
简介
临床特征。由胱硫醚β-合成酶(CBS)缺乏引起的高半胱氨酸尿症的特征是眼部受累(眼角膜外翻和/或严重近视),骨骼系统(身高过高,四肢长,脊柱侧弯和胸腔凹陷),血管系统(血栓栓塞) 和CNS(发育,迟缓/智力障碍)。可以涉及全部四个系统或只有一个系统; 表现谱多样。以前没有症状的个体,在成年人中仅表现出脑血管的血栓栓塞性事件并不罕见。两种表型变异,B6反应性高半胱氨酸尿症和B6非反应性高半胱氨酸尿症。 B6反应性高半胱氨酸尿症通常比非反应性症状轻。
血栓栓塞是早期死亡和发病的主要原因。未经治疗的高半胱氨酸尿症患者的智商IQ范围从10到138。在B6反应性中,平均智商为79,而B6无应答者中的智商为57。可能发生的其他特征包括:癫痫发作,精神病,锥体束外征象(例如肌张力障碍),皮肤和头发的色素沉着不足,黄斑潮红,网状网纹和胰腺炎。
诊断/测试。高半胱氨酸尿症的主要生化特征包括血浆总高半胱氨酸和蛋氨酸的浓度显著增加。可以通过在CBS中检测双等位基因的致病变异来证实该诊断,CBS是编码胱硫醚β-合成酶的基因。
管理。表现治疗:治疗旨在纠正生化异常,尤其是控制血浆同型半胱氨酸浓度并防止血栓形成。同型半胱氨酸尿症的并发症应适当处理;例如,通过外科手术治疗眼外翻。
预防主要症状:使用维生素B6(吡哆醇)治疗(如果显示对B6有反应),蛋氨酸限制饮食以及叶酸和维生素B12补充剂治疗个体,使其血浆总同型半胱氨酸浓度保持在正常或接近正常水平。甜菜碱疗法通常被添加到治疗方案中。在青少年和成人中,甜菜碱可能是主要的治疗方式,但最好保持终生代谢饮食。
监视:应定期监测受影响的个体,以发现可能出现的任何临床并发症,饮食依从性以及血浆总同型半胱氨酸和氨基酸的含量。
应避免的药物/情况:受累的女性避免口服避孕药。可能的话进行手术。如果需要进行手术,应给予0.5%生理盐水和5%葡萄糖的静脉输液,维持体液的1.5倍,并持续进行,直到随意口服液为止,并密切监测以避免液体超载。
有风险的亲属的评估:出生后立即测量高危同胞的总同型半胱氨酸和氨基酸,可确保通过早期诊断和治疗降低发病率和死亡率。如果已知该家族中的CBS致病变异,则可以使用分子遗传学检测来阐明同胞的遗传状态
怀孕管理:对于具有典型高半胱氨酸尿症的女性:饮食治疗和甜菜碱,以及对那些对B6有反应的用维生素B6,并在整个怀孕期间进行仔细的生化监测。建议在妊娠中期和产后用低分子量肝素进行预防性抗凝治疗,以减少血栓栓塞的风险。
遗传咨询。高半胱氨酸尿症以 常染色体隐性遗传的方式遗传。 受累的患者的每个同胞都有25%的机会受到感染,有50%的机会成为无症状的携带者,有25%的机会不受影响也不是携带者。如果在受影响的家庭成员中发现了CBS致病变异,则可以对高危家庭成员进行携带者检测,以及对高危妊娠进行产前检测。
诊断
欧洲已经制定了经典高半胱氨酸尿症的诊断和治疗指南[Morris et al 2017]。这些欧洲指南建议将总同型半胱氨酸(tHcy)连同血浆氨基酸分析一起作为诊断的一线测试;当tHcy高于100 umol / L但偶尔降低(正常值:<15 umol / L)时,伴有高或临界高蛋氨酸,则很有可能确立诊断。对于经典的高半胱氨酸尿症诊断,必须同时获得血浆总高半胱氨酸浓度和蛋氨酸浓度(通过血浆氨基酸分析确定)。
经典的高半胱氨酸尿症是由缺乏胱硫醚β-合成酶(CBS)(一种吡哆醇维生素B6依赖性酶)引起的。由于高半胱氨酸在蛋氨酸代谢周期中位于转硫和蛋氨酸再甲基化之间的分支点,因此CBS的阻滞限制了转硫作用,并导致高半胱氨酸和蛋氨酸的增加,后者是由再甲基化增强引起的
(Figure 1).
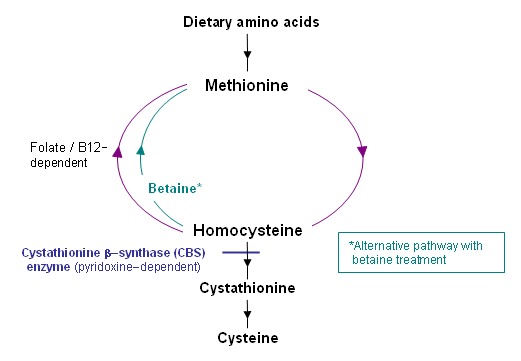
Figure 1.
Methionine metabolic pathway
提示性发现
新生儿筛查异常的新生儿蛋氨酸含量升高和临床发现范围从婴儿或儿童早期开始的多器官疾病到仅在成人的早期到中年血栓栓塞的个体,应怀疑由CBS缺乏引起的同型半胱氨酸尿症(典型的同型半胱氨酸尿症)。
新生儿筛查 通过筛查新生儿血斑样本中的高蛋氨酸血症,可以在一些(并非全部)受累的个体中检测到典型的高半胱氨酸尿症。
新生儿筛查用于测定蛋氨酸的方法是串联质谱法(MS / MS)。参见National Newborn Screening Status Report (pdf).。
如果最初的筛选测试结果超过了蛋氨酸的临界水平,则需要进行后续测试。这可能是:
- 1.
- 重复将干血标本提交给 新生儿筛查程序;要么
- 2.
- 血浆定量氨基酸分析和血浆总同型半胱氨酸分析,如American College of Medical Genetics的新生儿筛查ACT片和蛋氨酸的确认算法中所推荐(请参阅 ACMG ACT Sheet and ACMG Algorithm)。
干血标本和血浆分析之间的选择基于筛查程序的建议,该程序通常取决于初始筛查中蛋氨酸增加的程度。
如果选择上述1),并且结果证实高蛋氨酸血症,则应进行血浆总高半胱氨酸分析和蛋氨酸浓度的血浆氨基酸分析,以确认或排除经典高半胱氨酸尿症的诊断 (Table 1)。
注意:
- 至少有一个新生儿筛查程序对所有蛋氨酸含量较高的新生儿标本中的同型半胱氨酸进行第二级检测,以减少假阳性结果的发生率[Turgeon et al 2010].
- 新生儿筛查是针对蛋氨酸而不是高半胱氨酸。因此,由于这些疾病中蛋氨酸的水平降低了(或正常),因此可能无法检测到其他总高半胱氨酸升高的原因,例如再甲基化异常(例如亚甲基四氢叶酸还原酶缺乏症和钴胺素缺陷;参见 Differential Diagnosis)。一些 新生儿筛查实验室可能标记低蛋氨酸 [Tortorelli et al 2010]。
- 实际上,通过新生儿筛查程序检测到的所有具有经典高半胱氨酸尿症的婴儿均具有吡哆醇(维生素B6)无反应性高半胱氨酸尿症(请参见Clinical Description)。对于吡哆醇有反应的婴儿,在获得新生儿筛查标本后的头两到三天内,蛋氨酸的增加可能非常罕见。
- 卡塔尔使用总同型半胱氨酸作为主要的新生儿筛查 标记物进行测量,卡塔尔报道同型半胱氨酸尿症的发生率最高 [Gan-Schreier et al 2010]。
临床发现 经典同型半胱氨酸尿症的主要临床发现:
- 晶状体异位(眼晶状体脱位)和/或严重近视
- 骨骼异常(例如,身高过高,四肢长而狭窄,脊柱侧弯,胸廓凹陷)可能给人以马凡综合症的临床印象,但没有关节活动过度
- 以血栓栓塞为特征的血管异常
- 发育迟缓/智力障碍
建立诊断
通过测量血浆总同型半胱氨酸(tHcy)和血浆中的氨基酸(参见Plasma Homocysteine and Amino Acids)和/或通过鉴定CBS中 双等位基因的的致病变异,可以在先证者中建立经典同型半胱氨酸尿症的诊断。分子遗传学检测(请参阅Table 2)。如果未鉴定出致病性变异,可以进行胱硫醚β-合酶(CBS)活性的酶分析。
血浆同型半胱氨酸和氨基酸 血浆同型半胱氨酸浓度必须在两周不补充吡哆醇(包括多种维生素)的情况下测定。
Table 1总结了主要的生化特征,包括血浆总同型半胱氨酸(tHcy)和蛋氨酸的浓度显著增加。
Table 1.
同型尿毒症诊断的主要生化发现
| 分析物 | 标本 | 预期结果 | ||
|---|---|---|---|---|
| 高半胱氨酸尿症新生儿 | 未经治疗的高胱氨酸尿症成年人 | 对照 | ||
| 总同型半胱氨酸 1 (tHcy) | 血浆 2 | 50 to >100 µmol/L | >100 µmol/L | <15 µmol/L |
| 蛋氨酸(氨基酸分析) | 血浆 | 200-1500 µmol/L (3-23 mg/dL) | >50 µmol/L (>0.7 mg/dL) | 10-40 µmol/L (0.2-0.6 mg/dL) |
- 1.
单击 here(pdf)以获取用于描述硫氨基酸的术语
- 2.
血浆tHcy测定是确保准确诊断高半胱氨酸尿症的有效方法。 即使在储存一周后没有脱蛋白质化,实际上所有tHcy仍可以通过包括还原剂(如二硫苏糖醇)的制备方法回收 [Smith et al 1998].
分子遗传学测试 分子遗传学检测方法可以包括单基因检测或表型靶向检测的使用。
单基因测试
- 如果仅发现或未发现致病性变异,则首先进行CBS的序列分析,然后进行基因靶向的 deletion/duplication analysis。
- 靶向分析仅在卡塔尔人群中进行,其中93%的受累的人口中存在单一 致病性变异 (p.Arg336Cys; c.1006C>T) [Gan-Schreier et al 2010]。
可以考虑使用包含CBS和其他感兴趣基因的表型靶向检测方法(请参阅 Differential Diagnosis)。注意:(1)套餐中包含的基因和每种基因所用测试的诊断敏感性随实验室和时间而变化。 (2)一些多基因套餐可能包含与GeneReview中讨论的病症无关的基因;因此,临床医生需要确定哪个多基因套餐能够以最合理的成本提供最佳机会,以鉴定疾病的遗传原因,同时限制无关发现。 (3)套餐中使用的方法可能包括 序列分析, deletion/duplication analysis和/或其他基于非序列的测试。
Table 2.
胱硫醚β-合成酶缺乏引起的高半胱氨酸尿症的分子遗传学检测
| 基因 1 | 测试方法 | 具有致病性变异的先证者所占比例 2 可通过此方法检测 |
|---|---|---|
| CBS | Sequence analysis 3 | 95%-98% 4 |
| Gene-targeted deletion/duplication analysis 5 | <5% 6 | |
| Unknown 7 | NA | |
- 1.
- 2.
有关在该 基因中检测到的等位基因变异的信息,请参见Molecular Genetics。
- 3.
- 4.
- 5.
- 6.
迄今为止,已经报道了九个具有25个或更多个核苷酸的缺失或重复的个体 [Kraus Lab; CBS Mutation Database].
培养的成纤维细胞中的CBS酶活性 当分子分析未能鉴定出CBS中的两种致病变异时,可以进行CBS酶活性分析。可以在成纤维细胞[Smith et al 2012, Mendes et al 2014]或血浆[Krijt et al 2011, Alcaide et al 2015]中测量CBS酶的活性。然而,在轻度情况下,活性可能是正常的,尤其是在那些对B6有反应的人中 [Mendes et al 2014, Alcaide et al 2015]。酶分析不能可靠地区分B6反应性个体和B6非反应性个体。
注意:酶活性测试过去在分子检测基因检测结果不能诊断时用于确认同型半胱氨酸尿症的诊断,在美国不再可用。
诊断建立后进行测试
吡哆醇(B6)激发试验。经典高半胱氨酸尿症的两种表型变异-B6反应性和B6无反应性高半胱氨酸尿症-可能有不同的natural history和management。一旦确定了由胱硫醚β-合成酶(CBS)缺乏引起的高半胱氨酸尿症的诊断,就可以使用吡id哆醇激发来测量维生素B6的反应性,以确定存在哪种表型。
- 在新生儿中。 在继续正常饮食的同时,获取血浆用于总同型半胱氨酸和氨基酸的基线测量。
受累的者每天连续两天服用100 mg吡哆醇; 首次给药后48小时测量血浆总同型半胱氨酸和氨基酸的浓度。- 血浆总同型半胱氨酸和/或血浆蛋氨酸浓度降低30%或更多表明B6反应性。
- 如果未发生明显变化,则连续两天口服200 mg吡哆醇,并在48小时后以该剂量重复进行血浆总高半胱氨酸和氨基酸分析。
- 如果仍未发生变化,则给新生儿服用300 mg吡哆醇。 如果在最后一次剂量吡哆醇后血浆高半胱氨酸和蛋氨酸的浓度没有显著降低,那么可以得出结论,该个体对B6无反应。
- 超过新生儿期。 对于经临床鉴定的个体,继续正常饮食并提供5毫克叶酸补充剂(适用于儿童和成人); 纠正B12缺乏症(如果存在)。 获得血浆用于总同型半胱氨酸和氨基酸的基线测量。连续两天每天给受累的人服用100 mg吡哆醇,并在首次给药后48小时再次测量血浆总高半胱氨酸和氨基酸的浓度。
- 血浆总同型半胱氨酸和/或血浆蛋氨酸浓度降低30%或更多表明B6反应。
- 如果未发生明显变化,则连续两天口服200 mg吡哆醇,并在48小时后以该剂量重复进行血浆总高半胱氨酸和氨基酸分析。
- 如果仍未发生变化,则给儿童或成人口服500 mg吡哆醇。 如果在最后一次剂量吡哆醇后血浆高半胱氨酸和蛋氨酸的浓度没有显著降低,那么可以得出结论,该个体对B6无反应
注意:(1)婴儿不应接受超过300毫克的吡哆醇。 每天服用500 mg吡哆醇的几名婴儿出现呼吸衰竭并需要呼吸支持。 停用吡哆醇后,呼吸道症状得以缓解[Shoji et al 1998, Mudd et al 2001]。 (2)周围神经病变被认为是吡哆醇每天剂量超过900 mg的不良反应[Morris et al 2017]。
临床特征
临床表现
高半胱氨酸尿症的特征是累及眼睛,骨骼系统,血管系统和中枢神经系统。可以涉及所有四个或仅一个系统。对于所有临床体征,表达谱是广泛的。以前没有症状的个体在成人年仅出现脑血管血栓栓塞事件并不罕见 [Yap 2003, Skovby et al 2010]。
经典高半胱氨酸尿症的两个表型变异是B6反应性和B6非反应性高胱氨酸尿症。 B6反应性高半胱氨酸尿症通常(但不总是)比无反应性 轻。维生素B6反应性是通过吡i哆do醇激发试验确定的(请参阅 Testing Following Establishment of the Diagnosis)。
眼睛 近视眼随后晶体异位通常发生在一年后。在大多数未经治疗的个体中,晶体异位是在八岁时发生的。晶体异位通常在对B6无反应的 受累的患者中比对B6有反应的人更早发生。罕见的是,晶体异位在婴儿期发生的 [Mulvihill et al 2001]。
在没有晶体异位的情况下可能出现高度近视。
骨骼系统。患病个体通常身材瘦高 ,有马凡征表现。
高半胱氨酸尿症的个体容易发生骨质疏松症,尤其是椎骨和长骨。 50%的人到了十几岁就显示出骨质疏松的迹象。骨质疏松可以通过腰椎侧面或骨密度研究的影像学检查来检测。 DXA骨密度分析通常显示腰椎和臀部密度降低 [Weber et al 2016]。
脊柱侧凸,漏斗胸,鸡胸,膝外翻及弓形足也很常见。
血管系统。血栓栓塞是发病和早期死亡的主要原因[Yap 2003]。它会影响任何血管。尽管在年轻人中通常会出现问题,但已有婴儿发生脑血管意外的报道 [Yap et al 2001a, Kelly et al 2003]。
在对B6有反应的个体中,青春期或成年期的血管事件通常是高半胱氨酸尿症的表现特征[Magner et al 2011, Sarov et al 2014]。脑静脉窦血栓形成已成为儿童期的征兆 [Karaca et al 2014, Saboul et al 2015]。
怀孕会增加血栓栓塞的风险,尤其是在产后时期 [Novy et al 2010]。然而,大多数怀孕并不复杂。请参阅Pregnancy Management。
CNS。发育迟缓通常是高半胱氨酸尿症患者的第一个异常体征。高半胱氨酸尿症个体的智商范围为10到138。B6应答个体比B6无应答性高半胱氨酸尿症的个体在认知上完整或仅轻度受累的可能性更大。 B6反应性未治疗个体的平均智商为79,而B6反应性未治疗个体的平均智商为57。在新生儿筛查中鉴定出的B6无反应个体,接受早期治疗并具有良好依从性(维持血浆游离高半胱氨酸<11μmol/ L),平均智商为105 [Yap等,2001b]。
未经治疗的人中有21%发生癫痫发作。
许多人都有精神病问题,包括人格障碍,焦虑,抑郁,强迫行为和精神病发作。青春期可能表现精神病[Hidalgo Mazzei et al 2014].。
可能出现锥体束外症状,例如肌张力障碍。
其他特征包括皮肤和头发的色素沉着不足,黄斑潮红, 胰腺炎。
基因型-表型相关
单个 p.Gly307Ser 等位基因的存在可预测B6无反应性,而 p.Ile278Thr等位基因的存在通常可预测B6反应性[Gaustadnes et al 2002, Kruger et al 2003, Skovby et al 2010]。其他致病性等位基因与B6反应性或非反应性相关 [Kraus et al 1999]。
命名法
“高半胱氨酸尿症”因尿中过量的高半胱氨酸而得名,尽管现在主要是通过血浆中总高半胱氨酸的增加来检测到的。同型半胱氨酸尿症可能是由基因缺陷导致的胱硫醚β-合成酶(CBS)活性不足或多种遗传问题引起的,这些遗传问题最终会干扰同型半胱氨酸向蛋氨酸的转化(例如,亚甲基四氢叶酸还原酶缺乏症以及钴胺素转运或代谢异常)。有关后一种情况的详细信息,请参阅Watkins & Rosenblatt [2014]。 (另请参见Disorders of Intracellular Cobalamin Metabolism)
非遗传学上确定的严重饮食中钴胺素的缺乏(维生素B12缺乏症)也可能导致“高半胱氨酸尿症”[Mudd et al 2000]。
为了在使用“同型半胱氨酸尿症”一词时获得最大的特异性,可以添加特定的问题缺陷。例如“ CBS缺乏引起的高半胱氨酸尿症” [Mudd et al 2000],也被称为“经典高半胱氨酸尿症”。
此GeneReview中定义的经典高半胱氨酸尿症是由胱硫醚β-合成酶(CBS)的缺乏引起的,CBS是一种吡哆醇(维生素B6)依赖性酶。
患病率
目前患病率尚未确定;由于未发现病例,新生儿筛查和临床确诊均低估了患病率[Skovby et al 2010]。据报道患病率为1:200,000至1:335,000。
- 卡塔尔患病率估计为1:1800,可能是世界上最高的[Gan-Schreier et al 2010]。
- 爱尔兰。据报道患病率高达1:65,000 [Naughten et al 1998]。
- 德国。正常人群的分子遗传学筛查估计经典高半胱氨酸尿症的患病率为1:17,800[Linnebank et al 2001]。
- 挪威。基于杂合率,新生儿采用六个致病性变异进行分子遗传学筛查,估计经典高半胱氨酸尿症的患病率为1:6400 [Refsum et al 2004]。
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,没有其他表型与CBS中的致病变异相关。
鉴别诊断
最类似经典同型半胱氨酸尿症的临床症状的是Marfan syndrome:四肢、手指、脚趾细长不匀称,身高明显超出常人,伴有心血管系统异常, 晶体异位和近视的特征。 尽管在亚硫酸盐氧化酶缺乏症(OMIM 272300)的早期也可能发生眼角膜外翻,但这种情况在临床上不同于高半胱氨酸尿症。 亚硫酸氧化酶缺乏症和马凡氏综合征患者的血浆同型半胱氨酸和蛋氨酸浓度正常。
同型半胱氨酸或蛋氨酸的浓度升高也发生在生物化学遗传性疾病中,该疾病通常分为两组(见Figure 2 和Table 3),并且可以继发于其他疾病或营养畸变:
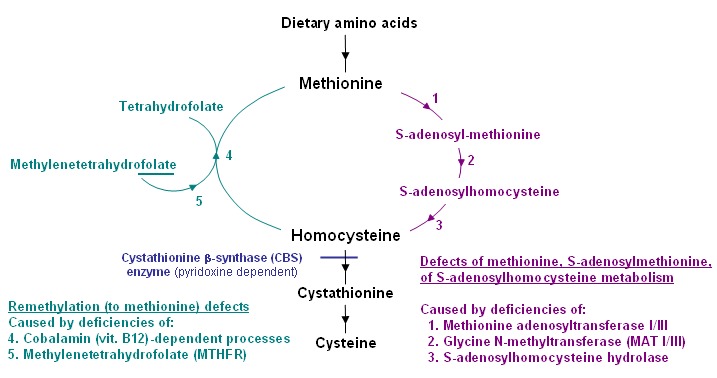
Figure 2.
在高半胱氨酸尿症疾病的途径的生化鉴别诊断
- 蛋氨酸,S-腺苷甲硫氨酸或S-腺苷同型半胱氨酸代谢的缺陷,通常蛋氨酸浓度增加,但总同型半胱氨酸浓度正常或仅略有增加。 此类别包括几种高甲硫氨酸血症疾病,例如甲硫氨酸腺苷转移酶I / III缺乏症,甘氨酸N-甲基转移酶缺乏症和S-腺苷同型半胱氨酸水解酶缺乏症 [Mudd 2011, Barić et al 2017].
- 蛋氨酸再甲基化缺陷,血浆总同型半胱氨酸水平升高,但蛋氨酸浓度低。 因为新生儿筛查是基于蛋氨酸(不是高半胱氨酸)的检测,所以由于这些疾病的血浆蛋氨酸浓度降低了(或正常),因此可能无法检测到甲基化障碍(例如亚甲基四氢叶酸还原酶缺乏症和钴胺素缺陷)。 这些疾病通常取决于叶酸或维生素B12 [Huemer et al 2017].
- 继发性高蛋氨酸血症,总同型半胱氨酸正常或仅轻度升高,可能发生在与tyrosinemia type I [Grompe 2001] 或 galactosemia相关的肝脏疾病中,以及高蛋白饮食或富含甲硫氨酸的婴儿配方食品中蛋氨酸摄入过多的情况下[Mudd et al 2003]
Table 3.
影响蛋氨酸代谢紊乱的生化方面
| 缺陷类型 | 疾病 | 总同型半胱氨酸 | 蛋氨酸 |
|---|---|---|---|
| 蛋氨酸 甲基化 | MAT I/III deficiency 1 | ↑ (normal, slight) | ↑↑ |
| GNMT deficiency 2 | |||
| S-adenosylhomocysteine hydrolase deficiency | |||
| 转硫 | Homocystinuria | ↑↑ | ↑↑ |
| 再甲基化 | MTHFR deficiency | ↑↑ | ↓↓ (rarely normal) |
| Cobalamin defects |
- 1.
MAT = 蛋氨酸腺苷转移酶
- 2.
GNMT = 甘氨酸N-甲基转移酶
处理
初步诊断后的评估
为了确定所有诊断为由胱硫醚β-合成酶缺乏症引起的同型半胱氨酸尿症的患者的疾病程度和需求,建议采取以下措施:
- 咨询临床遗传学家/医学生化遗传学家以获取其他测试和治疗计划
- 在开始治疗前进行吡哆醇(维生素B6)试验(请参阅Testing Following Establishment of the Diagnosis,吡哆醇(B6) 测试)
- 与遗传咨询师进行遗传咨询和 再发风险咨询。
表现治疗
治疗应由生化遗传学家和代谢营养师管理,旨在预防高半胱氨酸尿症的主要表现。有关已发布的管理指南,请参见 Morris et al [2017]。
并发症应得到适当处理(例如,手术晶体异位 [Neely & Plager 2001]。
预防主要表现
治疗的原则是纠正生化异常,尤其是尽可能地控制血浆同型半胱氨酸浓度的升高,预防或至少减少同型半胱氨酸尿症的并发症[Yap & Naughten 1998],并防止进一步的并发症,例如血栓形成 [Morris et al 2017]。
据报道,通过 新生儿筛查鉴定并在出生后不久接受治疗的个体中,其血浆游离高半胱氨酸浓度保持在11 µmol / L以下(最好是≤5µmol / L)以下是最好的结果 [Yap et al 2001b]。这对应于血浆总同型半胱氨酸浓度低于120 µmol / L或最好低于100 µmol / L [Morris et al 2017]。对于B6反应个体,血浆总同型半胱氨酸的目标是低于50 µmol / L [Morris et al 2017]。
当可以获得非常长期的数据时,可能需要修订这些目标。
用于控制血浆总同型半胱氨酸浓度的措施包括维生素B6(吡哆醇)治疗(如果显示对B6有反应),蛋氨酸限制饮食以及叶酸和维生素B12补充。甜菜碱疗法通常被添加到治疗方案中。在青少年和成人中,甜菜碱可能是主要的治疗方式,但最好保持终生代谢饮食。在已经发生血管事件的患者中,单独使用甜菜碱治疗可以预防复发事件[Lawson-Yuen&Levy 2010]。
有关治疗各个方面的详细信息如下。
维生素B6(吡哆醇)治疗。在那些显示出对B6有反应的患者中,吡哆醇的剂量约为200 mg /天或产生最大生化益处的最低剂量(即最低血浆同型半胱氨酸和蛋氨酸浓度),这是通过测量给出总同型半胱氨酸确定的和氨基酸水平 。
尽管有B6无反应的证据,也可将吡哆醇纳入治疗,通常每天100-200 mg(尽管某些成年人每天接受500-1000 mg)。
饮食治疗。 B6无反应的新生儿或仅对吡哆醇反应较差的新生儿需要限制蛋氨酸的饮食,并经常进行代谢监测。这种饮食应无限期继续。对于临床诊断的个体,应考虑饮食治疗,但如果从儿童期开始或以后开始,则通常不易耐受饮食治疗。
大多数对B6有反应的个体还需要限制蛋氨酸的饮食来控制代谢。
高半胱氨酸尿症的饮食非常复杂,必须利用经验丰富的代谢营养师的技能。饮食疗法通过限制天然蛋白质的摄入量来减少蛋氨酸的摄入量。但是,为了防止蛋白质营养不良, 提供其他氨基酸(以及半胱氨酸,可能是CBS缺乏症的必需氨基酸)的无蛋氨酸配方。可以与不含蛋氨酸的氨基酸婴儿配方食品一起继续母乳喂养 [MacDonald et al 2006]。所需的蛋氨酸的量由代谢营养师计算,并以天然食品和特殊低蛋白食品的形式提供,并基于总同型半胱氨酸和蛋氨酸的血浆浓度进行监测。
叶酸和维生素B12的补充。叶酸和维生素B12可通过蛋氨酸合酶优化高半胱氨酸向蛋氨酸的转化,从而有助于降低血浆高半胱氨酸浓度。当红细胞叶酸浓度和血清B12浓度降低时,每天口服5 mg叶酸。维生素B12以羟基钴胺素的形式每月1 mg肌注。
甜菜碱治疗。甜菜碱治疗提供了另一种重新甲基化途径,将过量的高半胱氨酸转化为蛋氨酸(见Figure 1),可能有助于预防并发症,尤其是血栓形成 [Yap et al 2001a, Lawson-Yuen & Levy 2010]。通过将同型半胱氨酸转化为蛋氨酸,甜菜碱可降低血浆总同型半胱氨酸浓度,但可提高蛋氨酸的血浆浓度。
对于儿童,甜菜碱的初始剂量是每天两次,每次50 mg / kg,根据反应进行调整(每周增加50 mg / kg的增量)。对于成年人,初始剂量是3 g每天两次。根据生化反应调节剂量和频率。超过150-200 mg / kg /天的剂量似乎没有任何好处[Morris et al 2017]。
甜菜碱可以在饮食治疗依从性差的个体中添加到治疗方案中,或者可能成为饮食不耐受者的主要治疗方式。补充甜菜碱后,不能通过饮食获得代谢控制的吡哆醇无反应个体显著降低了血浆同型半胱氨酸浓度[Singh et al 2004]。
甜菜碱的副作用很少。 (1)一些受累的人会产生可检测到的体味,从而导致依从性下降。 (2)甜菜碱产生的蛋氨酸的增加通常是无害的;然而,当高蛋氨酸血症严重(> 1000 µmol / L)时,就会发生脑水肿 [Yaghmai et al 2002, Devlin et al 2004, Tada et al 2004, Braverman et al 2005]。消除甜菜碱可导致高蛋氨酸血症的快速减少和脑水肿的消退。
注意:在高半胱氨酸尿症的鼠模型中,甜菜碱治疗的效果会随时间显著降低[Maclean et al 2012]。
监视
应定期监测受影响的个体,以发现可能出现的任何临床并发症,评估饮食依从性,并测量血浆总同型半胱氨酸和蛋氨酸的浓度。在出生后的头六个月应每月对婴儿进行一次监测,直到一年岁时每两个月监测一次,然后每三个月监测一次,直到三岁为止。 建议整个儿童期的半年度监测以及青春期和成年期的年度监测。并发症应及时采取适当的治疗措施。
应监测所有接受甜菜碱的人的血浆总同型半胱氨酸和蛋氨酸浓度(请参见Prevention of Primary Manifestations,甜菜碱治疗)。
应监测维生素B12和叶酸的水平。
定期的眼科评估可以识别眼部并发症,例如进行性近视和晶体异位,并允许早期治疗和预防进一步的并发症,例如视网膜脱离。
青春期后应每三到五年进行DXA扫描以监测骨质疏松症 [Morris et al 2017]。
避免的药物/情况
具有高半胱氨酸尿症的女性应避免口服避孕药,这可能会增加凝血能力并可能引起血栓栓塞。
如果可能的话,也应避免手术,因为手术期间,尤其是手术后血浆同型半胱氨酸浓度的增加会增加发生血栓栓塞事件的风险。如果需要进行手术,则应在手术前,手术中和手术后以0.5 N盐水中含5%葡萄糖的1.5倍体液的维持静脉内输液,直到可以口服为止。如果维持1.5倍的体液由于体液超负荷而导致心血管疾病的风险,则应在仔细的临床观察下进行基本体液维持。
评估处于危险中的亲戚
出生后应尽快在高危同胞中测定血浆总同型半胱氨酸和氨基酸的浓度,以便通过早期诊断和治疗降低发病率和死亡率。
如果已知该家族中的CBS致病变异,则可以使用分子遗传学检测来阐明同胞的遗传状态。
有关为遗传咨询目的而对高危亲戚进行测试的相关问题,请参见Genetic Counseling。
怀孕管理
由于具有高半胱氨酸尿症的女性血栓栓塞的风险可能高于平均水平,尤其是在产后,因此建议在妊娠中期和产后进行预防性抗凝治疗。通常的方案是在妊娠的最后两周和产后的前六周注射低分子量肝素 [Pierre et al 2006]。在整个怀孕期间也服用了小剂量的阿司匹林。
与母体苯丙酮尿症不同(见Phenylalanine Hydroxylase Deficiency),母体高胱氨酸尿症似乎没有重大的致畸潜力而需要进一步的咨询,或者就胎儿而言,不需更严格的管理[Levy et al 2002, Vilaseca et al 2004]。尽管如此,在怀孕期间应继续使用吡哆醇或蛋氨酸限制饮食或两者同时进行治疗 [Morris et al 2017]。甜菜碱也可以继续存在并且似乎不是致畸性的[Yap et al 2001b, Levy et al 2002, Vilaseca et al 2004, Pierre et al 2006]。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分介绍了遗传风险评估以及家族史和遗传测试的使用,以阐明家庭成员的遗传状况。 本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代与遗传学专业人员的咨询。 —编者。
遗传方式
由胱硫醚β合成酶缺乏症引起的高半胱氨酸尿症(经典高半胱氨酸尿症)以常染色体隐性遗传的方式遗传。
家庭成员的风险
先证者的父母
- 受累的患者的未受影响的父母是肯定杂合子(即,至少一种CBS 致病性变异的携带者)。
- 杂合子(携带者)无症状,没有发展成高半胱氨酸尿症的风险。
- 由于父母有可能(尽管不太可能)患有典型的高半胱氨酸尿症但仍无症状,因此有必要获取详细的病史并在父母双方中进行检查以及血浆高半胱氨酸和氨基酸分析。如果母亲考虑将来怀孕,这将变得更加必要,因为受累的妇女在怀孕期间发生血栓栓塞事件的风险增加。
先证者的后代。由于经典的高半胱氨酸尿症是可以治疗的,受累的人可以得到有效治疗,因此他们在身体和智力上都可能是正常的并且可以生育。
携带者(杂合子)检测
分子遗传学测试。对有风险的家庭成员进行携带者测试需要事先确定该家庭中的两种CBS致病变异。
生化检测。单一的生化测试无法区分CBS杂合子与对照。
- CBS缺乏症的杂合子具有正常的空腹血浆总同型半胱氨酸浓度。
- 蛋氨酸负荷后血浆总高半胱氨酸浓度响应(100 mg蛋氨酸/ kg [671 µmol / kg])在73%吡哆醇无反应性高半胱氨酸尿症的杂合子和33% 吡哆醇反应性高半胱氨酸尿症的杂合子中异常[Guttormsen et al 2001]。
注意:进行蛋氨酸负荷试验时应谨慎,因为已经报告了不良反应[Cottington et al 2002, Krupková-Meixnerová et al 2002]。
相关的遗传咨询问题
有关评估以早期诊断和治疗为目的的高风险亲戚的信息,请参阅《管理,Evaluation of Relatives at Risk 》。
家庭计划
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。因为将来测试方法和我们对基因,等位基因变异和疾病的理解可能会有所改善,所以应考虑受累的患者DNA银行。
产前检查和植入前遗传学诊断
一旦在一个受累的家庭成员中发现了CBS致病变异,就可以对风险增加的妊娠进行孕前检测,并进行植入前遗传诊断。
如果CBS致病变体未知,则在理论上可以测量在培养的羊水中测定的CBS酶活性。但是,确定实验室进行这项测试可能具有挑战性。目前在美国尚无此类测试。
在医疗专业人员之间以及在家庭内部,关于使用产前检查的观点可能存在差异,特别是如果考虑将检查用于终止妊娠而不是早期诊断的时候。尽管大多数中心将有关产前检查的决定视为父母的选择,但对这些问题的讨论是适当的。
资源
GeneReviews工作人员选择了以下特定疾病和/或雨伞支持组织和/或注册表,以保护患有该疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击here.
- Homocystinuria: Genetic Fact Sheet for ParentsScreening, Technology And Research in Genetics (STAR-G)
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- National Organization for Rare Disorders (NORD)
- Association for Neuro-Metabolic Disorders (ANMD)5223 Brookfield LaneSylvania OH 43560-1809Phone: 419-885-1809; 419-885-1497Email: volk4olks@aol.com
- Children Living with Inherited Metabolic Diseases (CLIMB)United KingdomPhone: 0800-652-3181Email: info.svcs@climb.org.uk
分子遗传
分子遗传学和OMIM表中的信息可能与GeneReview中其他地方的信息不同:表可能包含最新信息。 -编者
Table A.
胱硫醚β合成酶缺乏症引起的高半胱氨酸尿症:基因和数据库。
| 基因 | 染色体定位 | 蛋白 | 数据库 | HGMD |
|---|---|---|---|---|
| CBS | 21q22 | Cystathionine beta-synthase | CBS database | CBS |
胱硫醚β-合成酶缺乏症引起的高半胱氨酸尿症的OMIM条目 (View All in OMIM)
基因结构 。CBS有23个外显子,长度为25-30 kb,并根据组织的不同表达为剪接的 mRNA 异型体,大小在2.5到3.7 kb之间。有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
致病变异。在目前确定的164种致病变异中,CBS缺乏的个体中有67%是错义变异,其中绝大多数是私有的。在其他致病变异中,只有五个是nonsense变异,其余是各种缺失,插入和剪接变异(有关当前致病变异的数据库,请参见CBS Mutation Database)[Urreizti et al 2003, Linnebank et al 2004, Miles & Kraus 2004, Moat et al 2004]。
在外显子8中发现了两个最常见的CBS致病变异 p.Ile278Thr和p.Gly307Ser。
- p.Ile278Thr是泛种族;总体而言,它占所有致病变异的近25%,包括29%的英国变异和的18%美国变异 [Moat et al 2004]。在某些国家/地区(例如丹麦),它可能是导致致病性变异的主要原因[Skovby et al 2010]。
- p.Gly307Ser是爱尔兰高半胱氨酸尿症的主要原因(71%的致病变异)。在“凯尔特人”血统的美国和澳大利亚受累的中也经常发现它,包括爱尔兰,苏格兰,英国,法国和葡萄牙血统的家庭。在英国和美国,它占致病变异的21%,在美国占8%[Moat et al 2004]。
- p.Arg336Cys存在于卡塔尔人口的93%受累的人中[Gan-Schreier et al 2010]。
Table 4
GeneReview讨论的CBS致病变异
| DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
|---|---|---|
| c.833T>C | p.Ile278Thr | NM_000071 NP_000062 |
| c.919G>A | p.Gly307Ser | |
| c.1006C>T | p.Arg336Cys |
有关变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会(varnomen
- .hgvs.org )的标准命名约定。有关命名法的说明,请参见Quick Reference。
参考文献
发布的准则/共识声明
- Morris AAM, Kožich V, Santra S, Andria G, Ben-Omran TIM, Chakrapani AB, Crushell E, Henderson MJ, Hochuli M, Huemer M, Janssen MCH, Maillot F, Mayne PD, McNulty J, Morrison TM, Ogier H, O’Sullivan S, Pavlíková M, Talvares de Almeida I, Terry A, Yap S, Blom HJ, Chapman KA. Guidelines for the diagnosis and management of cystathionine β-synthase deficiency. Available online. 2017. Accessed 5-17-17.
Literature Cited
- Alcaide P, Krijt J, Ruiz-Sala P, Ješina P, Ugarte M, Kožich V, Merinero B. Enzymatic diagnosis of homocystinuria by determination of cystathionine-ß-synthase activity in plasma using LC-MS/MS. Clin Chim Acta. 2015;438:261 - 5. [PubMed: 25218699]
- Barić I, Staufner C, Augoustides-Savvopoulou P, Chien YH, Dobbelaere D, Grünert S, Opladen T, Petković Ramadža D, Rakić B, Wedell A, Blom H. Consensus recommendations for the diagnosis, treatment and follow-up of inherited methylation disorders. J Inherit Metab Dis. 2017;40:5 - 20. [PMC free article: PMC5203850] [PubMed: 27671891]
- Braverman NE, Mudd SH, Barker PB, Pomper MG. Characteristic MR imaging changes in severe hypermethioninemic states. AJNR Am J Neuroradiol. 2005;26:2705 - 6. [PubMed: 16286426]
- Bublil EM, Majtan T, Park I, Carrillo RS, Hůlková H, Krijt J, Kožich V, Kraus JP. Enzyme replacement with PEGylated cystathionine β-synthase ameliorates homocystinuria in murine model. J Clin Invest. 2016;126:2372 - 84. [PMC free article: PMC4887166] [PubMed: 27183385]
- Cottington EM, LaMantia C, Stabler SP, Allen RH, Tangerman A, Wagner C, Zeisel SH, Mudd SH. Adverse event associated with methionine loading test: a case report. Arterioscler Thromb Vasc Biol. 2002;22:1046 - 50. [PubMed: 12067919]
- Cozar M, Urreizti R, Vilarinho L, Grosso C, Dodelson de Kremer R, Asteggiano CG, Dalmau J, García AM, Vilaseca MA, Grinberg D, Balcells S. Identification and functional analyses of CBS alleles in Spanish and Argentinian homocystinuric patients. Hum Mutat. 2011;32:835 - 42. [PubMed: 21520339]
- Devlin AM, Hajipour L, Gholkar A, Fernandes H, Ramesh V, Morris AA. Cerebral edema associated with betaine treatment in classical homocystinuria. J Pediatr. 2004;144:545 - 8. [PubMed: 15069409]
- Gan-Schreier H, Kebbewar M, Fang-Hoffmann J, Wilrich J, Abdoh G, Ben-Omran T, Shahbek N, Bener A, Al Khal AL, Lindner M, Zschocke J, Hoffmann GF. Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J Pediatr. 2010;156:427 - 32. [PubMed: 19914636]
- Gaustadnes M, Wilcken B, Oliveriusova J, McGill J, Fletcher J, Kraus JP, Wilcken DE. The molecular basis of cystathionine β-synthase deficiency in Australian patients: genotype-phenotype correlations and response to treatment. Hum Mutat. 2002;20:117 - 26. [PubMed: 12124992]
- Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21:563 - 71. [PubMed: 11745044]
- Guttormsen AB, Ueland PM, Kruger WD, Kim CE, Ose L, Folling I, Refsum H. Disposition of homocysteine in subjects heterozygous for homocystinuria due to cystathionine β-synthase deficiency: relationship between genotype and phenotype. Am J Med Genet. 2001;100:204 - 13. [PubMed: 11343305]
- Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF, Burlina A, Cerone R, Couce ML, Garcia-Cazorla A, la Marca G, Pasquini E, Vilarinho L, Weisfeld-Adams JD, Kožich V, Blom H, Baumgartner MR, Dionisi-Vici C. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40:21 - 48. [PMC free article: PMC5203859] [PubMed: 27905001]
- Hidalgo Mazzei D, Martín Rodriguez S, Pérez Moltó H, Ruíz Izquierdo J, Baeza I. A forgotten lethal psychosis: a case report. Eur Child Adolesc Psychiatry. 2014;23:235 - 8. [PubMed: 23812867]
- Karaca M, Hismi B, Ozqul RK, Karaca S, Yilmaz DY, Coskun T, Sivri HS, Tokatli A, Dursun A. High prevalence of cerebral venous sinus thombosis (CVST) as presentation of cystathionine β-synthase deficiency in childhood: molecular and clinical findings of Turkish probands. Gene. 2014;534:197 - 203. [PubMed: 24211323]
- Kelly PJ, Furie KL, Kistler JP, Barron M, Picard EH, Mandell R, Shih VE. Stroke in young patients with hyperhomocysteinemia due to cystathionine β-synthase deficiency. Neurology. 2003;60:275 - 9. [PubMed: 12552044]
- Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M. Cystathionine β-synthase mutations in homocystinuria. Hum Mutat. 1999;13:362 - 75. [PubMed: 10338090]
- Krijt J, Kopecká J, Hnízda A, Moat S, Kluijtmans LAJ, Mayne P, Kožich V. Determination of cystathionine beta-synthase activity in human plasmas by LC-MS/MS: potential use in diagnosis of CBS deficiency. J Inherit Metab Dis. 2011;34:49 - 55. [PMC free article: PMC3026677] [PubMed: 20821054]
- Kruger WD, Wang L, Jhee KH, Singh RH, Elsas LJ 2nd. Cystathionine β-synthase deficiency in Georgia (USA): correlation of clinical and biochemical phenotype with genotype. Hum Mutat. 2003;22:434 - 41. [PubMed: 14635102]
- Krupková-Meixnerová L, Veselá K, Vitová A, Janosíková B, Andel M, Kozich V. Methionine-loading test: evaluation of adverse effects and safety in an epidemiological study. Clin Nutr. 2002;21:151 - 6. [PubMed: 12056788]
- Lawson-Yuen A, Levy HL. A betaine treatment for the homocystinurias. In: Theone J, ed. Small Molecule Therapy for Genetic Disease. New York, NY: Cambridge University Press; 2010:173-81.
- Levy HL, Vargas JE, Waisbren SE, Kurczynski TW, Roeder ER, Schwartz RS, Rosengren S, Prasad C, Greenberg CR, Gilfix BM, MacGregor D, Shih VE, Bao L, Kraus JP. Reproductive fitness in maternal homocystinuria due to cystathionine β-synthase deficiency. J Inherit Metab Dis. 2002;25:299 - 314. [PubMed: 12227460]
- Linnebank M, Homberger A, Junker R, Nowak-Goettl U, Harms E, Koch HG. High prevalence of the I278T mutation of the human cystathionine β-synthase detected by a novel screening application. Thromb Haemost. 2001;85:986 - 8. [PubMed: 11434706]
- Linnebank M, Janosik M, Kozich V, Pronicka E, Kubalska J, Sokolova J, Linnebank A, Schmidt E, Leyendecker C, Klockgether T, Kraus JP, Koch HG. The cystathionine β-synthase (CBS) mutation c.1224-2A>C in Central Europe: Vitamin B6 nonresponsiveness and a common ancestral haplotype. Hum Mutat. 2004;24:352 - 3. [PubMed: 15365998]
- MacDonald A, Depondt E, Evans S, Daly A, Hendriksz C, Chakrapani A A, Saudubray JM. Breast feeding in IMD. J Inherit Metab Dis. 2006;29:299 - 303. [PubMed: 16763891]
- Maclean KN, Jiang H, Greiner LS, Allen RH, Stabler SP. Long-term betaine therapy in a murine model of cystathionine β-synthase deficient homocystinuria: decreased efficacy over time reveals a significant threshold effect between elevated homocysteine and thrombotic risk. Mol Genet Metab. 2012;105:395 - 403. [PubMed: 22192524]
- Magner M, Krupková L, Honzík T, Zeman J, Hyánek J, Kožich V. Vascular presentation of cystathionine β-synthase deficiency in adulthood. J Inherit Metab Dis. 2011;34:33 - 7. [PMC free article: PMC3026685] [PubMed: 20567906]
- Mendes MIS, Colaҫo HG, Smith DEC, Ramos RJJF, Pop A, Van Dooren SJM, Tavares De Almeida I, Kluijtmans LAJ, Janssen MCH, Rivera I, Salomons GS, Leandro P, Blom HJ. Reduced response of cystathionine beta-synthase (CBS) to s-adenosylmethionine (SAM): identification and functional analysis of CBS gene mutations in homocystinuria patients. J inherit Metab Dis. 2014;37:245 - 54. [PubMed: 23974653]
- Miles EW, Kraus JP. Cystathionine β-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem. 2004;279:29871 - 4. [PubMed: 15087459]
- Moat SJ, Bao L, Fowler B, Bonham JR, Walter JH, Kraus JP. The molecular basis of cystathionine β-synthase (CBS) deficiency in UK and US patients with homocystinuria. Hum Mutat. 2004;23:206. [PubMed: 14722927]
- Morris AAM, Kožich V, Santra S, Andria G, Ben-Omran TIM, Chakrapani AB, Crushell E, Henderson MJ, Hochuli M, Huemer M, Janssen MCH, Maillot F, Mayne PD, McNulty J, Morrison TM, Ogier H, O’Sullivan S, Pavlíková M, Talvares de Almeida I, Terry A, Yap S, Blom HJ, Chapman KA. Guidelines for the diagnosis and management of cystathionine β-synthase deficiency. J Inherit Metab Dis. 2017;40:49 - 74. [PMC free article: PMC5203861] [PubMed: 27778219]
- Mudd SH. Hypermethioninemias of genetic and non-genetic origin. A review. Am J Med Genet C Semin Med Genet. 2011;157C:3 - 32. [PubMed: 21308989]
- Mudd SH, Finkelstein JD, Refsum H, Ueland PM, Malinow MR, Lentz SR, Jacobsen DW, Brattstrom L, Wilcken B, Wilcken DE, Blom HJ, Stabler SP, Allen RH, Selhub J, Rosenberg IH. Homocysteine and its disulfide derivatives: a suggested consensus terminology. Arterioscler Thromb Vasc Biol. 2000;20:1704 - 6. [PubMed: 10894806]
- Mudd SH, Glenn B, Wagner C, James SJ, Schulze A, Stabler S, Zeisel S, Fumic K, Baric I. S-Adenosylhomocysteine (AdoHcy) hydrolase deficiency in a Croatian boy. J Inherit Metab Dis. 2003;26 Suppl 1:15A.
- Mudd SH, Levy HL, Kraus JP. Disorders of transulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw Hill; 2001:2016-40.
- Mulvihill A, Yap S, O'Keefe M, Howard PM, Naughten ER. Ocular findings among patients with late-diagnosed or poorly controlled homocystinuria compared with a screened, well-controlled population. J AAPOS. 2001;5:311 - 5. [PubMed: 11641642]
- Naughten ER, Yap S, Mayne PD. Newborn screening for homocystinuria: Irish and world experience. Eur J Pediatr. 1998;157 Suppl 2:S84 - 7. [PubMed: 9587032]
- Neely DE, Plager DA. Management of ectopia lentis in children. Ophthalmol Clin North Am. 2001;14:493 - 9. [PubMed: 11705149]
- Novy J, Ballhausen D, Bonafe L, Cairoli A, Angelillo-Scherrer A, Bachmann C, Michel P. Recurrent postpartum cerebral sinus vein thrombosis as a presentation of cystathionine-β-synthase deficiency. Thromb Haemost. 2010;103:871 - 3. [PubMed: 20174770]
- Pierre G, Gissen P, Chakrapani A, McDonald A, Preece M, Wright J. Successful treatment of pyridoxine-unresponsive homocystinuria with betaine in pregnancy. J Inherit Metab Dis. 2006;29:688 - 9. [PubMed: 16972179]
- Refsum H, Fredriksen A, Meyer K, Ueland PM, Kase BF. Birth prevalence of homocystinuria. J Pediatr. 2004;144:830 - 2. [PubMed: 15192637]
- Saboul C, Darteyre S, Ged C, Fichtner C, Gay C, Stephan JL. Inaugural cerebral sinovenous thrombosis revealing homocystinuria in a 2-year-old boy. J Child Neurol. 2015;30:107 - 12. [PubMed: 24598125]
- Sarov M, Not A, de Baulny HO, Masnou P, Vahedi K, Bousser MG, Denier C. A case of homocystinuria due to CBS gene mutations revealed by cerebral venous thrombosis. J Neurol Sci. 2014;336:257 - 9. [PubMed: 24169224]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274 - 84. [PMC free article: PMC311435] [PubMed: 14722619]
- Shoji Y, Takahashi T, Sato W, Shoji Y, Takada G. Acute life-threatening event with rhabdomyolysis after starting on high-dose pyridoxine therapy in an infant with homocystinuria. J Inherit Metab Dis. 1998;21:439 - 40. [PubMed: 9700609]
- Singh RH, Kruger WD, Wang L, Pasquali M, Elsas LJ 2nd. Cystathionine β-synthase deficiency: effects of betaine supplementation after methionine restriction in B6-nonresponsive homocystinuria. Genet Med. 2004;6:90 - 5. [PubMed: 15017331]
- Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuria due to cystathionine β-synthase deficiency. Mol Genet Metab. 2010;99:1 - 3. [PMC free article: PMC2795104] [PubMed: 19819175]
- Smith DE, Mendes MI, Kluijtmans LA, Janssen MC, Smulders YM, Blom HJ. A liquid chromatography mass spectrometry method for the measurement of cystathionine beta-synthase activity in cell extracts. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;911:186 - 191. [PubMed: 23217323]
- Smith KL, Bradley L, Levy HL, Korson MS. Inadequate laboratory technique for amino acid analysis resulting in missed diagnoses of homocystinuria. Clin Chem. 1998;44:897 - 8. [PubMed: 9554515]
- Tada H, Takanashi J, Barkovich AJ, Yamamoto S, Kohno Y. Reversible white matter lesion in methionine adenosyltransferase I/III deficiency. AJNR Am J Neuroradiol. 2004;25:1843 - 5. [PubMed: 15569761]
- Tortorelli S, Turgeon CT, Lim JS, Baumgart S, Day-Salvatore DL, Abdenur J, Bernstein JA, Lorey F, Lichter-Konecki U, Oglesbee D, Raymond K, Matern D, Schimmenti L, Rinaldo P, Gavrilov DK. Two-tier approach to the newborn screening of methylenetetrahydrofolate reductase deficiency and other remethylation disorders with tandem mass spectrometry. J Pediatr. 2010;157:271 - 5. [PubMed: 20394947]
- Turgeon CT, Magera MJ, Cuthbert CD, Loken PR, Gavilov DK, Tortorelli S, Raymond KM, Oglesbee D, Rinaldo P, Matern D. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem. 2010;56:1686 - 95. [PubMed: 20807894]
- Urreizti R, Balcells S, Rodes M, Vilarinho L, Baldellou A, Couce ML, Munoz C, Campistol J, Pinto X, Vilaseca MA, Grinberg D. Spectrum of CBS mutations in 16 homocystinuric patients from the Iberian Peninsula: high prevalence of T191M and absence of I278T or G307S. Hum Mutat. 2003;22:103. [PubMed: 12815602]
- Vilaseca MA, Cuartero ML, Martinez de Salinas M, Lambruschini N, Pinto X, Urreizti R, Balcells S, Grinberg D. Two successful pregnancies in pyridoxine-nonresponsive homocystinuria. J Inherit Metab Dis. 2004;27:775 - 7. [PubMed: 15617186]
- Watkins D, Rosenblatt DS. Inherited disorders of folate and cobalamin transport and metabolism. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 155. New York, NY: McGraw-Hill. Available online. 2014. Accessed 5-15-17.
- Weber DR, Coughlin C, Brodsky JL, Lindstrom K, Ficicioglu C, Kaplan P, Freehauf CL, Levine MA. Low bone mineral density is a common finding in patients with homocystinuria. Mol Genet Metab. 2016;117:351 - 4. [PMC free article: PMC4788514] [PubMed: 26689745]
- Yaghmai R, Kashani AH, Geraghty MT, Okoh J, Pomper M, Tangerman A, Wagner C, Stabler SP, Allen RH, Mudd SH, Braverman N. Progressive cerebral edema associated with high methionine levels and betaine therapy in a patient with cystathionine β-synthase (CBS) deficiency. Am J Med Genet. 2002;108:57 - 63. [PubMed: 11857551]
- Yap S. Classical homocystinuria: vascular risk and its prevention. J Inherit Metab Dis. 2003;26:259 - 65. [PubMed: 12889665]
- Yap S, Boers GH, Wilcken B, Wilcken DE, Brenton DP, Lee PJ, Walter JH, Howard PM, Naughten ER. Vascular outcome in patients with homocystinuria due to cystathionine β-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol. 2001a;21:2080 - 5. [PubMed: 11742888]
- Yap S, Naughten E. Homocystinuria due to cystathionine β-synthase deficiency in Ireland: 25 years' experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis. 1998;21:738 - 47. [PubMed: 9819703]
- Yap S, Rushe H, Howard PM, Naughten ER. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine β-synthase deficiency. J Inherit Metab Dis. 2001b;24:437 - 47. [PubMed: 11596648]
Suggested Reading
- Sahai I, Marsden D. Newborn screening. Crit Rev Clin Lab Sci. 2009;46:55 - 82. [PubMed: 19255915]
本章节注解
作者备注
Authors’ website: New England Consortium of Metabolic Programs