
摘要
临床特征.
EPB42相关性遗传性球形红细胞增多症(EPB42-HS)是一种慢性非免疫性溶血性贫血,贫血通常为轻-中度。EPB42-HS可以表现为在早期如出生24小时内出现黄疸,也可以表现为在晚期,如儿童期出现溶血危像或再障危像(通常与病毒感染相关)导致的贫血。除了溶血表现,严重的并发症包括脾肿大和胆石症,脾肿大在儿童早期即表现得很明显,胆石症通常在二三十岁时才变得明显。EPB42-HS典型的实验室异常包括贫血(血红蛋白[Hgb]水平下降)和网织红细胞增多(网织红细胞百分比升高),红细胞平均血红蛋白浓度(MCHC)升高,外周血涂片中出现球形红细胞,结合珠蛋白明显下降或缺乏,红细胞渗透脆性轻度增加,激光衍射法测得的最大变形指数(DImax)下降和Omin(50%的红细胞溶血时的渗透压)升高。
诊断/检测.
EPB42-HS的诊断建立在EPB42上双等位基因致病性变异的确定。
处理.
对症治疗: 轻度EPB42-HS(Hgb 11-15 g/dL,网织红细胞 3%-8%)的治疗包括补充叶酸(400 µg 1x 每天直到1岁;之后1 mg 1x 每天),当发生溶血或再障危像的时候,必要时进行红细胞输注。由于疾病严重程度通常为轻度或中度,EPB42-HS很少需要进行脾切除术。但是对于那些大于5岁的中重度EPB42-HS患者(Hgb 6-8 g/dL,网织红细胞 ≥10%),当生活质量受到影响时,推荐进行脾切除术。虽然是一种治疗措施,但是脾切除需要经过一段长期的潜在致命感染风险增加的时间,因此在手术前需要进行完整的免疫接种,手术后需要预防性应用抗生素。既往有胆石症的患者在进行脾切除的时候需要行胆囊切除术。
原发表现的预防: 见对症治疗中脾切除的信息。
继发性并发症的预防:常规的免疫接种来预防能引发溶血或再障危像的感染。铁过载是一种风险,特别是在需要频繁输注红细胞的时候;在经过大约10次输注之后,通常需要开始使用铁螯合剂来进行治疗(相关的血清铁蛋白浓度大约为1000 ng/mL)。
监测: 新生儿在生后1周需要监测血清胆红素浓度,2-4个月的婴儿需要监测血红蛋白浓度以防出现严重贫血。对于那些依赖于频繁输血以及接受铁螯合剂治疗的患者需要监测血清铁蛋白浓度。当溶血严重的时候,不管是否出现胆石症的表现都要进行腹部超声检查,直到10-12岁,之后每5到10年检查一次。
需要避免的药物/环境: 任何包含铁的配制品;然而,如果检查发现铁缺乏的时候,需要在密切监测下进行补铁治疗,当铁储备恢复的时候即停止。对于那些脾肿大的患者推荐避免进行接触性运动;需要注意的是,急性或过度的脾肿大其风险远大于慢性轻度脾肿大。
有风险的亲属的评价: 当其中一个家系成员被诊断为EPB42-HS时,对于有风险的同胞推荐进行以下措施:(1)对于存在风险的新生儿,在生后1周需要监测血清胆红素浓度,这样能及时对高胆红素血症进行治疗;(2)对于存在风险的生后2-4个月的婴儿需要监测严重贫血,这需要进行红细胞输注和开始进行补充叶酸治疗。对有风险的亲属进行实验室评价(CBC和网织红细胞计数,血涂片,红细胞渗透脆性或激光衍射法)和/或家系中EPB42致病性变异(如果知道的话)的分子遗传学检测是必要的。
妊娠处理:需要补充叶酸(800-1000 µg每天);推荐通过CBC和网织红细胞计数来监测贫血是否加重。
遗传咨询.
EPB42-HS以常染色体隐性遗传方式进行遗传。在怀孕的时候,受累个体的每个同胞都有25%的机会是患者,50%的机会是无症状携带者,25%的机会既不是患者也不是携带者。如果在一个家系中确定了致病性变异,对有风险的亲属进行携带者检测和对风险增加的妊娠进行产前检测是可能的。
诊断
提示性发现
当患者出现以下任何临床和支持性的实验室发现的时候,应该怀疑EPB42-相关性遗传性球形红细胞增多症(EPB42-HS):
临床表现
- 由于贫血导致的苍白和/或疲乏,通常为轻到中度
- 黄疸
- 通常为间歇性的,由过度溶血导致的高未结合胆红素血症所致
- 在一些罕见的病例,是由于胆道阻塞导致的高结合胆红素血症所致
- 脾肿大
- 在二三十岁时出现胆石症
- 家族史符合常染色体隐性遗传方式注意:EPB42-HS家族史的缺乏不能排除诊断。
支持性的实验室发现
- 全血细胞计数 符合:
- 慢性,非免疫性溶血性贫血(血红蛋白下降伴网织红细胞增多),通常为轻到中度
- 平均红细胞血红蛋白浓度(MCHC)升高。通常正常水平为31-37 g/dL。HS中的值通常为35.5-37.5 g/dL。
- 直接抗球蛋白试验(DAT)阴性(即正常)注意:DAT通常用于评价一个新诊断的溶血性贫血患者,看其是否是由于急性免疫介导的(获得性)溶血性贫血。
- 外周血涂片 证实是否存在球形红细胞,有时候会发现一些卵圆形和椭圆形红细胞注意:球形红细胞指的是形状为球形的红细胞(表面积/体积比下降),其拥有红细胞细胞骨架病的特征(见鉴别诊断)。
- 结合珠蛋白严重下降或缺乏。 6个月后结合珠蛋白的正常值为16-200 mg/dL。在HS中,结合珠蛋白一般检测不到;然而,当并发炎症的时候结合珠蛋白水平可能正常(因为它是一种急性时相反应物)。
- 最大变形指数(DImax)下降和经激光衍射法测得的Omin(50%红细胞溶血时的渗透压)增加,得出一个典型的HS曲线[Clark et al 1983, Hammill et al 2011]
表 1.
遗传性球形红细胞增多症的严重程度
| 严重程度 | Hgb (g/dL) | 网织红细胞 (%) | 脾切除 |
|---|---|---|---|
| 轻度 | 11-15 | 3-8 | 没有必要 |
| 中度 | 8-11.5 | >8 | 如果活动能力&生活质量下降可以考虑 |
| 中重度 | 6-8 | ≥10 | 年龄>5岁需要进行 |
| 重度 | <6 | ≥10 | 年龄>3岁需要进行 |
| 正常 1 | 11.7-15.7 (成人女性) 13.3-17.7 (成人男性) | 0.5-1.5 2 |
确定诊断
当在一个先证者中确定了EPB42上双等位基因的致病性变异,即可确定诊断为EPB42-相关性遗传性球形红细胞增多症(见表 2)。分子遗传学检测方法可能包含单-基因检测或使用
- 单-基因检测. 进行EPB42的测序分析。
表 2.
EPB42-相关性遗传性球形红细胞增多症应用到的分子遗传学检测
| 基因 1 | 检测方法 | 用这种方法检测到致病性变异 2的先证者的比例 |
|---|---|---|
| EPB42 | 测序分析 3 | 16/16 4 |
| 基因靶向性缺失/重复分析 5 | 未知 6, 7 |
染色体位点和蛋白质见表 A. 基因和数据库。
2.
3.
测序分析检测那些是良性、可能良性、意义不确定、可能致病性或致病性的变异。致病性的变异可能包含小的基因内的缺失/插入和错义、无义和剪接位点变异;通常情况下,外显子或全-基因缺失/重复无法检测到。对于序列分析结果解读中需考虑的问题,点击这里。
4.
大多数为病例报导,对于没有EPB42致病性变异的遗传性球形红细胞增多症患者没有数量信息[Kanzaki et al 1997, Toye et al 2008]。
5.
基因靶向性的缺失/重复分析检测基因内的缺失或重复。可能用到的方法包括:定量PCR、长片段PCR、多重连接依赖性探针扩增技术(MLPA)和被设计用来检测单个外显子缺失或重复的基因靶向性微阵列。
6.
7.
至今为止报导过的唯一一个大的缺失是一个32个碱基对的缺失[Hammill et al 2011],其能被序列分析检测到。
临床特征
临床描述
EPB42-相关性遗传性球形红细胞增多症(EPB42-HS)的儿童通常在出生24小时内发病,表现为需要进行光照治疗的黄疸,或者,罕见的,需要进行换血治疗以避免核黄疸。他们也可以在较晚的儿童期发病,表现为贫血,贫血通常是由病毒感染相关的溶血危像或再障危像导致的。遗传性球形红细胞增多症的幼儿偶尔被发现有年龄相关性的缺铁性贫血;然而,贫血不能通过补铁治疗完全缓解,网织红细胞增多持续存在。就所有形式的轻或中度遗传性球形红细胞增多症而言(定义见表 1),在生命的前4-6个月EPB42-HS可能更加严重,需要进行规律的红细胞输注治疗。因此,生命前几个月频繁的输注治疗并不与之后疾病的严重程度有必然的联系。EPB42-HS,如果在婴儿期或儿童早期没有被识别,可能在较晚的时候被诊断为轻(Hgb 11-15 g/dL)到中度(Hgb 8-11.5 g/dL)慢性溶血性贫血(见表 1),伴黄疸,脾肿大和比较年轻的时候出现胆石症[Eber & Lux 2004]。
基因型-表型相关性
p.Ala142Thr的纯合子在日本人中最常见,被报导能导致中重度HS,Hgb能够低至6.1 g/dL [Bouhassira et al 1992]。一个非日本的受累的个体在意大利被报导,具有相同的基因型;她的表型包括从出生开始的中度溶血性贫血和脾肿大[Perrotta et al 1999]。p.Ala142Thr或p.Asp175Tyr的纯合子导致不典型HS,在血涂片中除了发现少数几个球形红细胞外还可见卵形和口形红细胞。溶血可能为轻到中重度,在脾切除后改善
[Bouhassira et al 1992, Kanzaki et al 1995]。p.Ala142Thr与另一个EPB42致病性变异的复合杂合子导致典型的HS,在血涂片中能发现小球形红细胞和红细胞渗透脆性增加[Takaoka et al 1994, Kanzaki et al 1995]。其他EPB42致病性变异患者的病例报导也提示轻到中度HS,只是偶尔地需要进行输血治疗[Hayette et al 1995, van den Akker et al 2010, Hammill et al 2011]。一个患者是无效c.950delG变异(导致转录本的提前终止,缺乏任何可见的红细胞膜蛋白带4.2蛋白-在最新的文献中被称作蛋白4.2)的纯合子,在胃肠道出血进行多次红细胞输注后出现了针对蛋白4.2的强烈的抗体反应,导致了同种免疫性溶血性贫血[Beauchamp-Nicoud et al 2000]。在其他EPB42致病性变异的患者中,还没有报导在输注红细胞后出现抗体,虽然在大多数的EPB42-相关性HS案例中没有检测到红细胞膜上的蛋白4.2[Satchwell et al 2009]。
患病率
遗传性球形红细胞增多症是北欧血统中最常见的遗传性贫血,患病率为1:2000或贫血程度非常轻的(通常没有被诊断出来)HS也被包含进来的时候患病率更高。世界范围内的患病率更低。
EPB42-相关性遗传性球形红细胞增多症占日本人遗传性球形红细胞增多症的40%-50%,健康人群中p.Ala142Thr的携带者频率可高达3%[Yawata 1994, Yawata et al 2000]。在其他人群中,EPB42-HS占HS的5%或更少[Eber & Lux 2004, Perrotta et al 2008]。
遗传相关(等位基因)疾病
除了在本GeneReview中讨论的表型之外,没有其他表型被认为与EPB42的致病性变异相关。
鉴别诊断
对于一个溶血性贫血患者的初始评估通常包括:全血细胞计数(CBC)和网织红细胞计数;复查血涂片;直接抗球蛋白试验(DAT)和间接抗球蛋白试验来评价自身免疫性(或者,对于一个婴儿,同种免疫性)溶血性贫血;血红蛋白电泳;和G6PD酶活性测定(特别是男性)。红细胞渗透脆性试验和/或激光衍射法聚焦在红细胞膜病的诊断上。图 1显示了典型的遗传性球形红细胞增多症激光衍射法的结果。基于遗传学病因的遗传性球形红细胞增多症的分类如表 3所示。
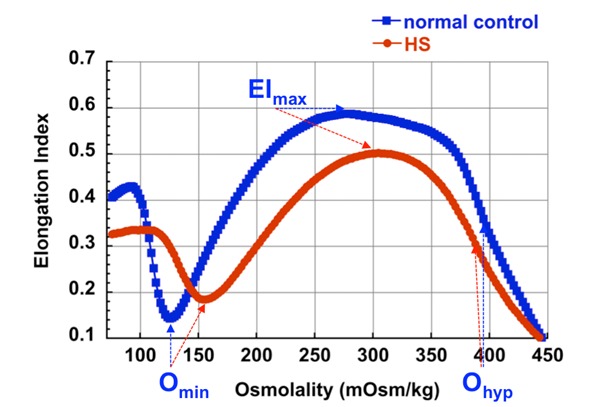
对于非免疫性溶血性贫血的患者,鉴别诊断包括其他原因导致的遗传性溶血性贫血,包括其他类型的遗传性球形红细胞增多症:
- 其他红细胞膜疾病
- 遗传性球形红细胞增多症(见表 3)
- 遗传性椭圆形红细胞增多症、口形红细胞增多症或东南亚卵形红细胞症,虽然不管是EPB42上p.Ala142Thr的纯合子还是p.Asp175Tyr的纯合子都报导过在血涂片上也有卵形红细胞和口形红细胞。注意:在遗传性口形红细胞增多症(水分过多或脱水/干瘪细胞增多症)的患者中脾切除与严重的和危及生命的栓塞事件有关;因此,当考虑进行脾切除的时候,需要在遗传性口形红细胞增多症和EPB42-相关性HS(EPB42-HS)之间进行鉴别。
- 血红蛋白病。EPB42-HS通常很容易与β-地中海贫血或血红蛋白H病相鉴别,后两者都以小细胞低色素贫血为特征。由于不稳定血红蛋白链导致的慢性轻度溶血性贫血可能需要进一步的评估,包括用电泳或高效液相色谱法对血红蛋白进行分析和/或珠蛋白基因测序。
- 红细胞酶病,例如葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症(OMIM)或丙酮酸激酶(PK)缺乏症(OMIM);通常与EPB42-HS的鉴别通过球形红细胞形态的缺乏和激光衍射法或红细胞渗透脆性正常。一些红细胞酶病(例如,磷酸丙糖异构酶[TPI]缺乏症[OMIM]或磷酸甘油酸激酶1[PGK1]缺乏症[OMIM])也有神经系统和肌肉骨骼系统表现。酶活性分析和/或基因测序能够确定诊断。
- 先天性红细胞生成异常性贫血,特别是II型,表现为轻度表型(即轻度贫血、网织红细胞增多症[虽然不理想]、黄疸和脾肿大)(见先天性红细胞生成异常性贫血I型)。
表 3.
遗传性球形红细胞增多症的分类
| 位置 | 基因 | 蛋白 | 遗传方式 | 严重程度 1 | 说明 | OMIM |
|---|---|---|---|---|---|---|
| SPH1 | ANK1 | 锚蛋白-1 | AD | 轻-中度 | 182900 | |
| AR | 中重度-重度 | 通常依赖输血 | ||||
| SPH2 | SPTB | 红细胞血影蛋白β链 | AD | 轻-中度 | 616649 | |
| AR | 重度 | 一例婴儿致死性病例已被报导 | ||||
| SPH3 | SPTA1 | 红细胞血影蛋白α链 1 | AR | 重度 | 依赖输血 | 270970 |
| SPH4 | SLC4A1 | 带3(阴离子转运蛋白) | AD | 轻-中度 | 只有双等位基因的时候某些SLC4A1致病性变异导致疾病 | 612653 |
| SPH5 | EPB42 | 蛋白 4.2 2 | AR | 轻-中度 3 | 一例中重度病例已被报导 | 612690 |
AD = 常染色体显性遗传
AR = 常染色体隐性遗传
1.
在表 1中定义
2.
HS患者红细胞膜蛋白带4.2(在最新的文献中被简称为蛋白4.2)严重下降或缺乏可能继发于SLC4A1双等位基因的致病性变异(SPH4),要么是由于红细胞膜上带3蛋白的下降[Toye et al 2008],要么是影响到了带3蛋白上的蛋白4.2结合位点[Kanzaki et al 1997]。
3.
SPH5通常比其他类型的遗传性球形红细胞增多症轻,以AR方式进行遗传(即SPH1 [AR]和SPH3)。
处理
初诊后的评估
为了对诊断为EPB42-相关性遗传性球形红细胞增多症(EPB42-HS)的患者确定疾病的严重程度和治疗措施,建议进行以下方面的评估:
- 血红蛋白浓度和网织红细胞计数来评价疾病的严重程度
- 血清胆红素浓度
- 输血史
- 血清铁蛋白浓度来评估铁负荷状态
- 进行腹部超声检查来评估:
- 脾脏大小,当由于身体习性体格检查不是很可靠或考虑进行接触性运动的时候
- 当症状出现的时候寻找胆石症的证据。如果溶血很严重,即使没有症状,在10-12岁之后也可能需要考虑筛查性的超声检查。
- 咨询临床遗传医师和/或遗传咨询师
对症治疗
遗传性球形红细胞增多症(HS)详细的治疗指南已经发表了[Eber & Lux 2004, Bolton-Maggs et al 2012]。对于轻度EPB42-HS (Hgb 11-15 g/dL,网织红细胞 3-8%)保守性的治疗建议(表 1)包括以下部分:
- 补充叶酸(400 µg 1x 每天直到1岁;然后接下来的日子1 mg 1x 每天)
- 避免补铁治疗除非检查中发现同时存在铁缺乏,在这样的病例中补铁治疗需密切监测,在铁储备恢复的时候应停止补铁以免铁超负荷
注意:遗传性球形红细胞增多症(像所有的慢性溶血性贫血一样),即使是口服补铁治疗,其铁超负荷的风险也增加(见继发性并发症的预防)。 - 如果需要的话,对溶血或再障危像进行红细胞(RBC)输注
在EPB42-HS中很少进行脾切除,因为疾病的严重程度通常为轻度或中度。然而,当疾病为中度(见表 1)和活动正常或生活质量受影响的时候,在5岁后可进行脾切除,因为那时候遗传性口形红细胞增多症已经被排除了(见鉴别诊断)。注意:对于5岁以下的儿童不推荐进行全脾切除,即使对于中重度HS需要反复输血(在EPB42-HS中罕见)。虽然脾切除是一种治疗方法,但是它增加了危及生命的病毒感染的长期潜在风险,因此,在进行脾切除之前需全面权衡风险和收益[Casale & Perrotta 2011]。理想情况下,在进行脾切除前需完成以下免疫接种:
- 23价肺炎球菌多糖疫苗(PPSV23)的肺炎链球菌免疫接种和至少在脾切除前2周接种针对脑膜炎双球菌的脑膜炎球菌结合疫苗,其能抵抗A、C、W和Y(MenACWY)血清型。两个剂量的初始系列MenACWY推荐隔8-12周进行接种[Committee on Infectious Diseases 2011]。
- 在婴儿期依照一般儿科免疫指南进行肺炎球菌疫苗-13®和b型流感嗜血杆菌疫苗的接种
脾切除术后败血症的发病率在不同的研究中存在差异。虽然总的发病率低,败血症的风险,是一种危及生命的并发症,比一般人群高[Iolascon et al 1998]。为了减少脾切除后感染的风险,建议进行以下措施:
- 第一次接种5年后进行PPSV23的加强免疫接种。不推荐进行多于2次的PPSV23剂量[Pekka Nuorti & Whitney 2010]。
- 如果初始的两个剂量的脑膜炎球菌疫苗是在2-6岁之间接种的,在初始接种后的三年进行加强免疫接种,如果2个剂量的初始接种或加强剂量是在7岁及以后接种的,则以后每5年接种一次[Cohn et al 2013]。
- 对于有脾切除史的10岁和大于10岁的患者推荐进行血清B群脑膜炎球菌疫苗接种。
对于脾切除后是否持续使用抗生素进行预防存在争议:一些血液科医生建议在脾切除后的前三年预防性使用抗生素,其他的建议终生使用[Eber & Lux 2004]。推荐的抗生素为青霉素V-K 250 mg每天两次或对那些对青霉素过敏的患者使用红霉素。对于任何病例,脾切除术后发热的患者需立即就医和迅速静脉使用抗生素进行治疗,抗生素需对有荚膜的微生物很好的覆盖(一般为头孢曲松,剂量为治疗脑膜炎的剂量:100 mg/kg/天加量到2 g/天 一天一次)。
部分脾切除 脾切除后败血症的风险较小,溶血的风险持续下降(虽然没有消失),如果外科医生对手术有经验的话,更适合于较小的儿童[Bader-Meunier et al 2001]。一项正在进行中的前瞻性的观察,包括多个机构登记的多于100名先天性溶血性贫血的儿童,可能能更好地阐明每个操作的风险和收益[Rice et al 2012, Rice et al 2015]。如果经痘痕计数(痘痕红细胞百分比)或脾脏摄取的放射性胶体评估脾脏免疫功能正常,部分性脾切除后1年预防性抗生素可以停止[Eber & Lux 2004]。
胆囊切除术
- 对于有胆石症病史的受累的个体,在进行脾切除的时候胆囊也应被切除。
- 对于需进行胆囊切除术的儿童,不再推荐同时自动进行脾切除。脾切除的需要应该每个病例具体分析,独立调整切除指征[Bolton-Maggs et al 2012, Ruparel et al 2014]。
原发表现的预防
在EPB42-HS中很少需要进行脾切除,因为疾病严重程度通常为轻度或中度。注意:当有指征的时候,脾切除是有疗效的;然而,它可能出现潜在的危及生命的并发症(见表现的治疗)。注意:对于小于5岁的儿童不推荐进行全脾切除术,即使是中重度的HS需要频繁的输血治疗(在EPB42-HS中罕见)。
继发性并发症的预防
推荐进行规律的免疫接种以及每年接种一次流感疫苗来预防感染,感染通常能触发溶血或再障危像。对于任何慢性溶血性贫血,存在
铁超负荷和其相关的慢性器官衰竭的风险,特别是需要频繁输血的时候。
- 需要应用铁螯合剂进行治疗,一般是在大约10次输血后(对应的血清铁蛋白的浓度大约为1000 ng/mL)。
- 应该对螯合作用的有效性进行监测,通过T2*-加权MRI或FerriScan®评价肝脏铁来进行监测,以便对铁螯合剂的剂量进行合理调整。
监测
HS的新生儿在生后一周内需要监测血清胆红素浓度,以便能够针对高胆红素血症立即进行治疗避免出现如核黄疸之类的并发症。HS的婴儿在生后的前2-4个月需要监测严重贫血,其可能需要输注红细胞。对于那些依赖频繁输血的患者至少需要每年一次铁蛋白浓度的检测。如果太小的儿童不能进行脾切除术,需要对继发于频繁输血的铁螯合治疗,合理地监测螯合治疗的毒性和有效性是必要的[Musallam et al 2013]。当溶血很严重的时候,需要到10到12岁的时候进行超声检查评估胆石症,之后每5-10年一次。
避免的药物/环境
应该避免任何含铁的制剂(见表现的治疗)。对于那些脾肿大的患者接触性运动是不可取的;值得注意的是,急性或过度脾肿大相对于慢性轻度脾肿大来说是一个更大的风险。
有风险的亲属的评估
在一个家系中,一个受累的个体经分子遗传学检测存在EPB42致病性变异,对表面上无症状的更大或更年轻的同胞进行表型(CBC和网织红细胞计数、血涂片、红细胞渗透脆性或激光衍射法)的实验室评估和确定遗传状态是合理的,这样能够尽可能早地让患者获益,迅速开始进行治疗和实施预防措施。
- 新生儿需要在生后1周内监测血清胆红素浓度,以便迅速治疗高胆红素血症,避免核黄疸等并发症。
- 婴儿需要在生后前1-4个月监测严重贫血,其可能需要进行输红细胞治疗和开始补充叶酸。
妊娠处理
对于慢性溶血性贫血的孕妇,如EPB42-HS补充叶酸(800-1000 µg 每天)是必要的。对于HS的孕妇,推荐用CBC和网织红细胞计数来监测贫血的加剧,因为曾经报导过妊娠期出现的溶血危像和持续性的贫血,特别是对于没有进行过脾切除的孕妇 Pajor et al 1993]。
在研中的治疗方法
对于各种各样疾病和情况的临床研究信息搜索ClinicalTrials.gov。注意:对于这种病可能没有临床试验。
遗传咨询
遗传咨询是向患者及家庭提供遗传性疾病的性质、遗传方式和影响相关信息的过程,以便他们做出比较完备的医学和个人决策。以下部分涉及到遗传风险评估以及使用家族史和遗传学检测来确定家系成员的遗传状态。这一部分不是为了解决患者可能会面对的所有个人的、文化的或伦理学的问题,也不能代替遗传学专家的遗传咨询。—ED.
遗传方式
EPB42-相关性遗传性球形红细胞增多症(EPB42-HS)以常染色体隐性遗传方式进行遗传。
家系成员的风险
先证者的父母
- 杂合子(携带者)无症状,没有发展成疾病的风险。
先证者的同胞
- 杂合子(携带者)无症状,没有发展成疾病的风险。
携带者(杂合子)检测
对于有风险的亲属的携带者检测需要先在家系中确定EPB42致病性变异。
遗传咨询相关问题
为了早期诊断和治疗,对有风险的亲属进行评估的相关信息见有风险亲属的评估、处理 。
生育计划
- 确定遗传风险、明确携带者状态和讨论是否进行产前检测的最佳时间是妊娠前。
DNA银行 是把DNA(一般从白细胞中提取)存储起来以便未来能够使用。因为可能检测方法以及我们对基因、等位基因变异和疾病的理解在未来都会有所进步,因此需要考虑把受累的个体的DNA储存起来。
产前检测和胚胎植入前遗传学诊断
一旦EPB42致病性变异在一个受累的家系成员中被确定,对EPB42-H风险增加的妊娠进行产前检测和植入前遗传诊断是可能的选择。关于是否使用产前检测,医疗专业人员之间以及家系成员内部可能存在分歧,特别是当检测是用来考虑终止妊娠而不是早期诊断的时候。虽然大多数中心关于产前检测的决定考虑的是父母的选择,对这些问题进行讨论是合理的。
资源
GeneReviews工作人员选择了以下的疾病特异性和/或伞状支持组织和/或登记机构,以使这种病的患者及其家庭获益。GeneReviews 对其他组织提供的信息不负有责任。对于选择标准的信息,点击这里。
- National Library of Medicine Genetics Home Reference
分子遗传学
分子遗传学和OMIM表的信息可能与GeneReview其他地方的信息不同:表可能包含最近的信息。-ED.
表 A.
EPB42-相关性遗传性球形红细胞增多症:基因和数据库
| 基因 | 染色体位置 | 蛋白 | 位点特异性数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| EPB42 | 15q15-.2 | 红细胞膜蛋白带4.2 | EPB42 database | EPB42 | EPB42 |
分子遗传机制
红细胞膜蛋白带4.2(也被称为蛋白4.2),由EPB42编码,是红细胞细胞骨架的主要成分,通过与其他关键红细胞蛋白的相互作用维持红细胞的稳定性和伸缩性,许多的其他红细胞关键蛋白(以其致病形式)也能导致遗传性球形红细胞增多症。蛋白4.2是锚蛋白-带3复合物的一部分,连接带3蛋白(由SLC4A1编码;见表 3,SPH4)和CD47以及恒河猴蛋白复合物抗原。蛋白4.2在细胞骨架和膜脂质双层之间起到物理支撑的作用[Bruce et al 2003]。蛋白4.2与血影蛋白相互作用,血影蛋白为包含一个α亚单位(见表 3,SPH3)和β亚单位(见表 3,SPH2)的一个四聚体,是红细胞上最大的细胞骨架蛋白[Mandal et al 2002, Korsgren et al 2010]。
基因结构.EPB42 (以前称为ELB42)横跨大约23kb基因组的 DNA。比较长的转录本(NM_000119.2)有2554bp和13个外显子。选择性的剪接产生了一个比较短的转录变体(NM_001114134.1),其不包括外显子1的最后90个核苷酸。基因和蛋白信息的详细总结,见表 A,基因.
致病性等位基因变异. 至今为止,至少13个明确的EPB42 致病性变异已被报导,包含错义、无义、移码和剪接变异以及小的缺失。大于50%确定的致病性变异发生在日本人群中[Bouhassira et al 1992, Yawata et al 2000]。来自欧洲[Perrotta et al 1999, van den Akker et al 2010]和北美[Hammill et al 2011]的受累的个体也有被确定的。p.Ala142Thr变异在一个受累的个体中被发现为纯合状态。这个变异在日本血统的人群中携带者频率大约为3%,它也在一个没有日本血统的意大利中部患者中被发现
[Perrotta et al 1999]。虽然日本人群中的频率可以用奠基者效应来解释,这个变异在其他人群中很明显的随机发生可以用DNA反义链上CpG位点发生的G>A转换这一事实来解释[Elango et al 2008]。
表 4.
选择的在这个GeneReview中讨论的EPB42致病性变异
| DNA核苷酸改变 | 预测的蛋白改变 (别名 1) | 参考序列 |
|---|---|---|
| c.424G>A | p.Ala142Thr (4.2 日本) | NM_000119-.2 NP_000110-.2 |
| c.523G>T | p.Asp175Tyr (4.2 小松) | |
| c.920C>T | p.Thr307Ile (4.2 辛辛那提) | |
| c.929G>A | p.Arg310Gln (4.2 托泽尔) | |
| c.949C>T | p.Arg317Cys (4.2 志贺) | |
| c.950delG | 移码 (4.2 南希) |
变异分类中需注意的是:这个表中列出的变异由作者提供。GeneReviews工作人员没有再单独对变异分类进行查证。
命名中需注意的是:GeneReviews遵循人类基因组变异协会(varnomen-.hgvs.org)的标准命名规则来命名。命名的解释见快速参考。
1.
不遵循当前命名规则的变异名称
正常 基因产物. 长和短的EPB42转录变异体编码两种蛋白4.2亚型:721个氨基酸的74-kd的小亚型(NP_000110.2)和691个氨基酸的72-kd的大亚型(NP_001107606.1)。虽然为转谷氨酰胺酶的同源物,蛋白4.2缺乏转谷氨酰胺酶活性需要的3个催化亚基中的2个,因此是一个没有酶活性的蛋白质[Toye et al 2005]。蛋白4.2是红细胞细胞骨架的主要成分,通过与其他关键红细胞蛋白的相互作用维持红细胞的稳定性和伸缩性,这些其他红细胞蛋白中的许多(以其致病形式)也能导致遗传性球形红细胞增多症(见分子遗传机制和表 3)。由于胎儿血红蛋白的存在,新生儿期的溶血会加重,胎儿血红蛋白对2,3-二磷酸甘油酸(2,3-DPG)的亲水性差,导致游离的细胞内的2,3-DPG增加,破坏了与血影蛋白4.1的相互作用,加重溶血[Mentzer et al 1987]。蛋白4.2通过特异性的结构域与红细胞细胞骨架上的结合伴侣相互作用。蛋白4.2
187-211个氨基酸残基的长的亚型NP_000110.2被认为介导其与锚蛋白(见表 3,SPH1)和带3的结合,桥接这两种蛋白[Su et al 2006]以及连接带3蛋白和细胞骨架支架上的脂质双层。
异常 基因产物. 多于一半的已经确定的EPB42致病性变异被预测能够导致蛋白完全缺失,这被推定为其发病机制。至少6个不同的致病性的错义变异已经被发现。功能分析包括以下方面:
- 位于假定存在的带3结合结构域的p.Thr307Ile [Hammill et al 2011]和p.Arg317Cys [Kanzaki et al 1995]致病性变异导致蛋白4.2的完全缺失,大概是由于其在并入膜之前已经蛋白水解了。
- 在体外研究中p.Ala142Thr等位基因与带3蛋白的结合正常,提示这个致病性变异导致的4.2缺陷与带3蛋白无关[Toye et al 2005]。p.Ala142Thr变异的纯合状态(在4个不相关的日本人中被发现)导致蛋白4.2几乎完全缺陷的红细胞血影膜[Bouhassira et al 1992]。
注意,虽然蛋白4.2缺陷的红细胞带3蛋白水平没有改变,带3蛋白的萃取率和横向扩散却显著增加了[Rybicki et al 1996]。
参考资料
已发表的指南/共识声明
- Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Available online. 2012. Accessed 11-7-16.
- Bolton-Maggs PH, Stevens RF, Dodd NJ, Lamont G, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis. Available online. 2004. Accessed 11-7-16.
- Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Available online (login or institutional access required). 2004. Accessed 11-7-16. [PubMed: 15071790]
引用文献
- Bader-Meunier B, Gauthier F, Archambaud F, Cynober T, Mielot F, Dommergues JP, Warszawski J, Mohandas N, Tchernia G. Long-term evaluation of the beneficial effect of subtotal splenectomy for management of hereditary spherocytosis. Blood. 2001;97:399-403. [PubMed: 11154215]
- Beauchamp-Nicoud A, Morle L, Lutz HU, Stammler P, Agulles O, Petermann-Khder R, Iolascon A, Perrotta S, Cynober T, Tchernia G, Delaunay J, Baudin-Creuza V. Heavy transfusions and presence of an anti-protein 4.2 antibody in 4. 2(-) hereditary spherocytosis (949delG). Haematologica. 2000;85:19-24. [PubMed: 10629586]
- Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012;156:37-49. [PubMed: 22055020]
- Bouhassira EE, Schwartz RS, Yawata Y, Ata K, Kanzaki A, Qiu JJ, Nagel RL, Rybicki AC. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood. 1992;79:1846-54. [PubMed: 1558976]
- Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, Tanner MJ. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180-8. [PubMed: 12531814]
- Casale M, Perrotta S. Splenectomy for hereditary spherocytosis: complete, partial or not at all? Expert Rev Hematol. 2011;4:627-35. [PubMed: 22077527]
- Clark MR, Mohandas N, Shohet SB. Osmotic gradient ektacytometry: comprehensive characterization of red cell volume and surface maintenance. Blood. 1983;61:899-910. [PubMed: 6831052]
- Cohn AC, MacNeil JR, Clark TA, Ortega-Sanchez IR, Briere EZ, Meissner HC, Baker CJ, Messonnier NE. Prevention and Control of Meningococcal Disease: Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morbidity and Mortality Weekly Report. Available online. 2013. Accessed 11-8-16. [PubMed: 23515099]
- Committee on Infectious Diseases. Meningococcal conjugate vaccines policy update: booster dose recommendations. Pediatrics. 2011;128:1213-8. [PubMed: 22123893]
- Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118-41. [PubMed: 15071790]
- Elango N, Kim SH, Vigoda E, Yi SV. Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comput Biol. 2008;4:e1000015. [PMC free article: PMC2265638] [PubMed: 18463707]
- Hammill AM, Risinger MA, Joiner CH, Keddache M, Kalfa TA. Compound heterozygosity for two novel mutations in the erythrocyte protein 4.2 gene causing spherocytosis in a Caucasian patient. Br J Haematol. 2011;152:780-3. [PMC free article: PMC3105174] [PubMed: 21275958]
- Hayette S, Dhermy D, dos Santos ME, Bozon M, Drenckhahn D, Alloisio N, Texier P, Delaunay J, Morlé L. A deletional frameshift mutation in protein 4.2 gene (allele 4.2 Lisboa) associated with hereditary hemolytic anemia. Blood. 1995;85:250-6. [PubMed: 7803799]
- Iolascon A, Miraglia del Giudice E, Perrotta S, Alloisio N, Morle L, Delaunay J. Hereditary spherocytosis: from clinical to molecular defects. Haematologica. 1998;83:240-57. [PubMed: 9573679]
- Kanzaki A, Hayette S, Morle L, Inoue F, Matsuyama R, Inoue T, Yawata A, Wada H, Vallier A, Alloisio N, Yawata Y, Delaunay J. Total absence of protein 4.2 and partial deficiency of band 3 in hereditary spherocytosis. Br J Haematol. 1997;99:522-30. [PubMed: 9401060]
- Kanzaki A, Yawata Y, Yawata A, Inoue T, Okamoto N, Wada H, Harano T, Harano K, Wilmotte R, Hayette S. Band 4.2 Komatsu: 523 GAT-->TAT (175 Asp-->Tyr) in exon 4 of the band 4.2 gene associated with total deficiency of band 4.2, hemolytic anemia with ovalostomatocytosis and marked disruption of the cytoskeletal network. Int J Hematol. 1995;61:165-78. [PubMed: 8547605]
- Korsgren C, Peters LL, Lux SE. Protein 4.2 binds to the carboxyl-terminal EF-hands of erythroid alpha-spectrin in a calcium- and calmodulin-dependent manner. J Biol Chem. 2010;285:4757-70. [PMC free article: PMC2836081] [PubMed: 20007969]
- Mandal D, Moitra PK, Basu J. Mapping of a spectrin-binding domain of human erythrocyte membrane protein 4.2. Biochem J. 2002;364:841-7. [PMC free article: PMC1222634] [PubMed: 12049649]
- Mentzer WC Jr, Iarocci TA, Mohandas N, Lane PA, Smith B, Lazerson J, Hays T. Modulation of erythrocyte membrane mechanical stability by 2,3-diphosphoglycerate in the neonatal poikilocytosis/elliptocytosis syndrome. J Clin Invest. 1987;79:943-9. [PMC free article: PMC424243] [PubMed: 3818955]
- Mohandas N, Clark MR, Health BP, Rossi M, Wolfe LC, Lux SE, Shohet SB. A technique to detect reduced mechanical stability of red cell membranes: relevance to elliptocytic disorders. Blood. 1982;59:768-74. [PubMed: 7059678]
- Musallam KM, Angastiniotis M, Eleftheriou A, Porter JB. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol. 2013;130:64-73. [PubMed: 23485589]
- Pajor A, Lehoczky D, Szakacs Z. Pregnancy and hereditary spherocytosis. Report of 8 patients and a review. Arch Gynecol Obstet. 1993;253:37-42. [PubMed: 8328819]
- Pekka Nuorti J, Whitney CG. Prevention of Pneumococcal Disease Among Infants and Children -- Use of 13-Valent Pneumococcal Conjugate Vaccine and 23-Valent Pneumococcal Polysaccharide Vaccine. Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morbidity and Mortality Weekly Report. Available online. 2010. Accessed 11-8-16. [PubMed: 21150868]
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-26. [PubMed: 18940465]
- Perrotta S, Iolascon A, Polito R, d'Urzo G, Conte ML, Miraglia del Giudice E. 4.2 Nippon mutation in a non-Japanese patient with hereditary spherocytosis. Haematologica. 1999;84:660-2. [PubMed: 10406914]
- Rice HE, Crary SE, Langer JC, Kemper AR. Consortium ftSiCHA. Comparative effectiveness of different types of splenectomy for children with congenital hemolytic anemias. J Pediatr. 2012;160:684-9.e13. [PubMed: 22050869]
- Rice HE, Englum BR, Rothman J, Leonard S, Reiter A, Thornburg C, Brindle M, Wright N, Heeney MM, Smithers C, Brown RL, Kalfa T, Langer JC, Cada M, Oldham KT, Scott JP, St Peter S, Sharma M, Davidoff AM, Nottage K, Bernabe K, Wilson DB, Dutta S, Glader B, Crary SE, Dassinger MS, Dunbar L, Islam S, Kumar M, Rescorla F, Bruch S, Campbell A, Austin M, Sidonio R, Blakely ML. Splenectomy in Congenital Hemolytic Anemia C. Clinical outcomes of splenectomy in children: report of the splenectomy in congenital hemolytic anemia registry. Am J Hematol. 2015;90:187-92. [PMC free article: PMC4333061] [PubMed: 25382665]
- Ruparel RK, Bogert JN, Moir CR, Ishitani MB, Khan SP, Rodriguez V, Zarroug AE. Synchronous splenectomy during cholecystectomy for hereditary spherocytosis: is it really necessary? J Pediatr Surg. 2014;49:433-5. [PubMed: 24650472]
- Rybicki AC, Schwartz RS, Hustedt EJ, Cobb CE. Increased rotational mobility and extractability of band 3 from protein 4.2-deficient erythrocyte membranes: evidence of a role for protein 4.2 in strengthening the band 3-cytoskeleton linkage. Blood. 1996;88:2745-53. [PubMed: 8839871]
- Satchwell TJ, Shoemark DK, Sessions RB, Toye AM. Protein 4.2: a complex linker. Blood Cells Mol Dis. 2009;42:201-10. [PubMed: 19269200]
- Su Y, Ding Y, Jiang M, Jiang W, Hu X, Zhang Z. Associations of protein 4.2 with band 3 and ankyrin. Molecular and cellular biochemistry. 2006;289:159-66. [PubMed: 16718373]
- Takaoka Y, Ideguchi H, Matsuda M, Sakamoto N, Takeuchi T, Fukumaki Y. A novel mutation in the erythrocyte protein 4.2 gene of Japanese patients with hereditary spherocytosis (protein 4.2 Fukuoka). Br J Haematol. 1994;88:527-33. [PubMed: 7819064]
- Toye AM, Ghosh S, Young MT, Jones GK, Sessions RB, Ramauge M, Leclerc P, Basu J, Delaunay J, Tanner MJ. Protein-4.2 association with band 3 (AE1, SLCA4) in Xenopus oocytes: effects of three natural protein-4.2 mutations associated with hemolytic anemia. Blood. 2005;105:4088-95. [PubMed: 15692067]
- Toye AM, Williamson RC, Khanfar M, Bader-Meunier B, Cynober T, Thibault M, Tchernia G, Dechaux M, Delaunay J, Bruce LJ. Band 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild-type band 3 and glycophorin A. Blood. 2008;111:5380-9. [PMC free article: PMC2605348] [PubMed: 18174378]
- van den Akker E, Satchwell TJ, Pellegrin S, Flatt JF, Maigre M, Daniels G, Delaunay J, Bruce LJ, Toye AM. Investigating the key membrane protein changes during in vitro erythropoiesis of protein 4.2 (-) cells (mutations Chartres 1 and 2). Haematologica. 2010;95:1278-86. [PMC free article: PMC2913075] [PubMed: 20179084]
- Yawata Y. Red cell membrane protein band 4.2: phenotypic, genetic and electron microscopic aspects. Biochim Biophys Acta. 1994;1204:131-48. [PubMed: 8142452]
- Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. 2000;71:118-35. [PubMed: 10745622]
推荐读物
- Cohen CM, Dotimas E, Korsgren C. Human erythrocyte membrane protein band 4.2 (pallidin). Semin Hematol. 1993;30:119-37. [PubMed: 8480187]
- Dotimas E, Speicher DW. GuptaRoy B, Cohen CM. Structural domain mapping and phosphorylation of human erythrocyte pallidin (band 4.2). Biochim Biophys Acta. 1993;1148:19-29. [PubMed: 8499466]
- Korsgren C, Cohen CM. Associations of human erythrocyte band 4.2. Binding to ankyrin and to the cytoplasmic domain of band 3. J Biol Chem. 1988;263:10212-8. [PubMed: 2968981]
修订历史
- 2016年11月10日 在网页上发布实时全面更新
- 2014年3月13日 内容发布到实时网页上
- 2013年10月20日 原创投稿