
概要
临床特征
Floating-Harbor综合征(FHS) 表现典型的颅面特征:出生低体重、头围正常、身材矮小;在6至12岁内出现骨龄延 迟;骨骼异常(短指、杵状指、指弯曲、短拇指、关节突出、锁骨畸形);严重的语言表达功能损害;鼻音过重、声调过高;轻到中度的智力残疾。儿童期性格、行 为异常,成年期改善。其它特征还包括:远视、斜视、传导性耳聋、癫痫、胃食管反流、肾脏异常(肾盂积水、肾盂扩张、肾囊肿、肾发育不良)及生殖异常(尿道 下裂、隐睾)。
疾病管理
对 症治疗:早期干预计划、特殊教育以及针对发育障碍的专门训练;手语交流康复或其它替代性方法;行为专家或心理专家进行行为矫治,必要时给予药物。 内分泌医师给予生长激素治疗,然而,在FHS中相关数据有限。屈光不正及斜视、听力损害、癫痫、胃食管反流及肾、生殖异常的标准治疗。
监测:密切监测生长发育,尤其在一周岁以内。每年一次眼科、听力筛查、血压监测以及肾功能评估。肾囊肿在青少年和成年期可以通过彩超检查评估。
诊断
FHS的诊断基于典型的临床表现(尤其是特征性面容)及 SRCAP基因致病性的变异 杂合的 致病性变异.的确认。
头面部表现参见图一 ( Figure 1)
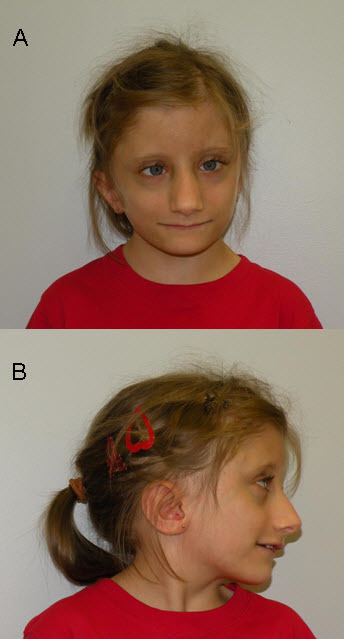
- 三角脸
- 眼窝深陷
- 短人中
- 宽嘴伴朱砂红上唇
- 长鼻、窄鼻梁、宽鼻根伴悬鼻柱
- 低耳位
其它特征
- 显著骨龄延迟(-2 SD或更多),六至12岁之间可见正常。
- 骨骼异常:指过短、宽指尖、表现为杵状指、指弯曲、短拇指、关节突出、锁骨异常,参见图2( Figure 2)
- 成人身材矮小:140-155 cm (see Figure 3)
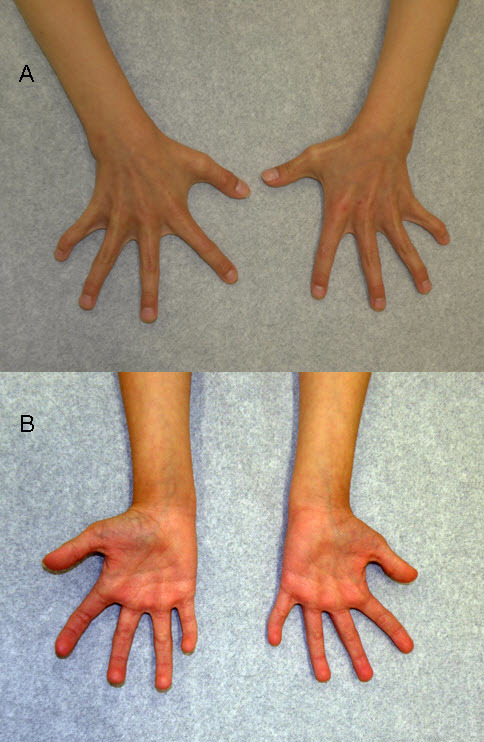
图2.
图1的手掌正(A)反(B)面。表现为指弯曲、宽指尖和关节突出。

图3
图1患者正面观。成比例的身材矮小,小于3个百分位。
表达和语言
- 构音障碍、词汇运用障碍伴发音失准。
- 重鼻音
- 声调高
- 严重的语言接受与表达功能受损
智力残疾所有患者都有不同程度的智力残疾或学习障碍,从正常临界值到中度智力残疾。
临床特征
临床描述
在 Hood et al [2012]等报道FHS分子特征之前,已有一些FHS病例报道,本综述仅涵盖19名经分子诊断的患者(即有检出SRCAP基因致病性杂合变异) [ Hood et al 2012, Le Goff et al 2013]。包括6名女性和13名男性,年龄范围在11个月到32岁。
FHS常在儿童早期被察觉,由于典型的面部特征(图1) ( Figure 1)。婴幼儿常被评估为生长落后或发育(语言发育显著落后)延迟。
智力尽管粗大动作和精细动作都在正常范围,受累者 受累的具有典型的轻到中度智力残疾。
语言能力出现严重障碍。大部分交流方面能力都将受累,语言表达持续受累,部分患者失去语言功能。
大部分受累的儿童接受主流教育同时辅以个体化教育计划。
技能退化不典型。
行为Many 许多FHS患者伴有脾气、性格的差异。婴儿期可见情绪化、学龄期可有多动症(deficit-hyperactivity disorder ,ADHD)。
可有侵略性和暴力。
可有强迫症Obsessive compulsive disorder (OCD) 和焦虑。
行为问题在成人期改善。
生长身材矮小是FHS主要表现。
大部分FHS患者存在低体重( -3 SD to 0 SD),头围正常(-2 SD to 0 SD)。
出生后第一年线性体重增加困难。
成人平均身高为140-155 cm.
青春期 青春期提前已被报道,不同性别的发生率暂无数据。
眼3/19 名患者已被报道有远视,1/13伴有斜视。1名患者眼前房异常。
听力3/19 名患者有传导性耳聋,1/19名患者有耳蜗异常。
神经病学癫痫见于3/19 名患者。
胃肠道反流严重,部分患者需胃管喂食,便秘和结肠狭窄已被报道。1/19名患者有乳糜泻,2名麸质不耐受。
泌尿生殖器男性可有泌尿生殖器异常包括尿道下裂、隐睾、囊肿、精索静脉曲张及后尿道瓣膜症。 肾盂积水/肾盂扩张、肾囊肿、肾发育不良也见报道。1名患者存在多囊肾和晚期肾病。
外形体态矮壮伴宽胸短颈。
其它特征包括手部异常,如指弯曲、指过短、短拇指、及宽指尖,表现为杵状指。 ( Figure 2).
锁骨异常包括假关节和锁骨发育不良、短掌骨、11对肋骨、脊柱后凸、髋关节发育不全及脱位。
牙齿一些FHS患者伴有牙齿问题(例如龋齿、小牙、乳牙缺如)以及牙齿畸形(如上颌后移、反颌)。
心脏心脏畸形不是FHS主要表现,19名患者中有1名伴有主动脉狭窄,1名左上腔静脉永存、1名房间隔缺损。
发病率
FHS发病率仍未知,已有19名携带SRCAP基因变异的患者被报道 [ Hood et al 2012, Le Goff et al 2013]。
大部分被报道的FHS患者为欧洲人,非白种人患FHS的出现率是否更低或已观察到的区别还有其它因素导致仍是未知。
基因相关疾病
本综述仅讨论已知致病性的SRCAP基因变异。
鉴别诊断
FHS以儿童早期出现典型的面容、骨龄延迟、言语障碍为特征,随着年龄增长,症状表现逐渐不明显。怀疑FHS时还应考虑以下病症。
宽大成角拇指和底鼻柱见于 Rubinstein-Taybi syndrome(RSTS).RSTS 以典型的面容特征包括下倾的眼裂、低鼻柱、高上颚、笑脸、和鹰爪型牙阜。RSTS的诊断可以通过验证仅已知的相关基因,CREBBP或EP300基因杂合致病性变异确认。遗传模式为常染色体显性 常染色体显性遗传,多数患者携带新发突变 新生的。
Russell-Silver syndrome(RSS)可见宫内发育迟缓伴随出生后生长缓慢。受累的患者呈成比例的身材矮小、正常头围、第五指弯曲、三角脸、宽前额、窄下巴以及营养不良导致的四肢长度不对成。生长速度在儿童期正常,男性成年平均身高151.2cm,女性成年平均身高139.9cm。RSS患者伴有显著的发育落后(运动和认知)及学习障碍风险,RSS具有遗传异质性,大多数患者表现一种表型而不是特定的障碍。
3-M syndrome以严重的产前或出生后的发育迟滞(低于5-6 SD,最终身高相当于120-130cm ),典型面容,大头、三角脸、中脸发育不全、浓眉、肉质鼻尖、长 人中、宽嘴厚唇、下巴突出、智力正常。其它特征性表现还包括短宽颈、斜方肌突出、胸骨畸形、短胸腔、方肩、翼状肩胛骨、脊柱前凸、第五指短小、足跟突出以 及关节松弛。骨龄轻度延迟。男性3M综合征患者可有生殖发育不良,可见尿道下裂。影像学检查可与FHS综合征相鉴别。已知CUL7、OBSL1、CCDC8基因双等位基因变异可导致3M综合征。遗传模式为常染色体隐性 常染色体隐性遗传。
疾病管理
初步诊断后的评估
为建立FHS的患病程度和患者了解患者需求,建议给予以下评估:
- 多学科的发育评估包括粗大、精细运动能力、语言表达能力、认知能力及特殊性的能表现语言延迟和异常的职业能力评估。
- 生长的测量以及生长参数的描绘。
注意:综合征图表目前已不适用于携带SRCAP致病性变异的儿童。 - 眼科检查
- 听力检查(评估细节参见 Deafness and Hereditary Hearing Loss Overview)
- 肾脏超声检查和血压测量。
- 男性隐睾的检查。
- 髋关节、锁骨发育不良整形外科的评估。
- 牙科评价。
- 临床遗传咨询。
对症治疗
治疗措施包括:
- 早期干预计划、特殊教育、针对发育残疾进行职业训练。
- 手语学习或替代方法进行沟通交流康复。
- 行为管理包括向行为专家或心理医师及根据需求选择药物治疗。
- 推荐家人给支持组织或其它相关资源性机构。
- 出现以下情况需针对性治疗:
- 屈光不正或斜视。
- 听力丧失
- 癫痫
- 肾病
- 隐睾
- 骨科并发症
- 牙科问题
- 推荐给内分泌专家进行生长激素治疗。生长激素治疗已被报道在3名儿童FHS患者具有中等效应,但仍需谨慎用药。
- 需注意乳糜泻的临床表现。
监测
检测措施如下:
- 密切监测生长发育,尤其是出生后的第一年。
- 每年一次:
- 眼科检查
- 听力筛查,如有反复发作的中耳炎需加大监测频率。
- 血压检测与肾功能评估。
- 肾脏异常的标准监测。
- 骨龄的监测以评估青春期提前尤其是生长激素的使用。
- 肾脏彩超监测肾囊肿
家属患病风险的评估
参见遗传咨询部分 Genetic Counseling 。
研发阶段的治疗措施
查询 ClinicalTrials.gov 以获取更多临床研究信息。注:针对该病症可能暂无临床试验。
遗传咨询
遗传咨询是一个给患者及家属提供关于遗传性疾病本质、遗传特性以及影响并帮助他们做出知情的医疗决定的过程。下列段落描述遗传风险的评估以及根据家族史和 基因检测判断家族成员遗传状态。本段落描述不适用于解决患者实际面对的个人、文化或伦理问题,也不能代替专业的遗传咨询。—ED.
遗传模式
Floating-Harbor syndrome(FHS) 为常染色体显性遗传模式,大多数患者携带一个新发致病性的变异。
家庭成员患病风险
先证者的父母
- FHS先证者通常由新发突变导致,因此多数受累者表现为家系中单发 单发的。
- 目前有一例母——婴遗传的FHS被报道 [ Hood et al 2012]。
- 建议先证者父母对FHS进行临床评估以及 SRCAP基因致病性变异进行检测,以确认父母其中之一是否为受累者而因轻度表型在此前被漏诊。
- 研究显示一个明显的父亲年龄效应,在13名携带 SRCAP基因致病性变异的患者中,患者父亲平均年龄为36.9岁(从29至44岁)。
先证者的同胞
- 同胞患病风险取决于父母遗传状态。
- 极少数病例父母受累,先证者同胞患病风险为50%。
- 当父母无临床受累时,同胞患病风险低。
- 若致病性变异在先证者白细胞DNA中检出,其同胞的患病风险低,但仍高于一般人群,由于理论上生殖系嵌合 胚系嵌合的可能性。
先证者子代FHS患者的每一个子代有50%概率遗传致病性的变异。
其他家系成员其他家系成员的风险根据先证者父母遗传状态决定,若父母受累,父母的家系成员可能处于患病风险中。
相关的遗传咨询问题
携带明显的新发致病性变异的家系需考虑的问题。当先证者父母没有临床表现以及未携带致病性变异时需考虑新发突变的情况,然而,可能的非医学的解释包括替代性的父母 非生物学父亲(例如辅助生殖)或未公开的领养情况也需考虑到。
家庭计划
- 决定判断遗传风险及施行产前诊断的最佳时机是怀孕前。
- 年轻的成人应进行遗传咨询(包括对其子代的风险预估及生育的选择)。
基因银行DNA的保存(提取自血液中白细胞的最常见)可以备将来所用,考虑到检测手段以及我们对基因、等位基因变异及疾病的理解认识在未来将进一步提升,受累的患者可以保存基因。
产前诊断和胚胎植入前遗传学诊断
一旦 SRCAP基因致病性变异在一个家系中检出,该家系成员妊娠增加FHS风险,产前诊断和胚胎植入前遗传学诊断是可能的选择。
资源
工作人员已经筛选了以下专科疾病和患者帮扶组织,注册登记能使患者及其家庭获益。GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处 here.
- Human Growth Foundation (HGF)997 Glen Cove AvenueSuite 5Glen Head NY 11545Phone:800-451-6434 (toll-free)Fax:516-671-4055Email:hgf1@hgfound.org
- MAGIC Foundation6645 West North AvenueOak Park IL 60302Phone:800-362-4423 (Toll-free Parent Help Line); 708-383-0808Fax:708-383-0899Email:ContactUs@magicfoundation.org
分子遗传学
分子遗传学检测的信息和OMIM相关列表可能同其它GeneReview信息会有不同。(可能有更新原因)—ED.
表A
Floating-Harbor Syndrome:基因和数据库
| 基因 | 染色体位置 | 蛋白 | 位置相关数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| SRCAP | 16p11 .2 | Helicase SRCAP | SRCAP @ LOVD | SRCAP | SRCAP |
表B
OMIM 收录 Floating-Harbor Syndrome信息参见( View All in OMIM)
基因结构SRCAP基因编码SRCAP多蛋白染色体重构核心催化组件 。 (NM_006662.2).SRCAP基因大小为154 kb,包含34个外显子,外显子1和2非编码,基因和蛋白详细信息参见表A。
致病性等位基因变异Pathogenic allelic variants.13名FHS患者被报道,致病性变异都在34号外显子。所有的致病性变异(框移或无义突变)都导致蛋白的截短,并簇集在密码子2407至2517之间。常见的变异见表2 Table 2[ Hood et al 2012]。
表2
SRCAP基因致病性的变异
| DNA核酸改变 | 蛋白氨基酸改变 | 参考序列 |
|---|---|---|
| c.7303C>T | p.Arg2435Ter | NM_006662 .2 NP_006653 .2 |
| c.7330C>T | p.Arg2444Ter |
变异的分级:列表中变异由作者提供,GeneReviews 工作人员未对变异进行独立分级。
命名: GeneReviews 遵循人类基因组变异协会(Human Genome Variation Society (varnomen .hgvs.org))原则。命名的解释参见 Quick Reference 。
正常基因产物基因产物.SRCAP解旋酶是一个大小为400-kd核蛋白,包含3230个氨基酸,调控细胞间细胞转到及染色体重构。编码蛋白为ATP酶,为组蛋白变体H2A.Z并入核小体必需,为 CREB结合蛋白互作蛋白(CREBBP, 又名CBP),SRCAP为CBP介导转录的强力活化剂,还参与Notch信号转导和 类固醇受体介导转录。
异常的基因产物基因产物.FHS相关 SRCAP基因生殖系变异可能导致655末端片段反式激活的缺失。该致病性的变异可能是显性抑制 显性负效的致病机制。
参考文献
文献引用
- Hood RL, Lines MA, Nikkel SM, Schwartzentruber J, Beaulieu C, Nowaczyk MJ, Allanson J, Kim CA, Wieczorek D, Moilanen JS, Lacombe D, Gillessen-Kaesbach G, Whiteford ML, Quaio CR, Gomy I, Bertola DR, Albrecht B, Platzer K, McGillivray G, Zou R, McLeod DR, Chudley AE, Chodirker BN, Marcadier J., FORGE Canada Consortium. Majewski J, Bulman DE, White SM, Boycott KM. Mutations in SRCAP, encoding SNF2-related CREBBP activator protein, cause Floating-Harbor syndrome. Am J Hum Genet.2012; 90:308–13.[ PMC free article : PMC3276662] [ PubMed : 22265015]
- Le Goff C, Mahaut C, Bottani A, Doray B, Goldenberg A, Moncla A, Odent S, Nitschke P, Munnich A, Faivre L, Cormier-Daire V. Not all Floating-Harbor syndromecases are due to mutations in exon 34 of SRCAP. Hum Mutat.2013; 34:88–92.[ PubMed : 22965468]
推荐阅读
- Eissenberg JC, Wong M, Chirivia JC. Human SRCAP and Drosophila melanogaster DOM are homologs that function in the notch signaling pathway. Mol Cell Biol.2005; 25:6559–69.[ PMC free article : PMC1190335] [ PubMed : 16024792]
- Feingold M. Thirty-two year follow-up of the first patient reported with the Floating-Harbor syndrome. Am J Med Genet A.2006; 140:782–4.[ PubMed : 16523514]
- García RJ, Kant SG, Wit JM, Mericq V. Clinical and genetic characteristics and effects of long-term growth hormone therapy in a girl with Floating-Harbor syndrome. J Pediatr Endocrinol Metab.2012; 25:207–12.[ PubMed : 22570979]
- Johnston H, Kneer J, Chcackalaparampil I, Yaciuk P, Chrivia J. Identification of a novel SNF2/SWI2 protein family member, SRCAP, which interacts with CREB-binding protein. J Biol Chem.1999; 274:16370–6.[ PubMed : 10347196]
- Pelletier G, Feingold M. Case report 1. In: Bergsma D, ed. Syndrome Identification. White Plains, NY: National Foundation-March of Dimes; 1973:8-9.
- Reschen M, Kini U, Hood RL, Boycott KM, Hurst J, O'Callaghan CA. Floating-Harbor syndromeand polycystic kidneys associated with SRCAP mutation. Am J Med Genet A.2012; 158A:3196–200.[ PubMed : 23165645]
- Robinson PL, Shohat M, Winter RM, Conte WJ, Gordon-Nesbitt D, Feingold M, Laron Z, Rimoin DL. A unique association of short stature, dysmorphic features, and speech impairment ( Floating-Harbor syndrome). J Pediatr.1988; 113:703–6.[ PubMed : 3171794]
- Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP, Conaway RC, Conaway JW, Chrivia JC. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone varian H2Z.A in to nucleosomes. Biochemistry.2006; 45:5671–7.[ PubMed : 16634648]
- Wong MM, Cox LK, Chrivia JC. The chromatin remodeling protein SCRAP, is critical for deposition of the histone variant H2Z.A at promoters. J Biol Chem.2007; 282:26132–9.[ PubMed : 17617668]
Chapter Notes
Revision History
- 24 January 2013 (cd) Revision: prenatal testing available clinically
- 29 November 2012 (me) Review posted live
- 26 June 2012 (mjmn) Original submission