
摘要
临床特征.
Pallister-Hall综合征 (在本篇文章中被称为PHS) 的特征是一系列异常,包括多指(趾)畸形,无症状的会厌分叉,以及轻度末端的下丘脑错构瘤,以及严重末端的新生儿致死性喉气管裂。患有轻微PHS的个体可能被错误的诊断孤立的轴后性多指畸形A型。PHS患者可能有垂体功能不全,进而可能在新生儿时死于未确诊和未治疗的肾上腺功能不全。
管理.
对症处理: 对内分泌异常,尤其是皮质醇缺乏进行紧急治疗;根据异常情况和呼吸损害程度的不同,对会厌异常进行管理。会厌分叉,是最常见的异常,通常不需要治疗。对肛门闭锁或狭窄进行标准治疗;对癫痫发作进行对症治疗;对多指畸形进行选择性修复;对发育迟缓进行发展性干预或者特殊教育。
预防继发性并发症: 活检或切除下丘脑错构瘤可能导致并发证和终生需要激素替代;由于使用兴奋剂治疗注意力缺陷障碍,癫痫发作可能出现或恶化。
监测: 在儿童时期,每年进行一次年度发育评估和年度医学评估,以评估生长和监测性早熟迹象。
遗传咨询.
Pallister-Hall综合征以常染色体显性遗传方式遗传。患有PHS的个体可能有受累的父母,或者由于新生 致病性变异而患有该疾病。大约25%的人有新生致病性变异。与有PHS家族史的人相比,新生致病性变异的人通常受到更严重的影响。受影响个体后代的风险为50%。如果已知该家族的致病性变异,就可以对风险增加的妊娠进行产前检测。超声检查对产前诊断 的可靠性尚不清楚。
GeneReview 范围
| 包括的表型 |
|---|
|
对于同义词和过时的名称,请参阅术语。
诊断
提示性结论
对于有以下特征的个体,应怀疑Pallister-Hall综合征 (PHS) :
- 下丘脑错构瘤, 在是交叉后第三脑室底部的肺增强性肿块,在MRI的T1和T2脉冲序列上与灰质等强度信号,但在FLAIR上可能具有不同的强度。注: 颅CT检查和颅超声检查均不适用于下丘脑错构瘤的诊断。
- 多指(趾)(即插入或中央)畸形, 指有六个或更多形状良好的手指(趾)与掌骨或跖骨成“Y”形。
- 轴后性多指(趾)畸形(PAP)A型和B型。PAP-A是指在肢体的尺侧或腓骨侧有一个成形良好的手指。PAP-B是指在同一位置存在一个发育未完全的手指或小瘤。轴后性多指(趾)畸形可能比中轴性多指(趾)畸形更常见;然而,在中非人后裔中,轴后性多指(趾)畸形的非特异性和轴后性多指(趾)畸形B型的高频率要求谨慎使用它作为诊断特征。
- 会厌分叉, 会厌的中线前后裂,至少占会厌叶的三分之二。它对于临床诊断有用的特征,因为它在PHS以外的综合征中似乎非常罕见,并且作为孤立的畸形也很少见。
- 其他. 肛门闭锁,肾脏异常,包括囊性畸形,肾发育不良,输尿管异位着床,包括子宫阴道积水在内的泌尿生殖器异常,包括双侧肺在内的肺分割异常,以及短肢在内的非多指(趾)骨骼异常。
诊断建立
PHS的临床诊断是在下丘脑错构瘤和多指(趾)畸形的先证者中建立的。
通过分子遗传学检测鉴定GLI3的 杂合的 致病性变异证实了PHS的诊断(见 表1)。
sub-PHS的临床诊断是在这样的先证者中建立的:
- 以下之一:
- 多指(趾)畸形
- 下丘脑错构瘤
- 少指(趾)畸形
- 轴后性多指(趾)畸形; 以及
- 以下之一:
- 会厌分叉
- 肛门闭锁
- 小指甲
- 垂体功能减退症
- 生长激素缺乏症
- 生殖器发育不全
通过分子遗传学检测鉴定GLI3的杂合的 致病性变异证实了PHS的诊断(见 表1)。
注: 对于错构瘤中的GLI3致病变体,有几个患有非综合征性下丘脑错构瘤和体细胞嵌合的个体报告 [Wallace et al 2008].虽然这些个体不符合PHS敏感性的临床诊断标准,但可以认为它们具有部分形式的PHS,并且应该考虑评估这些个体该疾病的其他表现。
分子遗传学检测
分子遗传学检测方法可以包括单基因测试,表型靶向检测,的使用,以及更全面的基因组的测试:
Table 1.
用于Pallister-Hall综合征的分子遗传学诊断
| 基因 1 | 测试方法 | 通过该方法可检测到具有致病性变异2的先证者们的比例 |
|---|---|---|
| GLI3 | 序列分析 3 | 20/22 4,5 |
| 基因定位删除/重复分析 6 | 未知7 | |
| 未知8 | NA |
- 1.
- 2.
有关在该基因中检测到的等位基因变体的信息,请参阅 Molecular Genetics 。
- 3.
- 4.
综合数据来自于 Johnston et al [2005], Johnston et al [2010], 以及 Démurger et al [2015]。
- 5.
在20名患有sub-PHS的人中,有8名(40%)确定了GLI3致病性变异;这个致病性变异类似于PHS患者中鉴定到的变异 [Johnston et al 2010]。
- 6.
- 7.
- 8.
在5%具有PHS临床特征的个体中,未发现GLI3中的致病性变异,表明至少一个额外的基因 位点 [Démurger et al 2015]或GLI3中的隐蔽变体(包括深内含子的或镶嵌致病变体)。
临床特征
临床描述
Pallister-Hall综合征(PHS)表现出广泛的严重程度。文献经常反映PHS严重且格列格头多发性神经综合征轻微的假设。这显然是不正确的,因为少数患有PHS的个体显示出多个严重的异常,并且大多数患有PHS的个体轻度的受累于多指,无症状的会厌分叉和下丘脑错构瘤。如果没有仔细的临床评估,这些人可能会被错误地诊断为轴后性多指畸形A型(PAP-A)。
下丘脑错构瘤. 下丘脑错构瘤是一种畸形,而不是肿瘤。下丘脑错构瘤的生长速度等于或者比周围的脑组织慢。下丘脑错构瘤可能很大(最大尺寸≤4cm);下丘脑错构瘤的大小与症状的存在或严重程度之间几乎没有相关性。患有下丘脑错构瘤的个体可能有神经系统症状,尽管大多数是无症状的。下丘脑错构瘤的切除是不需要的,常常导致医源性垂体功能不全或其他并发症。
内分泌表现. 下丘脑错构瘤的内分泌表现范围从孤立的生长激素缺乏或孤立的性早熟到泛垂体功能减退,这可能危及生命。皮质醇缺乏可能发生在患有非家族性PHS上午个体中,但在家族性PHS患者中似乎很少见。
神经系统发现. 最好描述的下丘脑错构瘤的神经系统并发症是痴笑性癫痫,一部分复杂的癫痫发作表现为胸部和膈肌的阵挛运动引起的模拟发笑。其他类型的癫痫发作可能由下丘脑错构瘤引起。与患有PHS的个体的下丘脑错构瘤相关的癫痫发作通常较轻并且对治疗有反应,与非综合征型下丘脑错构瘤患者相反,后者通常具有难治性癫痫发作[Boudreau et al 2005]。
多指(趾)畸形. 在患有PHS的个体中,轴后性多指(趾)畸形可能比多指(趾)畸形更常见。轴后性多指(趾)畸形(PAP)A型是在肢体的尺骨或腓骨侧存在成形良好的手指(足趾)。PAP B型是在相同位置存在发育未完全的手指(足趾)或小肿块。多指(即插入或中央)畸形是存在六个或更多个与掌骨或跖骨成“Y”形的形成良好的手指(足趾)。
注: 在中非人后裔中,轴后性多指(趾)畸形的非特异性和轴后性多指(趾)畸形B型的高频率要求谨慎使用它作为诊断特征。
会厌异常. 会厌分叉几乎总是无症状的;然而,PHS患者报告的喉部裂隙越严重,越可能导致严重的气道症状。轴后喉裂可能是致命的。
精神病学和神经心理学研究发现. 一些患有PHS的人具有个例行为表现包括一些具有严重智力残疾和行为障碍的人[Ng et al 2004]。对这种疾病的行为表现的一项更大规模的研究尚无定论,反映了在罕见疾病中评估轻度行为表型的困难[Azzam et al 2005]。
泌尿生殖系统异常. 肾脏异常包括囊性畸形,肾发育不良和输尿管异位着床;包括子宫阴道积水在内的泌尿生殖器异常。泌尿生殖系统异常的发病机制已经被描述过[Blake et al 2016]。
其他 研究结果包括肛门闭锁,包括双侧肺在内的肺分割异常,以及包括短肢在内的非多指(趾)骨骼异常。
具有PHS且没有已知的PHS家族史的个体的预后基于个体中存在的畸形。文献调查对于确定预后是无用的,因为报告的人表现出倾向于对更严重的参与的确定。尽管PHS已被归类为CAVE(脑血管早期致死性)疾病组的成员,但很少受累的的个体具有早期致死性表型。这种早期致死性表性最有可能归因于有垂体或下丘脑发育不良或眼中起到畸形(如喉气管裂隙)引起的泛垂体功能减退症。此外,如果不及时识别肛门闭锁可能会导致严重的并发症。因此,在没有危机生命的畸形的情况下,预后应该被认为是有益于非家族性PHS的个体。对于有家族史的个体,预后基于家族中存在的严重程度。
Sub-PHS
Sub-PHS是应用于具有PHS特征但不符合PHS临床诊断标准的个体的描述符(参见 Establishing the Diagnosis)。在一项研究中,20名受累于sub-PHS的人中有8名(40%)患有GLI3的致病性变异,其与PHS患者的致病变异相似[Johnston et al 2010 ]。
基因型-表型相关性
参见 图 1。GCPS和PHS的突变光谱大多不同。GCPS有所有类型的致病性变异引起,而PHS仅由截断变异和一个产生移码和截断的剪接变异引起。在移码变异 类别中,已经在两个层面上证明了基因型-表型相关性:
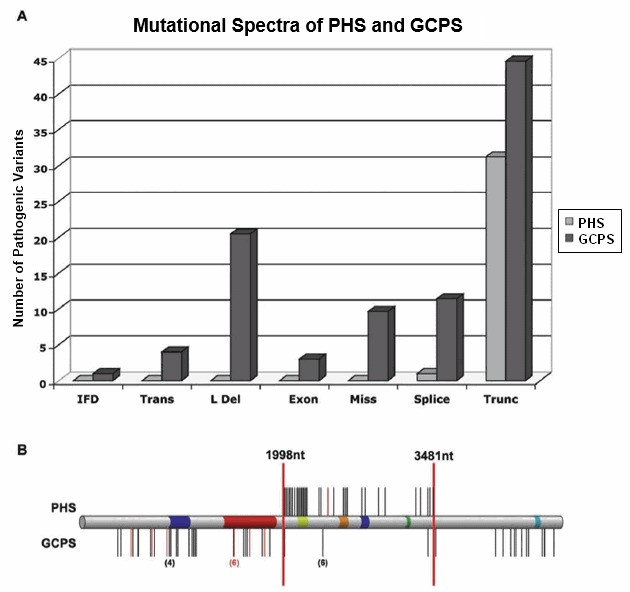
图 1.
GCPS和PHS的突变光谱大多不同。
- 变异种类. 所有类别的致病变异都可以引起格列格头多发性神经综合征(GCPS)。而引起等位基因疾病PHS的大多朱致病变异是移码变异。GLI3的单倍体不足引起GCPS,而GLI3的锌指 结构域3'的截断变异通常引起PHS[Kang et al 1997] (图 1A )。
术语
使用的其他描述符包括:
- CAVE (脑血管早期致死性) 复合体. 这种命名是不恰当的,因为大多数人只是轻微的 受累,并没有表现出早期的致命性。
- Hall-Pallister 综合征
注: 缩写“PHS”是用于Hermansky-Pudlak 综合征。
流行
PHS是罕见的。流行情况不明。提交人已知超过100名受累的人(Biesecker,个人观察)并且已报告了许多其他个体 (参见, 例如, Démurger et al [2015])。怀疑许多轴后性多指(趾)畸形和无症状的下丘脑错构瘤或会厌分叉的个体可能被误诊为患有非综合征性PAP-A。
PHS是泛民族的。
基因相关(等位)疾病
GLI3. 其他表型与GLI3中的致病变体相关。
格列格头多发性神经综合征(GCPS)包括多指(趾)畸形,通常是轴前性的,也可能是轴中性的。多指畸形通常与皮肤相关。在GCPS中看到的颅面特征包括眼睛间隔宽,前额宽阔和巨头畸形。掌骨的中间多指骨和骨性掌骨不是GCPS的一部分。
大多数具有GCPS的个体具有引起GLI3 单倍剂量不足 的致病性变体,尽管已经描述了少数具有单碱基变异(SNV)的个体。然而,尚未证实这些SNV具有稳定的mRNA或蛋白质。如果这些分子的稳定性降低,则SNV将导致功能性单倍体不足。据报道,至少有一名患有前外型多指型IV型(PPDIV)的个体患有GLI3 致病性变异 [Radhakrishna et al 1999] — 这一发现与临床怀疑一致,即PPDIV是一种轻度形式的GCPS,其中肢体方面没有发现颅面特征。然而,对于非综合征性PPDIV和轻度GCPS之间的区别,颅面发现是微妙的,存在争议[Biesecker 2006]。
Sub-GCPS 是应用于具有GCPS特征但不符合GCPS诊断临床标准的个体的描述符。sub-GCPS的临床标准包括:
- 以下之一:
- 轴前性多指(趾)畸形
- 宽大拇指或脚趾
- 皮肤并指畸形
- 巨头畸形
- 宽眼间距; 或者
- 以下两种情况:
- 轴后性多指(趾)畸形
- 胼胝体发育不全
在一项研究中,符合这些标准的2个个体中有8个(29%)在GLI3中具有致病性变异[Johnston et al 2010]。
孤立的轴后性多指畸形A型(PAP-A)已经在具有PAP-A的个体中鉴定了GLI3中的致病变体 [Radhakrishna et al 1999]。PAP-A表型也显示出来自其他基因的致病性变异。 Isolated postaxial polydactyly type A (PAP-A).
孤立的轴前性多指畸形IV型包括在没有其他畸形的情况下手和/或脚的轴前性多指畸形。PPDIV的严重程度变化很大[Everman 2006]。由于巨头畸形发生在一般人群中并且在GCPS中很常见,因此很难解释具有明显 孤立的 PPDIV的人中巨头畸形的存在。
口-面-指重叠综合症是用于具有与OFD和PHS重叠的特征的个体的描述符。这种疾病的标准:
- 多指(趾)畸形; 以及
- 以下之一:
- 口腔系带
- 口腔错构瘤
- 唇裂
- 腭裂
- 小脑蚓部发育不全
- 胫骨发育不全
在一项研究中,该类别21个个体中的6个(29%)在GLI3中具有致病性变异 [Johnston et al 2010]。
鉴别诊断
中央多指畸形
- Holzgreve综合征 (OMIM 236110)包括中央多指(趾)畸形,腭裂,心脏缺陷。
轴后性多指(趾)畸形
- McKusick-Kaufman综合征(MKS) 的特征是女性的三联征和男性生殖畸形,轴后性多指(趾)畸形(PAP)或中央性多指(趾)畸形和 先天的 心脏病(CHD)。 MKKS在MKS中的致病变体在 Amish人群中已发现。遗传方式是常染色体隐性遗传 。
- Bardet-Biedl综合征(BBS)以视杆-视锥萎缩,躯干肥胖,轴后性多指(趾)畸形,认知障碍,男性低促性腺激素性性腺功能减退,复杂的女性泌尿生殖道畸形,肾功能不全为特征,这是发病和死亡的主要原因。下丘脑错构瘤和会厌分叉是BBS的罕见表现 [Stevens & Ledbetter 2005]。至少19个基因与BBS有关,如:BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12, MKS1, CEP290, WDPCP, SDCCAG8, LZTFL1, BBIP1和 IFT27。遗传通常是 常染色体隐性遗传 。
- Holt-Oram 综合征(HOS) 的特征是:上肢畸形累及桡骨,鱼际骨或腕骨;先天的心脏畸形的个人和/或家族史(最常见的是窦房间隔缺损和室间隔缺损,尤其是肌肉小梁间隔缺损);和/或心脏传导疾病。在符合严格的HOS诊断标准的个体中,超过70%的人发现TBX5致病变异体。遗传方式是 常染色体显性遗传 。
- Smith-Lemli-Opitz综合征(SLOS)是由7-脱氢胆固醇还原酶缺乏引起的胆固醇代谢异常引起的 先天的 多发性异常综合征。其特征在于产前和产后生长迟缓,小头畸形,中度至重度智力残疾,以及包括轴后性多指(趾)畸形在内的多种主要和轻微畸形。DHCR7是唯一已知与SLOS相关的 基因 。遗传方式是 常染色体隐性遗传。
下丘脑错构瘤. 非综合征或 孤立的 下丘脑错构瘤可能导致内分泌紊乱(最常见的是生长激素缺乏或性早熟)或严重的神经图示顽固性癫痫发作,行为问题和认知衰退。可能与痴笑性癫痫有关。已经在非综合征性下丘脑错构瘤中鉴定出体细胞GLI3致病变体[Wallace et al 2008]。
管理
初次诊断后的评估
为了确定被诊断为Pallister-Hall综合征(PHS)的个体的疾病和需求程度,如果还未完成,建议进行以下评估:
- 评估皮质醇缺乏症。这必须在没有PHS家族史的个体和患有PHS及皮质醇缺乏的家庭成员的个体中紧急进行。值得注意的是,肾上腺危象对于未经过适当评估和肾上腺功能不全治疗的婴儿可能是致命的。
- 在评估和治疗ATCH缺乏症后,有内分泌专家会诊,进行包括早期生长激素分泌,FSH和LH分泌以及婴儿早期血清甲状腺激素水平的评估
- 颅MRI检查确定错构瘤的位置和范围
- 神经系统检查排除颅内高压的迹象,这不是典型的下丘脑错构瘤
- 通过喉镜观察会厌;当存在吸入体征或症状时,由耳鼻喉科医生对喉气管裂隙进行紧急评估;由耳鼻喉科医生在无症状个体中进行选择性评估,以及确定诊断或确定异常程度
- 肢体X线片以区分轴后性多指(趾)畸形和中央性多指(趾)畸形
- 由外科医生评估纠正多指(趾)畸形的时机和手术方法。请注意,轴中性多指(趾)畸形的手术矫正通常比轴后性多指(趾)畸形更复杂,应由专业外科医生进行
- 肾脏超声检查评估肾脏异常
- 肛门闭锁或肛门狭窄(如果有)的外科咨询
- 发展评估
- 咨询临床遗传学专家和/或遗传咨询师
治疗的指征
内分泌异常与普通人群一样,皮质醇缺乏症的治疗是最紧迫的。
肛门闭锁或狭窄应以标准方式进行。
会厌异常的管理取决于异常的类型和呼吸危害的程度,并且与一般人群相同。会厌分叉通常是无症状的,大多数不需要治疗,除非有明确的梗阻证据或其他异常相关,如气管狭窄。
癫痫发作是对症治疗。与PHS相关的癫痫发作通常对抗癫痫药物(AEDs)有反应,而非综合征性下丘脑错构瘤相关的癫痫发作更常见于AED难治。
多指(趾)畸形的修复应该在选择性修复的基础上进行。
如果发现发育迟缓,则指示干预发展和/或特殊教育。
对于手部灵巧度的职业治疗可能是必要的,因为一些具有轴中性多指(趾)畸形的个体具有手指对不齐的现象。
继发性并发症的预防
只有在最不寻常的情况下才能切除下丘脑错构瘤甚至活检,因为术后并发症和术后终身激素补充剂的需求通常会超过益处。
对于易患癫痫发作的CNS病变(如下丘脑错构瘤)患者,应慎重考虑使用兴奋剂治疗注意力缺陷障碍。
儿童时期:
- 每年进行医学评估,以评估生长和监测性早熟的迹象
- 每年筛查发育迟缓或学习障碍
避免因素/情况
如预防继发性并发症所述,一些兴奋剂(通常用于注意力缺陷和多动症)可能加剧癫痫发作。
亲属风险评估
评估有风险的亲属是合适的,以便尽早确定那些将从起始治疗和预防措施中受益的人
- 如果家族中的 致病性变异 未知,则可以使用多指(趾)畸形的临床检查,会厌分叉的喉镜检查或下丘脑错构瘤的MRI来阐明有风险的亲属的遗传状态。
妊娠管理
受累于PHS的女性的妊娠管理应该与疾病的具体表现相一致。例如,对需要服用抗惊厥药物的患有痴笑性癫痫的孕妇进行管理具有挑战性。由于没有针对PHS的指南,作者建议遵循妊娠期抗惊厥药的一般指南[Borthen & Gilhus 2012]。
一般而言,癫痫患者或任何原因引起的癫痫发作的女性在孕期死亡的风险比没有癫痫发作的孕妇更大;在怀孕期间使用抗癫痫药物降低了这种风险。然而暴露于抗癫痫药物可能会增加胎儿不良结局的风险(取决于使用的药物,剂量和服用药物的妊娠阶段。)然而,抗癫痫药物暴露导致胎儿不良结果的风险通常低于暴露于未治疗的母体癫痫发作的风险。因此,通常推荐使用抗癫痫药物来治疗妊娠期间的母体癫痫发作。关于在怀孕期间使用抗癫痫药物的风险和益处的讨论应该在受孕前进行。在怀孕前过渡到风险较低的药物是可能的[Sarma et al 2016]。
在PHS引起的垂体功能低下的个体中,生育能力和妊娠的管理(不常见于垂体功能低下的个体)同样具有挑战性,而且建议遵循一般的指导方针[Kübler et al 2009]。
请参阅MotherToBaby关于妊娠期间药物使用的进一步信息。
在研治疗
在ClinicalTrials.gov中搜索有关各种疾病和病症的临床研究信息。注:这种疾病可能没有临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。以下部分涉及遗传风险评估以及使用家族史和基因检测来明确家庭成员的遗传状况。本部分并不意味着可以解决个体可能面临的所有个人,文化和伦理问题。也不是为了替代遗传专业人士的咨询。—ED
遗传模式
Pallister-Hall综合征以常染色体显性遗传方式遗传。
家族成员风险
先证者父母
- 被诊断患有PHS的个体中约有75%有 受累的 父母。
先证者 同胞
先证者后代
其他家庭成员。其他家庭成员的风险取决于 先证者 父母的身份:如果父母 受累或者患有GLI3 致病性变异 。他或她的家庭成员将面临风险。
相关遗传咨询问题
有关评估风险亲属的信息以便早期诊断和治疗,请参阅危险亲属评估,管理。S
具有明显的 新生 致病性变异 的家庭的考虑。当具有 常染色体显性遗传 病症的 先证者 的亲本都不具有该病症的致病变异或临床证据时,先证者可能具有新生致病性变异。正如 家庭成员风险 中所述,已经描述了先证者的同胞,胚系嵌合现象和同胞复发的一个例子。然而,这种现象的实际风险无法从单一案例中估算出来,但显然并不常见。还可以探讨其他可能的非医学解释,包括 非生物学父亲 或母亲(例如辅助生育)和未披露的收养。
家庭计划
- 确定遗传风险的最佳时间和产前检测可用性的讨论是在妊娠前。
DNA库是DNA的存储(通常从白细胞中提取),以备将来使用。因为测试方法和我们对基因,等位基因变体和疾病的理解将来可能会有所改善,所以应该考虑 受累的 个体的DNA库。
产前检查和植入前遗传学诊断
分子遗传学诊断。一旦在 受累的 家庭成员中鉴定出GLI3 致病性变异 ,就可以对风险增加的妊娠进行产前检测,并对PHS进行 植入前遗传诊断 。
超声检查。在50%风险的胎儿中,产前超声检查可能会检测到多指畸形。然而,正常的超声检查并未消除胎儿中PHS的可能性。
资源
GeneReviews的工作人员选择了以下针对特定特定疾病和/或综合支持组织和/或注册中心,以造福有这种疾病的患者和他们的家庭。GeneReviews对其他组织提供的信息概不负责。有关选择标准的信息,请单击 此处 。
- 国家医学遗传图书馆主页参考
- 美国癫痫学会 (AES)
- 癫痫基金会8301 Professional Place EastSuite 200Landover MD 20785-7223Phone: 800-332-1000 (toll-free)Email: ContactUs@efa.org
- Medline Plus
分子诊断
Molecular Genetics 和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能含有更多信息。- ED。
Table A.
Pallister-Hall综合征: 基因和数据库
| 基因 | 染色体位点 | 蛋白 | 位点特异数据库 | HGMD |
|---|---|---|---|---|
| GLI3 | 7p14 | Transcriptional activator GLI3 | GLI3 @ LOVD | GLI3 |
Table B.
Pallister-Hall综合征的OMIM词条 (View All in OMIM)
基因结构.GLI3延伸超过276kb并包含15个外显子。 mRNA 约为8kb,参考 cDNA 为8,209bp (NM_000168.3, NP_000159.3), 开放阅读框架 为4,740bp。有关基因和蛋白质信息的详细摘要,请参见 表 A ,基因。
良性变异. GLI3中存在许多假定的良性变异;见 表2 (pdf)。大多数变异已经在多个不相关的人身上看到,并且不被认为与任何表型效应相关,尽管他们没有经过严格的细微分析。如果它们位于 外显子 内或者它们位于外显子25bp内的 内含子 中,则包含在表 2中。读者应参考ClinVar中的dbSNP参考编号和信息,表A(特定位点和HGMD)以及用于确认和其他数据的主要文献。
致病性变异. 在具有PHS的个体中报告的选定致病性变异在 表 3 中(pdf)。多种新生致病性变异已经被Johnston et al [2005], Johnston et al [2010], Démurger et al [2015]鉴定出。
正常 基因产物. 该基因编码1,580个氨基酸的蛋白质。
注:由于 cDNA 测序错误的结果,较早的引文描述了一个较长的开放阅读框架,预测了一个1,596个氨基酸的蛋白质;该错误已在GenBank的条目NM_000168.3中得到纠正。
异常基因产物. 已经证明截短形式的GLI3蛋白抑制转录[Blake et al 2016]。
参考文献
Literature Cited
- Azzam A, Lerner DM, Peters KF, Wiggs E, Rosenstein DL, Biesecker LG. Psychiatric and neuropsychological characterization of Pallister-Hall syndrome. Clin Genet. 2005;67:87 - 92. [PubMed: 15617553]
- Biesecker LG. What you can learn from one gene: GLI3. J Med Genet. 2006;43:465 - 9. [PMC free article: PMC2564530] [PubMed: 16740916]
- Blake J, Hu D, Cain JE, Rosenblum ND. Urogenital development in Pallister-Hall syndrome is disrupted in a cell-lineage-specific manner by constitutive expression of GLI3 repressor. Hum Mol Genet. 2016;25:437 - 47. [PMC free article: PMC4731018] [PubMed: 26604140]
- Borthen I, Gilhus NE. Pregnancy complications in patients with epilepsy. Curr Opin Obstet Gynecol. 2012;24:78 - 83. [PubMed: 22327733]
- Boudreau EA, Liow K, Frattali CM, Wiggs E, Turner JT, Feuillan P, Sato S, Patsalides A, Patronas N, Biesecker LG, Theodore WH. Hypothalamic hamartomas and seizures: distinct natural history of isolated and Pallister-Hall syndrome cases. Epilepsia. 2005;46:42 - 7. [PubMed: 15660767]
- Démurger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide AL, Quélin C, Manouvrier S, Baujat G, Fradin M, Pasquier L, Megarbané A, Faivre L, Baumann C, Nampoothiri S, Roume J, Isidor B, Lacombe D, Delrue MA, Mercier S, Philip N, Schaefer E, Holder M, Krause A, Laffargue F, Sinico M, Amram D, André G, Liquier A, Rossi M, Amiel J, Giuliano F, Boute O, Dieux-Coeslier A, Jacquemont ML, Afenjar A, Van Maldergem L, Lackmy-Port-Lis M, Vincent-Delorme C, Chauvet ML, Cormier-Daire V, Devisme L, Geneviève D, Munnich A, Viot G, Raoul O, Romana S, Gonzales M, Encha-Razavi F, Odent S, Vekemans M, Attie-Bitach T. New insights into genotype-phenotype correlation for GLI3 mutations. Eur J Hum Genet. 2015;23:92 - 102. [PMC free article: PMC4266745] [PubMed: 24736735]
- Everman D. The polydactylies. In: Stevenson RE, Hall JG, eds. Human Malformations and Related Anomalies. 2 ed. Oxford, UK: Oxford University Press; 2006:937-53.
- Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS, Bamshad MJ, Booth C, Curry CJ, David A, Dinulos MB, Flannery DB, Fox MA, Graham JM, Grange DK, Guttmacher AE, Hannibal MC, Henn W, Hennekam RC, Holmes LB, Hoyme HE, Leppig KA, Lin AE, Macleod P, Manchester DK, Marcelis C, Mazzanti L, McCann E, McDonald MT, Mendelsohn NJ, Moeschler JB, Moghaddam B, Neri G, Newbury-Ecob R, Pagon RA, Phillips JA, Sadler LS, Stoler JM, Tilstra D, Walsh Vockley CM, Zackai EH, Zadeh TM, Brueton L, Black GC, Biesecker LG. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet. 2005;76:609 - 22. [PMC free article: PMC1199298] [PubMed: 15739154]
- Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, Murphy H, Raygada M, Robin NH, Rope AF, Rosenbaum KN, Schaefer GB, Shealy A, Smith W, Soller M, Sommer A, Stalker HJ, Steiner B, Stephan MJ, Tilstra D, Tomkins S, Trapane P, Tsai AC, Van Allen MI, Vasudevan PC, Zabel B, Zunich J, Black GC, Biesecker LG. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 2010;31:1142 - 54. [PMC free article: PMC2947617] [PubMed: 20672375]
- Kang S, Graham JM Jr, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet. 1997;15:266 - 8. [PubMed: 9054938]
- Kübler K, Klingmüller D, Gembruch U, Merz WM. High-risk pregnancy management in women with hypopituitarism. J Perinatol. 2009;29:89 - 95. [PubMed: 19177043]
- Ng D, Johnston JJ, Turner JT, Boudreau EA, Wiggs EA, Theodore WH, Biesecker LG. Gonadal mosaicism in severe Pallister-Hall syndrome. Am J Med Genet. 2004;124A:296 - 302. [PubMed: 14708104]
- Radhakrishna U, Bornholdt D, Scott HS, Patel UC, Rossier C, Engel H, Bottani A, Chandal D, Blouin JL, Solanki JV, Grzeschik KH, Antonarakis SE. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type- A/B; No phenotype prediction from the position of GLI3 mutations. Am J Hum Genet. 1999;65:645 - 55. [PMC free article: PMC1377970] [PubMed: 10441570]
- Sarma AK, Khandker N, Kurczewski L, Brophy GM. Medical management of epileptic seizures: challenges and solutions. Neuropsychiatr Dis Treat. 2016;12:467 - 85. [PMC free article: PMC4771397] [PubMed: 26966367]
- Stevens CA, Ledbetter JC. Significance of bifid epiglottis. Am J Med Genet A. 2005;134:447 - 9. [PubMed: 15782417]
- Wallace RH, Freeman JL, Shouri MR, Izzillo PA, Rosenfeld JV, Mulley JC, Harvey AS, Berkovic SF. Somatic mutations in GLI3 can cause hypothalamic hamartoma and gelastic seizures. Neurology. 2008;70:653 - 5. [PubMed: 18057317]
Suggested Reading
- Biesecker LG, Abbott M, Allen J, Clericuzio C, Feuillan P, Graham JM Jr, Hall J, Kang S, Olney AH, Lefton D, Neri G, Peters K, Verloes A. Report from the workshop on Pallister-Hall syndrome and related phenotypes. Am J Med Genet. 1996;65:76 - 81. [PubMed: 8914745]
- Johnston JJ, Biesecker LG. Pallister Hall syndrome (PHS). Atlas of Genetics and Cytogenetics Oncology and Haematology. Available online. 2007. Accessed 5-10-17.
- Vortkamp A, Gessler M, Grzeschik KH. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature. 1991;352:539 - 40. [PubMed: 1650914]
Chapter Notes
Author Notes
The author is a board-certified clinical geneticist and pediatrician. He performs clinical and molecular research on PHS and related disorders at the NIH.
Revision History
- 18 May 2017 (sw) Comprehensive update posted live
- 18 December 2014 (me) Comprehensive update posted live
- 13 September 2012 (me) Comprehensive update posted live
- 15 June 2010 (cd) Revision: deletion/duplication analysis no longer available clinically
- 3 February 2009 (cd) Revision: deletion/duplication analysis available clinically
- 18 March 2008 (me) Comprehensive update posted to live Web site
- 2 June 2006 (cd) Revision: 产前诊断 clinically available
- 6 June 2005 (me) Comprehensive update posted to live Web site
- 1 May 2003 (me) Comprehensive update posted to live Web site
- 25 May 2000 (me) Review posted to live Web site
- 20 January 2000 (lb) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview ‘Pallister-Hall Syndrome’ is in the public 结构域 in the United States of America.