
摘要
临床表现.
HFE相关的遗传性血色病(HFE-HH)的特征在于胃肠粘膜的过量吸收铁。目前HFE-HH公认的表型谱包括:
- 有HFE-HH的临床患者,表现为铁储存的终末靶器官继发损伤;
- HFE-HH生化指标中,铁负荷过高的唯一迹象是铁离子浓度的增加和血清铁蛋白浓度的增加;
- 非表达的p.Cys282Tyr纯合子, 既不存在HFE-HH的临床表现,也不存在铁负荷过量。
临床HFE-HH的特征是在肝脏,皮肤,胰腺,心脏,关节和睾丸中铁储存过量。在未经治疗的个体中,早期症状可能包括:腹痛,虚弱,嗜睡和体重减轻;当血清铁蛋白高于1,000 ng/mL时,肝硬化的风险显着增加;其它发现可能包括皮肤色素沉着,糖尿病,充血性心力衰竭和/或心律失常,关节炎和性腺功能减退症的逐渐增加。临床HFE-HH在男性中比女性更常见。
诊断/实验室检查.
结合临床症状作出HFE-HH临床诊断,HFE-HH诊断生化指标主要是铁饱和度大于或等于45%,血清铁蛋白含量高于正常值上限(即男性>300 ng/mL ,女性>200 ng/mL),且HFE分子遗传学检测有两种致病性变异。虽然血清铁蛋白浓度可能随着时间的推移在HFE-HH未经治疗的个体中逐渐增加,但这并不是HFE-HH特有的,因此不能单独用于HFE-HH的诊断。
管理.
治疗的表现:临床HFE-HH:通过静脉切开术(放血)治疗以维持血清铁蛋白浓度≤50 ng/mL。生化HFE-HH:当血清铁蛋白浓度>500 ng/mL时开始静脉放血。p.Cys282Tyr 纯合突变:放血不是必需的,因为这些患者没有铁负荷过高的情况出现。
预防继发性并发症:预防甲型和乙型肝炎疫苗:
- 临床HFE-HH: 血清铁蛋白浓度<50 ng/mL,每3-4个月进行一次血清铁蛋白浓度的常规监测;对肝硬化个体的肝细胞癌(HCC)进行常规筛查。
- 生化HFE-HH和非表达p.Cys282Tyr纯合突变:当铁蛋白浓度超过正常水平时开始每年测量血清铁蛋白浓度。
药物/情况避免:药用铁、矿物质补充剂、过量的维生素C和未经烹煮过的海鲜;肝脏受累者饮酒。
亲属风险评估;提供分子遗传学检测的先证者纯合性为p.Cys282Tyr的成年同胞允许早期诊断和监测。
遗传咨询.
HFE-HH以常染色体隐性遗传方式遗传。
同胞风险研究:当一个患病个体父母均为杂合的HFE致病性变异时,同胞继承两个HFE致病性变异的风险是25%。然而,由于欧洲人HFE致病性变异携带者频率高(11%或1/9),有时受影响个体的父母中有一方有两个异常的HFE等位基因。在这种情况下,每个同胞继承两个HFE致病性变异的风险是50%。
后代风险研究:HFE-HH个体的后代从患病父母那里遗传了一个HFE致病性变异。由于另一个亲本是突变HFE等位基因的携带者的几率为1/9,所以后代继承两个HFE致病性变异的风险大约为5%。
产前检测:对于有25%或更高风险的疾病,虽然产前检测技术上可行,但是需要非常高的要求,因为HFE-HH是成人期发病,是可治疗且低临床外显率的疾病。
诊断
美国肝病研究协会(AASLD) 出版了诊断和治疗血色病的实践指南[Bacon et al 2011] (全文).
结果显示
HFE相关的遗传性血色病(HFE-HH)应该对疑似患病个体的铁负荷量过高相关的临床体征做进一步检测,其中包括:
- 肝肿大
- 肝硬化
- 原发性肝细胞癌
- 糖尿病
- 心肌病
- 性腺功能减退症
- 关节炎(尤其是掌指关节)
- 皮肤色素沉着逐渐增多
铁负荷过量的生化迹象表明血清转铁蛋白-铁饱和度升高(TS)和血清铁蛋白浓度升高。
- 转铁蛋白-铁饱和度(TS) 是HFE-HH铁负荷过量风险检测的早期的可靠指标;在成年人中,TS与年龄和临床表现的存在与否无关。
- 在没有其它已知导致转铁蛋白升高原因的情况下,约80%的HFE-HH患者出现两次或两次以上的空腹血清转铁蛋白饱和度高于60%(男性)或50%(女性)。
- 在未经治疗的临床HFE-HH患者中,血清铁蛋白浓度通常随时间逐渐增加;然而,血清铁蛋白浓度升高并不是铁负荷过量的具体原因,因为它是急性期反应物,并且也可能是由炎症或肿瘤引起的(特别是当血清TS正常时)。
一般公认的铁蛋白值的正常上限(来自HEIRS研究)中男性<300 ng/mL,女性<200 ng/mL[Adams et al 2005]。 - HFE-HH患者的铁蛋白值没有“典型”的范围:可能是从正常值到几千。
已经开发了用于筛选HFE相关血色病的算法(见 图 1)。
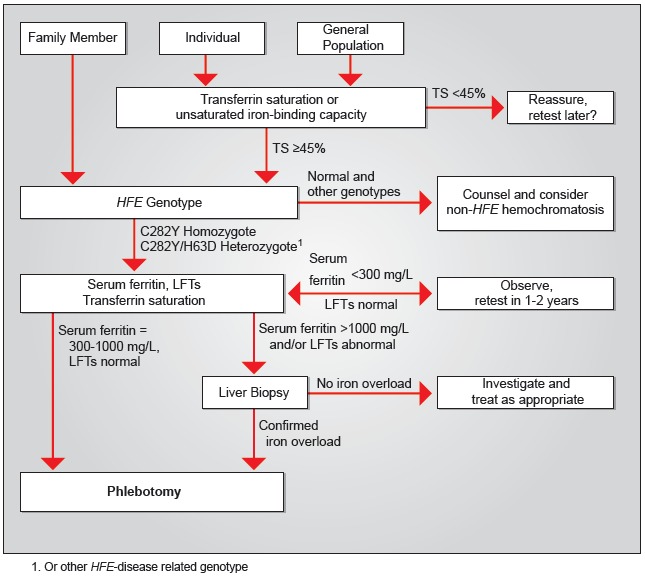
图 1.
使用LFT(肝功能测试)和TS(转铁蛋白饱和度)筛选HFE相关血色病的算法最初发表在 Eijkelkamp et al [2000];
建立诊断
HFE-HH的诊断可通过先证者HFE中鉴定到双等位基因的致病性变异进行确诊(见 表 1)。
分子测试方法包括:
- 针对p.Cys282Tyr和p.His63Asp位点的HFE致病性变异分析;
- 如果发现少于两个致病性变异,则对HFE的序列进行分析,然后进行基因-靶向缺失/重复分析。
注意:目前发现,全HFE基因缺失是撒丁人群中最常见的致病性变种[Le Gac et al 2010]。
表 1.
HFE-HH分子遗传学检测总结
| 基因 1 | 测试方法 | 该方法可检测出致病性变异者的比例 | |
|---|---|---|---|
基因型 | % 个体HH百分比 2,3 | ||
| HFE | 针对特定致病性变异分析 4 | p.[Cys282Tyr]+[Cys282Tyr] | ~60%-90% 5 |
| p.[Cys282Tyr]+[His63Asp] | 3%-8% 5 | ||
| p.[His63Asp]+[His63Asp] | ~1% 6 | ||
序列分析 7 | 其它 8 | 罕见 8 | |
基因靶向 缺失/重复分析 9 | 其它 | 罕见 10 | |
2.欧洲血统的人群[Ramrakhiani & Bacon 1998]
3.Morrison et al [2003]
4.通常包括p.Cys282Tyr和p.His63Asp致病性变异。注:表格中所包含的致病性变异可能因实验室而异。
5.Feder et al [1996]
6.虽然生化定义的异常可能存在于p.[His63Asp]+[His63Asp],但是特征性的临床表现是罕见的 [Gochee et al 2002]。
7.序列分析检测结果意义不确定,可能是恶性或良性结果,也可能是良性的变异。致病性变异可以包括小的基因内缺失/插入和错义, 无义,和剪接位点变异;通常不检测外显子或全-基因缺失/重复。 对于解释序列分析结果时需要考虑的问题,请点击这里。
8.通常复合杂合为p.Cys282Tyr 致病性变异和第二个罕见的致病性变异。描述了许多区域性的致病性变异,包括p.Glu168Ter和p.Trp169Ter,在意大利北部两个地区,血色病患者的等位基因频率分别为25%和8.4%[Piperno et al 2000]。
9.基因-靶向缺失/重复分析检测基因缺失或重复。可用方法包括:定量 PCR,长片段PCR,多重连接探针扩增(MLPA),和设计用于检测单外显子缺失或重复的基因-靶向微阵列。
10.HFE全基因缺失是撒丁裔人群血色病的最常见原因[Le Gac et al 2010]。
临床特征
临床描述
公认的三种HFE相关的遗传性血色病(HFE-HH)表型:
- 临床HFE-HH ((继发于铁储存的器官损伤个体[例如晚期肝硬化,心力衰竭,皮肤色素变化或糖尿病])
- 生化HFE-HH (仅通过转铁蛋白-铁饱和度和血清铁蛋白浓度确定具有铁负荷过量迹象的个体)
- 非表达p.Cys282Tyr纯合突变(无铁负荷过量的临床或生化迹象的p.cys282tyr纯合子[即正常的血清铁蛋白浓度])
HFE-HH患者可能因铁负荷过量相关的体征和症状(即临床HFE-HH)而被确诊;然而,症状出现之前可以通过检测铁异常的相关研究(即生化HFE-HH),或通过分子遗传学检测的方法进行评估家族成员是否患有HFE-HH(非表达p.Cys282Tyr纯合子)。
临床HFE-HH和生化HFE-HH之间的差异 可以在评估与HH有关的发病率的人群研究解释中得到答案。人群中的几项大规模筛查研究表明,大多数p.Cys282Tyr纯合性 致病性变异个体不具有临床HFE-HH特征;然而,这个基因型(特别是男性)的相当一部分个体具有生化HFE-HH特征。
影响疾病表现的因素
- 性别:有报道称p.Cys282Tyr纯合子中,男性比例高于女性(28% vs 1%),有明确的血色素沉着病的表现[Allen et al 2008].
- 筛选检测:当通过铁研究或筛查有风险的家庭成员时,75%-90%HFE-HH患者都是无症状的。
- 正常的血清铁蛋白浓度通常与缺乏症状发展有关 [Yamashita & Adams 2003]。
- 在临床受累的个体的高风险同胞中,临床疾病似乎更为常见。
临床HFE-HH
具有临床HFE-HH的个体通过胃肠粘膜过量吸收正常饮食中的铁,导致铁负荷过量,可能导致一些终末靶器官的损伤,并可能导致器官衰竭。
发病年龄:铁负荷过量相关症状通常出现在男性40-60岁之间以及女性更年期之后。HFE-HH偶尔会出现在较早的年龄,但肝纤维化或肝硬化很少在40岁之前出现。
早期的迹象:
临床HFE-HH的最初征兆通常是累及掌指关节的关节病(关节僵硬和疼痛),黑色素和铁沉积导致的皮肤色素沉着逐渐增加,由胰脏铁沉积物引起的糖尿病,和由心脏实质铁储存产生的心肌病。疾病早期可能会出现肝肿大;然而,无症状的个体偶尔也会有肝肿大。男性可能患有垂体功能减退症。女性性腺功能减退导致性欲减退,可能不育。腹痛,虚弱,嗜睡和体重减轻是常见的,但具有非特异性。
饮酒会使HFE-HH的症状恶化[Scotet et al 2003]。中度至重度饮酒使肝硬化的风险增加10% [Fletcher et al 2002]。
随着病情的发展,肝硬化可能并发门静脉高压、肝细胞癌和终末期肝病。[Kowdley et al 2005]。HEIRS研究发现男性肝脏疾病的比值为3.3,纯合性为p.Cys282Tyr [Adams et al 2005]。另外,如上所述,与饮酒少的人群相比,每天消耗超过60 g酒精的p.Cys282Tyr纯合子个体中,肝硬化更常见[Fletcher et al 2002]。当确认为肝硬化或肝功能衰竭时,约
50%的人患有糖尿病,的人有充血性心力衰竭或心律失常。
预期寿命:肝硬化发生前诊断和治疗的患者有正常的预期寿命;而在肝硬化发生后确诊的患者,即使是经过由于肝细胞癌的发展而导致的缺铁治疗,其预期寿命[Adams et al 2005]也会降低。
预后:肝硬化患者的治疗效果优于非肝硬化患者; 然而,在肝硬化患者成功的铁耗竭后,治疗并不能消除肝癌(HCC)和胆管癌,依然存在10%-30% 的患病风险。
经过18个月的治疗后,如果患者铁储存未能消耗,表明预后不良。 随着铁耗竭,一些受累的器官(肝脏和心脏)的功能障碍可以改善;然而,内分泌异常和关节病中只有20%的人得到治疗改善。
临床HFE-HH患者的死亡通常是由肝衰竭,癌症,充血性心力衰竭或心律失常引起的。
生化HFE-HH
对于没有临床HFE-HH但具有生化HFE-HH的个体是否铁负荷过量并发症的风险会增加,以及他们是否为静脉切开术治疗的候选人,专家之间存在争议(见 管理)。
铁蛋白水平诊断:Bardou-Jacquet及其团队的结论是,HFE Cys282Tyr纯合子诊断血清铁蛋白在正常上限与1000 μg/L之间时,与静脉切开术治疗的一般人群相比死亡率降低[Bardou-Jacquet et al 2015a, Bardou-Jacquet et al 2015b, Bardou-Jacquet et al 2015c]。 相反,澳大利亚团队相信由于没有随机研究的支持,静脉切开术对血清铁蛋白轻度升高的p.Cys282Tyr纯合突变患者是否能带了益处是未得到证实的[Delatycki et al 2015]。
一些前瞻性研究研究铁负荷过量是否是一个渐进过程,目前尚无明确结论。目前的证据表明,尽管血清铁蛋白浓度随着时间的推移可能在这些个体中升高,但终末靶器官损伤并不常见,但在p.Cys282Tyr纯合性突变的患者中,男性比女性更频繁的出现终末靶器官损伤[见 Yamashita & Adams 2003, Andersen et al 2004, Olynyk et al 2004, Allen et al 2008, Gurrin et al 2008]。
基因修饰因素:多年来,人们一直认为p.Cys282Tyr纯合突变的血色病的发生需要其他基因参与到疾病过程中来。一个国际团队最近发现了基因GNPAT,它在p.Cys282Tyr纯合性突变的男性患者中表现出了严重的铁负荷过量[McLaren et al 2015]。
非表达p.Cys282Tyr纯合突变
三项基于群体学的纵向调查研究表明,p.Cys282Tyr纯合突变的患者中38%-50%可能发生铁超载(即血清铁蛋白浓度升高) ,10%-33%可能最终会发生血色病相关症状[Whitlock et al 2006] (即可能包括疲劳和关节痛的非特异性症状)或终末靶器官损伤(例如糖尿病,肝硬化和/或心肌病)。绝大多数受到发展中器官损伤的是男性[欧洲肝脏研究协会 2010]。
因此,具有双等位基因的 p.Cys282Tyr突变的大部分男性可能发展为渐进性铁负荷过量或铁超载症状。[Yamashita & Adams 2003, Gurrin et al 2008, Gurrin et al 2009, Allen et al 2010, Gan et al 2011]。
杂合子
:带有HFE基因 p.Cys282Tyr或p.His63Asp杂合的致病性变异的个体,血清转铁蛋白饱和度(TS)升高,并且铁蛋白浓度超过正常值,但不会出现铁负荷过量并发症[Bulaj et al 1996, Allen et al 2008]。HFE-HH血清
TS检测,45%阈值比过去用的高阈值更敏感,但该阈值鉴定出的杂合子无临床风险[McLaren et al 1998]。
一项丹麦男性的大型研究中 Pedersen & Milman [2009]表明:
- 带有p.His63Asp/wt杂合子的个体,8%的人血清TS升高,12%的人铁蛋白升高,2%的人血清TS和铁蛋白都高。
外显率:HFE-HH的外显率(男女外显率不同)是指带有HFE致病性变异的纯合性或复合杂合的成年人表现出临床HFE-HH或生化HFE-HH表型的百分比:
- p.Cys282Tyr/p.His63Asp复合杂合子:这种基因型的外显率很低,只有大约0.5%-2.0%的此类个体出现了铁负荷过量的临床症状[Gurrin et al 2009]。
- p.Cys282Tyr/p.His63Asp复合杂合子的铁负荷过量临床症状可能由某种复杂因素(如脂肪肝,病毒性肝炎)导致铁超载。
- 在HEIRS研究中带有p.Cys282Tyr/p.His63Asp复合杂合子的男性有肝病史可能性高,中OR值为1.7 (p = 0.05)[Adams et al 2005]。
患病率
p.Cys282Tyr纯合性个体的患病率约为3:1000-5:1000,或1:200-1:400 [Adams et al 2005]:
带有p.His63Asp的纯合性或复合杂合 (p.[Cys282Tyr]+[His63Asp])的个体很少出现临床问题。但在大多数种群杂合子中临床问题相对普遍:北欧人25%,西班牙人18%,非洲裔美国人6%,亚洲人8.5%。
考虑到p.Cys282Tyr和p.His63Asp杂合子频率高,约有三分之一北欧人中这两个变异等位基因中的一个是杂合的。
遗传相关(等位基因)疾病
虽然有许多研究HFE 致病性变异与其他疾病之间的关系,但除本GeneReview中讨论的那些表型以外没有其他与HFE的致病变异相关表型 [DuBois & Kowdley 2004]。
鉴别诊断
HFE相关的遗传性血色病(HFE-HH) (也称1型HH)需要区别于几种更为罕见的原发性铁负荷过量疾病以及继发性铁过载疾病。
原发性铁负荷过量症的特征在于正常饮食中铁的吸收增加。以下血色病,前两种是由于铁调素缺乏引起的,临床表现与p.Cys282Tyr 血色病相同,但情况更严重:
- 青少年遗传性血色病 (也称为2型HH)的发病年龄较早,临床表现比1型HH更为严重。肝细胞癌尚未见报道,可能是因为这种疾病的寿命短。在两个基因中鉴定出致病性变异会导致两种临床上难以区分的“亚型”:2A型,由编码血凝素的HJV基因致病性变异引起;2B型,由HAMP基因的致病性变异引起。遗传方式是常染色体隐性遗传 [Roetto et al 1999, Camaschella et al 2000, De Gobbi et al 2002].
- TFR2相关的遗传性血色病 (也称3型HH)与HFE-HH有相似的表现。发病年龄较早,进展速度慢于青少年HH。它是由编码转铁蛋白受体2的TFR2基因的致病性变异引起的。TFR2相关的遗传性血色病很少见,主要在意大利报道过。遗传方式是常染色体隐性遗传 [Mattman et al 2002].
- 铁转运蛋白(SLC40A1)相关的铁负荷过量(铁转运蛋白疾病,4型血色病,HFE4) (OMIM 606069)也是铁负荷过量导致的疾病,但与青少年型和HFE-HH型不同,铁负载发生在巨噬细胞。HFE4发病较晚,与其它血色病相反,铁储存影响内皮细胞而不是实质细胞[Montosi et al 2001, Njajou et al 2001]。HFE成年期发病,是由编码铁转运蛋白的SLC40A1基因致病性变异引起的。遗传方式是常染色体显性遗传 [Pietrangelo 2004]。
- 非洲裔铁负荷过量(OMIM 601195)易感者由于过量摄入铁而使病情加重。在非洲人群中,喝用非镀锌钢桶酿造的传统啤酒是很普遍的。其它未定义的铁相关基因的变异可能导致该病。SLC40A1 (NM_014585.5)编码铁转运蛋白的特定致病性变异 (p.Gln248His)与非洲人和非洲裔美国人的铁负荷过量倾向有关[McNamara et al 2005, Rivers et al 2007]。在一个轻度铁负荷过量的非洲裔美国人中发现SLC40A1另一个可能的致病性变异(p.Asp270Val)[Lee et al 2012].
- 新生儿血色病是一种严重的、通常致命的铁负荷过量综合征,通常在刚出生时发病。胎儿在子宫内发生铁负荷过量。该病遗传方式未知,推测为常染色体隐性遗传和线粒体遗传 ,没有发现相关 位点。最近的一篇论文指出,编码脱氧鸟苷激酶的DGUOK基因中的致病性变异可能导致类似这种病的表型[Pronicka et al 2011]。见DGUOK相关的线粒体DNA缺失综合征,肝脑型。
继发性铁负荷过量症
- 与肝脏实质疾病相关的肝病包括酒精性肝病、急性病毒性肝炎或慢性丙型肝炎、肿瘤、迟发性皮肤卟啉病和炎症性疾病,如类风湿性关节炎等。
- 常见的肝脏疾病,非酒精性脂肪肝(NAFLD) (OMIM 613282)常可导致血清铁蛋白水平升高,可能与肝脏铁储存增加有关[Nelson et al 2011, Kowdley et al 2012]。
见血色病: OMIM 表型系列,在OMIM中查看与此表型相关的基因。
管理
美国肝脏研究协会(AASLD)已经出版了诊断和治疗血色病的实践指南[Bacon et al 2011] (全文)。 欧洲肝脏研究协会(EASL)出版了关于血色病管理的临床实践指南[欧洲肝脏研究协会 2010] (全文)。
初次诊断后的评估
为了确定诊断为HFE相关遗传性血色病(HFE-HH)的个体的疾病程度和需求,建议在初始诊断时进行以下评估:
- 血清铁蛋白浓度对疾病状态及预后的影响(见 图 2)
- 对于p.Cys282Tyr纯合子:
- 肝活检仍然是确定或排除肝硬化的金标准。
- MRI利用铁的顺磁性来估计实质铁含量:
- 专业的MRI技术由于敏感性好,已经被FDA批准了用于临床评估肝脏铁浓度[St Pierre et al 2005]。这种准确定量MRI方法能够测定肝脏铁含量,可测肝脏铁浓度范围较宽。与肝活检相比,MRI-R2具有无创性并显著降低了活检取样误差[Fischer & Harmatz 2009]。 此外,肝铁的T2* MRI测量现在已经广泛使用[Brittenham et al 2003, Cheong et al 2005, Ptaszek et al 2005]。
- 生物磁式肝敏感器(BLS)通过测量磁通量的变化来估计肝脏铁量,当血色病患者移动通过稳定的磁场时,在SQUID传感器中产生电压[Fischer & Harmatz 2009]。由于BLS尚未广泛使用,因此不在血色病的诊断或管理指南中。
- 咨询医学遗传学专家和/或遗传咨询师。
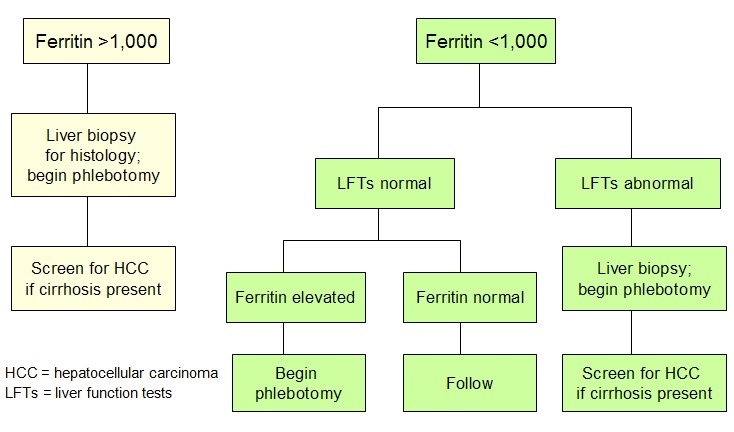
图 2.
使用血清铁蛋白浓度来帮助直接管理
临床表现的处理
美国肝脏研究协会(AASLD)已经出版了诊断和治疗血色病的实践指南[Bacon et al 2011] (全文)。 欧洲肝脏研究协会(EASL)出版了关于血色病管理的临床实践指南[欧洲肝脏研究协会 2010] (全文)。
临床HFE-HH
静脉切开治疗:在出现铁负荷过量症状或终末靶器官损伤的迹象时用(如晚期肝硬化、心力衰竭、皮肤色素改变或糖尿病):
- 定期静脉切开术是一种简单、廉价、安全和有效的治疗方法。
- 血细胞比容为40%的每400-500 mL血液单位中含有约160-200 mg 的铁。
- 每毫升红细胞中含有1毫克的铁。
- 常规治疗是每周静脉放血(去除一个单位的血液)去除过量的铁,直到血清铁蛋白浓度为50 ng/mL或者更低。每周两次静脉放血可能有时会加速铁消耗。
- 每周行静脉切开术直到血细胞比容降到基线血细胞比容的75%。
- 如果血细胞比容显着降低,血清铁蛋白浓度≥50 ng/mL,进行静脉切开的间隔需要进一步增大。一旦血清铁蛋白浓度≤100 ng/mL,每增加一到两次治疗后,所有受累的个体应检测血清铁蛋白浓度[Barton et al 1998]。
- 血清铁蛋白浓度是监测放血治疗最可靠和最廉价的方法。
- 维持治疗旨在维持血清铁蛋白浓度<50 ng/mL,转铁蛋白铁饱和度<50%。平均男性需要去除的血液单位是女性的两倍。为了防止再次铁储存,后续放血治疗可以每三四个月进行一次,而女性可以一年进行一次或两次。
- 不推荐铁螯合疗法,除非个体血清铁蛋白浓度升高并伴有贫血而不能采用治疗性放血。然而,这种情况在HFE-HH的个体中是不常见的。
- 除肝硬化外,治疗性静脉切开术可改善肝纤维化[Powell et al 2006]。
原位肝移植是失代偿期肝硬化导致的终末期肝病的唯一治疗方法。然而,未经治疗的HFE-HH患者,肝移植后存活率较低[Crawford et al 2004, Kowdley et al 2005]。虽然有一项研究表明肝移植后患者病况可能有所改善,但该报告没有用文件记录HFE-HH个体信息,而是数据库资料[Yu & Ioannou 2007]。
生化HFE-HH
EASL和AASLD指南都建议对生化HFE-HH患者(即患者无铁负荷过量临床表现,但体内铁储存量增加)进行治疗性静脉切开术。见[欧洲肝脏研究协会 2010] (全文)和 Bacon et al [2011] (全文)。可进行治疗性放血的确切血清铁蛋白浓度尚不清楚,欧洲肝脏研究协会建议血清铁蛋白浓度高于500 ng/mL时进行静脉切开术。
非表达 p.Cys282Tyr 纯合子
这些人没有铁负荷过量,因此不需要静脉切开治疗。
主要预防的表现
见治疗表现。
预防继发性并发症
建议铁负荷过量的个体不要食用贝类或生鱼。建议接种甲型肝炎和乙型肝炎的疫苗[Tavill 2001]。
监控
临床 HFE-HH
如果血清蛋白浓度<50ng/ml,每3到4个月监测一次血清铁蛋白浓度。
有心脏受累或已知心脏铁沉积病史的随访患者进行T2* MRI以评估心脏铁是合理的。
肝硬化是血色沉着病自然史中的一个关键阶段,由于疾病管理的变化,肝硬化的诊断比较重要。肝硬化患者应定期随访并筛查肝细胞癌(HCC) [Tavill 2001]。AASLD实践指南建议每六个月进行一次超声检查[Sherman 2010],可同时测量甲胎蛋白。无创性方法检测血清标志物值(例如HepaScore®和FibroMeter™)也可用于评估肝纤维化的阶段[Martínez et al 2011]。
尽管诊断肝硬化的金标准是活检,但已有结果显示FibroScan® 检测肝硬化,敏感性和特异性良好[Tsochatzis et al 2011]。FibroScan(也称为瞬时弹性成像)以非侵入方式超声定量肝纤维化[Tsochatzis et al 2011]。Meta分析的结果提示诊断肝硬化临界值是13.01 kPa [Friedrich-Rust et al 2008]。FibroScan瞬时弹性成像(TE)值正常的个体不需要活检,而TE值中等或高的个体应进行活检以确认肝硬化。同时参考临床和生化指标。
注:AASLD指南建议肝硬化患者不管是否缺铁均接受监测[Bacon et al 2011] (全文)。
生化 HFE-HH
当铁蛋白浓度超过正常水平时,开始每年测量血清铁蛋白浓度[欧洲肝脏研究协会 2010] (全文)。
非表达 p.Cys282Tyr 纯合子
当铁蛋白浓度超过正常水平时,开始每年测量血清铁蛋白浓度[欧洲肝脏研究协会 2010]。
避免药物/环境不良刺激
避免以下情况:
- 服用药用铁,矿物质补充剂,过量的维生素C和食用未经烹煮的海鲜
- 肝脏受累者饮酒
家庭成员风险评估
应评估p.Cys282Tyr无明显症状的成年同胞以及纯合性后代的风险,确定那些能够通过迅速进行治疗和预防措施而获益的人。
以下策略比较合适:
p.Cys282Tyr 纯合性个体的成年同胞提供分子遗传学检测。
2.
为p.Cys282Tyr 纯合性个体的同胞检测铁转铁蛋白饱和度和血清铁蛋白浓度。
3.
如果血清铁蛋白浓度超过正常值上限,并且先证者有临床HFE-HH,则开始静脉放血治疗。
注:临床HFE-HH先证者的同胞似乎比通过筛查检测到的无症状者的临床HFE-HH外显率高。
注:基因型检测阴性的预测价值高,对大多数人有成本效益。然而,其阳性预测值较低,因为许多带有p.Cys282Tyr纯合子和复合杂合子的个体不会患该病[El-Serag et al 2000, Beutler et al 2002]。
参见
遗传咨询中的亲属遗传风险的评估检测。
妊娠期管理
无相关指南。常见的做法是在怀孕期间不进行放血治疗。
治疗研究
血色病患者口服铁螯合剂地拉罗司 (deferasirox,Exjade®)研究试验已进入临床I / II期。该试验的最终结果表明,地拉罗司可有效减少铁负载,安全性可接受[Phatak et al 2010]。但该药尚未被批准用于治疗血色病。
搜索ClinicalTrials.gov获取疾病和病症更大范围的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供关于遗传疾病的性质、遗传方式和后果信息的过程,以帮助他们做出明智的医疗和个人决策。以下部分涉及遗传风险评估以及利用家族史和基因检测澄清家庭成员的遗传状态。本部分并不意味着解决个体可能面临的所有个人、文化或伦理问题,或代替遗传学专业人士的咨询。—ED.
遗传咨询的相关问题
参见管理,亲属风险评估,了解亲属的相关风险信息,为早期诊断和治疗准备。
T18岁以下无病症未成年个体进行检测并不合适, 主要是因为它否定了孩子的自主权,也没有带来特别益处。此外,还有人担心此类信息可能对家庭造成潜在的不良影响,未来可能有歧视和耻辱的风险,而且此类信息可能引起焦虑。
在确诊有HFE-HH的家庭,有症状的个体进行检测无需考虑年龄。
更多信息参见全国遗传咨询师协会关于未成年人通过基因检测成人发病风险的立场声明 ,美国儿科学会、美国医学遗传学和基因组学协会政策声明基因检测和儿童基因筛查的伦理和政策问题》。
生育规划
- 怀孕前是遗传风险评估,明确携带者的情况和产前检查可行性分析的最佳时间。
DNA 库:储存DNA(通常从白血细胞中提取),以备将来使用。因为在将来我们对基因、等位基因变异和疾病的认识以及测试方法很有可能得到改善,所以应该考虑建立受累的个体的DNA库。
人群筛查
鉴于HFE-HH发病率高,疾病早期临床难以发现,症状出现后缺乏特异性临床所见,诊断费用低,早期诊治相对简单有效,晚期诊断确诊后治疗成本高,成功率低的情况,因此考虑人群筛查。但是操作流程方面的原因,仍然不建议进行人群筛查。
HFE-HH不推荐人群基因筛查,因为该病外显率可能较低,无法预测未经治疗的个体的疾病发展。参见欧洲肝脏研究协会[2010] (全文)和Bacon et al [2011] (全文)。
生化筛查, 年龄超过30岁且北欧血统的男性人群尤其应该考虑进行血清铁标志物转铁蛋白铁饱和度和铁蛋白的血清浓度筛查[Phatak et al 2008, 欧洲肝脏研究协会2010, Bacon et al 2011]。
产前检查
如果受累的家族成员已经鉴定出HFE致病性变异,可以在提供该基因或定制产前检测致病变异的临床实验室进行高风险怀孕产前检查。对于产前诊断作用的认识,医疗专家和家庭成员之间可能存在分歧,特别是产前诊断用于终止妊娠而不是为早期诊断时。虽然大多数检测中心会考虑产前检查的决定是由父母选择,但这些问题也可讨论。
一些已经鉴定为HFE致病性变异的家族成员可选择胚胎植入前基因诊断(PGD)。
资源
为该病患者及其家庭成员方便,GeneReviews的工作人员选择了以下针对该病,或包括其他疾病的支持组织和患者注册管理机构。GeneReviews不对以下组织提供的信息承担责任。请点击此处了解选择标准。
- American Hemochromatosis Society, Inc.4044 West Lake Mary Boulevard#104, PMB 416Lake Mary FL 32746-2012Phone: 1-888-655鈥揑RON; 1-888-655-4766; 407-829-4488Fax: 407-333-1284Email: mail@americanhs.org
- Haemochromatosis AustraliaAustraliaPhone: 1300 019 028
- My46 Trait Profile
- National Digestive Diseases Information Clearinghouse (NDDIC)2 Information WayBethesda MD 20892-3570Phone: 800-891-5389 (toll-free); 866-569-1162 (TTY)Fax: 703-738-4929Email: nddic@info.niddk.nih.gov
- National Human Genome Research Institute (NHGRI)
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
分子遗传学
分子遗传学和OMIM表中的信息可能与GeneReview中的其他信息不同:表格中可能包含更新的信息。-ED.
表 A.
HFE相关的遗传性血色病:基因和数据库
基因结构:HFE基因长约13 kb,包含7个外显子[Feder et al 1996, Albig et al 1998];HFE产生了至少11个转录物,编码4-7个外显子。有关基因和蛋白质信息详细概述,见表 A,基因。
致病等位基因变异:至少28种不同的致病性变异已被报道,大多数是错义或nonsense。两种错义变异包含了人群中绝大多数致病等位基因:
- p.Cys282Tyr去除与β-2-微球蛋白形成分子间二硫键的高度保守的半胱氨酸残基,从而阻止蛋白质在细胞表面表达。
- p.His63Asp可能改变pH依赖的分子内盐桥,可能影响HFE蛋白与转铁蛋白受体的相互作用。
另外,在带有p.Cys282Tyr的铁负荷过量个体中发现p.Ser65Cys[Bacon et al 2011]。与常见杂合的致病变异不同,带有p.Ser65Cys/wt 杂合子个体血清TS和铁蛋白不升高。
表 2.
| DNA 核苷酸变化 | 蛋白质氨基酸变化 | 参考序列 |
|---|---|---|
| c.187C>G | p.His63Asp | NM_000410-.3 NP_000401-.1 |
| c.193A>T | p.Ser65Cys | |
| c.502G>T | p.Glu168Ter | |
| c.506G>A | p.Trp169Ter | |
| c.845G>A | p.Cys282Tyr |
变异分类说明:表中列出的变异是由作者提供的。GeneReviews工作人员未独立核对变异分类。
命名注释:GeneReviews遵循人类基因组变异协会的标准命名惯例(varnomen-.hgvs.org)的标准命名约定,参见快速引用术语的注释。
正常基因产物.预测最大初级翻译产物是348个氨基酸,切除信号序列后产生约321个氨基酸的成熟蛋白质。 HFE蛋白在初级结构[Feder et al 1996]和三级结构[Lebrón et al 1998]与HLA I类分子相似。成熟蛋白在细胞表面上表达,与β-2-微球蛋白形成的异源二聚体,并且这种相互作用对于细胞表面上的正常信号呈递是必需的。正常的HFE蛋白与细胞表面上的转铁蛋白受体1结合并可以减少细胞铁吸收,HFE蛋白质调节铁吸收的确切方法尚不清楚[Fleming et al 2004]。
异常
基因产物. p.Cys282Tyr 致病性变异破坏了与β-2-微球蛋白结合的二硫键所需的关键半胱氨酸残基,导致HFE蛋白质不能正常成熟,从而陷入内质网和高尔基体,导致细胞表面表达下降。其他HFE致病变异的表型效应的机制目前还不清楚。
参考资料
已发布指南/共识声明
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Available online. 2011. Accessed 9-14-15.
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 9-14-15. [PubMed: 23428972]
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. Available online. 2010. Accessed 9-14-15.
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset disorders. Available online. 2012. Accessed 9-14-15.
参考文献
- Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, Leiendecker-Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769-78. [PubMed: 15858186]
- Albig W, Drabent B, Burmester N, Bode C, Doenecke D. The haemochromatosis candidate gene HFE (HLA-H) of man and mouse is located in syntenic regions within the histone gene cluster. J Cell Biochem. 1998;69:117-26. [PubMed: 9548560]
- Allen KJ, Bertalli NA, Osborne NJ, Constantine CC, Delatycki MB, Nisselle AE, Nicoll AJ, Gertig DM, McLaren CE, Giles GG, Hopper JL, Anderson GJ, Olynyk JK, Powell LW, Gurrin LC. HealthIron Study Investigators. HFE Cys282Tyr homozygotes with serum ferritin concentrations below 1000 microg/L are at low risk of hemochromatosis. Hepatology. 2010;52:925-33. [PMC free article: PMC2932765] [PubMed: 20583211]
- Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, Bahlo M, Nisselle AE, Vulpe CD, Anderson GJ, Southey MC, Giles GG, English DR, Hopper JL, Olynyk JK, Powell LW, Gertig DM. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-30. [PubMed: 18199861]
- Andersen RV, Tybjaerg-Hansen A, Appleyard M, Birgens H, Nordestgaard BG. Hemochromatosis mutations in the general population: iron overload progression rate. Blood. 2004;103:2914-9. [PubMed: 15070663]
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS., American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-43. [PMC free article: PMC3149125] [PubMed: 21452290]
- Bardou-Jacquet E, Lainé F, Deugnier Y. Reply to: "Reduced mortality due to phlebotomy in moderately iron-loaded HFE Haemochromatosis? The need for clinical trials. J Hepatol. 2015a;63:283-4. [PubMed: 25841362]
- Bardou-Jacquet E, Morcet J, Manet G, Lainé F, Perrin M, Jouanolle AM, Guyader D, Moirand R, Viel JF, Deugnier Y. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. 2015b;62:682-9. [PubMed: 25450707]
- Bardou-Jacquet E, de Tayrac M, Mosser J, Deugnier Y. GNPAT Variant associated with severe iron overload in HFE hemochromatosis. Hepatology. 2015c Apr 18; Epub ahead of print. [PubMed: 25891252]
- Barton JC, McDonnell SM, Adams PC, Brissot P, Powell LW, Edwards CQ, Cook JD, Kowdley KV. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med. 1998;129:932-9. [PubMed: 9867745]
- Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G--> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211-8. [PubMed: 11812557]
- Brittenham GM, Badman DG., National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Workshop. Noninvasive measurement of iron: report of an NIDDK workshop. Blood. 2003;101:15-9. [PubMed: 12393526]
- Bulaj ZJ, Griffen LM, Jorde LB, Edwards CQ, Kushner JP. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. N Engl J Med. 1996;335:1799-805. [PubMed: 8943161]
- Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14-5. [PubMed: 10802645]
- Cheong B, Huber S, Muthupillai R, Flamm SD. Evaluation of myocardial ironoverload by T2* cardiovascular magnetic resonance imaging. Tex Heart Inst J. 2005;32:448-9. [PMC free article: PMC1336733] [PubMed: 16392242]
- Crawford DH, Fletcher LM, Hubscher SG, Stuart KA, Gane E, Angus PW, Jeffrey GP, McCaughan GW, Kerlin P, Powell LW, Elias EE. Patient and graft survival after liver transplantation for hereditary hemochromatosis: Implications for pathogenesis. Hepatology. 2004;39:1655-62. [PubMed: 15185307]
- De Gobbi M, Roetto A, Piperno A, Mariani R, Alberti F, Papanikolaou G, Politou M, Lockitch G, Girelli D, Fargion S, Cox TM, Gasparini P, Cazzola M, Camaschella C. Natural history of juvenile haemochromatosis. Br J Haematol. 2002;117:973-9. [PubMed: 12060140]
- Delatycki MB, Gurrin LC, Ong SY, Ramm GA, Anderson GJ, Olynyk JK, Allen KJ, Nicoll AJ, Powell LW. Reduced mortality due to phlebotomy in moderately iron-loaded HFE haemochromatosis? The need for clinical trials. J Hepatol. 2015;63:282-3. [PubMed: 25839407]
- DuBois S, Kowdley KV. Review article: targeted screening for hereditary haemochromatosis in high-risk groups. Aliment Pharmacol Ther. 2004;20:1-14. [PubMed: 15225165]
- Eijkelkamp EJ, Yapp TR, Powell LW. HFE-associated hereditary hemochromatosis. Can J Gastroenterol. 2000;14:121. [PubMed: 10694284]
- El-Serag HB, Inadomi JM, Kowdley KV. Screening for hereditary hemochromatosis in siblings and children of affected patients. A cost-effectiveness analysis. Ann Intern Med. 2000;132:261-9. [PubMed: 10681280]
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3-22. [PubMed: 20471131]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399-408. [PubMed: 8696333]
- Fischer R, Harmatz P. Non-invasive assessment of tissue iron overload. Hematology Am Soc Hematol Educ Program. 2009:215-21. [PubMed: 20008201]
- Fleming RE, Britton RS, Waheed A, Sly WS, Bacon BR. Pathogenesis of hereditary hemochromatosis. Clin Liver Dis. 2004;8:755-73. [PubMed: 15464654]
- Fletcher LM, Dixon JL, Purdie DM, Powell LW, Crawford DH. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology. 2002;122:281-9. [PubMed: 11832443]
- Friedrich-Rust MF, Martens S, Sarrazin C, Bojunga J, Zeuzem S, Herrmann E. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology. 2008;134:960-74. [PubMed: 18395077]
- Gan EK, Powell LW, Olynyk JK. Natural history and management of HFE-hemochromatosis. Semin Liver Dis. 2011;31:293-301. [PubMed: 21901659]
- Gochee PA, Powell LW, Cullen DJ, Du Sart D, Rossi E, Olynyk JK. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002;122:646-51. [PubMed: 11874997]
- Gurrin LC, Bertalli NA, Dalton GW, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, Delatycki MB, Nicoll AJ, Southey MC, Hopper JL, Giles GG, Anderson GJ, Olynyk JK, Powell LW, Allen KJ. HealthIron Study Investigators. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-relatedmorbidity. Hepatology. 2009;50:94-101. [PMC free article: PMC3763940] [PubMed: 19554541]
- Gurrin LC, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, Delatycki MB, Southey MC, Hopper JL, Giles GG, Anderson GJ, Olynyk JK, Powell LW, Allen KJ. HealthIron Study Investigators. The natural history of serum ironindices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology. 2008;135:1945-52. [PubMed: 18848943]
- Kowdley KV, Brandhagen DJ, Gish RG, Bass NM, Weinstein J, Schilsky ML, Fontana RJ, McCashland T, Cotler SJ, Bacon BR, Keeffe EB, Gordon F, Polissar N. Survival after liver transplantation in patients with hepatic iron overload: the national hemochromatosis transplant registry. Gastroenterology. 2005;129:494-503. [PubMed: 16083706]
- Kowdley KV, Belt P, Wilson LA, Yeh MM, Neuschwander-Tetri BA, Chalasani N, Sanyal AJ, Nelson JE., NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:77-85. [PMC free article: PMC3245347] [PubMed: 21953442]
- Le Gac G, Congiu R, Gourlaouen I, Cau M, Férec C, Melis MA. Homozygous deletion of HFE is the common cause of hemochromatosis in Sardinia. Haematologica. 2010;95:685-7. [PMC free article: PMC2857203] [PubMed: 20007136]
- Lebrón JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111-23. [PubMed: 9546397]
- Lee PL, Gaasterland T, Barton JC. Mild iron overload in an African American man with SLC40A1 D270V. Acta Haematol. 2012;128:28. [PubMed: 22584997]
- Martínez SM, Crespo G, Navasa M, Forns X. Noninvasive assessment of liver fibrosis. Hepatology. 2011;53:325-35. [PubMed: 21254180]
- Mattman A, Huntsman D, Lockitch G, Langlois S, Buskard N, Ralston D, Butterfield Y, Rodrigues P, Jones S, Porto G, Marra M, De Sousa M, Vatcher G. Transferrin receptor 2 (TfR2) and HFE mutational analysis in non-C282Y iron overload: identification of a novel TfR2 mutation. Blood. 2002;100:1075-7. [PubMed: 12130528]
- McLaren CE, Emond MJ, Subramaniam VN, Phatak PD, Barton JC, Adams PC. Goh JB3, McDonald CJ, Powell LW, Gurrin LC, Allen KJ, Nickerson DA, Louie T, Ramm GA, Anderson GJ, McLaren GD. Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload. Hepatology. 2015;62:429-39. [PMC free article: PMC4508230] [PubMed: 25605615]
- McLaren CE, McLachlan GJ, Halliday JW, Webb SI, Leggett BA, Jazwinska EC, Crawford DH, Gordeuk VR, McLaren GD, Powell LW. Distribution of transferrin saturation in an Australian population: relevance to the early diagnosis of hemochromatosis. Gastroenterology. 1998;114:543-9. [PubMed: 9496946]
- McNamara L, Gordeuk VR, MacPhail AP. Ferroportin (Q248H) mutations in African families with dietary iron overload. J Gastroenterol Hepatol. 2005;20:1855-8. [PubMed: 16336444]
- Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108:619-23. [PMC free article: PMC209405] [PubMed: 11518736]
- Morrison ED, Brandhagen DJ, Phatak PD, Barton JC, Krawitt EL, El-Serag HB, Gordon SC, Galan MV, Tung BY, Ioannou GN, Kowdley KV. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627-33. [PubMed: 12693884]
- Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, Kowdley KV., Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology. 2011;53:448-57. [PMC free article: PMC3058264] [PubMed: 21274866]
- Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, Snijders PJ, Rutten WP, Sandkuijl LA, Oostra BA, van Duijn CM, Heutink P. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet. 2001;28:213-4. [PubMed: 11431687]
- Olynyk JK, Hagan SE, Cullen DJ, Beilby J, Whittall DE. Evolution of untreated hereditary hemochromatosis in the Busselton population: a 17-year study. Mayo Clin Proc. 2004;79:309-13. [PubMed: 15008603]
- Pedersen P, Milman N. Genetic screening for HFE hemochromatosis in 6,020 Danish men: penetrance of C282Y, H63D, and S65C variants. Ann Hematol. 2009;88:775-84. [PubMed: 19159930]
- Phatak PD, Bonkovsky HL, Kowdley KV. Hereditary hemochromatosis: time for targeted screening. Ann Intern Med. 2008;149:270-2. [PubMed: 18711158]
- Phatak P, Brissot P, Wurster M, Adams PC, Bonkovsky HL, Gross J, Malfertheiner P, McLaren GD, Niederau C, Piperno A, Powell LW, Russo MW, Stoelzel U, Stremmel W, Griffel L, Lynch N, Zhang Y, Pietrangelo A. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52:1671-779. [PMC free article: PMC3034044] [PubMed: 20814896]
- Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131-8. [PubMed: 14757427]
- Piperno A, Arosio C, Fossati L, Viganò M, Trombini P, Vergani A, Mancia G. Two novel nonsense mutations of HFE gene in five unrelated Italian patients with hemochromatosis. Gastroenterology. 2000;119:441-5. [PubMed: 10930379]
- Powell LW, Dixon JL, Ramm GA, Purdie DM. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294-301. [PubMed: 16476869]
- Pronicka E, Węglewska-Jurkiewicz A, Taybert J, Pronicki M, Szymańska-Dębińska T, Karkucińska-Więckowska A, Jakóbkiewicz-Banecka J, Kowalski P, Piekutowska-Abramczuk D, Pajdowska M, Socha P, Sykut-Cegielska J, Węgrzyn G. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure. J Appl Genet. 2011;52:61-6. [PMC free article: PMC3026684] [PubMed: 21107780]
- Ptaszek LM, Price ET, Hu MY, Yang PC. Early diagnosis of hemochromatosis-related cardiomyopathy with magnetic resonance imaging. J Cardiovasc Magn Reson. 2005;7:689-92. [PubMed: 16136860]
- Ramazzotti A, Pepe A, Positano V, Rossi G, De Marchi D, Brizi MG, Luciani A, Midiri M, Sallustio G, Valeri G, Caruso V, Centra M, Cianciulli P, De Sanctis V, Maggio A, Lombardi M. Multicenter validation of the magnetic resonance T2* technique for segmental and global quantification of myocardial iron. J Magn Reson Imaging. 2009;30:62-8. [PubMed: 19557847]
- Ramrakhiani S, Bacon BR. Hemochromatosis: advances in molecular genetics and clinical diagnosis. J Clin Gastroenterol. 1998;27:41-6. [PubMed: 9706768]
- Rivers CA, Barton JC, Gordeuk VR, Acton RT, Speechley MR, Snively BM, Leiendecker-Foster C, Press RD, Adams PC, McLaren GD, Dawkins FW, McLaren CE, Reboussin DM. Association of ferroportin Q248H polymorphism with elevated levels of serum ferritin in African Americans in the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Blood Cells Mol Dis. 2007;38:247-52. [PMC free article: PMC3727273] [PubMed: 17276706]
- Roetto A, Totaro A, Cazzola M, Cicilano M, Bosio S, D'Ascola G, Carella M, Zelante L, Kelly AL, Cox TM, Gasparini P, Camaschella C. Juvenile hemochromatosis locus maps to chromosome 1q. Am J Hum Genet. 1999;64:1388-93. [PMC free article: PMC1377875] [PubMed: 10205270]
- Scotet V, Merour MC, Mercier AY, Chanu B, Le Faou T, Raguenes O, Le Gac G, Mura C, Nousbaum JB, Ferec C. Hereditary hemochromatosis: effect of excessive alcohol consumption on disease expression in patients homozygous for the C282Y mutation. Am J Epidemiol. 2003;158:129-34. [PubMed: 12851225]
- Sherman M. The radiological diagnosis of hepatocellular carcinoma. Am J Gastroenterol. 2010;105:610-2. [PubMed: 20203642]
- St Pierre TG, Clark PR, Chua-anusorn W, Fleming AJ, Jeffrey GP, Olynyk JK, Pootrakul P, Robins E, Lindeman R. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105:855-61. [PubMed: 15256427]
- Tavill AS. Diagnosis and management of hemochromatosis. Hepatology. 2001;33:1321-8. [PubMed: 11343262]
- Tsochatzis EA, Gurusamy KS, Ntaoula S, Cholongitas E, Davidson BR, Burroughts AK. Elastography for the diagnosis of severity of fibrosis in chronic liver disease: A meta-analysis of diagnostic accuracy. J Hepatol. 2011;54:650-9. [PubMed: 21146892]
- Whitlock EP, Garlitz BA, Harris EL, Beil TL, Smith PR. Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2006;145:209-23. [PubMed: 16880463]
- Yamashita C, Adams PC. Natural history of the C282Y homozygote for the hemochromatosis gene (HFE) with a normal serum ferritin level. Clin Gastroenterol Hepatol. 2003;1:388-91. [PubMed: 15017658]
- Yu L, Ioannou GN. Survival of liver transplant recipients with hemochromatosis in the United States. Gastroenterology. 2007;133:489-95. [PubMed: 17681170]
章节注解
作者简历
Robin L Bennett, MS; University of Washington (2000-2015)
Kris V Kowdley, MD; Virginia Mason Medical Center (2000-2015)
Arno G Motulsky, MD; University of Washington (2000-2015)
Lawrie Powell, AC, MD, PhD (2015-present)
Rebecca Seckington (2015-present)
Jonathan F Tait, MD, PhD; University of Washington School of Medicine (2000-2011)
更历史新
- 17 September 2015 (me) Comprehensive update posted live
- 19 April 2012 (me) Comprehensive update posted live
- 4 December 2006 (me) Comprehensive update posted to live Web site
- 13 September 2004 (kk) Author revisions
- 7 October 2003 (me) Comprehensive update posted to live Web site
- 3 April 2000 (me) Review posted to live Web site
- October 1998 (kk) Original submission