
概要
临床特征.
X-linked 先天性静止性夜盲(stationary night blindness ,CSNB)以非进展性视网膜性视力下降,视力下降幅度在20/30到20/200范围,暗适应差,屈光不正,伴典型的高度近视 (-0.25 diopters [D] to -4.75 D) 至 (≥-10.00 D) ,偶伴远视,眼球震颤,斜视,视觉正常,眼底镜检查正常,分为两个临床表现相近但又不同的表型:
- 完全夜盲(Complete CSNB (CSNB1A)),由NYX基因致病性变异导致(45%)
- 不完全夜盲(Incomplete CSNB (CSNB2A)),由CACNA1F基因致病性变异导致(55%)
诊断/检查
诊断建立在临床症状、特异表征、视网膜电图 electroretinography (ERG),】家族史以及对X-linked CSNB目前已知的NYX和CACNA1F两个基因的分子遗传学检测。
疾病管理
对症治疗:配镜以纠正屈光不正(近视或远视),常规斜视手术以改善双视和头部姿势。
预防继发并发症:斜视手术偶能提高休止眼位的功能范围。
监测: 每年进行眼科检查,尽早对近视进行干预。
药物/环境禁忌:减少夜间用眼,禁开车或考驾照。
遗传咨询
X-linked CSNB为X连锁遗传模式,受累的 男性的父亲非X-linked CSNB,也非致病性变异携带者,如果先证者先证者母亲是携带者,每次妊娠都有50%概率将致病性变异向子代传递,遗传了致病性变异的男性子代将受累,女性子代则为携带者,一般不发病。男性患者可将致病性变异遗传给女性子代,但不会遗传给男性子代。对于有携带致病性变异的家系,有患病风险的家系成员可选择携带者筛查以及产前诊断。
诊断
临床诊断
受累的男性
X-linked CSNB诊断可基于:
- 视力下降 所有受累的男性伴有视力下降,范围从20/30 (6/9; log MAR 0.1)到20/200 (6/60; log MAR 1.0)。
- 暗适应差 夜盲是主观症状,ERG检查可辅助进一步分型
- CSNB1A ( NYX基因致病完全型)一般伴有严重夜盲。
- CSNB2A (CACNA1F基因致病不完全型)夜盲较轻。
- 近视 近视屈光度数在 (-0.25 diopters [D] to -4.75 D) 到 (≥ -10.00 D) [Boycott et al 2000, Allen et al 2003],少数病例有远视。
- 眼球震颤和斜视
- 50%-70%受累的个体伴有眼球震颤和斜视 [Boycott et al 2000, Allen et al 2003]。
- 一项大规模门诺派人群伴不完全型X-linked CSNB的研究表明,72%的病例至少有以下一项症状不会出现:近视、眼球震颤、或夜盲 [Boycott et al 2000]。
- 视觉正常 然而,严重的 X-linked CSNB患者可有轻度色觉损害。
- 眼底镜检查正常 然而,严重近视可导致近视性眼底变性。
- 家族史表现为X连锁遗传模式。
- ERG检查的特征性表现
- ERG用以评估视网膜对光的电生理改变,双极细胞去极化诱发b波,可感受光刺激,依赖于感受器细胞对双极细胞的突出传导。
- X-linked CSNB患者在暗适应后的闪光刺激下b波下降(Figure 1),ERG波形图尤为平淡。(a波明显高于b波) [Miyake et al 1986],该波形也被称为Schubert-Bornschein波形[Schubert & Bornschein 1952]。
- ERG可辅助鉴别X-linked CSNB类型:
- ERG检查结果可以评价视网膜功能以及鉴别分型(Table 1) ,还可以大约评估涉及的致病性基因 (see Testing Strategy)。
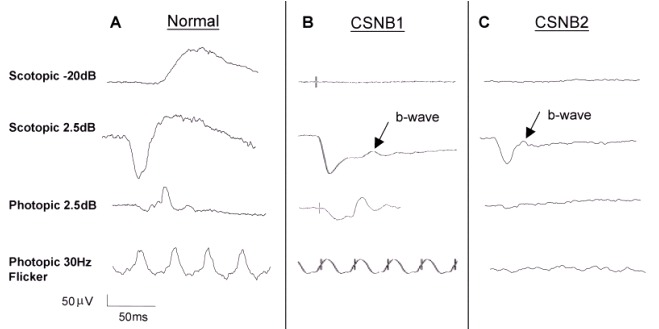
Figure 1.
男性全视野ERG报告峰图:A. 未受累的35岁男性。B. 受累的66岁男性(NYX基因变异)。C.受累的35岁男性(CACNA1F基因变异)箭头指示b波,比a波低平。 (参见更多more...)
Table 1.
完全型和不完全型X-Linked CSNB对比
| ERG表现 | 完全型 (CSNB1A) | 不完全型 (CSNB2A) |
|---|---|---|
| 暗杆b波 | 严重下降或缺少 | 下降 |
| 混合暗a波 | 正常 | 轻度下降 |
| 混合暗b波 | 下降 | 下降 |
| 暗OP | 缺少 | 轻度下降 |
| 明a波 | 正常,轻度下降,齿(方)形 | 下降 |
| 明b波 | 轻度下降 | 下降 |
| 明OP | 除OP4,均缺失 | 缺失 |
| 30-Hz 闪烁 | 正常/轻度下降 | 下降伴双峰 |
OP = oscillatory potential震荡电位
注意:根据瞳孔反应来分型在文献或论著中有不同观点(在暗适应下瞳孔会扩大),该方法早于基因分型,有学者描述在17名5至51岁的 X-linked CSNB患者均未记录到矛盾的瞳孔反射,但根据该现象用以分型仍需更多瞳孔测量性研究。
携带者女性
In general一般而言,携带者女性不表现X-linked CSNB临床症状,然而,偶见报道CACNA1F基因纯合变异报道,表现同男性患者 [Bech-Hansen et al 1998]。
X-linked CSNB携带者可有ERG改变:
- 杆细胞震荡电位下降[Rigaudière et al 2003]
- 个案可有明b波下降,振幅闪烁[Rigaudière et al 2003]
分子遗传学监测
基因NYX 和 CACNA1F 是目前已知的两个致病基因(染色体位点和蛋白参见Table A )
Table 2.
X-linked CSNB分子遗传学检测概述
| 基因 1 / CSNB 表型 | 基因致病占比 2 | 检测方法 | 变异检测 3 |
|---|---|---|---|
| NYX / CSNB1A 4 | 45% | 序列分析 5 | 序列变异 6, 7 |
| 缺失/重复 analysis 8 | 外显子及全基因缺失 7 9 | ||
| CACNA1F / CSNB2A 10 | 55% | 靶向分析 | c.3167_3168dupC 11 |
| 序列分析 5 | 序列变异 6, 7 |
- 1.
- 2.
- 3.
更多等位基因信息参见Molecular Genetics
- 4.
- 5.
- 6.
- 7.
序列分析不能检测女性X连锁基因单纯缺失或单个、多个外显子缺失。
- 8.
序列分析不能检测外显子侧翼区域以及内含子区域,可选择其它检测方法:quantitative PCR、长片段PCR、多重连接依赖探针扩增(MLPA)以及染色体芯片 (CMA)。
- 9.
- 10.
- 11.
见于荷兰-德国门诺派人群报道[Bech-Hansen et al 1998, Boycott et al 2000]。
检测策略
先证者诊断的确立 男性患者伴有视力下降、近视、眼球震颤、斜视以及视觉正常需考虑X-linked CSNB。一般而言,X-linked CSNB可通过眼科检查(ERG)及X连锁遗传家族史建立。
- 方法1 序列分子遗传学检测适用于家族史明确的个体,ERG可用于初步判断疾病分型并提示对应的基因检测。注意:荷兰-德国门诺派人群CSNB2A可优先检测CACNA1F基因建立者变异。
- 方法2 多基因检测适用于家族史不明确的个体,可选多基因Panel,包含NYX和CACNA1F基因,其它CSNB基因参见(参见鉴别诊断Differential Diagnosis)。注意:多基因Panel根据检测方法、基因以及CSNB个体 表型差异而有不同的选择。更多关于多基因Panel信息参见here。
患病风险亲属携带者筛查 可对家系患者检出致病性变异进行检测。
注意:杂合携带者也可表现某些症状。
产前诊断和植入前遗传诊断 (PGD) 适用于家系患病风险的妊娠。
临床特征
临床描述
X-linked CSNB为进展性视网膜疾患,以夜盲、视力下降、近视、眼球震颤以及斜视(参见临床诊断 Clinical Diagnosis)。
G基因型-表型关联
CSNB1A,完全型X-linked CSNB,由NYX基因变异导致 [Bech-Hansen et al 2000, Pusch et al 2000]。
CSNB2A,不完全型X-linked CSNB,由CACNA1F基因变异导致 [Bech-Hansen et al 1998, Strom et al 1998]。
外显率
CSNB1A 和CSNB2A外显率约在100%,但表现度各异[Boycott et al 2000];如果不做ERG检查,也可见因临床症状轻而漏诊的病例。
命名
X-linked CSNB曾称Schubert-BornscheinCSNB,是以特异性ERG波形图命名[Schubert & Bornschein 1952]。
CSNB1和CSNB2有时用于描述完全型和不完全型缩写而不考虑遗传模式,该术语也最早用于描述 X-linkedCSNB的两个类型。
发病率
X-linked CSNB发病率未知。
CSNB2A已有报道在荷兰-德国门诺派人群存在建立者效应,[Bech-Hansen et al 1998, Boycott et al 1998, Boycott et al 2000],NYX 基因常见致病性变异在比利时佛兰德 CSNB1A人群也有检出 [Leroy et al 2009]。
基因相关障碍
NYX 该基因变异关联表型:
CACNA1F CACNA1F基因变异还关联其它 X-linked4种表型:
- Åland Island eye disease (AIED),也名Forsius-Eriksson syndrome,是一类视网膜疾患,表现为眼底色素减退,视力下降,眼球震颤,散光,红色盲,进展性近视,暗适应缺陷,ERG提示明、暗功能异常。AIED 与 CSNB2A表型相似。 [Jalkanen et al 2007]在 AIED病例报道1个新的CACNA1F基因致病性变异 。
- X-linked 视锥-视杆营养不良(CORDX3) 该表型以中度进展性光感受器功能障碍及部分CSNB2A表现[Jalkanen et al 2006]。
- X-linked 视网膜障碍 [Hope et al 2005]报道一个毛利大家系,病例有CSNB2A样临床表现和ERG,同时还伴有智力残疾,女性携带者也有表型,由CACNA1F基因功能获得性 错义突变引起[Hemara-Wahanui et al 2005])。
- 视网膜和视神经萎缩 见于日本一对同胞病例,主要表现进展性视力下降[Nakamura et al 2003]。
鉴别诊断
参见基因关联OMIM词条Night blindness, congenital stationary: OMIM Phenotypic Series 。
正常眼底
X-linked CSNB 眼底正常,仅有少数疾病与CSNB需鉴别:
CSNB (非X-linked) 家族史不同,见于常染色体显性遗传和常染色体隐性遗传模式,(参见 OMIM Phenotypic Series).
锥细胞蓝色盲 (OMIM 303700),为X连锁遗传,以视力差、眼球震颤为特征,可与X-linked CSNB在以下方面鉴别:
- 色觉测试正常。Color vision testing is abnormal.
- ERG提示完全明视缺失,暗视正常或轻度受累。
- 尽管眼底检查在年轻男性患者正常,但部分男性成年后发展为黄斑萎缩。
锥细胞蓝色盲是由调控红绿色素细胞基因变异导致的。
X连锁运动性眼球震颤 该疾病可与X-linkedCSNB通过ERG鉴别,由FRMD7基因致病性变异导致[Tarpey et al 2006]。
异常眼底
一些伴有眼底异常的X连锁遗传的疾病可与X-linked CSNB混淆。
- X-linked ocular albinismX连锁黄斑白化病 临床特征为虹膜透明、中央凹营养不良和眼底发育不良,而X-linkedCSNB无异常,X连锁黄斑白化病无选择性b波下降,视觉诱发电位(VEP)提示视交叉纤维增多,致病基因为OA1。
- X-linked juvenile retinoschisisX连锁青少年视网膜劈裂症 视力下降同X-linked CSNB,眼底检查提示视网膜中央凹裂开,约50%男性患者眼底见可外周视网膜劈裂,而X-linkedCSNB无异常,ERG提示选择性b波下降,致病基因为RS1。
以下常染色体隐性遗传疾病也表现为异常眼底,在此讨论是由于这类疾病也表现为非进展性,部分表型可与X-linked CSNB相似:
- Oguchi disease(小口氏病) 在日本被报道为CSNB的一种类型,致病基因是SAG,编码抑制蛋白,以及GRK1基因,编码视紫红质激酶,该症眼底观察色素异常,暗适应时间延长可表现为正常(the Mizuo phenomenon) [Dryja 2000]。
- Fundus albipunctatus(洛伯病,眼底白点状病),也被认为是CSNB的一种类型, 致病基因是RDH5,编码视黄醇脱氢酶,眼底提示散在白点,ERG提示选择性b波下降,暗适应时间延长可表现为正常[Dryja 2000]。
疾病管理
对症治疗
对于高度近视或远视可配戴眼睛或隐性眼睛。
一些病例中,斜视手术可提高改善双视和头部姿势。
继发并发症的预防
X-linked CSNB男性患儿可能会表现功能上笨拙的头部姿势,以减弱特定凝视位置(所谓的“零点”)的眼球震颤程度。通过精心设计的斜视手术,注视休止眼位的位置可以转移到一个更好的功能范围。
监测
建议每年例行眼科检查以评估近视进展。
药物/环境禁忌
减少夜间用眼,禁开车或考驾照。
亲属患病风险评估
对于婴幼儿,若伴有高度近视,不寻常的头部姿势或眼球震颤以及有CSNB家族史,眼科检查和d 分子遗传学检测可确认疾病诊断,而无需麻醉下进行神经学检查或临床电生理检查。
参见Genetic Counseling 以获取更多关于家系亲属患病风险遗传咨询内容。
研究中治疗方法
参见ClinicalTrials.gov 以获取更多关于临床研究相关信息。注意:该病症可能暂无临床试验。
遗传咨询
遗传咨询是一个给患者及家属提供关于遗传性疾病本质、遗传特性以及影响并帮助他们做出知情的医疗决定的过程。下列段落描述遗传风险的评估以及根据家 族史和基因检测判断家族成员遗传状态。本段落描述不适用于解决患者实际面对的个人、文化或伦理问题,也不能代替专业的遗传咨询。—ED.
遗传模式
X-linkedCSNB为X连锁遗传模式。
家系患病风险
先证者父母
- 如果先证者母亲携带致病变异,先证者同胞将有50%概率遗传到致病变异,男性同胞遗传到致病变异将受累,女性同胞遗传到变异将为携带者且通常不受累,在极少数病例中,女性受累,遗传到两个致病变异,一个来自于母亲,一个来自于父亲。
- 如果先证者母亲DNA未能检出变异,先证者同胞患病风险极低,但较普通人群高( 胚系嵌合)。
先证者子代 男性X-linked CSNB可能将致病 等位基因遗传给儿子或女儿。
先证者其他家系成员 先证者母系家族阿姨及其子代可能为携带者,子代是否患病取决于性别。
携带者检测
对于已检出致病性变异家系的有患病风险的成员可选择携带者检测。
遗传咨询相关问题
参见疾病管理Evaluation of Relatives at Risk,以评估家系患病风险及临床早诊断和治疗措施。
家庭计划
- 明确遗传患病风险的最佳时间是怀孕前。
- 建议对受累的成年或携带者提供遗传咨询(可评估子代患病风险和生育计划)。
基因库 储存DNA(通常从白细胞中提取),以备将来使用。我们对检测技术以及对基因、等位基因变异和疾病的理解在未来将深入,受累的个体可考虑基因库。
产前诊断和胚胎植入前遗传学检测
一旦检出NYX 或CACNA1F 致病性变异 ,在受累的家系中,产前诊断和植入前遗传诊断可予以考虑。
对不影响智力或寿命的疾病(如x连锁CSNB)进行产前检查的要求并不常见。在使用产前检查方面,医学专业人员和家庭内部可能存在着不同的看法,特别是在考虑进行产前检查的目的是终止妊娠而不是早期诊断的情况下。虽然大多数中心认为产前检查的决定是父母的选择,但该问题仍需更多讨论。
资源
GeneReviews工作人员已经筛选了以下专科疾病和患者帮扶组织 ,注册登记能使患者及其家庭获益。 GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处 ( here)。
- My46 Trait Profile
- Foundation Fighting Blindness11435 Cronhill DriveOwings Mills MD 21117-2220Phone: 800-683-5555 (toll-free); 800-683-5551 (toll-free TDD); 410-568-0150Email: info@fightblindness.org
- Foundation Fighting Blindness - Canada890 Yonge Street12th FloorToronto Ontario M4W 3P4CanadaPhone: 800-461-3331 (toll-free); 416-360-4200Fax: 416-360-0060Email: info@ffb.ca
- National Eye Institute31 Center DriveMSC 2510Bethesda MD 20892-2510Phone: 301-496-5248Email: 2020@nei.nih.gov
- eyeGENE - National Ophthalmic Disease Genotyping Network RegistryPhone: 301-435-3032Email: eyeGENEinfo@nei.nih.gov
分子遗传学
分子遗传学检测的信息和OMIM相关列表可能同其它GeneReview信息会有不同。(可能有更新原因)—ED.
Table A.
X-linked CSNB:基因和数据库
Table B.
OMIM收录X-linked CSNB词条 (View All in OMIM)
分子遗传发病机制
X-linked CSNB致病基因编码特异表达蛋白在视网膜上:夜盲蛋白和压敏电阻器l型钙通道亚基alpha-1F (Cav1.4 /α1F)的表达异常导致完全型和不完全型的CSNB,在这些基因中发现的致病变异会影响从光感受器(杆状体和锥状体)到视网膜内细胞的突触传递。
NYX
基因结构 NYX 基因约28 kb,有3个外显子,详见Table A。
致病性变异 变异类型较广,包括错义, 无义, 和剪接位点变异,缺失,插入,超过50%的致病性变异为错义突变。 [Zeitz et al 2005, Zeitz 2007]. 特定人群常见变异信息参见 (参见发病率Prevalence)。
正常基因产物 NYX 基因编码夜盲蛋白,包含481个氨基酸,为亮氨酸富集蛋白家族,该蛋白包含1个单肽,1个亮氨酸富集重复区域和1个糖基磷脂酰肌醇锚定蛋白(GPI)序列[Bech-Hansen et al 2000, Pusch et al 2000, Bech-Hansen et al 2005]。
异常基因产物 Pathogenic variants in NYX 基因致病性变异导致夜盲蛋白功能缺陷,包括改变其结构,丢失GPI区域,蛋白缺失等[Zeitz 2007]。
CACNA1F
基因结构 CACNA1F约28 kb,包含28个外显子,详见Table A。
致病性变异 参见Table 3 变异类型较广,包括错义, 无义, 和剪接位点变异,缺失,插入,超过50%的致病性变异为错义突变。[Zeitz et al 2005, Zeitz 2007], 建立者变异信息参见(参见发病率Prevalence)。
Table 3.
本文讨论的CACNA1F 致病性变异
| DNA 核酸改变 (Alias 1) | 预测蛋白改变 (Alias 1) | 参考序列 |
|---|---|---|
| c.3167_3168dupC (3166dupC) | p.Leu1056ProfsTer11 (Leu991insC) | NM_005183 |
变异的分级:列表中变异由作者提供, GeneReviews工作人员未对变异进行独立分级。
命名: GeneReviews遵循人类基因组变异协会(Human Genome Variation Society ( varnomen .hgvs.org))原则。命名的解释参见 Quick Reference。
- 1.
变异命名与现行命名规则不一致
正常 基因产物 CACNA1F 编码蛋白(Cav1.4/α1F),存在剪接异型体,包含 1966个氨基酸 [Bech-Hansen et al 1998, Strom et al 1998]。
异常基因产物 表达研究表明,一些CACNA1F 致病性错义突变改变Cav1.4/α1F钙离子通道[McRory et al 2004, Hemara-Wahanui et al 2005, Hoda et al 2005],另一些错义突变影响突出前膜[Hoda et al 2006]。无义突变和移码突变导致通道蛋白或光感受器突触功能丧失。
参考文献
文献引用
- Allen LE, Zito I, Bradshaw K, Patel RJ, Bird AC, Fitzke F, Yates JR, Trump D, Hardcastle AJ, Moore AT. Genotype-phenotype correlation in British families with X linked congenital stationary night blindness. Br J Ophthalmol. 2003;87:1413 - 20. [PMC free article: PMC1771890] [PubMed: 14609846]
- Bech-Hansen NT, Cockfield J, Liu D, Logan CC. Isolation and characterization of the leucine-rich proteoglycan nyctalopin gene (cNyx) from chick. Mamm Genome. 2005;16:815 - 24. [PubMed: 16261423]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:264 - 7. [PubMed: 9662400]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Sparkes RL, Koop B, Birch DG, Bergen AA, Prinsen CF, Polomeno RC, Gal A, Drack AV, Musarella MA, Jacobson SG, Young RS, Weleber RG. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26:319 - 23. [PubMed: 11062471]
- Boycott KM, Pearce WG, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can J Ophthalmol. 2000;35:204 - 13. [PubMed: 10900517]
- Boycott KM, Pearce WG, Musarella MA, Weleber RG, Maybaum TA, Birch DG, Miyake Y, Young RS, Bech-Hansen NT. Evidence for genetic heterogeneity in X-linked congenital stationary night blindness. Am J Hum Genet. 1998;62:865 - 75. [PMC free article: PMC1377021] [PubMed: 9529339]
- Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130:547 - 63. [PubMed: 11078833]
- Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, Sharp DM, Lundon-Treweek P, Clover GM, Hoda JC, Striessnig J, Marksteiner R, Hering S, Maw MA. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc Natl Acad Sci U S A. 2005;102:7553 - 8. [PMC free article: PMC1140436] [PubMed: 15897456]
- Hoda JC, Zaghetto F, Koschak A, Striessnig J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels. J Neurosci. 2005;25:252 - 9. [PubMed: 15634789]
- Hoda JC, Zaghetto F, Singh A, Koschak A, Striessnig J. Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type Ca2+ channel function and expression. J Neurochem. 2006;96:1648 - 58. [PubMed: 16476079]
- Hope CI, Sharp DM, Hemara-Wahanui A, Sissingh JI, Lundon P, Mitchell EA, Maw MA, Clover GM. Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin Experiment Ophthalmol. 2005;33:129 - 36. [PubMed: 15807819]
- Jalkanen R, Bech-Hansen NT, Tobias R, Sankila EM, Mantyjarvi M, Forsius H, de la Chapelle A, Alitalo T. A novel CACNA1F gene mutation causes Aland Island eye disease. Invest Ophthalmol Vis Sci. 2007;48:2498 - 502. [PubMed: 17525176]
- Jalkanen R, Mantyjarvi M, Tobias R, Isosomppi J, Sankila EM, Alitalo T, Bech-Hansen NT. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J Med Genet. 2006;43:699 - 704. [PMC free article: PMC2564595] [PubMed: 16505158]
- Leroy BP, Budde BS, Wittmer M, De Baere E, Berger W, Zeitz C. A common NYX mutation in Flemish patients with X linked CSNB. Br J Ophthalmol. 2009;93:692 - 6. [PubMed: 18617546]
- McRory JE, Hamid J, Doering CJ, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle AM, Feldcamp L, Zamponi GW, Snutch TP. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004;24:1707 - 18. [PubMed: 14973233]
- Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch Ophthalmol. 1986;104:1013 - 20. [PubMed: 3488053]
- Nakamura M, Ito S, Piao CH, Terasaki H, Miyake Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch Ophthalmol. 2003;121:1028 - 33. [PubMed: 12860808]
- Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, Maurer J, Jacobi FK, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, Berger W, Meindl A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet. 2000;26:324 - 7. [PubMed: 11062472]
- Rigaudière F, Roux C, Lachapelle P, Rosolen SG, Bitoun P, Gay-Duval A, Le Gargasson JF. ERGs in female carriers of incomplete congenital stationary night blindness (I-CSNB). A family report. Doc Ophthalmol. 2003;107:203 - 12. [PubMed: 14661912]
- Schubert G, Bornschein H. Analysis of the human electroretinogram. Ophthalmologica. 1952;123:396 - 413. [PubMed: 14957416]
- Simonsz HJ, Florijn RJ, van Minderhout HM, Bergen AA, Kamermans M. Nightblindness-associated transient tonic downgaze (NATTD) in infant boys with chin-up head posture. Strabismus. 2009;17:158 - 64. [PubMed: 20001510]
- Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Ruther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:260 - 3. [PubMed: 9662399]
- Tarpey P, Thomas S, Sarvananthan N, Mallya U, Lisgo S, Talbot CJ, Roberts EO, Awan M, Surendran M, McLean RJ, Reinecke RD, Langmann A, Lindner S, Koch M, Jain S, Woodruff G, Gale RP, Degg C, Droutsas K, Asproudis I, Zubcov AA, Pieh C, Veal CD, Machado RD, Backhouse OC, Baumber L, Constantinescu CS, Brodsky MC, Hunter DG, Hertle RW, Read RJ, Edkins S, O'Meara S, Parker A, Stevens C, Teague J, Wooster R, Futreal PA, Trembath RC, Stratton MR, Raymond FL, Gottlob I. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet. 2006;38:1242 - 4. [PMC free article: PMC2592600] [PubMed: 17013395]
- Zeitz C. Molecular genetics and protein function involved in nocturnal vision. Exp Rev Ophthalmol. 2007;2:467 - 85.
- Zeitz C, Minotti R, Feil S, Matyas G, Cremers FP, Hoyng CB, Berger W. Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness. Mol Vis. 2005;11:179 - 83. [PubMed: 15761389]
- Zhang Q, Xiao X, Li S, Jia X, Yang Z, Huang S, Caruso RC, Guan T, Sergeev Y, Guo X, Hejtmancik JF. Mutations in NYX of individuals with high myopia, but without night blindness. Mol Vis. 2007;13:330 - 6. [PMC free article: PMC2642916] [PubMed: 17392683]
推荐阅读
- Dryja TP. Retinitis pigmentosa and stationary night blindness. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 235. New York, NY: McGraw-Hill. Available online. Accessed 2-2-16.
- Miyake Y. Congenital stationary blindness. In: Heckenlively JR, Arden GB, eds. Principles and Practice of Clinical Electrophysiology of Vision. 2 ed. Cambridge, MA: MIT Press; 2006:829-39.
章节注释
致谢
The authors would like to thank Linda MacLaren and Karen McElligott for years of service to the Mennonite community 受累的 with CSNB2A.
作者历史
N Torben Bech-Hansen, PhD; University of Calgary, Canada (2007-2012)
Kym M Boycott, PhD, MD (2007-present)
Ian M MacDonald, MD, CM (2007-present)
Yves Sauvé, PhD (2007-present)
修订历史
- 26 April 2012 (me) Comprehensive update posted live
- 16 January 2008 (me) Review posted to live Web site
- 9 August 2007 (im) Original submission