
脑白质营养不良的定义
脑白质营养不良连同相关的名词如髓鞘形成障碍、脱髓鞘与脑白质病适用于一大组疾病。 在GeneReview本章中用于以下定义:脑白质营养不良是一组累及中枢神经系统白质伴或不伴周围神经系统异常的遗传性髓鞘病。表现
如下:
:
- 胶质细胞或者髓鞘异常,目前已经明确这类神经病通过累及少突胶质细胞、星形胶质细胞与其它非神经元细胞类型为主要特征。特别注意的是,许多脑白质营养不良基础的致病机制不清楚。
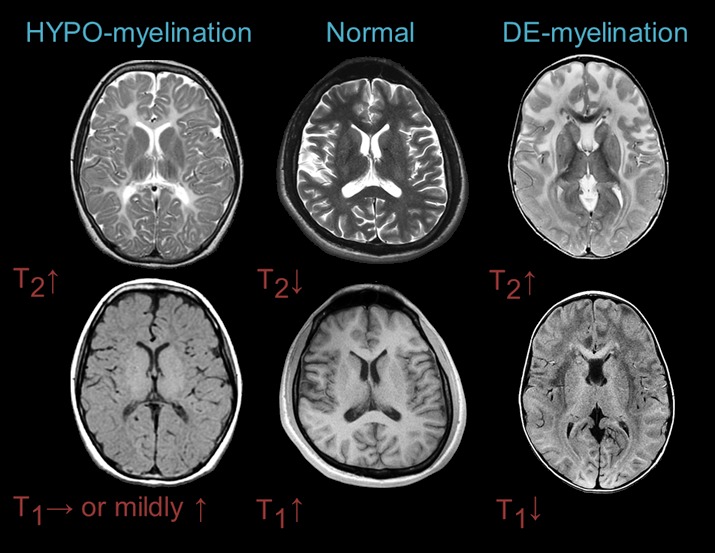
- MRI表现 (见 图1)
- 必须具有脑白质T2加权像高信号表现。
- T1加权像可以表现不同:等信号或者高信号见于髓鞘形成不良性脑白质营养不良;低信号则存在于脱髓鞘性脑白质营养不良。
脑白质营养不良的临床表现
大多数脑白质营养不良符合GeneReview本章中的定义见表 1. 由于白质纤维束的广泛所累所致儿童早期肌张力低下的表现,随着时间推移逐渐进展为痉挛性截瘫,这些均可能导致从轻度痉挛性双侧瘫痪到严重痉挛性截瘫的程度不同运动受限的功能障碍。此外,运动功能障碍有可能显著损伤吞咽、咀嚼以及呼吸(有些患者)等重要功能。痉挛可能来自骨科并发症如脊柱侧弯和大关节脱位。明确椎体功能障碍(例如痉挛)有时可能隐藏或者掩盖像肌张力障碍和/或运动障碍的椎体外系运动疾病的表现。
例如, 在 MCT8-特异性甲状腺激素细胞转运体缺乏 肌张力障碍是主要表现。某些脑白质营养不良以共济失调为主要表现以致功能丧失; 例如, 伴中枢神经系统髓鞘形成低下性儿童共济失调/消融性白质脑病 (CACH/VWM) 与伴促性腺功能减退与牙齿发育不全的髓鞘形成不良 (4H 综合征). 惊厥常常见于脑白质营养不良病程的晚期,而极少数例外的脑白质营养不良惊厥呈特征性表现
( 例如 亚历山大病 ) 。脑白质营养不良在儿童或者成人的认知发育迟缓或者认知功能改变远不如运动功能障碍更常见。
由于大多数脑白质营养不良进行性认知功能丧失呈缓慢过程,痴呆并不是早期特征性表现。
脑白质营养不良的流行病学
脑白质营养不良相关总发病率的流行病学数据非常有限 [Heim et al 1997, Bonkowsky et al 2010]. 并且很难从中推断出这些研究中特定的脑白质营养不良的发病率。已有脑白质营养不良流行病学的较好信息来源于常规的专科门诊与综合神经科诊所; 包括 亚历山大病 [van der Knaap et al 1999], X连锁肾上腺脑白质营养不良 [Bezman et al 2001] 与异染性脑白质营养不良. 由于一些仅特定在成年期发病的脑白质营养不良与髓鞘形成不良性脑白质营养不良等疾病越来越明确,进而对遗传性白质病的异质性也提高了认识。
值得注意的是由于这类疾病的异质性与复杂性,大约50%的脑白质病患者的特定病因并不明确 [Schiffmann & van der Knaap 2009]。
脑白质营养不良鉴别诊断
表 2 (pdf)列出了明显累及白质的遗传病但非脑白质营养不良,包括以下疾病::
- 大脑皮层或者其它灰质结构中原发受累的神经元中枢神经系统疾病 (这些病头颅MRI也表现明显白质异常). 典型表现为明显的脑病与惊厥。
- 不同类型疾病像GM1神经节苷脂沉积症, GM2 神经节苷脂沉积症, 与神经元蜡样脂褐质沉积症 可以表现明显的白质异常,由于这类疾病原发于神经元也影响髓鞘的正常过程,这类疾病晚发类型不可避免影响大脑白质; 轻度信号异常与轴突和髓鞘丢失的变性有关。
- 其它疾病如 MCT8-特异性甲状腺激素细胞转运子缺乏髓鞘化明显迟缓但随时间变化其能够基本完成髓鞘化进程。
- 先天性代谢缺陷 伴有明显继发性白质异常 像有机酸血症、氨基酸代谢病与许多其它病. 见表 2 (pdf).
伴有明显白质受累的其它疾病包括:
- 获得性中枢神经系统髓鞘疾病 可以导致脱髓鞘像多发性硬化与感染、感染后与自身免疫病因的疾病. 虽然不表现孟德尔遗传方式, 但这些疾病中一些呈 多因素的 病因学包括遗传易感性。
- 像急性播散性脑脊髓膜炎、多发性硬化与视神经脊髓炎这类疾病与遗传性白质病典型鉴别通过起病急与多相表现。 脑MRI异常也表现为多灶与片状伴随时间、治疗的变化甚至好转[Schiffmann & van der Knaap 2009]。
- 髓鞘毒性损伤 如见于海洛因滥用或者甲氨蝶呤相关的毒性。
- 中枢神经系统损伤, 特别在围产期能够导致明显的白质信号异常,典型呈不规则,也可以引起白质体积的减少。
- 非遗传性血管损伤
脑白质营养不良诊断策略
患者疑诊脑白质营养不良,下述诊断方法可以鉴别特异性脑白质营养不良,有助于预后评估与遗传咨询. 对于疑诊患者进行特异性脑白质营养不良(表1)诊断通常包括病史和详细家族史;进行体格检查与神经系统体检;查阅头MRI表现;以及特异实验室检查,通常包括
分子遗传学检测. 注意:
: 虽然遵循本节讨论的诊断方法,但在临床上大多数脑白质营养不良患者的特异诊断尚未建立 [van der Knaap et al 1999, Schiffmann & van der Knaap 2009].
病史
明确的临床特征病史有助于确定特异的脑白质营养不良;然而,对于大多数病例仅仅表现非特异性功能丧失 (主要为运动与单纯依靠病史并不能提供深入的特异性诊断。例如,可以为髓鞘形成低下性脑白质营养不良提供诊断线索如下所列:
- 先天性白内障: 髓鞘低下性脑白质营养不良与先天性白内障 (HCC)
- 牙齿发育不全和/或低促性腺素性功能减退症: POLR3相关性脑白质营养不良
- 严重惊厥: 唾液酸贮积病
可以为脱髓鞘性脑白质营养不良提供诊断线索如下所列::
- 反复呕吐: 亚历山大病
- 肾上腺功能障碍:X连锁肾上腺脑白质营养不良 (X-ALD)
- 早发自主神经功能障碍: 常显成人发病的脑白质营养不良 (ADLD)
- 慢性脑脊液淋巴细胞增多或(更常见)复发“无菌性脑膜炎”: Aicardi-Goutières综合征
- 发烧或跌倒后功能突然丧失: 伴中枢神经系统髓鞘形成低下性儿童共济失调/消融性白质病 (与其它可能的脑白质营养不良)
家族史
详细的三代家族史特别关注肌张力低下、痉挛、肌张力障碍、癫痫发作、共济失调和/或认知发发育迟缓与随时间推移的认知功能改变的个体。
- 需要亲属评估和/或查阅病历。
体格检查与神经系统检查
在大多数情况下,体检结果并不能提示特异诊断;然而,某些表现可能引导读者进一步探索特定的病因:
- 巨颅; 见 表 6 (pdf)
- 牙齿排列异常: POLR3相关性脑白质营养不良 或者眼齿指发育不良 (ODDD)
- 成年腭肌肌阵挛: 亚历山大病
- 黄色瘤: 脑腱黄瘤病 (CTX)
- 皮肤色素异常沉着: X连锁肾上腺脑白质营养不良 (X-ALD)
- 鱼鳞病: Sjögren-Larsson综合征
- 视网膜血管异常:伴钙化和囊肿脑视网膜微血管病变(CRMCC)
- 视网膜检查樱桃红斑:像 GM1 与 GM2神经节苷酯沉积症
脑MRI表现
步骤1
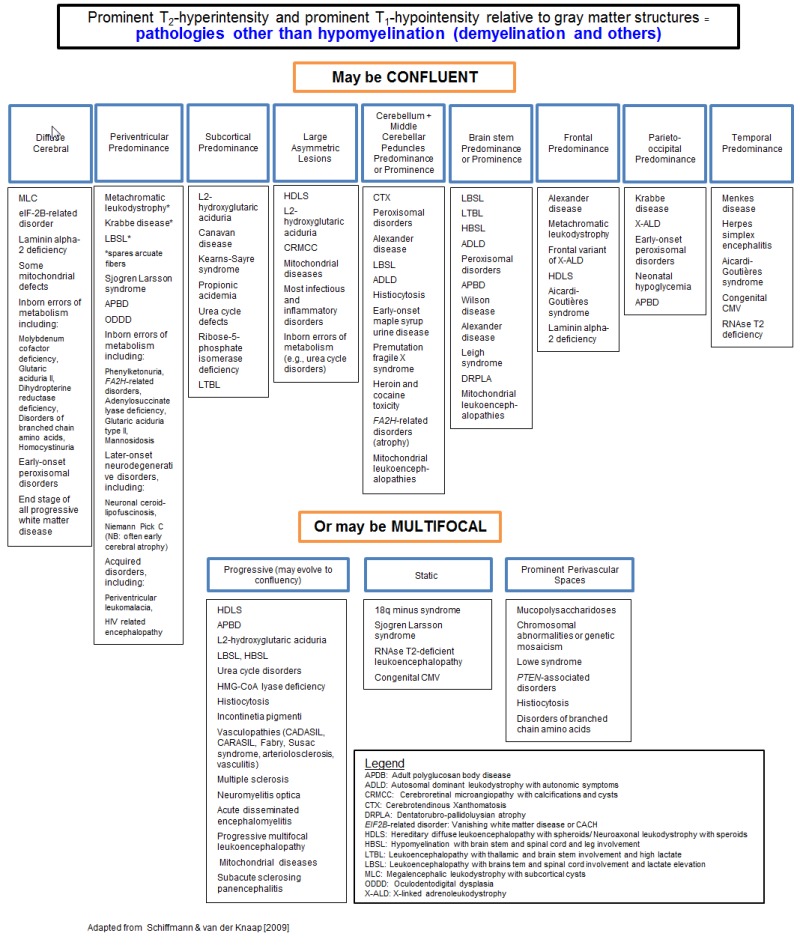
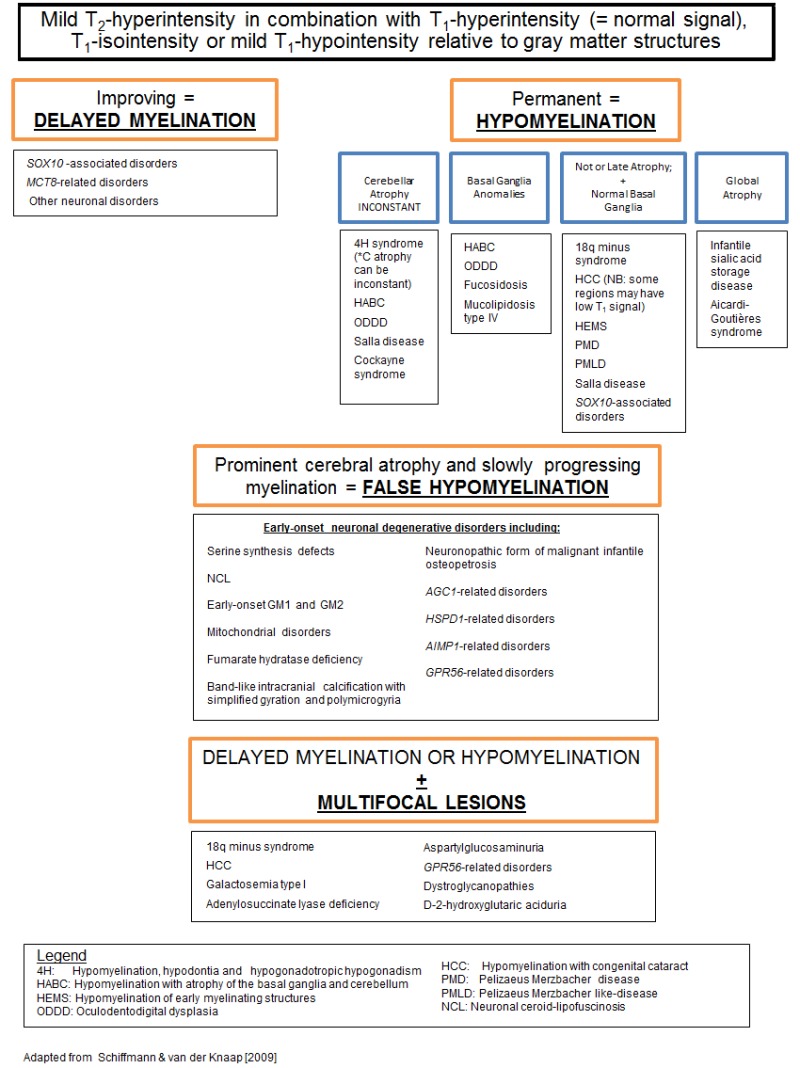
确定脑MRI异常模式是否与髓鞘形成低下性脑白质营养不良或脱髓鞘性脑白质营养不良一致。 (图 1) [van der Knaap et al 1999, van der Knaap & Valk 2005, Schiffmann & van der Knaap 2009].
步骤2
如果所累及模式提示特异诊断则确定髓鞘形成低下性或者脱髓鞘性脑白质营养不良。
髓鞘形成低下性脑白质营养不良。 注意神经影像表现的特异模式如下所列:(图 2):
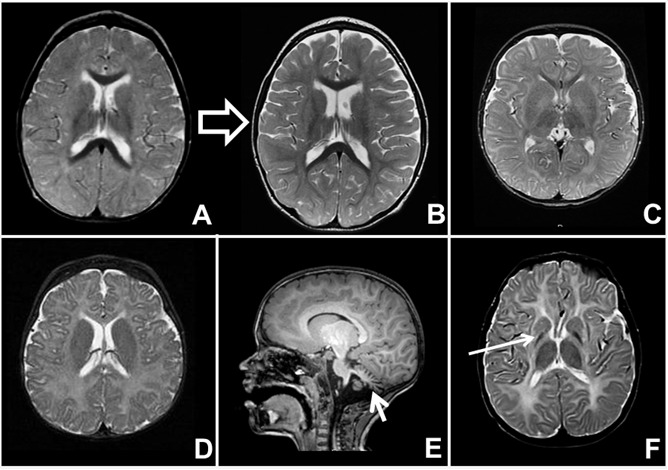
- 随时间推移髓鞘改善 (例如,见 MCT8特异性甲状腺激素细胞转运缺陷;见 图 2A→B)
- 严重皮层灰质萎缩(见于原发神经元疾病以及数量有限的经典性脑白质营养不良;见 图 2C)
- 小脑萎缩 (例如, POLR3相关性脑白质营养不良 [4H综合征]; 见 图 2E)
- 基底节受累 ((例如, HABC综合征; 见 图 2F)
脱髓鞘性脑白质营养不良
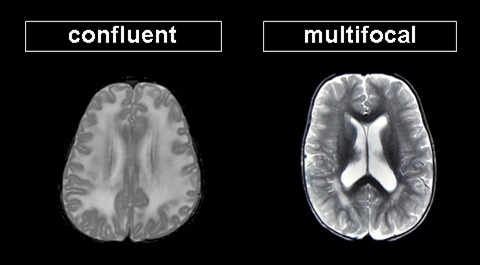
- 确定白质异常主要区域。
- 融合性白质病变呈广泛的大脑重要部位白质异常,常影响特定区域或通路,不一定完全对称。
- 多灶性白质病变更为散在分布,常不对称,累及范围有限。多灶性异常具有特异性鉴别诊断。
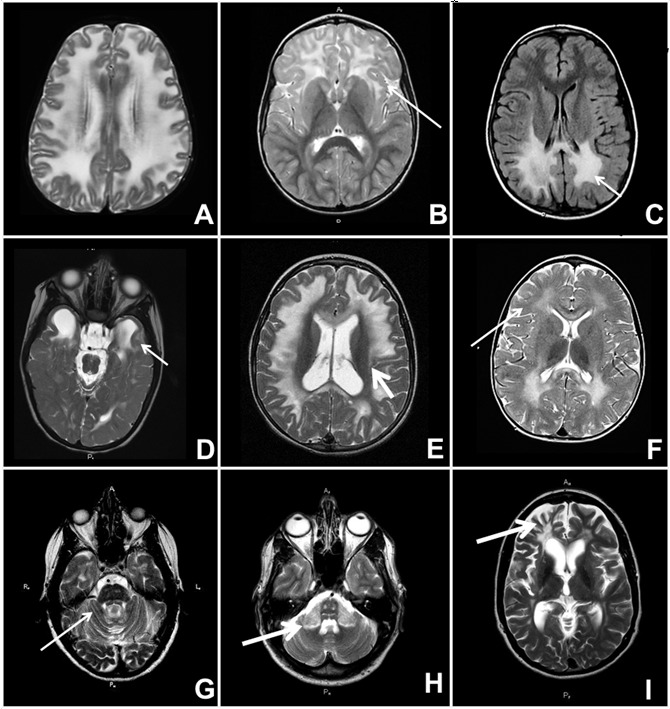
脱髓鞘性脑白质营养不良与其它疾病
髓鞘形成低下性脑白质营养不良
步骤 3
寻找以下相关特征,除了脑MRI表现模式外关注以下相关特征有助于识别特定的脑白质营养不良:

特异实验室检测 (包括分子遗传学检测)
如果脑MRI表现与特异性脑白质营养不良一致则考虑相应疾病的生物化学或者分子遗传学检测 (表 1). 注意: 基于评价过程中获得的分子遗传学检测信息可作为在阶梯式单 基因检测, 如果可行,也可以进行表型靶向检测.
表 1.
严格符合脑白质营养不良诊断标准
| 过氧化物酶体的生物合成障碍, Zellweger综合征谱系 (PBD, ZSS) 7 | AR | PEX | 血浆VLCFA,植烷与降植烷酸,血浆及尿哌啶酸和胆汁酸浓度有助于区分不同类型的过氧化物酶体病 |
| Pol III相关性脑白质营养不良 8 | AR | POLR3A POLR3B | |
| RNAse T2缺乏脑白质病 | AR | RNASET2 | |
| 过氧化物酶体脂肪酸β-氧化的单酶缺陷9 | AR | 双功能蛋白缺陷: HSD17B4 | 血浆VLCFA,植烷与降植烷酸,血浆及尿哌啶酸和胆汁酸浓度有助于区分不同类型的过氧化物酶体病 |
| 过氧化物酶体酰基辅酶A氧化酶缺乏: ACOX1 | |||
| SCPx缺陷: SCP2 | |||
| Sjögren-Larsson 综合征 | AR | ALDH3A2 | 尿中代谢产物白三烯B4异常 体外培养的皮肤成纤维细胞,白细胞:脂肪醛脱氢酶活性不足(FALDH)和/或脂肪醇:NAD氧化还原酶(FAO) |
| SOX10-相关疾病 | AD | SOX10 | |
| X连锁肾上腺脑白质营养不良 (X-ALD) | XL | ABCD1 | 血浆VLCFA测定法: C26:0, ↑ C24:0 / C22:0, ↑C26:0 /C22:0 |
AD = 常染色体显性遗传
AR = 常染色体隐性遗传
XL = X-linked
VLCFA = 极长链脂肪酸
按字母顺序排列疾病;作为genereviews命名
1.
2.
目前这种疾病表现为了区别Coats加编码保守端粒维持元件1的CTC1致病突变致病。
3.
包括Salla病;婴儿唾液酸贮积病,中间型
4.
PSAP缺陷引起已经报道GALC 活动必须的SapA-d活化蛋白缺陷。
PSAP致病突变导致ARSA 活动必须的活化蛋白SapB-d缺陷。
也被称为伴神经轴突球体和色素细胞成年发病的脑白质营养不良神经轴突球体和色素细胞;可能包括遗传性弥漫型;伴色素胶质细胞色素正色脑白质营养不良的(POLD)
7.
包括新生儿肾上腺脑白质营养不良;婴儿Refsum病
包括髓鞘形成低下、牙齿发育不良、性腺功能减退(4H综合征);共济失调,出牙延迟和髓鞘形成低下(ADDH);震颤-共济失调伴中枢髓鞘髓鞘形成低下(TACH);伴少牙的脑白质营养不良(LO);小脑萎缩和与胼胝体发育不全髓鞘形成低下(HCAHC)。
9.
包括D-双功能蛋白(DBP)不足; 固醇 携带者 蛋白-2 (SCPx) 缺乏; 过氧化物酶体酰基辅酶A氧化酶缺乏
遗传咨询
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
遗传咨询相关问题
DNA库 是储存DNA为将来可能的用途。DNA储存 (经典从白细胞中提取) 有利于将来在测试方法以及对基因的了解,等位基因变异与疾病方面具有较大进展,应当建立个人银行 受累的个体的DNA库。
来源
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- 髓鞘病生物注册项目Email: myelindisorders@cnmc.org
管理
治疗管理
虽然脑白质营养不良的基础机制是多种多样的,但这组疾病具有许多类似表现。在大多数情况下,虽然目前没有对因治疗方法,但对症治疗可以改善这些复杂疾病患者的舒适和照顾。理想状态下,对脑白质营养不良儿童或成人的治疗管理应该由脑白质营养不良医疗方面有经验的专家组成的多学科进行。
痉挛.药物用于治疗肌张力与神经信号到肌肉的传导阻滞(化学去神经法)。强化物理治疗用于改善运动能力与功能。
椎体外系表现.肌张力障碍与运动障碍的药物治疗能够明显促进功能的改善。共济失调.尽管康复措施可以有很大的帮助,但对于已经存在共济失调并没有特异治疗。癫痫发作. 癫痫发作应用典型的抗惊厥药物治疗,难治性癫痫很少发生,除了在生命的终点。认知发育迟缓/脑病. 重要的是倡导学校与工作中脑白质营养不良患者避免运动障碍相关的限制。 增强交流可以用于言语障碍.根据需要为认知落后与运动障碍提供便利条件。整形手术. 应注意预防和治疗骨科问题,如髋关节脱位和脊柱侧凸。喂养. 吞咽障碍和因吸入的风险增加引起肺部问题在疾病进展中很常见。 营养摄入减少和发育停滞也可能发生,为营养放置胃管的决定基于个人的整体健康状况、预期疾病病程与家庭和患者的意愿。
原发病的预防
少数的脑白质营养不良对原发病进行预防:例如X连锁肾上腺脑白质营养不良, Krabbe病,与异染性脑白质营养不良, 疾病早期进行造血干细胞移植(HSCT)或骨髓移植(BMT)可能是有益的。对于考虑HSCT 或者 BMT的患者建议到专科中心。[Eichler et al 2009]
监测
标准的监测包括以下方面:
- 体重与身高的常规测量以评估生长与营养状态
- 体格检查和/或系列髋与脊柱X线检查以监测骨科并发症
- 常规复查病史与癫痫发作症状
有些疾病需要专门的监测,例如监测亚历山大病脑积水的发展.
避免情况
大量脑白质营养不良证据表明轻微头部外伤与感染可以使得临床表现阵发性恶化。虽然这种情况仅 在伴中枢神经髓鞘形成低下性儿童共济失调/消融性脑白质病清晰证据, 但避免其触发的可能性。
研究中的治疗
搜索 ClinicalTrials.gov 获取大范围疾病的临床研究信息。
References
Literature Cited
- Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, Kass NE, Moser HW. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49:512鈥�7. [PubMed: 11310629]
- Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R. The burden of inherited leukodystrophies in children. Neurology. 2010;75:718鈥�25. [PMC free article: PMC2931652] [PubMed: 20660364]
- Eichler F, Grodd W, Grant E, Sessa M, Biffi A, Bley A, Kohlschuetter A, Loes DJ, Kraegeloh-Mann I. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am J Neuroradiol. 2009;30:1893鈥�7. [PubMed: 19797797]
- Heim P, Claussen M, Hoffmann B, Conzelmann E, Gärtner J, Harzer K, Hunneman DH, Köhler W, Kurlemann G, Kohlschütter A. Leukodystrophy incidence in Germany. Am J Med Genet. 1997;71:475鈥�8. [PubMed: 9286459]
- Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750鈥�9. [PMC free article: PMC2677542] [PubMed: 19237705]
- van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999;213:121鈥�33. [PubMed: 10540652]
- van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. Berlin, Germany: Springer. 2005.
Chapter Notes
Revision History
- 6 February 2014 (me) Review posted live
- 14 February 2012 (av) Original submission