中的致病性变异是持续性中度甲基丙二酸尿症的一种非常罕见的原因。在具有 MCEE 突变的婴儿/儿童中的发现的表现范围从完全没有症状到严重的代谢性酸中毒,初始表现时尿液中 MMA 和 2-甲基柠檬酸和酮增加[Dobson et al 2006, Gradinger et al 2007]。症状包括共济失调、构音障碍、张力减退、轻度痉挛性截瘫和癫痫发作;然而,许多受累的人来自近亲婚配的婚配——包括第一个被确诊的个体,他们也患有由 SPR 的纯合性致病变异引起的多巴反应性肌张力障碍,SPR基因编码 sepiaterin 还原酶 [Bikker et al 2006].
次要并发症。尽管对孤立的甲基丙二酸血症和早期症状诊断的认识有所增加,但孤立性甲基丙二酸血症仍然与大量发病率和死亡率相关 [de Baulny et al 2005, Dionisi-Vici et al 2006, Kölker et al 2015b],由潜在缺陷导致[Hörster et al 2007]。 mut0型和 cblB 亚型个体的死亡率和神经系统并发症的发生率高于 mut–型和 cblA 个体。
主要的继发并发症包括:
- 智力残疾 即使患有严重疾病的人也可能存在或不存在智力障碍。在一项基于调查的回顾性研究中,大约 50% 的 mut0 型个体和 25% 的 cblA/cblB 酶亚型个体的智商低于 80 且有明显的神经功能障碍[Baumgarter & Viardot 1995].
在另一项研究中,大约 50% 的 mut0、85% 的 mut–、48% 的 cblA 和 70% 的 cblB 的 IQ 超过 90[Hörster et al 2007]。
在最近的一项自然史研究中,所有患有孤立的甲基丙二酸血症的个体(n = 37)的平均 FSIQ 为 85.0 ± 20.68,处于较低的平均范围(80 ≤ IQ ≤89)。 cblA (n = 6) 和 mut 的个体在产前诊断或通过 新生儿筛查 (n = 3) 的 FSIQ 在平均范围内 (90 ≤ IQ ≤09)。 发病年龄、诊断时存 在严重的高氨血症以及癫痫病史与更严重的损伤相关 [O'Shea et al 2012]。 [O'Shea et al 2012].
- 肾功能进行性损害的肾小管间质性肾炎。所有患有孤立的甲基丙二酸血症的个体,即使是轻度 受累的或接受过同种异体肝移植的人 [Nyhan et al 2002],都有发生肾功能不全的风险[Walter et al 1989, Kruszka et al 2013]。终末期肾病 (ESRD) 在 mut0型 (61%) 和 cblB (66%) 型的个体中很常见,而在 cblA (21%) 型的个体中发生率较低 [Hörster et al 2007]。
甲基丙二酸的继发性线粒体功能障碍多于直接的肾毒性。已在人类和小鼠研究中显示主要在近端小管中的细胞特异性线粒体病理与细胞 色素 c 氧化酶缺乏和尿液和血浆中氧化应激标志物增加有关[Atkuri et al 2009, Mc Guire et al 2009, Manoli et al 2013, Zsengellér et al 2014].
在代谢失代偿的情况下可见的急性肾综合征 [Stokke et al 1967],需要进一步的临床研究。此外,肾小管功能障碍表现为尿液浓缩能力和酸化降低、低肾素血症性低醛固酮增多症、4 型肾小管酸中毒和高钾血症已在许多受累的个体中报告,并得到小鼠研究的支持 [Walter et al 1989, D'Angio et al 1991, Pela et al 2006, Manoli et al 2013]. - 神经系统发现 有些人在急性代谢失代偿期间会出现“代谢性中风”或基底节梗死(典型的苍白球),这会导致运动障碍 [Korf et al 1986, Heidenreich et al 1988]。不同研究组中报告的发病率为 17%-30%[Baumgarter & Viardot 1995, Hörster et al 2007]。苍白球的不同部分(有时是大脑脚中的黑质)是受累的,这表明对梗塞机制的不均匀的细胞特异敏感性[Baker et al 2015]。髓鞘形成延迟、鳃盖不完全、皮层下白质改变以及脑干和小脑改变已有报道[Harting et al 2008, Radmanesh et al 2008].值得注意的是,接受肝和/或肾移植的个体可以在没有明显代谢失代偿的情况下发展为急性损伤,这表明大脑中的酶缺乏保持不变,并且尽管有其他部位的移植后改善,但在 CNS 隔室中的有毒代谢物依然存在且可能会导致损伤[Chakrapani et al 2002, Kaplan et al 2006, Vernon et al 2014].
- 胰腺炎。孤立的甲基丙二酸血症的胰腺炎发病率未知,但它是公认的并发症 [Kahler et al 1994]。它可以急性或慢性发生。胰腺炎可能未被充分认识,因为它可以非特异性地表现为呕吐和腹痛。
- 成长落后。生长落后是常见的,而且是多因素的。这是严重慢性疾病的结果,也可能因慢性肾功能衰竭导致的相对蛋白质营养不良,许多婴儿的身高和体重都低于正常值的三个标准差。一些儿童有生长激素 (GH) 缺乏症的记录,但对 GH 治疗的反应可能会有所不同(见Management)。
- 功能性免疫损伤。这导致对严重感染的易感性增加,尤其是真菌和革兰氏阴性菌 [Oberholzer et al 1967, Wong et al 1992].
- 骨髓衰竭。在代谢失代偿发作期间,患者可能表现出全血细胞减少症,伴有骨髓发育不全和/或发育异常,在支持治疗下最常恢复正常。
- 视神经萎缩。与急性视力丧失相关的迟发性视神经萎缩,类似于线粒体疾病 Leber hereditary optic neuropathy (LHON) 的表现,已在孤立的甲基丙二酸血症中[Wasserstein et al 1999, Williams et al 2009, Pinar-Sueiro et al 2010, Traber et al 2011]以及丙酸血症有报道 [Williams et al 2009, Martinez Alvarez et al 2016].
- 肝母细胞瘤。在 mut MMA 患者的肝脏或供体肝脏中报告了孤立的肝母细胞瘤病例;然而,这些患者的总体癌症发病率尚不清楚 [Cosson et al 2008, Chan et al 2015]
近年孤立的甲基丙二酸血症的存活率有所提高[Matsui et al 1983, van der Meer et al 1994, Baumgarter & Viardot 1995, Nicolaides et al 1998, Kölker et al 2015a].
在mut0 型的患者中,1 岁时的存活率从 1970s的 65% 提高到 1990s的 90% 以上;五年生存率从 1970s的 33% 提高到 1990s的 80% 以上。
在一个系列研究中,比较了近年 mut0型患者的中位死亡年龄: 1970s100% 的中位年龄为 1.6 岁, 1980s中位年龄为 7.6 岁的为 50%,以及在 1990s,20% 的人死于中位年龄 2.2 岁。 mut0型(中位死亡年龄 2 岁)的总死亡率约为 50%,而 cblB 型(中位死亡年龄 2.9 岁)为 50%,mut–型为 40%(中位死亡年龄4.5 岁),cblA 型约占 5%(出生14 天后死亡 1 人) [Hörster et al 2007]。
早期器官移植对总生存期的影响尚未得到系统研究。
新生儿筛查效果。新生儿筛查 (NBS) 检测到的婴儿数量有限且随访时间短,因此无法就 NBS 对甲基丙二酸血症长期结局的影响得出结论 [Leonard et al 2003, Dionisi-Vici et al 2006]。此外,必须强调的是,在 NBS 结果可用之前,大量具有 mut0型的婴儿可能会出现临床症状。对 cblA型同胞的有限观察表明,从新生儿期开始接受治疗的个体的智商明显优于在症状发作后被诊断出的年长受累的同胞 [Hörster et al 2007].
值得注意的是,通过新生儿筛查 ,cblA 和 cblA的儿童在出现脑病和苍白斑损伤之前,被 NBS 检测到且症状出现之前治疗,理论上可以避免这种情况。
基因型-表型相关性
精确的genotype-phenotype correlations很难确定,因为大多数受累的个体都是复合杂合子。
纯合性MMUT 致病性变异 p.Asn219Tyr 通常与严重的变位酶缺乏相关(即mut0 型)[Acquaviva et al 2001]. p.Arg108Cys,也与mut0 型相关,在西班牙裔个体中更常见[Worgan et al 2006]。
临床表型取决于许多因素, 基因型无法准确预测,包括全身酶活性、体内对钴胺素的反应、环境因素,以及替代丙酰辅酶 A 处理途径的效率和激活。通过基于稳定同位素研究估计全身残余代谢能力,可以更好地了解孤立的甲基丙二酸血症的临床相关性 [Leonard 1997]。
患病率
一些研究估计了孤立的甲基丙二酸血症的患病率 [Sniderman et al 1999]。在魁北克对孤立性甲基丙二酸血症进行尿液筛查后,在大约 1:80,000 的筛查新生儿中发现了“症状性甲基丙二酸尿症” [Sniderman et al 1999],观察与Chace et al [2001]在美国通过质谱筛查的 908,543 名新生儿中发现的10例孤立性甲基丙二酸血症的观察结果相似。
在日本,患病率可能高达 1:50,000 [Shigematsu et al 2002]。
因此,孤立的甲基丙二酸血症的患病率似乎在 1:50,000 和 1:100,000 之间;然而,准确数字需要更大规模的研究。
遗传相关(等位基因)疾病
除本 GeneReview 中讨论的表型外,没有已知表型与 MMUT、MMAA、MMAB 或 MCEE 中双等位基因的致病变异相关。
据报道,一名患有mut0MMA 和胰岛素依赖型糖尿病的个体是由父系6号染色体同源引起的[Abramowicz et al 1994]。
MMADHC 双等位基因的致病性变异也与 cblD-复合型(甲基丙二酸血症/酸尿症和高同型半胱氨酸血症/同型半胱氨酸尿症)和 cblD-同型半胱氨酸尿症(高同型半胱氨酸血症/同型半胱氨酸尿症)相关,这些在Disorders of Intracellular Cobalamin Metabolism中进行了讨论。
鉴别诊断
非典型甲基丙二酸血症与尿中甲基丙二酸排泄增加(通常是轻度)有关。罕见的缺陷,如琥珀酸辅酶A连接酶缺乏症、丙二酸和甲基丙二酸尿症、cblX缺乏症、转钴胺素受体缺陷和甲基丙二酸半醛脱氢酶缺乏症,可导致甲基丙二酸血症/酸尿症,尽管大多数患者会有额外的生化检查发现。
唯一已知的与细胞内钴胺素代谢途径相关的 X-linked疾病是 cblX 缺乏症,由 HCFC1 突变引起,合并甲基丙二酸血症和高同型半胱氨酸血症、严重智力障碍、复杂性癫痫发作和其他神经系统发现相关。 cblX 缺乏症是最近描述的一种谱系未知的疾病,但可能包括 X 连锁发育迟缓,无生化异常或者孤立的甲基丙二酸异常升高。
“良性”甲基丙二酸血症。魁北克省的新生儿筛查发现有轻度至中度尿甲基丙二酸排泄的婴儿。随访显示超过 50% 的儿童消退,一些儿童出现明显的良性、持续性、低中度甲基丙二酸血症 [Ledley et al 1984, Sniderman et al 1999]。已经报道了其他具有相对良性甲基丙二酸血症类型的个体 [Coulombe et al 1981, Martens et al 2002]。对这些个体进行随访时需要谨慎,因为有些个体可能属于中度 mut–型,具有发生急性代谢危机的显著风险 [Shapira et al 1991]。
这些个体的长期结果和临床表型有待进一步关注。值得注意的是,一个亚组具有丙二酸和甲基丙二酸血症的联合生化表型,因此可能是由 ACSF3 缺乏引起的丙二酸合并甲基丙二酸尿症 (CMAMMA)。
ACSF3 缺乏引起的丙二酸合并甲基丙二酸尿症 (CMAMMA)。 CMAMMA 患者的尿液或血浆中丙二酸 (MA) 和甲基丙二酸 (MMA) 水平较高,MMA 排泄量通常高于 MA 排泄量 (MMA/MA >5)。由于 C3(丙酰肉碱)不升高,CMAMMA婴儿不是通过新生儿筛查确诊,后者基于干血点 M的丙酰肉碱分析。
表型谱很广,从完全无症状的个体到患有神经系统表现(癫痫发作、记忆问题、精神疾病和/或认知能力下降)的成人或具有广泛表现的儿童,如昏迷、酮症酸中毒、低血糖、生长落后、转氨酶升高、小头畸形、肌张力障碍、肌张力减退和/或发育迟缓。这种疾病的完整自然史仍有待阐明。
ACSF3 的突变(编码甲基丙二酰和丙二酰辅酶 A 合成酶,产生第一个底物丙二酰辅酶 A,用于线粒体内脂肪酸合成)是致病原因[Alfares et al 2011, Sloan et al 2011].
丙二酸甲酯半醛脱氢酶缺乏症(MMDSH)。在缬氨酸降解途径的最后酶促步骤中,3-羟基异丁酸脱氢酶将 3-羟基异丁酸转化为 (S)-甲基丙二酸半醛 (MMSA),甲基丙二酸半醛脱氢酶 (MMDSH) 将 (S)-甲基丙二酸半醛转化为丙酰辅酶 A)。值得注意的是,相同的酶催化由胸腺嘧啶代谢产生的 (R)-甲基丙二酸半醛氧化脱羧为丙酰辅酶 A。
少数具有编码 MMSDH 酶的 ALDH6A1 致病性变异的患者具有极其不同的生化表型:一些患者表现出 3-羟基异丁酸尿症[Chambliss et al 2000, Sass et al 2012],而另一些患者也表现出短暂的甲基丙二酸酸血症/酸尿 [Marcadier et al 2013]。他们还具有极其多变的临床表型,包括与严重的脑髓鞘形成缺陷相关的严重智力障碍。
转钴胺素受体缺陷 (TCblR/CD320)。有关病例和另外四名受累的个体是在 NBS 中发现的无症状新生儿,其 C3 和 C3/C2 比值升高。血浆和尿液 MMA增加以及正常的血清维生素 B12水平;四人中有两人同型半胱氨酸升高。
在有关病例中,生化异常通过单次羟钴胺素注射恢复正常,并在 9 个月内保持正常[Quadros et al 2010]。成纤维细胞显示出对转钴胺素的摄取减少。
近亲婚配的父母所生的患有视网膜动脉阻塞的7周大男孩中也发现了 CD320 致病性变异 [Karth et al 2012]。所有报告的受累的个体均为 NM_016579.3:c.262_264del (p.Glu88del) 的 纯合性变异。在爱尔兰研究队列中,TCblR 中的多态性与神经管缺陷风险增加有关 [Pangilinan et al 2010].
合并甲基丙二酸血症hyperhomocysteinemia/homocystinuria。 干扰钴胺素细胞内代谢的疾病会导致腺苷钴胺素和/或甲基钴胺素合成受到干扰。 然而,这些情况通常伴有临床上显著的高同型半胱氨酸血症。 下列疾病包括在这组疾病中:
- 钴胺素C缺乏症 (cblC) 可能是intracellular cobalamin metabolism最常见的先天性错误。 患有这种疾病的个体几乎总是血浆同型半胱氨酸和甲基丙二酸浓度升高,蛋氨酸水平低,发病年龄差异很大。 受影响的个体经常出现发育迟缓并发展为色素性视网膜病变和“牛眼”黄斑病变。 cblC 是由 MMACHC 中双等位基因的致病性变异引起的,该变异编码参与细胞内钴胺素加工和运输的蛋白质。 致病性变异c.271dupA;p.Arg91LysfsTer14 约占等位基因的 40% [Lerner-Ellis et al 2006].
- 互补群 cblD、cblF 和 cblJ 的缺陷是极为罕见的常染色体隐性遗传疾病。
- cblD 缺乏症 具有生化异质性[Suormala et al 2004],Coelho et al [2008]确定 MMADHC(以前称为 C2ORF25)的突变是导致 cblD 和确定基因型/表型相关性的原因。 MMADHC 具有多个翻译起始密码子 (ATG),并编码远距离多肽。 因此, 致病性变异的位置和性质决定了患者是否会出现甲基丙二酸尿、高胱氨酸尿或两种代谢异常:注意:具有互补 cblD-高半胱氨酸尿症 [Coelho et al 2008]、cblE(蛋氨酸合成还原酶)和 cblG(蛋氨酸合成酶)异常的个体没有甲基丙二酸血症,而是由甲基钴胺素合成受损引起的孤立的高胱氨酸尿症/高同型半胱氨酸血症。
- cblF 缺乏是由 LMBRD1 的突变引起的,LMBRD1 编码溶酶体钴胺素输出蛋白[Rutsch et al 2009],影响甲基丙二酰辅酶 A 变位酶(由 MMUT 编码)和甲基四氢叶酸酶的辅助因子的合成:同型半胱氨酸甲基转移酶,也称为蛋氨酸合成酶 (MS)(由 MTR 编码)。
- 由 ABCD4 突变引起的 cblJ 缺陷,一种 ATP 结合盒 (ABC) 转运蛋白,影响 Cbl 溶酶体释放到细胞质中,类似于 cblF,表现为肌张力减退、嗜睡、喂养不良、骨髓抑制、大红细胞性贫血和先天的一些患者的心脏病[Coelho et al 2012]。
值得注意的是,一些患有 cblF 或 cblJ 的人的血清维生素 B12水平可能会降低,这表明溶酶体在肠道摄取摄入的钴胺素中起作用。 - cblX 缺陷是由X-linked 基因HCFC1 的突变引起的,HCFC1 是一种影响 MMACHC 表达的转录共调节因子。迄今为止,所有描述受累的男性都患有 MMA 血症和 MMAuria,并且大多数在研究时都表现出高同型半胱氨酸尿症和甲基丙二酸血症。临床表型特征为顽固性癫痫和严重的神经认知障碍,没有 cblC 缺乏症的特异性牛眼黄斑病变[Yu et al 2013];然而,表型需要进一步研究。
维生素B12缺乏。缺乏维生素B12的人可能有甲基丙二酸血症和高胱氨酸尿症。
母体维生素B12缺乏可导致婴儿出现甲基丙二酸血症综合征,从严重脑病到血清丙酰肉碱(C3) 浓度升高,可通过新生儿筛查 [Chace et al 2001]。这种代谢异常可能发生在纯素食母亲的母乳喂养婴儿、患有亚临床恶性贫血的母亲所生的婴儿[Marble et al 2008]以及接受过胃手术的母亲所生的婴儿 [Grange & Finlay 1994, Celiker & Chawla 2009]。母亲的维生素B12血清浓度不一定非常低。使维生素B12血清浓度正常化的肌肉注射维生素 B12替代疗法可逆转代谢异常。
甲基丙二酸升高的线粒体脑肌病。轻度甲基丙二酸尿已在琥珀酸连接酶 α 亚基缺陷(由双等位基因的SUCLG1 致病变异引起)和琥珀酸连接酶 ADPβ 亚基缺陷(由双等位基因 SUCLA2 致病变异引起)中描述,与线粒体 DNA缺陷相关,并伴有严重的乳酸酸中毒和脑肌病。
琥珀酰辅酶A连接酶(SUCL)催化琥珀酰辅酶A和ADP或GDP可逆转化为琥珀酸和ATP或GTP,并且包含由SUCLG1编码的α亚基和由SUCLA2或SUCLG2编码的β亚基。
SUCLG1双等位基因致病变异导致严重的表型,与乳酸酸中毒和生命第一周的早期死亡有关。 (参见 SUCLG1-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria.)
双等位基因 SUCLA2 致病性变异与肌张力减退、大约 3 到 6 个月出现肌肉萎缩(mtDNA缺陷、肌肉中复合体 I、III 和 IV 缺乏)、多动症、癫痫发作、严重听力障碍和生长障碍有关。患者会出现 Leigh 综合征样疾病、皮质和基底节萎缩以及肌张力障碍。一些 受累的人在婴儿期死亡;其他人则活到了 20 多岁。 (参见SUCLA2-Related mtDNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria.)。
这些个体的甲基丙二酸尿症表现为 10 至 200 mmol/mol 肌酐,并伴有乳酸、柠檬酸甲酯、3-羟基丙酸和 3-羟基异戊酸、丙酰肉碱和 C4-二羧酸肉碱 (C4DC) 的血浆浓度升高 [Elpeleg et al 2005, Carrozzo et al 2007, Ostergaard et al 2007, Morava et al 2009].
雷氏样综合征。面对轻度并发感染时出现肝肿大和反应迟钝的 Reye 样综合征可被视为许多先天性代谢障碍的未被识别的表现,包括孤立的甲基丙二酸血症 [Chang et al 2000].
其他疾病。尽管甲基丙二酰辅酶A变位酶活性正常,但仍可显示甲基丙二酸血症的其他疾病包括:
- “非典型”甲基丙二酸血症伴进行性神经退行性疾病、小头畸形和白内障(2 位同胞) [Strømme et al 1995]或伴有线粒体耗竭综合征/复杂 IV 缺乏症以及丙酸和甲基丙二酸血症合并症(1 人) [Yano et al 2003].这些病例与SUCLA2突变引起的表型有相似之处。
- 伴有远端肾小管酸中毒的良性甲基丙二酸血症(一家同胞)[Dudley et al 1998]
- 丙二酰辅酶A脱羧酶缺乏症,通常与甲基丙二酸和丙二酸尿症有关,丙二酸水平明显高于甲基丙二酸水平 [Brown et al 1984]
- 孤立的甲基丙二酸尿症和正常血浆浓度的甲基丙二酸血症(2 个家族)[Sewell et al 1996, Martens et al 2002]
处理
初步诊断后的评估
为了确定诊断为孤立的甲基丙二酸血症的个体的疾病程度和需求,建议进行以下评估:
- 血生化(Na+、K+、CI-、葡萄糖、尿素、肌酐、碳酸氢盐、AST、ALT、碱性磷酸酶、胆红素 [T/U]、甘油三酯和胆固醇);带分类的全血细胞计数;动脉或静脉血气;血浆氨和乳酸浓度;标准的尿液分析和酮体测量;定量血浆氨基酸;通过气相色谱和质谱 (GC-MS) 进行尿液有机酸分析
- 如果可能,测量甲基丙二酸、柠檬酸甲酯、游离和总肉碱的血浆浓度,以及记录丙酰肉碱(C3 种类)浓度的酰基肉碱谱
- 测量血清维生素B12浓度以确定患者和可能的母亲(新生儿)是否存在营养缺乏
- 生化遗传学咨询
治疗表现
对于甲基丙二酸血症的急性和慢性并发症的治疗,各个代谢研究中心之间没有达成共识。最近由 12 个欧洲国家和美国的专业人士根据严格的文献评估和专家组会议制定的指南概述了当前的管理建议和进一步研究的领域[Baumgartner et al 2014].
危重病人的稳定
- 用等渗溶液进行扩容
- 所有 IV 溶液都应含有葡萄糖,最好是D10或D12.5,如果出现高血糖,可能需要输注胰岛素。
- 应通过重复电解质和静脉或动脉血气测量连续跟踪,并根据需要通过液体和碳酸氢盐补充进行校正 [Baumgartner et al 2014]。必须提供足够的千卡。可能需要中枢或外周全胃肠外营养 (TPN),它通常包含葡萄糖和氨基酸,在某些情况下还包含脂质。总蛋白给药通常完全停止不超过 24-48 小时,并根据患者的酸碱平衡和测试值(包括氨、乳酸和血浆氨基酸等)逐渐恢复。
- 对于胰腺炎的风险,必须谨慎使用脂质输注。
- 肉碱可以 50-100 mg/kg/d bid-qid 静脉内给药。
- 需要监测尿量和血清钠和钾浓度。
- 考虑到临床情况,应尽快在肠内重新引入膳食蛋白质,并且可能需要进一步增加 TPN。应强烈考虑鼻饲或口饲,以便立即重新引入肠 内营养。
- N-氨基甲酰谷氨酸(NCG,Carbaglu®)可考虑用于高氨血症。 NCG 变构激活 CPS1(氨甲酰磷酸合成酶 1),这是尿素循环的第一步。它可以有效地使缺乏 NAGS(N-乙酰谷氨酸合酶)的患者的血氨浓度正常化,并且还可以使一些丙酸血症和可能的甲基丙二酸血症患者受益 [Tuchman et al 2008, Ah Mew et al 2010].
- 如果治疗失败(无法控制的酸中毒和/或高氨血症),可能需要进行血液透析或血液滤过。
给家属备用一封信,急诊提交给急诊科的医生,说明推荐的急性管理方案应该是标准护理。
Medic Alert® 手环和概述液体和电解质疗法的紧急治疗方案应适用于所有受累的个人。
其他
除了危重病发作外,病毒感染等并发疾病的患者或因各种原因接受手术的患者应进行积极的液体、代谢和营养管理。
物理治疗、物理治疗和职业治疗方面的专家可以帮助解决患者和家属面临的复杂挑战,最大限度地提高功能并提高生活质量 [Ktena et al 2015b].
在该患者群体中麻醉剂选择的可能特殊考虑[Ktena et al 2015a, Ruzkova et al 2015].
大多数人需要“病假”管理方案,通常包括减少或消除蛋白质摄入量以及增加液体和葡萄糖以确保提供足够的卡路里并阻止脂肪分解。如果体征提示并发感染,通常需要立即住院治疗。
尽管脆弱的个体可能需要上述所有治疗方法,但它们仍可能无法避免死亡、代谢失代偿的严重后遗症(例如,基底神经节的代谢性卒中)或肾脏疾病。需要对治疗模式和预后进行相关性识别,以便为患有孤立的甲基丙二酸血症的个体制定更有效的管理方案。
许多受累的人因为厌食和呕吐需要胃造口术/胃空肠造口术管喂养,以确保热量和液体摄入并促进生长。
在极少数情况下代谢失代偿发作期间的骨髓衰竭需要粒细胞集落刺激因子 (GCSF)。
贫血是慢性肾功能衰竭的一种常见并发症,可通过促红细胞生成素和最终的肾移植治疗 [Inoue et al 1981, Guerra-Moreno et al 2003, MacFarland & Hartung 2015].
一些儿童有生长激素 (GH) 缺乏症的记录;然而,由于对 GH 治疗的反应可能不同,因此需要仔细调整饮食和 GH 替代剂量 [Bain et al 1995, Al-Owain et al 2004]. GH 替代疗法的适应症及反应需要进一步研究。
预防主要表现
饮食管理
营养. 病情稳定后,营养管理至关重要。这通常包括制定低蛋白、高热量的饮食。如果可用,准确评估静息能量消耗可以指导饮食和热量处方并避免过度喂养 [Hauser et al 2011].
天然蛋白质的输入需要仔细计算以供正常生长,同时避免过量的丙酸氨基酸(异亮氨酸、缬氨酸、蛋氨酸和苏氨酸) 。这些患者终生需要根据临床和实验室发现调整膳食全(完全)蛋白质摄入量。
FAO/WHO/UNU report报告 [2007] 建议,每个年龄组的安全水平应该是天然蛋白质摄入的目标 [Baumgartner et al 2014];然而,规定的个体蛋白质量将取决于生长参数、代谢稳定性、肾衰竭阶段和其他因素。现已经提供了一些缺乏氨基酸的配方奶粉(例如 Propimex®-1/2、XMTVI-1/2、OA-1/2)和不含蛋白质的配方奶粉(例如 Phree®, Duocal®),给患者提供额外的液体和卡路里。随着婴儿的成长,总蛋白质负荷根据生长、血浆氨基酸浓度以及血浆和尿液甲基丙二酸浓度缓慢降低。
值得注意的是,在蛋白质耐受性低的患者中,严格限制丙酸氨基酸前体(异亮氨酸、缬氨酸、蛋氨酸和苏氨酸)可产生营养缺乏状态。此外,通过 MMA 配方摄入相对较高的亮氨酸可诱导医源性必需氨基酸缺乏,这会对长期生长和可能的其他结果产生负面影响[Manoli et al 2016b].药用食品应适量食用,必需蛋白质与促生氨基酸缺乏配方的相对摄入量不超过1:1。应避免单独补充缬氨酸或异亮氨酸。
这些饮食指南不适用于患有CblC deficiency的患者,这是该途径中的另一种单独疾病 [Manoli et al 2016a].
羟钴胺注射液。对维生素B12有反应的个体通常需要每天至隔天注射 1.0 毫克。B12注射方案需要根据患者的年龄和可能的体重进行单独调整。
肉碱可以以 50-100 毫克/公斤/天的剂量给药,最高约 300 毫克/公斤/天。作为膳食补充剂,肉碱可替代游离肉碱池,增强丙酰肉碱的结合和排泄。然而,丙酰肉碱排泄对总丙酸盐负荷的贡献很小。细胞内 CoA 积聚的缓解可能是肉碱补充剂有益于某些个体的机制。
抗生素.可以使用多种抗生素方案来减少肠道菌群中丙酸盐的产生:
- 口服新霉素,250 mg,每天 4 次,是Snyderman et al [1972]报道的原始方案。
- 10-15 mg/kg/天的甲硝唑也有报道。
受累个体接受治疗的间隔可能会有所不同,但典型的疗程是每1到3个月治疗1周到10天。
尽管口服抗生素可以减少受累个体肠道菌群产生的丙酸盐负荷,但长期抗生素治疗并非无害;它引入了具有抗药性菌群的个体重新繁殖的风险。这可能会造成严重的感染威胁,并且对患有孤立性甲基丙二酸血症的人尤其危险,因为大多数死亡与代谢失代偿有关,通常由感染引起。
应通过定时收集尿液或血浆甲基丙二酸浓度与该个体的基线值相比降低来确定接受抗生素治疗的患者对抗生素给药的反应。
受累的个体接受治疗的间隔可能会有所不同,但典型的疗程是每 1 到 3 个月治疗 1 周到 10 天。
尽管口服抗生素可以减少受累的个体肠道菌群产生的丙酸盐负荷,但长期抗生素治疗并非无害;它引入了具有抗药性菌群重新繁殖的风险。这可能会造成严重的感染威胁,并且对患有孤立的甲基丙二酸血症的人尤其危险,因为大多数死亡与代谢失代偿有关,通常由感染引起。
应通过定时收集尿液或血浆甲基丙二酸浓度与该个体的基线值相比来确定接受抗生素治疗的患者对抗生素给药的反应。
某些人可能会考虑轮换抗生素治疗方案。
抗氧化剂.一名患有孤立的甲基丙二酸血症的个体在严重代谢危机后被证明缺乏谷胱甘肽,对抗坏血酸治疗有反应 [Treacy et al 1996]。最近的几项研究记录了甲基丙二酸血症mut0型患者的氧化应激增加、谷胱甘肽消耗和特定呼吸链复合物缺乏症 [Schwab et al 2006, Atkuri et al 2009, Chandler et al 2009, de Keyzer et al 2009, Manoli et al 2013],表明在这些患者中使用抗氧化剂或其他线粒体靶向疗法治疗的潜在益处。
辅酶 Q10 和维生素 E 方案已被证明可预防 MMA 患者急性视神经受累的进展[Pinar-Sueiro et al 2010],并显示可减轻 MMA 小鼠模型中肾脏疾病的进展 [Manoli et al 2013]。
器官移植
接受肝和/或肾移植的人数、对潜在代谢紊乱的详细影响以及接受此手术的人的预后总体结果尚未确定 [Sloan et al 2015].。在移植患者的病例系列中包含酶和基因型信息将允许更好地比较结果和基因型-表型关联,这可以为个别病例的移植适应症和时机的决定提供信息。
肝移植.因为丙酸盐的大部分代谢转化发生在肝脏中,更换肝脏可以提供足够的酶活性来避免代谢失代偿。肝移植已被证明在很大程度上可以防止代谢不稳定,但不能治愈,患有 MMA 孤立的MMA 的个体仍面临肾病、基底节损伤和神经系统并发症等长期并发症的风险 [Chakrapani et al 2002, Nyhan et al 2002, Kaplan et al 2006, Vernon et al 2014]。迄今为止,超过 35 名患有孤立性甲基丙二酸血症的个体接受了活体供体或尸体、原位或部分肝移植或肝肾联合移植(>20 名患者)[van't Hoff et al 1998, van't Hoff, McKiernan et al 1999, Kayler et al 2002, Nyhan et al 2002, Hsui et al 2003, Kasahara et al 2006, Morioka et al 2007, McGuire et al 2011, Niemi et al 2015]。
- 尽管存在持续的代谢异常,但在接受肝移植的个体中,潜在的生化参数和代谢失代偿的频率显著改善 [Nyhan et al 2002, Kaplan et al 2006, Niemi et al 2015],可能是由于肝外甲基丙二酸的产生增加,主要来自骨骼肌[Chandler et al 2007]。
- 肝移植后,一些人继续出现进行性肾功能衰竭以及脑脊液中甲基丙二酸的高浓度 [Nyhan et al 2002, Kaplan et al 2006]。
- 移植后的神经系统并发症,包括苍白球损伤 [Chakrapani et al 2002, Cosson et al 2008, McGuire et al 2011]表明移植后应继续进行适当的蛋白质限制和支持性护理。随着手术技术和预后的改善,尤其是对于mut0患者(非常脆弱)的早期移植正在获得支持 [Niemi et al 2015, Spada et al 2015]。对于家庭和治疗医生来说,选择移植手术的种类和时间仍然具有挑战性 [Sloan et al 2015]。从长远来看,有关肾脏疾病、视神经萎缩和神经系统并发症发展的细节将是最重要的。
肾移植. 有些人只接受了同种异体肾移植[Van Calcar et al 1998, Lubrano et al 2001, Coman et al 2006, Cosson et al 2008, Clothier et al 2011].
关于 mut0甲基丙二酸血症中 孤立的肾移植的首批报告之一声称提供足够的酶活性以使甲基丙二酸排泄正常化并允许增加膳食蛋白质耐受性;然而,后来确定该患者患有 cblA 缺乏症并对维生素B12有反应。因此,该患者病情较轻,不能代表严重 MMA 亚型(mut0 or cblB)患者的孤立肾移植结果 [Lubrano et al 2001, Lubrano et al 2007, Lubrano et al 2013]。
择期肾移植,甚至在肾病发作之前,已被提倡作为“细胞疗法”的一种形式,以帮助稳定患有 mut0 MMA 的个体 [Brassier et al 2013]。然而,一名患者在发生肝母细胞瘤、伴有脑脊液乳酸酸中毒的神经功能恶化和多器官衰竭后死亡;第二名患者出现进行性神经系统症状;另外两人在移植后出现代谢失代偿。需要进行长期随访以确定这是否是肝移植或肝肾移植的安全替代方案,尤其是对于患有严重mut0MMA 的人。
预防继发性并发症
必须经常监测血浆氨基酸以避免必需氨基酸(特别是异亮氨酸、缬氨酸和蛋氨酸)的缺乏,这是由于蛋白质过度限制和甲基丙二酸尿症中发生肢端皮炎-肠病样皮肤损伤的结果,就像在其他有机酸尿症(戊二酸尿症-I)和氨基酸疾病(maple syrup urine disease) [De Raeve et al 1994]。
低血浆氨基酸可反映天然蛋白质摄入量低、使用代谢配方导致支链氨基酸摄入不平衡或慢性酸中毒对支链氨基酸代谢的影响 [Manoli et al 2016b].
监视
在生命的第一年,婴儿可能需要像每周一样频繁地进行评估。尚未发布关于推荐的实验室测试或频率的指南。
如果患者病情不稳定并需要频繁改变管理,则应定期监测以下内容,每 6 个月至 1 年或更频繁:
- 血浆氨基酸
- 血浆和尿液 MMA 水平
- 血清酰基肉碱和游离和总肉碱水平
- 生化:Na+、K+、CI-、葡萄糖、尿素、肌酐、碳酸氢盐、AST、ALT、碱性磷酸酶、胆红素 (T/U)、甘油三酯和胆固醇
- 肝脏、肾脏和骨骼健康
- 骨髓指标
定期用肌酐、胱抑素-C 监测肾功能,如果可能的话,肾小球滤过率 (GFR) 研究(例如碘海醇血浆衰变),除了肾脏成像外,将有助于及早转诊至肾内科并进行适当的检查,需要时进行肾移植 [van't Hoff et al 1999, Kruszka et al 2013]。基于肌酐和胱抑素-C 的联合方程有望更准确地反映该患者群体的肾功能[Schwartz et al 2009]。
建议定期进行眼科和听力学评估,以筛查视神经变薄/苍白和听力损失[作者,未发表的观察结果]。
要避免的药物/情况
应避免以下情况:
- 禁食。在急性疾病期间,需要摄入足够的卡路里来阻止/防止失代偿。
- 压力
- 增加膳食蛋白质
- 补充单独的丙酸氨基酸缬氨酸和异亮氨酸,因为它们直接增加丙酸氧化紊乱患者的毒性代谢物负荷 [Nyhan et al 1973, Hauser et al 2011, Manoli et al 2016b]
评估有风险的亲属
根据先证者的基因型和表型,对有风险的同胞进行生化检测,如果同胞受累的,应尽快进行治疗。分子遗传学检测(如果家族中的致病变异已知)或细胞酶学通常可以进一步确认生化研究的结果。对有风险的同胞进行产前诊断可以在分娩时对受影响的新生儿进行及时治疗。
有关为遗传咨询目的而对高危亲属进行测试的相关问题,请参阅Genetic Counseling。
怀孕管理
怀有维生素B12反应性 MMA 胎儿的孕妇口服和肌肉注射维生素B12,导致母体 MMA 尿量减少 [Ampola et al 1975, van der Meer et al 1990]。尽管有这些观察结果,母体维生素B12对 孤立的MMA 的补充仍需进一步研究。
尽管母体 MMA 水平较高,但所有报道的 MMA 女性妊娠的胎儿生长和发育均正常[Wasserstein et al 1999, Deodato et al 2002]。
在 MMA 女性妊娠中观察到的并发症可能包括急性失代偿或高氨血症、肾功能恶化和产科并发症,包括先兆子痫、早产和剖宫产[Raval et al 2015].
正在研究的疗法
需要精心设计的临床研究来评估抗氧化方案对 MMA 患者的疗效。
基因治疗。人类肝细胞和甲基丙二酸血症动物模型的初步研究表明基因治疗具有潜在益处基因治疗 [Chandler & Venditti 2008, Carrillo-Carrasco et al 2010, Chandler & Venditti 2010, Chandler & Venditti 2012, Sénac et al 2012]。这种治疗方法对患者的影响,特别是对甲基丙二酸血症长期并发症的影响,仍有待在适当的临床研究中阐明。
搜索美国的ClinicalTrials.gov和欧洲的www.ClinicalTrialsRegister.eu,以获取有关各种疾病和病症的临床研究的信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。 本节并非旨在解决个人可能面临的所有个人、文化、道德问题,或替代咨询遗传学专业人士。 —编者。
遗传方式
孤立性甲基丙二酸血症(甲基丙二酰辅酶A变位酶完全或部分缺乏;甲基丙二酰辅酶A辅因子腺苷钴胺素的转运或合成缺陷;以及甲基丙二酰辅酶A差向异构酶缺乏)以常染色体隐性遗传 方式遗传。
家庭成员的风险
先证者的父母
先证者的后代。患有孤立的甲基丙二酸血症的个体的后代是 MMUT、MMAA、MMAB、MCEE 或 MMADHC 致病性变异的杂合子(携带者)。
其他家庭成员。 先证者父母的每个同胞有50%的风险成为 MMUT、MMAA、MMAB、MCEE 或 MMADHC 中 致病性变异的携带者。
携带者(杂合子)检测
对有风险的亲属进行携带者检测需要事先确定家族中的 MMUT、MMAA、MMAB、MCEE 或 MMADHC 致病变异。
分子遗传学检测检测携带者以外的方法不可靠。
遗传咨询相关问题
有关为早期诊断和治疗目的而对有风险的亲属进行检测的信息,请参阅管理,Evaluation of Relatives at Risk。
家庭计划
DNA 银行是存储 DNA(通常从白细胞中提取)以备将来使用。因为将来测试方法和我们对基因、等位基因变异和疾病的理解可能会有所改善,所以应该考虑储存受累的个体的 DNA。
产前检查
产前检测妊娠孤立的甲基丙二酸血症风险为25%,可通过以下方式进行:
- 如果在受累的家庭成员中发现了 MMUT、MMAA、MMAB、MCEE 或 MMADHC 致病变异,则进行分子遗传学检测。
注意:由于细胞生化检测的局限性且周转时间较长, 产前诊断的首选方法是分子遗传学检测。
- 使用母体血浆中胎儿游离 DNA [Gu et al 2014]。
- 生化检测。从历史上看,羊水监测和细胞生化分析都被使用过:
- 通常在约15至18周妊娠或绒毛膜绒毛取样 (CVS) 在约10至12周妊娠时进行羊膜穿刺术获得的培养的胎儿细胞的 14C丙酸盐和互补测定。对绒毛膜绒毛细胞的测试可能会出现假阴性,应随后对培养的羊水细胞进行研究测试[Morel et al 2005]。在进行产前检查之前,必须在 受累的 f家庭成员中通过相同的检测来确认诊断。注意:对于对通过羊膜穿刺术或 CVS 进行 产前诊断不感兴趣的孕妇,尿液有机酸检测可能会有所帮助,因为已证明怀有 受累的胎儿的女性会在其尿液中排泄 MMA [Ampola et al 1975, van der Meer et al 1990]。
植入前遗传学诊断 (PGD) 对于已鉴定出 MMUT、MMAA、MMAB、MCEE 或 MMADHC 致病变异的家族,植入前遗传学诊断 (PGD) 可能是一种选择。
资源
GeneReviews 工作人员选择了以下特定疾病和/或支持组织和/或登记处,以造福患有这种疾病的个人及其家人。 GeneReviews 不对其他组织提供的信息负责。 有关选择标准的信息,请单击 here.
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- Save Babies Through Screening Foundation, Inc.P. O. Box 42197Cincinnati OH 45242Phone: 888-454-3383Email: email@savebabies.org
- Organic Acidemia AssociationPhone: 763-559-1797Fax: 866-539-4060 (toll-free)Email: kstagni@oaanews.org; menta@oaanews.org
- European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD)
分子遗传学
Molecular Genetics 和 OMIM 表格中的信息可能与 GeneReview 中其他地方的信息不同:表格可能包含更新的信息。 —编者。
Table A
孤立的甲基丙二酸血症:基因和数据库
Table B
孤立的甲基丙二酸血症:基因和数据库(View All in OMIM)
MMAA
基因结构。MMAA包含七个外显子; 第一个是非编码[Dobson et al 2002b]。 有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病性变异。 已经描述了超过 20 种致病变异,包括错义, nonsense和剪接变异、缺失和插入[Dobson et al 2002a, Lerner-Ellis et al 2004, Yang et al 2004, Merinero et al 2008].
Table 3
本次 GeneReview 中讨论的 MMAA 致病性变异
| DNA改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.64C>T | p.Arg22Ter | NM_172250 NP_758454 |
| c.161G>A | p.Trp54Ter | |
| c.266T>C | p.Leu89Pro | |
| c.283C>T | p.Gln95Ter | |
| c.358C>T | p.Gln120Ter | |
| c.397C>T | p.Gln133Ter | |
| c.433C>T 1 | p.Arg145Ter 1 | |
| c.503delC 2 | p.Thr168MetfsTer9 2 | |
| c.562G>C | p.Gly188Arg | |
| c.650T>A | p.Leu217Ter | |
| c.653G>A | p.Gly218Glu | |
| c.733+1G>A | -- | |
| c.988C>T | p.Arg330Ter | |
| c.1076G>A | p.Arg359Gln | |
| c.592_595delACTG | p.Thr198SerfsTer6 |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
最常见的致病性变异; 占一项大型研究中发现的突变等位基因的 43% [Lerner-Ellis et al 2004]。 这种变异存在于一个常见的单倍型上,并且在西班牙个体中也有发现[Martínez et al 2005]。
- 2.
在日本,已观察到一种常见的致病性缺失 c.503delC [Yang et al 2004]。
正常基因产物。该基因编码418个氨基酸的蛋白质。基因产物具有线粒体前导序列,属于 G3E GTPases 的 ArgK 蛋白亚家族 [Leipe et al 2002]。虽然最初提出这种蛋白质在钴胺素进入线粒体中发挥作用 [Dobson et al 2002a],但它最近被认为是一种金属伴侣 GTP 酶,可在催化循环期间保护甲基丙二酰辅酶 A 变位酶免受氧化失活并促进辅因子腺苷钴胺素)结合[Korotkova & Lidstrom 2004, Hubbard et al 2007]。
异常的基因产物。 MMAA 基因产物的精确生化功能尚不清楚,但怀疑与细菌中的同源物相似。错义致病变异似乎落在进化上保守的残基或共有剪接位点。在这种情况下,环境、饮食和(可能)epigenetic遗传修饰剂可以定义表型,特别是因为具有纯合性致病变异的个体可能表现出不同的表型 [Lerner-Ellis et al 2004]。
MMAB
基因结构.MMAB 包含九个外显子。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病性变异。已经确定了几种错义, nonsense/移码和 剪接位点致病变异[Dobson et al 2002a, Yang et al 2004, Martínez et al 2005, Lerner-Ellis et al 2006]。超过一半发生在外显子 7 [Lerner-Ellis et al 2006]:
两个晚期发病(3 岁和 8 岁)的非裔美国人后裔都具有三种 MMAB 致病变异:c.403G>A、c.571C>T 和 c.656A>G [Lerner-Ellis et al 2006].
Table 4
本次 GeneReview 中讨论的 MMAB 致病性变异
| DNA 改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.287T>C | p.Ile96Thr | NM_052845.3 NP_443077.1 |
| c.291-1G>A | -- | |
| c.403G>A | p.Ala135Thr | |
| c.556C>T 1 | p.Arg186Trp 1 | |
| c.568C>T | p.Arg190Cys | |
| c.569G>A | p.Arg190His | |
| c.571C>T | p.Arg191Trp | |
| c.572G>A | p.Arg191Gln | |
| c.656A>G | p.Tyr219Cys | |
| c.197-1G>T | -- | |
| c.700C>T | p.Gln234Ter |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
大样本中最常见的致病性变异[Lerner-Ellis et al 2006],,占所有等位基因的 33%,仅见于欧洲血统的 受累的个体中,与症状的早期发作有关(年龄 <1 岁) )。
MMUT
正常基因产物。该基因编码 250 个氨基酸的 ATP 依赖性线粒体蛋白VitB12腺苷转移酶,该酶将腺苷基团从 ATP 转移到 VitB12 [Leal et al 2003]以形成腺苷钴胺素并转运该辅因子到 MUT 酶。该基因产物晶体结构已经确定[Saridakis et al 2004]。
异常的基因产物。报告的致病性错义变异属于进化上保守的残基 [Dobson et al 2002b]。其中一种 致病性变异会破坏一个 剪接位点 [Dobson et al 2002b, Martínez et al 2005]。几种致病变异已被生物化学确定[Saridakis et al 2004]。
MMUT
基因结构.MMUT 包含13个外显子。有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病性变异。已描述了190多种致病变异,包括 103 (54%) 种错义; 27 (14%) nonsense; 18 (9%) 剪接; 42 (22%) 个小的插入/删除;和一个大的外显子12的缺失,致病变异分布在整个编码序列中,除了外显子1,它是不翻译的[Crane et al 1992, Crane & Ledley 1994, Ogasawara et al 1994, Ledley & Rosenblatt 1997, Adjalla et al 1998, Fuchshuber et al 2000, Acquaviva et al 2001, Acquaviva et al 2005, Jung et al 2005, Martínez et al 2005, Worgan et al 2006, Gradinger et al 2007, Lempp et al 2007, Sakamoto et al 2007, Merinero et al 2008].
如需在不同人群中鉴定的致病变异列表,请单击 here (pdf)。
虽然一些个体的 致病性变异是纯合性,但大多数是复合杂合子。等位基因间互补现象使基因型/表型/酶活性的预测变得困难,因为一些具有两种致病变异的个体在复合状态下可能具有 mut– 型,但在纯合状态下可能具有mut0型 [Janata et al 1997, Ledley & Rosenblatt 1997, Acquaviva et al 2005].
具有两个截断致病变异的人通常具有 mut0型。
无义致病变异已在以下密码子中描述:7、18、23、31、54、84、117、121、135、152、156、161、224、228、284、342、403、413、414、426 、429、451、467、474、494、511、544、581、589、688 和 727。
只有少数经常报道的致病变异以纯合性形式出现; p.Arg108Cys、p.Asn219Tyr 和 p.Arg369His 在纯合时会导致 mut0型 [Acquaviva et al 2001, Worgan et al 2006],而 p.Gly717Val 和 p.Arg694Trp 在纯合时与mut– 型相关[Worgan et al 2006]。
已报道一例染色体 6号父系单亲二倍体导致mut0MMA 和胰岛素依赖型糖尿病[Abramowicz et al 1994].
已知mut– 型主要但不完全与 mut 蛋白的钴胺素结合结构域中的致病变异有关。mut–型致病性变异处于复合杂合子状态时, mut–型致病性酶变异通常起主导作用, OH-Cbl 反应在体外试验中证实 [Lempp et al 2007]。
Table 5
这篇 GeneReview 中讨论的 MMUT 致病性错义变异
| 变异酶的类型 (纯合) | DNA核酸改变 | 蛋白质改变 | 参考序列 |
|---|---|---|---|
| mut0 | c.19C>T | p.Gln7Ter | NM_000255 NP_000246 |
| mut0 | c.52C>T | p.Gln18Ter | |
| mut0 | c.91C>T | p.Arg31Ter | |
| mut0 | c.278G>A | p.Arg93His | |
| mut0 | c.284C>G | p.Pro95Arg | |
| mut0 | c.313T>C | p.Trp105Arg | |
| mut0 | c.322C>T 1 | p.Arg108Cys | |
| mut0 | c.521T>C | p.Phe174Ser | |
| mut0 | c.572C>A | p.Ala191Glu | |
| mut0 | c.607G>A | p.Gly203Arg | |
| mut0 | c.643G>A | p.Gly215Ser | |
| mut0 | c.655A>T | p.Asn219Tyr | |
| mut0 | c.935G>T | p.Gly312Val | |
| mut0 | c.1105C>T | p.Arg369Cys | |
| mut0 | c.1106G>A | p.Arg369His | |
| mut0 | c.1280G>A | p.Gly427Asp | |
| mut0 | c.1867G>A | p.Gly623Arg | |
| mut– | c.299A>G | p.Tyr100Cys | |
| mut– | c.691T>A | p.Tyr231Asn | |
| mut– | c.1097A>G | p.Asn366Ser | |
| mut0 | c.1553T>C | p.Leu518Pro | |
| mut0 | c.1867G>A | p.Gly623Arg | |
| mut– 2 | c.2054T>G | p.Leu685Arg | |
| mut– | c.2080C>T | p.Arg694Trp | |
| mut– | c.2099T>A | p.Met700Lys | |
| mut– | c.2150G>T | p.Gly717Val | |
| mut0 | c.2179C>T | p.Arg727Ter |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。mut0 = mut0 enzymatic subtype
mut– = mut– enzymatic subtype
NA = not applicable
- 1.
在西班牙裔个体中观察到。
- 2.
正常基因产物。甲基丙二酰辅酶A变位酶是一种位于线粒体中的核编码酶,以同源二聚体形式存在。该蛋白质包含 750 个氨基酸,并具有一个 N 端线粒体前导序列(残基 1-32),该序列被线粒体输入和加工机器去除。线粒体前导信号之后是参与亚基相互作用的 N 末端延伸片段(残基 33-87)。 N 末端桶是底物结合结构域(残基 88-422),并通过长接头区域(423-577)连接到 C 末端腺苷钴胺结合域(残基 578-750)。该蛋白质每摩尔亚基含有一摩尔腺苷钴胺素,并进行 1, 2 重排反应,将 L-甲基丙二酰辅酶 A 异构化为琥珀酰辅酶 A [Fenton et al 2001]。人类酶的晶体结构已得到解析 [Froese et al 2013]。
异常的基因产物。仅对已知的致病变异进行了酶学研究。甲基丙二酰辅酶A变位酶蛋白有几个功能域;每一个都有致病变异。
线粒体前导序列位于氨基末端。三个 nonsense致病变异属于这个结构域:p.Gln7Ter[Acquaviva et al 2005]和 p.Gln18Ter 和 p.Arg31Ter [Worgan et al 2006]。一份报告指出,可能从内部 AUG 翻译而来的截短蛋白质来自 p.Gln18Ter 变异。这种突变的蛋白质是“错误靶向的”并且没有功能。
酶亚基的二聚结构域与线粒体前导序列相邻但不同。
辅酶 A 跨越第二个 外显子的中间到第六个外显子的末端。位于该位置(氨基酸 86 和 423 之间)的致病变异可能会破坏底物结合,并且会通过多种机制阻碍催化。 例如 p.Arg93His,可以参与等位基因间互补。这种现象背后的机制尚不清楚[Worgan et al 2006]。
接头结构域跨越残基 424-577 将 C 端钴胺素结合域分开。在该域中鉴定的大多数致病性变异是剪接位点或nonsense变化,并且与甲基丙二酸血症的mut0型有关 [Acquaviva et al 2005],而唯一的致病性错义变异 (c.1553T>C) 位于该片段的中间影响一个高度保守的氨基酸[Worgan et al 2006].
大多数mut–型致病变异存在于钴胺素结合 结构域中,该结构域位于氨基酸 578 和 750 之间。该区域中的一些致病变异可以表现出纯粹的 Km效应,正如辅因子结合致病性变异那样变异,而其他影响 Km 和 Vmax[Janata et al 1997]。该区域还包含可以参与等位基因间互补的残基 [Ledley & Rosenblatt 1997]。
少数错义变异可提供详细的功能表征,这些变异导致 (a) 由于错误折叠导致蛋白质水平降低,(b) 耐热性增加,(c) 酶活性受损,以及 (d) 底物周转中的辅因子反应降低[Forny et al 2014]。
MCEE
基因结构.MCEE 包含四个外显子。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病性变异。在初步鉴定了两个甲基丙二酸尿症患者 p.Arg47Ter 纯合性致病性变异 [Bikker et al 2006, Dobson et al 2006] ,另外 229 位 MMA 升高且病因不明的患者中的4位被报道在 MCEE 中有致病性变异 [Gradinger et al 2007]。两个[14C]丙酸减少的人是外显子2中纯合子致病性nonsense变异c.139C>T的纯合子。在199名[14C]丙酸正常的人中,一个人是新发的纯合子致病性错义变异:在外显子2中c.178A>C,两个是 新发的在外显子3中c.427C>T杂合的致病性错义变异 。
Table 6
本次 GeneReview 中讨论的 MCEE 致病变异
| DNA核酸改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.139C>T | p.Arg47Ter | NM_032601 NP_115990 |
| c.178A>C | p.Lys60Gln | |
| c.427C>T | p.Arg143Cys |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。
正常基因产物。MCEE 编码 176 个氨基酸的甲基丙二酰辅酶 A 差向异构酶,将 D-甲基丙二酰辅酶 A 转化为 L-甲基丙二酰辅酶 A。
异常基因产物。 迄今为止描述的致病变异要么是错义,要么是nonsense,降低或消除产物功能。
MMADHC
基因结构.MMADHC(以前称为 C2orf25)包含 8 个外显子,跨度为 18 kb [Coelho et al 2008]。 有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病性变异。 Table 7列出了 C 末端区域(外显子 3、4 或 5)中导致 cblD 变异 2 表型(孤立的甲基丙二酸尿症)的致病性变异。
N 末端区域(外显子6和8)的致病性错义变异仅导致 cblD 变异1表型 (孤立的同型胱氨酸尿症),而外显子 5 和 8 以及内含子7中的截断致病变异导致经典的 cblD 表型(高胱氨酸尿症和甲基丙二酸尿症组合)[Coelho et al 2008, Miousse et al 2009]。
Table 7
本GeneReview 中讨论了孤立的MMA相关MMADHC 致病变异
| DNA核酸改变 (Alias 1) | 蛋白质改变 (Alias 1) | 参考序列 |
|---|---|---|
| c.57_64delCTCTTTAG | p.Ser20Ter (Cys19fsTer20) | NM_015702 NP_056517 |
| c.60_61insAT (60insAT) | p.Leu21IlefsTer2 (Leu20fsTer21) | |
| c.133dupG | p.Arg45GlyfsTer15 | |
| c.160C>T | p.Arg54Ter | |
| c.228dupG | p.Asn77GlufsTer5 | |
| c.307_324dup | p.Leu103_Ser108dup | |
| c.455dupC | p.Cys153MetfsTer10 (Thr152fsTer162) |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
不符合当前命名约定的变异名称
正常基因产物。 MMADHC 产物有 296个氨基酸,分子量为 32.8 kd。它与细菌 ABC 转运蛋白ATPase 成分显示出同源性。有一个 N 端线粒体前导序列和一个预测的B12结合序列[Coelho et al 2008]并且 MMADHC 在体外已定位于细胞质和线粒体 [Mah et al 2013]。
异常基因产物。 cblD-MMA 突变的等位基因(c.57_64delCTCTTTAG、c.60_61insAT、c.133dupG、c.160C>T 和 c.228dupG)在来自具有 cblD 组合表型的患者的永生化细胞系中表达,并且能够拯救 MeCbl 合成[Stucki et al 2012]。这项工作表明,Met62 和 Met116 上的额外重新启动密码子导致功能更少的 cblD 蛋白缺乏线粒体前导序列,但允许正常的甲基钴胺素合成 [Coelho et al 2008, Stucki et al 2012]。迄今为止报道的每位患有 cblD-MMA 的患者似乎至少有一个致病性变异,导致该酶的 N 末端过短。
参考文献
引用文献
- Abramowicz MJ, Andrien M, Dupont E, Dorchy H, Parma J, Duprez L, Ledley FD, Courtens W, Vamos E. Isodisomy of chromosome 6 in a newborn with methylmalonic acidemia and agenesis of pancreatic beta cells causing diabetes mellitus. J Clin Invest. 1994;1994;94:418 - 21. [PMC free article: PMC296325] [PubMed: 7913714]
- Acquaviva C, Benoist JF, Callebaut I, Guffon N, Ogier de Baulny H, Touati G, Aydin A, Porquet D, Elion J. N219Y, a new frequent mutation among mut(o) forms of methylmalonic acidemia in Caucasian patients. Eur J Hum Genet. 2001;9:577 - 82. [PubMed: 11528502]
- Acquaviva C, Benoist JF, Pereira S, Callebaut I, Koskas T, Porquet D, Elion J. Molecular basis of methylmalonyl-CoA mutase apoenzyme defect in 40 European patients affected by mut(o) and mut- forms of methylmalonic acidemia: identification of 29 novel mutations in the MUT gene. Hum Mutat. 2005;25:167 - 76. [PubMed: 15643616]
- Adjalla CE, Hosack AR, Matiaszuk NV, Rosenblatt DS. A common mutation among blacks with mut- methylmalonic aciduria. Hum Mutat. 1998 Suppl 1:S248 - 50. [PubMed: 9452100]
- Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126:e208 - 14. [PMC free article: PMC3297024] [PubMed: 20566609]
- Alfares A, Nunez LD, Al-Thihli K, Mitchell J, Melançon S, Anastasio N, Ha KC, Majewski J, Rosenblatt DS, Braverman N. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet. 2011;48:602 - 5. [PubMed: 21785126]
- Al-Owain M, Freehauf C, Bernstein L, Kappy M, Thomas J. Growth hormone deficiency associated with methylmalonic acidemia. J Pediatr Endocrinol Metab. 2004;17:239 - 43. [PubMed: 15055362]
- Ampola MG, Mahoney MJ, Nakamura E, Tanaka K. Prenatal therapy of a patient with vitamin-B12-responsive methylmalonic acidemia. N Engl J Med. 1975;293:313 - 7. [PubMed: 239344]
- Atkuri KR, Cowan TM, Kwan T, Ng A, Herzenberg LA, Herzenberg LA, Enns GM. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc Natl Acad Sci USA. 2009;106:3941 - 5. [PMC free article: PMC2656184] [PubMed: 19223582]
- Bain MD, Nussey SS, Jones M, Chalmers RA. Use of human somatotrophin in the treatment of a patient with methylmalonic aciduria. Eur J Pediatr. 1995;154:850 - 2. [PubMed: 8529687]
- Baker EH, Sloan JL, Hauser NS, Gropman AL, Adams DR, Toro C, Manoli I, Venditti CP. MRI characteristics of globus pallidus infarcts in isolated methylmalonic acidemia. AJNR Am J Neuroradiol. 2015;36:194 - 201. [PubMed: 25190203]
- Baumgarter ER, Viardot C. Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. J Inherit Metab Dis. 1995;18(2):138 - 42. [PubMed: 7564229]
- Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, Burlina A, Fowler B, Grünert SC, Grünewald S, Honzik T, Merinero B, Pérez-Cerdá C, Scholl-Bürgi S, Skovby F, Wijburg F, MacDonald A, Martinelli D, Sass JO, Valayannopoulos V, Chakrapani A. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. [PMC free article: PMC4180313] [PubMed: 25205257]
- Bikker H, Bakker HD, Abeling NG, Poll-The BT, Kleijer WJ, Rosenblatt DS, Waterham HR, Wanders RJ, Duran M. A homozygous nonsense mutation in the methylmalonyl-CoA epimerase gene (MCEE) results in mild methylmalonic aciduria. Hum Mutat. 2006;27:640 - 3. [PubMed: 16752391]
- Brassier A, Boyer O, Valayannopoulos V, Ottolenghi C, Krug P, Cosson MA, Touati G, Arnoux JB, Barbier V, Bahi-Buisson N, Desguerre I, Charbit M, Benoist JF, Dupic L, Aigrain Y, Blanc T, Salomon R, Rabier D, Guest G, de Lonlay P, Niaudet P. Renal transplantation in 4 patients with methylmalonic aciduria: a cell therapy for metabolic disease. Mol Genet Metab. 2013;110:106 - 10. [PubMed: 23751327]
- Brown GK, Scholem RD, Bankier A, Danks DM. Malonyl coenzyme A decarboxylase deficiency. J Inherit Metab Dis. 1984;7:21 - 6. [PubMed: 6145813]
- Carrillo-Carrasco N, Chandler RJ, Chandrasekaran S, Venditti CP. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum Gene Ther. 2010;21:1147 - 54. [PMC free article: PMC2936498] [PubMed: 20486773]
- Carrozzo R, Dionisi-Vici C, Steuerwald U, Lucioli S, Deodato F, Di Giandomenico S, Bertini E, Franke B, Kluijtmans LA, Meschini MC, Rizzo C, Piemonte F, Rodenburg R, Santer R, Santorelli FM, van Rooij A, Vermunt-de Koning D, Morava E, Wevers RA. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130:862 - 74. [PubMed: 17301081]
- Celiker MY, Chawla A. Congenital B12 deficiency following maternal gastric bypass. J Perinatol. 2009;29:640 - 2. [PubMed: 19710657]
- Chace DH, DiPerna JC, Kalas TA, Johnson RW, Naylor EW. Rapid diagnosis of methylmalonic and propionic acidemias: quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin Chem. 2001;47:2040 - 4. [PubMed: 11673377]
- Chakrapani A, Sivakumar P, McKiernan PJ, Leonard JV. Metabolic stroke in methylmalonic acidemia five years after liver transplantation. J Pediatr. 2002;140:261 - 3. [PubMed: 11865284]
- Chambliss KL, Gray RG, Rylance G, Pollitt RJ, Gibson KM. Molecular characterization of methylmalonate semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2000;23:497 - 504. [PubMed: 10947204]
- Chan R, Mascarenhas L, Boles RG, Kerkar N, Genyk Y, Venkatramani R. Hepatoblastoma in a patient with methylmalonic aciduria. Am J Med Genet A. 2015;167A:635 - 8. [PubMed: 25691417]
- Chandler RJ, Sloan J, Fu H, Tsai M, Stabler S, Allen R, Kaestner KH, Kazazian HH, Venditti CP. Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle. BMC Med Genet. 2007;8:64. [PMC free article: PMC2140053] [PubMed: 17937813]
- Chandler RJ, Venditti CP. Adenovirus-mediated gene delivery rescues a neonatal lethal murine model of mut(0) methylmalonic acidemia. Hum Gene Ther. 2008;19:53 - 60. [PMC free article: PMC2683146] [PubMed: 18052792]
- Chandler RJ, Venditti CP. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol Ther. 2010;18:11 - 6. [PMC free article: PMC2839224] [PubMed: 19861951]
- Chandler RJ, Venditti CP. Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol Genet Metab. 2012;107:617 - 9. [PMC free article: PMC3522145] [PubMed: 23046887]
- Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, Venditti CP. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009;23:1252 - 61. [PMC free article: PMC2660647] [PubMed: 19088183]
- Chang PF, Huang SF, Hwu WL, Hou JW, Ni YH, Chang MH. Metabolic disorders mimicking Reye's syndrome. J Formos Med Assoc. 2000;99:295 - 9. [PubMed: 10870312]
- Ciani F, Donati MA, Tulli G, Poggi GM, Pasquini E, Rosenblatt DS, Zammarchi E. Lethal late onset cblB methylmalonic aciduria. Crit Care Med. 2000;28:2119 - 21. [PubMed: 10890676]
- Clothier JC, Chakrapani A, Preece MA, McKiernan P, Gupta R, Macdonald A, Hulton SA. Renal transplantation in a boy with methylmalonic acidaemia. J Inherit Metab Dis. 2011;34:695 - 700. [PubMed: 21416195]
- Coelho D, Kim JC, Miousse IR, Fung S, du Moulin M, Buers I, Suormala T, Burda P, Frapolli M, Stucki M, Nürnberg P, Thiele H, Robenek H, Höhne W, Longo N, Pasquali M, Mengel E, Watkins D, Shoubridge EA, Majewski J, Rosenblatt DS, Fowler B, Rutsch F, Baumgartner MR. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet. 2012;44:1152 - 5. [PubMed: 22922874]
- Coelho D, Suormala T, Stucki M, Lerner-Ellis JP, Rosenblatt DS, Newbold RF, Baumgartner MR, Fowler B. Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med. 2008;358:1454 - 64. [PubMed: 18385497]
- Coman D, Huang J, McTaggart S, Sakamoto O, Ohura T, McGill J, Burke J. Renal transplantation in a 14-year-old girl with vitamin B12-responsive cblA-type methylmalonic acidaemia. Pediatr Nephrol. 2006;21:270 - 3. [PubMed: 16247646]
- Cosson MA, Touati G, Lacaille F, Valayannnopoulos V, Guyot C, Guest G, Verkarre V, Chrétien D, Rabier D, Munnich A, Benoist JF, de Keyzer Y, Niaudet P, de Lonlay P. Liver hepatoblastoma and multiple OXPHOS deficiency in the follow-up of a patient with methylmalonic aciduria. Mol Genet Metab. 2008;95:107 - 9. [PubMed: 18676166]
- Coulombe JT, Shih VE, Levy HL. Massachusetts Metabolic Disorders Screening Program. II. Methylmalonic aciduria. Pediatrics. 1981;67:26 - 31. [PubMed: 7243433]
- Crane AM, Jansen R, Andrews ER, Ledley FD. Cloning and expression of a mutant methylmalonyl coenzyme A mutase with altered cobalamin affinity that causes mut- methylmalonic aciduria. J Clin Invest. 1992;89:385 - 91. [PMC free article: PMC442864] [PubMed: 1346616]
- Crane AM, Ledley FD. Clustering of mutations in methylmalonyl CoA mutase associated with mut- methylmalonic acidemia. Am J Hum Genet. 1994;55:42 - 50. [PMC free article: PMC1918235] [PubMed: 7912889]
- D'Angio CT, Dillon MJ, Leonard JV. Renal tubular dysfunction in methylmalonic acidaemia. Eur J Pediatr. 1991;150:259 - 63. [PubMed: 2029917]
- de Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM. Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis. 2005;28:415 - 23. [PubMed: 15868474]
- de Keyzer Y, Valayannopoulos V, Benoist JF, Batteux F, Lacaille F, Hubert L, Chrétien D, Chadefeaux-Vekemans B, Niaudet P, Touati G, Munnich A, de Lonlay P. Multiple OXPHOS deficiency in the liver, kidney, heart and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. 2009;66:91 - 5. [PubMed: 19342984]
- De Raeve L, De Meirleir L, Ramet J, Vandenplas Y, Gerlo E. Acrodermatitis enteropathica-like cutaneous lesions in organic aciduria. J Pediatr. 1994;124:416 - 20. [PubMed: 8120711]
- Deodato F, Rizzo C, Boenzi S, Baiocco F, Sabetta G, Dionisi-Vici C. Successful pregnancy in a woman with mut- methylmalonic acidaemia. J Inherit Metab Dis. 2002;25:133 - 4. [PubMed: 12118529]
- Dietzen DJ, Rinaldo P, Whitley RJ, Rhead WJ, Hannon WH, Garg UC, Lo SF, Bennett MJ. National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clin Chem. 2009;55:1615 - 26. [PubMed: 19574465]
- Dionisi-Vici C, Deodato F, Röschinger W, Rhead W, Wilcken B. 'Classical' organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis. 2006;29:383 - 9. [PubMed: 16763906]
- Dobson CM, Gradinger A, Longo N, Wu X, Leclerc D, Lerner-Ellis J, Lemieux M, Belair C, Watkins D, Rosenblatt DS, Gravel RA. Homozygous nonsense mutation in the MCEE gene and siRNA suppression of methylmalonyl-CoA epimerase expression: a novel cause of mild methylmalonic aciduria. Mol Genet Metab. 2006;88:327 - 33. [PubMed: 16697227]
- Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet. 2002a;11:3361 - 9. [PubMed: 12471062]
- Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci U S A. 2002b;99:15554 - 9. [PMC free article: PMC137755] [PubMed: 12438653]
- Dudley J, Allen J, Tizard J, McGraw M. Benign methylmalonic acidemia in a sibship with distal renal tubular acidosis. Pediatr Nephrol. 1998;12:564 - 6. [PubMed: 9761355]
- Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081 - 6. [PMC free article: PMC1196446] [PubMed: 15877282]
- Fenton WA, Gravel RA, Rosenblatt DS. Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds) The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill. 2001:2165-92.
- Forny P, Froese DS, Suormala T, Yue WW, Baumgartner MR. Functional characterization and categorization of missense mutations that cause methylmalonyl-CoA mutase (MUT) deficiency. Hum Mutat. 2014;35:1449 - 58. [PMC free article: PMC4441004] [PubMed: 25125334]
- Fowler B, Leonard JV, Baumgartner MR. Causes and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis. 2008;31:350 - 60. [PubMed: 18563633]
- Froese DS, Forouhar F, Tran TH, Vollmar M, Kim YS, Lew S, Neely H, Seetharaman J, Shen Y, Xiao R, Acton TB, Everett JK, Cannone G, Puranik S, Savitsky P, Krojer T, Pilka ES, Kiyani W, Lee WH, Marsden BD, von Delft F, Allerston CK, Spagnolo L, Gileadi O, Montelione GT, Oppermann U, Yue WW, Tong L. Crystal structures of malonyl-coenzyme A decarboxylase provide insights into its catalytic mechanism and disease-causing mutations. Structure. 2013;21:1182 - 92. [PMC free article: PMC3701320] [PubMed: 23791943]
- Fuchshuber A, Mucha B, Baumgartner ER, Vollmer M, Hildebrandt F. mut0 methylmalonic acidemia: eleven novel mutations of the methylmalonyl CoA mutase including a deletion-insertion mutation. Hum Mutat. 2000;16:179. [PubMed: 10923046]
- Giorgio AJ, Trowbridge M, Boone AW, Patten RS. Methylmalonic aciduria without vitamin B12 deficiency in an adult sibship. N Engl J Med. 1976;295:310 - 3. [PubMed: 6909]
- Gradinger AB, Bélair C, Worgan LC, Li CD, Lavallée J, Roquis D, Watkins D, Rosenblatt DS. Atypical methylmalonic aciduria: frequency of mutations in the methylmalonyl CoA epimerase gene (MCEE). Hum Mutat. 2007;28:1045. [PubMed: 17823972]
- Grange DK, Finlay JL. Nutritional vitamin B12 deficiency in a breastfed infant following maternal gastric bypass. Pediatr Hematol Oncol. 1994;11:311 - 8. [PubMed: 8060815]
- Gu W, Koh W, Blumenfeld YJ, El-Sayed YY, Hudgins L, Hintz SR, Quake SR. Noninvasive prenatal diagnosis in a fetus at risk for methylmalonic acidemia. Genet Med. 2014;16:564 - 7. [PMC free article: PMC4079742] [PubMed: 24406457]
- Guerra-Moreno J, Barrios N, Santiago-Borrero PJ. Severe neutropenia in an infant with methylmalonic acidemia. Bol Asoc Med P R. 2003;95:17 - 20. [PubMed: 12898746]
- Harting I, Seitz A, Geb S, Zwickler T, Porto L, Lindner M, Kölker S, Hörster F. Looking beyond the basal ganglia: the spectrum of MRI changes in methylmalonic acidaemia. J Inherit Metab Dis. 2008;31:368 - 78. [PubMed: 18470632]
- Hauser NS, Manoli I, Graf JC, Sloan J, Venditti CP. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr. 2011;93:47 - 56. [PMC free article: PMC3001598] [PubMed: 21048060]
- Heidenreich R, Natowicz M, Hainline BE, Berman P, Kelley RI, Hillman RE, Berry GT. Acute extrapyramidal syndrome in methylmalonic acidemia: "metabolic stroke" involving the globus pallidus. J Pediatr. 1988;113:1022 - 7. [PubMed: 3193307]
- Hörster F, Baumgartner MR, Viardot C, Suormala T, Burgard P, Fowler B, Hoffmann GF, Garbade SF, Kölker S, Baumgartner ER. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut-, cblA, cblB). Pediatr Res. 2007;62:225 - 30. [PubMed: 17597648]
- Hsui JY, Chien YH, Chu SY, Lu FL, Chen HL, Ho MJ, Lee PH, Hwu WL. Living-related liver transplantation for methylmalonic acidemia: report of one case. Acta Paediatr Taiwan. 2003;44:171 - 3. [PubMed: 14521026]
- Hubbard PA, Padovani D, Labunska T, Mahlstedt SA, Banerjee R, Drennan CL. Crystal structure and mutagenesis of the metallochaperone MeaB: insight into the causes of methylmalonic aciduria. J Biol Chem. 2007;282:31308 - 16. [PubMed: 17728257]
- Iles RA, Chalmers RA, Hind AJ. Methylmalonic aciduria and propionic acidaemia studied by proton nuclear magnetic resonance spectroscopy. Clin Chim Acta. 1986;161:173 - 89. [PubMed: 3802528]
- Inoue S, Krieger I, Sarnaik A, Ravindranath Y, Fracassa M, Ottenbreit MJ. Inhibition of bone marrow stem cell growth in vitro by methylmalonic acid: a mechanism for pancytopenia in a patient with methylmalonic acidemia. Pediatr Res. 1981;15:95 - 8. [PubMed: 7254944]
- Janata J, Kogekar N, Fenton WA. Expression and kinetic characterization of methylmalonyl-CoA mutase from patients with the mut- phenotype: evidence for naturally occurring interallelic complementation. Hum Mol Genet. 1997;6:1457 - 64. [PubMed: 9285782]
- Jung JW, Hwang IT, Park JE, Lee EH, Ryu KH, Kim SH, Hwang JS. Mutation analysis of the MCM gene in Korean patients with MMA. Mol Genet Metab. 2005;84:367 - 70. [PubMed: 15781199]
- Kahler SG, Sherwood WG, Woolf D, Lawless ST, Zaritsky A, Bonham J, Taylor CJ, Clarke JT, Durie P, Leonard JV. Pancreatitis in patients with organic acidemias. J Pediatr. 1994;124:239 - 43. [PubMed: 8301430]
- Kaplan P, Ficicioglu C, Mazur AT, Palmieri MJ, Berry GT. Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl-CoA mutase deficiency. Mol Genet Metab. 2006;88:322 - 6. [PubMed: 16750411]
- Karth P, Singh R, Kim J, Costakos D. Bilateral central retinal artery occlusions in an infant with hyperhomocysteinemia. J AAPOS. 2012;16:398 - 400. [PubMed: 22819238]
- Kasahara M, Horikawa R, Tagawa M, Uemoto S, Yokoyama S, Shibata Y, Kawano T, Kuroda T, Honna T, Tanaka K, Saeki M. Current role of liver transplantation for methylmalonic acidemia: a review of the literature. Pediatr Transplant. 2006;10:943 - 7. [PubMed: 17096763]
- Kayler LK, Merion RM, Lee S, Sung RS, Punch JD, Rudich SM, Turcotte JG, Campbell DA Jr, Holmes R, Magee JC. Long-term survival after liver transplantation in children with metabolic disorders. Pediatr Transplant. 2002;6:295 - 300. [PubMed: 12234269]
- Kölker S, Cazorla AG, Valayannopoulos V, Lund AM, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Barić I, Karall D, Augoustides-Savvopoulou P, Aksglaede L, Arnoux JB, Avram P, Baumgartner MR, Blasco-Alonso J, Chabrol B, Chakrapani A, Chapman K. I Saladelafont EC, Couce ML, de Meirleir L, Dobbelaere D, Dvorakova V, Furlan F, Gleich F, Gradowska W, Grünewald S, Jalan A, Häberle J, Haege G, Lachmann R, Laemmle A, Langereis E, de Lonlay P, Martinelli D, Matsumoto S, Mühlhausen C, de Baulny HO, Ortez C, Peña-Quintana L, Ramadža DP, Rodrigues E, Scholl-Bürgi S, Sokal E, Staufner C, Summar ML, Thompson N, Vara R, Pinera IV, Walter JH, Williams M, Burgard P. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015a;38:1041 - 57. [PubMed: 25875215]
- Kölker S, Valayannopoulos V, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Barić I, Karall D, Arnoux JB, Avram P, Baumgartner MR, Blasco-Alonso J, Boy SP, Rasmussen MB, Burgard P, Chabrol B, Chakrapani A, Chapman K, Cortès I, Saladelafont E, Couce ML, de Meirleir L, Dobbelaere D, Furlan F, Gleich F, González MJ, Gradowska W, Grünewald S, Honzik T, Hörster F, Ioannou H, Jalan A, Häberle J, Haege G, Langereis E, de Lonlay P, Martinelli D, Matsumoto S, Mühlhausen C, Murphy E, de Baulny HO, Ortez C, Pedrón CC, Pintos-Morell G, Pena-Quintana L, Ramadža DP, Rodrigues E, Scholl-Bürgi S, Sokal E, Summar ML, Thompson N, Vara R, Pinera IV, Walter JH, Williams M, Lund AM. Garcia Cazorla A. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015b Nov;38(6):1059 - 74. [PubMed: 25875216]
- Korf B, Wallman JK, Levy HL. Bilateral lucency of the globus pallidus complicating methylmalonic acidemia. Ann Neurol. 1986;20:364 - 6. [PubMed: 3767321]
- Korotkova N, Lidstrom ME. MeaB is a component of the methylmalonyl-CoA mutase complex required for protection of the enzyme from inactivation. J Biol Chem. 2004;279:13652 - 8. [PubMed: 14734568]
- Kruszka PS, Manoli I, Sloan JL, Kopp JB, Venditti CP. Renal growth in isolated methylmalonic acidemia. Genet Med. 2013;15:990 - 6. [PMC free article: PMC4149057] [PubMed: 23639900]
- Ktena YP, Paul SM, Hauser NS, Sloan JL, Gropman A, Manoli I, Venditti CP. Delineating the spectrum of impairments, disabilities, and rehabilitation needs in methylmalonic acidemia (MMA). Am J Med Genet A. 2015a Sep;167A(9):2075 - 84. [PubMed: 25959030]
- Ktena YP, Ramstad T, Baker EH, Sloan JL, Mannes AJ, Manoli I, Venditti CP. Propofol administration in patients with methylmalonic acidemia and intracellular cobalamin metabolism disorders: a review of theoretical concerns and clinical experiences in 28 patients. J Inherit Metab Dis. 2015b;38:847 - 53. [PMC free article: PMC5577977] [PubMed: 25985870]
- la Marca G, Malvagia S, Casetta B, Pasquini E, Donati MA, Zammarchi E. Progress in expanded newborn screening for metabolic conditions by LC-MS/MS in Tuscany: update on methods to reduce false tests. J Inherit Metab Dis. 2008;31 Suppl 2:S395 - 404. [PubMed: 18956250]
- Leal NA, Park SD, Kima PE, Bobik TA. Identification of the human and bovine ATP:Cob(I)alamin adenosyltransferase cDNAs based on complementation of a bacterial mutant. J Biol Chem. 2003;278:9227 - 34. [PubMed: 12514191]
- Ledley FD, Levy HL, Shih VE, Benjamin R, Mahoney MJ. Benign methylmalonic aciduria. N Engl J Med. 1984;311:1015 - 8. [PubMed: 6148691]
- Ledley FD, Rosenblatt DS. Mutations in mut methylmalonic acidemia: clinical and enzymatic correlations. Hum Mutat. 1997;9:1 - 6. [PubMed: 8990001]
- Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41 - 72. [PubMed: 11916378]
- Lempp TJ, Suormala T, Siegenthaler R, Baumgartner ER, Fowler B, Steinmann B, Baumgartner MR. Mutation and biochemical analysis of 19 probands with mut0 and 13 with mut- methylmalonic aciduria: identification of seven novel mutations. Mol Genet Metab. 2007;90:284 - 90. [PubMed: 17113806]
- Leonard JV. Stable isotope studies in propionic and methylmalonic acidaemia. Eur J Pediatr. 1997;156:S67 - 9. [PubMed: 9266219]
- Leonard JV, Vijayaraghavan S, Walter JH. The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr. 2003;162:S21 - 4. [PubMed: 14586648]
- Lerner-Ellis JP, Dobson CM, Wai T, Watkins D, Tirone JC, Leclerc D, Dore C, Lepage P, Gravel RA, Rosenblatt DS. Mutations in the MMAA gene in patients with the cblA disorder of vitamin B12 metabolism. Hum Mutat. 2004;24:509 - 16. [PubMed: 15523652]
- Lerner-Ellis JP, Gradinger AB, Watkins D, Tirone JC, Villeneuve A, Dobson CM, Montpetit A, Lepage P, Gravel RA, Rosenblatt DS. Mutation and biochemical analysis of patients belonging to the cblB complementation class of vitamin B12-dependent methylmalonic aciduria. Mol Genet Metab. 2006;87:219 - 25. [PubMed: 16410054]
- Lindner M, Ho S, Kölker S, Abdoh G, Hoffmann GF, Burgard P. Newborn screening for methylmalonic acidurias--optimization by statistical parameter combination. J Inherit Metab Dis. 2008;31:379 - 85. [PubMed: 18563635]
- Lubrano R, Elli M, Rossi M, Travasso E, Raggi C, Barsotti P, Carducci C, Berloco P. Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature. Pediatr Nephrol. 2007;22:1209 - 14. [PubMed: 17401587]
- Lubrano R, Perez B, Elli M. Methylmalonic acidemia and kidney transplantation. Pediatr Nephrol. 2013;28:2067 - 8. [PubMed: 23793882]
- Lubrano R, Scoppi P, Barsotti P, Travasso E, Scateni S, Cristaldi S, Castello MA. Kidney transplantation in a girl with methylmalonic acidemia and end stage renal failure. Pediatr Nephrol. 2001;16:848 - 51. [PubMed: 11685586]
- MacFarland S, Hartung H. Pancytopenia in a patient with methylmalonic acidemia. Blood. 2015;125:1840. [PubMed: 25927084]
- Mah W, Deme JC, Watkins D, Fung S, Janer A, Shoubridge EA, Rosenblatt DS, Coulton JW. Subcellular location of MMACHC and MMADHC, two human proteins central to intracellular vitamin B(12) metabolism. Mol Genet Metab. 2013;108:112 - 8. [PubMed: 23270877]
- Manoli I, Myles JG, Sloan JL, Carrillo-Carrasco N, Morava E, Strauss KA, Morton H, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: cobalamin C deficiency. Genet Med. 2016a Apr;18(4):396 - 404. [PMC free article: PMC4752912] [PubMed: 26270766]
- Manoli I, Myles JG, Sloan JL, Shchelochkov OA, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet Med. 2016b Apr;18(4):386 - 95. [PMC free article: PMC4752925] [PubMed: 26270765]
- Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog K, Young S, Trivedi NS, Cheng J, Sloan JL, Chandler RJ, Abu-Asab M, Tsokos M, Elkahloun AG, Rosen S, Enns GM, Berry GT, Hoffmann V, DiMauro S, Schnermann J, Venditti CP. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc Natl Acad Sci U S A. 2013;110:13552 - 7. [PMC free article: PMC3746875] [PubMed: 23898205]
- Marble M, Copeland S, Khanfar N, Rosenblatt DS. Neonatal vitamin B12 deficiency secondary to maternal subclinical pernicious anemia: identification by expanded newborn screening. J Pediatr. 2008;152:731 - 3. [PubMed: 18410783]
- Marcadier JL, Smith AM, Pohl D, Schwartzentruber J, Al-Dirbashi OY., FORGE Canada Consortium. Majewski J, Ferdinandusse S, Wanders RJ, Bulman DE, Boycott KM, Chakraborty P, Geraghty MT. Mutations in ALDH6A1 encoding methylmalonate semialdehyde dehydrogenase are associated with dysmyelination and transient methylmalonic aciduria. Orphanet J Rare Dis. 2013;8:98. [PMC free article: PMC3710243] [PubMed: 23835272]
- Martens DH, Bakker JA, van der Meer SB, Spaapen LJ. Unexplained familial benign methylmalonic aciduria. Eur J Pediatr. 2002;161:219 - 20. [PubMed: 12014390]
- Martinez Alvarez L, Jameson E, Parry NR, Lloyd C, Ashworth JL. Optic neuropathy in methylmalonic acidemia and propionic acidemia. Br J Ophthalmol. 2016;100:98 - 104. [PubMed: 26209586]
- Martínez MA, Rincón A, Desviat LR, Merinero B, Ugarte M, Pérez B. Genetic analysis of three genes causing isolated methylmalonic acidemia: identification of 21 novel allelic variants. Mol Genet Metab. 2005;84:317 - 25. [PubMed: 15781192]
- Matern D, Tortorelli S, Oglesbee D, Gavrilov D, Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: the Mayo Clinic experience (2004-2007). J Inherit Metab Dis. 2007;30:585 - 92. [PubMed: 17643193]
- Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857 - 61. [PubMed: 6132336]
- Mc Guire PJ, Parikh A, Diaz GA. Profiling of oxidative stress in patients with inborn errors of metabolism. Mol Genet Metab. 2009;98:173 - 80. [PMC free article: PMC2915835] [PubMed: 19604711]
- McGuire MM, Jones BA, Hull MA, Misra MV, Smithers CJ, Feins NR, Jenkins RL, Lillehei CW, Harmon WE, Jonas MM, Kim HB. Combined en bloc liver-double kidney transplantation in an infant with IVC thrombosis. Pediatr Transplant. 2011;15:E142 - 4. [PubMed: 20412506]
- Merinero B, Pérez B, Pérez-Cerdá C, Rincón A, Desviat LR, Martínez MA, Sala PR, García MJ, Aldamiz-Echevarría L, Campos J, Cornejo V, Del Toro M, Mahfoud A, Martínez-Pardo M, Parini R, Pedrón C, Peña-Quintana L, Pérez M, Pourfarzam M, Ugarte M. Methylmalonic acidaemia: examination of genotype and biochemical data in 32 patients belonging to mut, cblA or cblB complementation group. J Inherit Metab Dis. 2008;31:55 - 66. [PubMed: 17957493]
- Miousse IR, Watkins D, Coelho D, Rupar T, Crombez EA, Vilain E, Bernstein JA, Cowan T, Lee-Messer C, Enns GM, Fowler B, Rosenblatt DS. Clinical and molecular heterogeneity in patients with the cblD inborn error of metabolism. J Pediatr. 2009;154:551 - 6. [PubMed: 19058814]
- Morava E, Steuerwald U, Carrozzo R, Kluijtmans LA, Joensen F, Santer R, Dionisi-Vici C, Wevers RA. Dystonia and deafness due to SUCLA2 defect; Clinical course and biochemical markers in 16 children. Mitochondrion. 2009;9:438 - 42. [PubMed: 19666145]
- Morel CF, Watkins D, Scott P, Rinaldo P, Rosenblatt DS. Prenatal diagnosis for methylmalonic acidemia and inborn errors of vitamin B12 metabolism and transport. Mol Genet Metab. 2005;86:160 - 71. [PubMed: 16150626]
- Morioka D, Kasahara M, Horikawa R, Yokoyama S, Fukuda A, Nakagawa A. Efficacy of living donor liver transplantation for patients with methylmalonic acidemia. Am J Transplant. 2007;7:2782 - 7. [PubMed: 17908273]
- Nagarajan S, Enns GM, Millan MT, Winter S, Sarwal MM. Management of methylmalonic acidaemia by combined liver-kidney transplantation. J Inherit Metab Dis. 2005;28:517 - 24. [PubMed: 15902554]
- Nicolaides P, Leonard J, Surtees R. Neurological outcome of methylmalonic acidaemia. Arch Dis Child. 1998;78:508 - 12. [PMC free article: PMC1717592] [PubMed: 9713004]
- Niemi AK, Kim IK, Krueger CE, Cowan TM, Baugh N, Farrell R, Bonham CA, Concepcion W, Esquivel CO, Enns GM. Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J Pediatr. 2015 Jun;166(6):1455 - 61.e1. [PubMed: 25771389]
- Nyhan WL, Fawcett N, Ando T, Rennert OM, Julius RL. Response to dietary therapy in B 12 unresponsive methylmalonic acidemia. Pediatrics. 1973;51:539 - 48. [PubMed: 4707869]
- Nyhan WL, Gargus JJ, Boyle K, Selby R, Koch R. Progressive neurologic disability in methylmalonic acidemia despite transplantation of the liver. Eur J Pediatr. 2002;161:377 - 9. [PubMed: 12111189]
- Oberholzer VG, Levin B, Burgess EA, Young WF. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis. Arch Dis Child. 1967;42:492 - 504. [PMC free article: PMC2019805] [PubMed: 6061291]
- Ogasawara M, Matsubara Y, Mikami H, Narisawa K. Identification of two novel mutations in the methylmalonyl-CoA mutase gene with decreased levels of mutant mRNA in methylmalonic acidemia. Hum Mol Genet. 1994;3:867 - 72. [PubMed: 7951229]
- O'Shea CJ, Sloan JL, Wiggs EA, Pao M, Gropman A, Baker EH, Manoli I, Venditti CP, Snow J. Neurocognitive phenotype of isolated methylmalonic acidemia. Pediatrics. 2012;129:e1541 - 51. [PMC free article: PMC3362903] [PubMed: 22614770]
- Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, Thorgrimsson S, Wibrand F, Christensen E, Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853 - 61. [PubMed: 17287286]
- Pangilinan F, Mitchell A, VanderMeer J, Molloy AM, Troendle J, Conley M, Kirke PN, Sutton M, Sequeira JM, Quadros EV, Scott JM, Mills JL, Brody LC. Transcobalamin II receptor polymorphisms are associated with increased risk for neural tube defects. J Med Genet. 2010;47:677 - 85. [PMC free article: PMC4112773] [PubMed: 20577008]
- Pela I, Gasperini S, Pasquini E, Donati MA. Hyperkalemia after acute metabolic decompensation in two children with vitamin B12-unresponsive methylmalonic acidemia and normal renal function. Clin Nephrol. 2006;66:63 - 6. [PubMed: 16878438]
- Pinar-Sueiro S, Martínez-Fernández R, Lage-Medina S, Aldamiz-Echevarria L, Vecino E. Optic neuropathy in methylmalonic acidemia: the role of neuroprotection. J Inherit Metab Dis. 2010;33 Suppl 3:S199 - 203. [PubMed: 20449661]
- Quadros EV, Nakayama Y, Sequeira JM. Targeted delivery of saporin toxin by monoclonal antibody to the transcobalamin receptor, TCblR/CD320. Mol Cancer Ther. 2010;9:3033 - 40. [PMC free article: PMC2978776] [PubMed: 20858723]
- Radmanesh A, Zaman T, Ghanaati H, Molaei S, Robertson RL, Zamani AA. Methylmalonic acidemia: brain imaging findings in 52 children and a review of the literature. Pediatr Radiol. 2008;2008;38:1054 - 61. [PubMed: 18636250]
- Raval DB, Merideth M, Sloan JL, Braverman NE, Conway RL, Manoli I, Venditti CP. Methylmalonic acidemia (MMA) in pregnancy: a case series and literature review. J Inherit Metab Dis. 2015;2015;38:839 - 46. [PMC free article: PMC4496322] [PubMed: 25567501]
- Rutsch F, Gailus S, Miousse IR, Suormala T, Sagné C, Toliat MR, Nürnberg G, Wittkampf T, Buers I, Sharifi A, Stucki M, Becker C, Baumgartner M, Robenek H, Marquardt T, Höhne W, Gasnier B, Rosenblatt DS, Fowler B, Nürnberg P. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nature Genet. 2009;41:234 - 9. [PubMed: 19136951]
- Ruzkova K, Weingarten TN, Larson KJ, Friedhoff RJ, Gavrilov DK, Sprung J. Anesthesia and organic aciduria: is the use of lactated Ringer's solution absolutely contraindicated? Paediatr Anaesth. 2015;25:807 - 17. [PubMed: 25943188]
- Sakamoto O, Ohura T, Matsubara Y, Takayanagi M, Tsuchiya S. Mutation and haplotype analyses of the MUT gene in Japanese patients with methylmalonic acidemia. J Hum Genet. 2007;52:48 - 55. [PubMed: 17075691]
- Saridakis V, Yakunin A, Xu X, Anandakumar P, Pennycooke M, Gu J, Cheung F, Lew JM, Sanishvili R, Joachimiak A, Arrowsmith CH, Christendat D, Edwards AM. The structural basis for methylmalonic aciduria. The crystal structure of archaeal ATP:cobalamin adenosyltransferase. J Biol Chem. 2004;279:23646 - 53. [PubMed: 15044458]
- Sass JO, Walter M, Shield JP, Atherton AM, Garg U, Scott D, Woods CG, Smith LD. 3-Hydroxyisobutyrate aciduria and mutations in the ALDH6A1 gene coding for methylmalonate semialdehyde dehydrogenase. J Inherit Metab Dis. 2012;35:437 - 42. [PubMed: 21863277]
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Dröse S, Brandt U, Hoffmann GF, Ter Laak H, Kölker S, Smeitink JA. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398:107 - 12. [PMC free article: PMC1525008] [PubMed: 16686602]
- Schwartz GJ, Muñoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20:629 - 37. [PMC free article: PMC2653687] [PubMed: 19158356]
- Sénac JS, Chandler RJ, Sysol JR, Li L, Venditti CP. Gene therapy in a murine model of methylmalonic acidemia using rAAV9-mediated gene delivery. Gene Ther. 2012;19:385 - 91. [PMC free article: PMC3382069] [PubMed: 21776024]
- Sewell AC, Poets CF, Degen I, Stöss H, Pontz BF. The spectrum of free neuraminic acid storage disease in childhood: clinical, morphological and biochemical observations in three non-Finnish patients. Am J Med Genet. 1996;63:203 - 8. [PubMed: 8723111]
- Shapira SK, Ledley FD, Rosenblatt DS, Levy HL. Ketoacidotic crisis as a presentation of mild ("benign") methylmalonic acidemia. J Pediatr. 1991 Jul;119(1 Pt 1):80 - 4. [PubMed: 2066863]
- Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Sakura N, Tajima T, Yamaguchi S. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776:39 - 48. [PubMed: 12127323]
- Sloan JL, Johnston JJ, Manoli I, Chandler RJ, Krause C, Carrillo-Carrasco N, Chandrasekaran SD, Sysol JR, O'Brien K, Hauser NS, Sapp JC, Dorward HM, Huizing M., NIH Intramural Sequencing Center Group. Barshop BA, Berry SA, James PM, Champaigne NL, de Lonlay P, Valayannopoulos V, Geschwind MD, Gavrilov DK, Nyhan WL, Biesecker LG, Venditti CP. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet. 2011;43:883 - 6. [PMC free article: PMC3163731] [PubMed: 21841779]
- Sloan JL, Manoli I, Venditti CP. Liver or combined liver-kidney transplantation for patients with isolated methylmalonic acidemia: who and when? J Pediatr. 2015;166:1346 - 50. [PubMed: 25882873]
- Sniderman LC, Lambert M, Giguere R, Auray-Blais C, Lemieux B, Laframboise R, Rosenblatt DS, Treacy EP. Outcome of individuals with low-moderate methylmalonic aciduria detected through a neonatal screening program. J Pediatr. 1999;134:675 - 80. [PubMed: 10356133]
- Snyderman SE, Sansaricq C, Norton P, Phansalkar SV. The use of neomycin in the treatment of methylmalonic aciduria. Pediatrics. 1972 Dec;50(6):925 - 7. [PubMed: 4636459]
- Spada M, Calvo PL, Brunati A, Peruzzi L, Dell'Olio D, Romagnoli R, Porta F. Liver transplantation in severe methylmalonic acidemia: The sooner, the better. J Pediatr. 2015;167:1173. [PubMed: 26362094]
- Stokke O, Eldjarn L, Norum KR, Steen-Johnsen J, Halvorsen S. Methylmalonic acidemia: a new inborn error of metabolism which may cause fatal acidosis in the neonatal period. Scand J Clin Lab Invest. 1967;20:313 - 28.
- Strømme P, Stokke O, Jellum E, Skjeldal OH, Baumgartner R. Atypical methylmalonic aciduria with progressive encephalopathy, microcephaly and cataract in two siblings--a new recessive syndrome? Clin Genet. 1995;48:1 - 5. [PubMed: 7586637]
- Stucki M, Coelho D, Suormala T, Burda P, Fowler B, Baumgartner MR. Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Hum Mol Genet. 2012;21:1410 - 8. [PubMed: 22156578]
- Suormala T, Baumgartner MR, Coelho D, Zavadakova P, Kozich V, Koch HG, Berghauser M, Wraith JE, Burlina A, Sewell A, Herwig J, Fowler B. The cblD defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J Biol Chem. 2004;279:42742 - 9. [PubMed: 15292234]
- Therrell BL Jr, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113:14 - 26. [PMC free article: PMC4177968] [PubMed: 25085281]
- Traber G, Baumgartner MR, Schwarz U, Pangalu A, Donath MY, Landau K. Subacute bilateral visual loss in methylmalonic acidemia. J Neuroophthalmol. 2011;31:344 - 6. [PubMed: 21873889]
- Treacy E, Arbour L, Chessex P, Graham G, Kasprzak L, Casey K, Bell L, Mamer O, Scriver CR. Glutathione deficiency as a complication of methylmalonic acidemia: response to high doses of ascorbate. J Pediatr. 1996;129:445 - 8. [PubMed: 8804337]
- Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Nissim I, Korson M, Burton B, Yudkoff M. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64:213 - 7. [PMC free article: PMC2640836] [PubMed: 18414145]
- Van Calcar SC, Harding CO, Lyne P, Hogan K, Banerjee R, Sollinger H, Rieselbach RE, Wolff JA. Renal transplantation in a patient with methylmalonic acidaemia. J Inherit Metab Dis. 1998;21:729 - 37. [PubMed: 9819702]
- van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C, et al. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr. 1994;125:903 - 8. [PubMed: 7996362]
- van der Meer SB, Spaapen LJ, Fowler B, Jakobs C, Kleijer WJ, Wendel U. Prenatal treatment of a patient with vitamin B12-responsive methylmalonic acidemia. J Pediatr. 1990;117:923 - 6. [PubMed: 2246694]
- van 't Hoff WG, Dixon M, Taylor J, Mistry P, Rolles K, Rees L, Leonard JV. Combined liver-kidney transplantation in methylmalonic acidemia. J Pediatr. 1998;132:1043 - 4. [PubMed: 9627602]
- van't Hoff W, McKiernan PJ, Surtees RA, Leonard JV. Liver transplantation for methylmalonic acidaemia. Eur J Pediatr. 1999;158 Suppl 2:S70 - 4. [PubMed: 10603103]
- Vernon HJ, Sperati CJ, King JD, Poretti A, Miller NR, Sloan JL, Cameron AM, Myers D, Venditti CP, Valle D. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. J Inherit Metab Dis. 2014;37:899 - 907. [PMC free article: PMC4373418] [PubMed: 24961826]
- Walter JH, Michalski A, Wilson WM, Leonard JV, Barratt TM, Dillon MJ. Chronic renal failure in methylmalonic acidaemia. Eur J Pediatr. 1989;148:344 - 8. [PubMed: 2707280]
- Wasserstein MP, Gaddipati S, Snyderman SE, Eddleman K, Desnick RJ, Sansaricq C. Successful pregnancy in severe methylmalonic acidaemia. J Inherit Metab Dis. 1999;22:788 - 94. [PubMed: 10518278]
- Williams ZR, Hurley PE, Altiparmak UE, Feldon SE, Arnold GL, Eggenberger E, Mejico LJ. Late onset optic neuropathy in methylmalonic and propionic acidemia. Am J Ophthalmol. 2009;147:929 - 33. [PubMed: 19243738]
- Wong SN, Low LC, Lau YL, Nicholls J, Chan MY. Immunodeficiency in methylmalonic acidaemia. J Paediatr Child Health. 1992;28:180 - 3. [PubMed: 1562372]
- Worgan LC, Niles K, Tirone JC, Hofmann A, Verner A, Sammak A, Kucic T, Lepage P, Rosenblatt DS. Spectrum of mutations in mut methylmalonic academia and identification of a common Hispanic mutation and haplotype. Hum Mutat. 2006;27:31 - 43. [PubMed: 16281286]
- Yang X, Sakamoto O, Matsubara Y, Kure S, Suzuki Y, Aoki Y, Suzuki Y, Sakura N, Takayanagi M, Iinuma K, Ohura T. Mutation analysis of the MMAA and MMAB genes in Japanese patients with vitamin B(12)-responsive methylmalonic acidemia: identification of a prevalent MMAA mutation. Mol Genet Metab. 2004;82:329 - 33. [PubMed: 15308131]
- Yano S, Li L, Le TP, Moseley K, Guedalia A, Lee J, Gonzalez I, Boles RG. Infantile mitochondrial DNA depletion syndrome associated with methylmalonic aciduria and 3-methylcrotonyl-CoA and propionyl-CoA carboxylase deficiencies in two unrelated patients: a new phenotype of mtDNA depletion syndrome. J Inherit Metab Dis. 2003;26:481 - 8. [PubMed: 14518828]
- Yu HC, Sloan JL, Scharer G, Brebner A, Quintana AM, Achilly NP, Manoli I, Coughlin CR 2nd, Geiger EA, Schneck U, Watkins D, Suormala T, Van Hove JL, Fowler B, Baumgartner MR, Rosenblatt DS, Venditti CP, Shaikh TH. An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am J Hum Genet. 2013;93:506 - 14. [PMC free article: PMC3769968] [PubMed: 24011988]
- Zsengellér ZK, Aljinovic N, Teot LA, Korson M, Rodig N, Sloan JL, Venditti CP, Berry GT, Rosen S. Methylmalonic acidemia: a megamitochondrial disorder affecting the kidney. Pediatr Nephrol. 2014;29:2139 - 46. [PubMed: 24865477]
- Zwickler T, Haege G, Riderer A, Hörster F, Hoffmann GF, Burgard P, Kölker S. Metabolic decompensation in methylmalonic aciduria: which biochemical parameters are discriminative? J Inherit Metab Dis. 2012;35:797 - 806. [PubMed: 22249333]
章节备注
作者备注
Manoli 医生是儿科医生、临床和生化遗传学家。 她是美国国立卫生研究院国家人类基因组研究所有机酸研究室的临床医生。
Venditti 医生是一名儿科医生、临床和生化遗传学家。 他是美国国家人类基因组研究所有机酸紊乱研究室主任和美国国立卫生研究院临床中心的主治医师。
更新历史
- 2016 年 12 月 1 日 (cpv) 修订:分子遗传学:MMAB 和 MMUT
- 2016 年 1 月 7 日(我)实时发布全面更新
- 2010 年 9 月 28 日(我)实时发布全面更新
- 2007 年 1 月 18 日 (cd) 修订:临床上可用的 MMAA 和 MMAB 突变检测
- 2005 年 8 月 16 日(我)综述发布
- 2004 年 5 月 11 日 (cpv) 原稿提交
注:根据美国版权法 17 USC 第 105 节,GeneReview“孤立的甲基丙二酸血症”在美国的公共结构域中。