
概要
临床特征:Smith-Magenis综合征(SMS)的临床症状包括独特的体格特征(尤其是伴年龄增长的特殊面容),发育迟缓,认知障碍,及行为异常。新生儿有喂养困难,生长停滞,肌张力降低,腱反射减弱,白天嗜睡或需唤醒喂养,昏睡。大部分患者有轻到中度智力障碍。行为 表型, 包括显著的睡眠紊乱,刻板行为, 适应性不良,自我伤害行为等一般在18月龄或者更大的时候被识别,且会持续变化至成年期。感觉整合困难。儿童及成人有典型的注意力缺陷,多动,易冲动,适 应性不良包括易怒,注意寻求,叛逆,攻击行为,大小便困难,自伤行为(SIB)包括撞头,自咬,扯皮,将外物插入身体有孔部位(插孔癖),剔指甲/脚甲(剔甲癖)。刻板行为中,间歇性上身挤压或“自我拥抱”似乎与SMS高度相关,舔手指及翻页 行为少见,潜在不同步发育,尤其是智力水平与情感成熟之间的不同步,也可能导致了SMS患者的适应不良行为。
诊断/检测 . SMS的诊断基于临床表现及分子遗传学检测手段证实存在17p11.2区域的间隙 缺失或RAI1基因分子检测结果所确立的。 染色体17p11.2的间隙缺失可通过分辨率大于550条带的常规G显带分析检测出。该缺失容易被忽视,尤其是当 细胞遗传学 提示不是SMS的情况。可使用 特异性DNA探针对SMS关键区域 进行FISH分析或 aCGH对疑难病例进行亚显微水平缺失的检测。RAI1基因的突变或缺失是导致大部分SMS临床表型的原因。
管理.
临床症状的治疗:儿童早期干预;特殊教育;后期的职业训练/支持;语言,体格,职业,行为,及感觉整合治疗。患者可通过精神类药物治疗提高注意力,降低多动症及改善睡眠问题。建议家庭成员进行短期护理及心理支持。
监测:对患者的发育情况进行每年一次的多学科评估,甲状腺功 能评估,空腹血脂测试,对是否存在隐蔽性尿道感染行常规尿检,监测脊柱侧弯,眼科检查,定期神经发育评估和/或儿童发育/儿科行为咨询、耳鼻喉科随访评 估,中耳炎及其他窦性异常的管理,听力评估以监测是否存在传导或感觉神经性听力损失。
诊断
SMS的诊断依据为包含RAI1基因的17p11.2缺失 或者RAI1基因发生致病性变异。
具以下复杂临床表现的患者,可怀疑为Smith-Magenis 综合征(SMS)
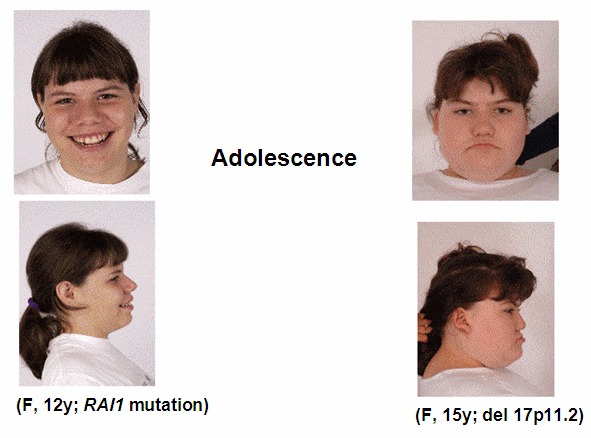
- 稍微特殊面容(见临床描述),且伴随年龄增长愈发明显(见 图 1, 图 2, 图 3 )
- 轻到中度的婴儿期肌张力降低,伴喂养困难及生长不旺。
小的微骨骼异常
- 身材矮小(青春期前)
- 短指
- 眼科异常
- 耳鼻喉异常
- 早期语言发育迟滞,伴或不伴听力缺失。
- 周围神经病变
- 某种程度的认知障碍及发育迟缓。
- 独特的神经行为症状包括睡眠紊乱,刻板行为,及适应不良行为[ Finucane et al 1994, Dykens & Smith 1998, Smith et al 1998a, Finucane et al 2001, Martin et al 2006]。慢性睡眠紊乱与昼夜节律的褪黑激素分泌异常有关[ Potocki et al 2000b, De Leersnyder et al 2001, Boone et al 2011]。
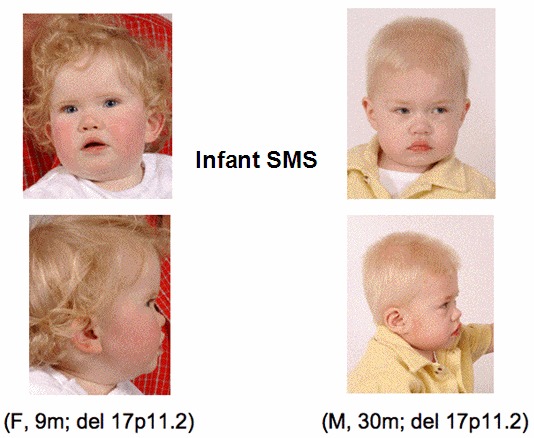

图2
图示为学龄早期的SMS患者,4岁男孩(左)及5岁女孩(右);图3为该女孩15岁时的照片。可见宽前额,眼窝深陷,面中部后缩。
肾脏异常及唇/腭裂的发生率小于25%。
婴儿及儿童早期的临床表现可以是轻微的,诊断通常会延迟到特征性异常面容及行为 症状更加明显的学龄期。
检测
细胞遗传学检测. SMS的典型诊断是通过G显带细胞遗传分析或 FISH检测出17p11.2的染色体内缺失。FISH的检测探针必须包含RAI1基因[Vlangos et al 2005]。染色体17p11.2的染色体内缺失可通过分辨率大于550条带的常规 G显带分析检测。研究表明大约90%的SMS患者通过FISH检测出缺失,其中约70%患者的缺失大小为3.5Mb左右[ Potocki et al 2003, Vlangos et al 2003, Girirajan et al 2006]。
注:该缺失容易被忽视,尤其是当细胞遗传学 提示不是SMS的情况。因此,对于先前常规细胞遗传学分析为正常但是强烈怀疑为SMS的患者,需重新进行细胞遗传学分析,包括 FISH或者aCGH检测。
分子遗传学检测
基因。RAI1基因为Smith-Magenis综合征唯一已知致病基因,该基因的突变或者缺失与Smith-Magenis综合征的临床表现相关。
临床检测
表1
应用于Smith-Magenis 综合征的分子遗传检测方法汇总。
基因 1 | 检测方法 | 变异 2 | 该方法检出的变异频率3 |
|---|---|---|---|
| RAI1 | FISH 4 | 包含RAI1基因的17p11.2缺失5 | ~95% |
| 测序分析 6 | 测序分析 | 5%-10% 7 | |
| 缺失/重复分析8 | 包含 RAI1基因的缺失3 | ~95% 9 |
- 1.
- 2.
等位变异信息可查询分子遗传学
- 3.
最佳检测方法对变异的检测能力。
- 4.
FISH探针包含RAI1基因或 D17S258。注:并非所有的FISH商业化探针都包含 RAI1基因。 [Vlangos et al 2005].
- 5.
- 4.
测序检测出的变异分为良性,可能良性 , 临床意义不明, 可疑致病性, 致病性. 致病性变异包括基因内缺失/插入, 错义突变, 无义突变, 及剪接位点突变; 通常, 外显子 或者是全- 基因的缺失/重复不能被检出 . 对 测序分析 结果解释有疑问, 请点击 这里.
- 7.
测序分析 ( 目前发现所有的致病性变异均位于3号 外显子 ) 可以检出那些通过 细胞遗传学 及 FISH 检测为 17p11.2 缺失 阴性的SMS患者中的RAI1致病性变异 [Slager et al 2003, Bi et al 2004, Girirajan et al 2005, Truong et al 2010, Vilboux et al 2011].
- 8.
- 9.
CMA检测范围宽泛,可提高前期疑似SMS病例的诊断率。
临床特征
临床描述
Smith-Magenis综合征(SMS)的临床 表型 包括体格、发育、及行为症状(表 2)。男女同等受累。特殊面容包括宽方形脸,短头畸形,突出前额,连眉,轻度眼睑上斜,深眼窝,宽鼻梁,面中部后缩(旧称面中部发育不良),小却尖圆鼻头并伴鼻骨低,婴儿期小颌畸形,与年龄相关的相对下颌前突,及嘴部明显的上唇下翻 。
随着年龄增长,异常面容逐渐明显及粗糙,可见面中部后缩,相对下颌前突,似“拳击手”外观的浓眉。牙齿异常机率增加,齿发育不全(尤其是 小臼齿)及牛齿症[Tomona et al 2006] 。
SMS有着不同程度的认知水平及适应能力,多数SMS患者有着轻至重度的智力障碍。
行为症状,包括睡眠紊乱,刻板行为,适应不良行为及自伤行为,通常要到18岁之后才能被识别,行为症状自儿童早期至成人期间会持续性变化[ Dykens & Smith 1998, Smith et al 1998a, Sarimski 2004, Gropman et al 2006]。睡眠紊乱包括间断性睡眠,夜间/清晨时的短周期睡眠,白天过度嗜睡[ Greenberg et al 1996, Smith et al 1998b, Potocki et al 2000b, De Leersnyder et al 2001, Smith & Duncan 2005]。报道发现总睡眠时间减少的间断性睡眠最早可发生于6月龄 [Duncan et al 2003, Gropman et al 2006] ,且睡眠问题会持续至成年期。夜间睡眠活动记录随不同的年龄段及不同时间点的觉醒模式而变化[ Gropman et al 2007].。
褪黑激素的昼夜节律异常(颠倒)是SMS的病征性(能确定诊断的)表型;这在95%(26/28)携带 缺失[Potocki et al 2000b, De Leersnyder et al 2001, Boudreau et al 2009], 及RAI1基因致病性变异 [Boone et al 2011]的患者中出现。最新数据[ Boudreau et al 2009] 表明睡眠紊乱不会由先前所报道的褪黑激素合成异常或降解所引起[ Potocki et al 2000b, De Leersnyder et al 2001, Chik et al 2010, Nováková et al 2012]。
表2
Smith-Magenis 综合征的临床表型
| 频率 | 系统 | 症状 |
|---|---|---|
>75%患者 | 颅面/骨骼 |
|
耳鼻喉 |
| |
| 神经行为 |
| |
常见(50%-75%患者) |
| |
少见(25%-50%患者) |
| |
偶见(<25%患者) |
|
婴儿期
体格特征。50%孕史表明胎动明显减少。SMS婴儿通常足月出生,有着正常的出生体重,身长,及头围。婴儿早期时身高及体重的增长逐渐减速。约20%的SMS儿童的头围小于相应年龄段的第三百分位 [Smith & Gropman 2010]。
婴儿期轻微的面容异常,通常表现为面中后缩,短鼻,上唇下翻,呈特征性“帐篷”形,小颌畸形,这些症状在婴儿早期可被识别(见 图 1)。喂养困难导致的生长迟滞常见,口腔运动功能障碍包括吞咽困难,对食物质地的反感,胃食管反流。几乎所有的SMS婴儿都有肌张力降低,包括反射减弱(84%)及自我满足及嗜睡等与唐氏综合征类似的症状。
神经行为症状. 生后的第一年出现粗大及精细运动发育迟缓。频繁出现与感统相关的问题[ Hildenbrand & Smith 2012]。
前瞻性评估显示对小于1岁龄婴儿普遍存在肌张力降低,口腔运动功能障碍,中耳异常及年龄相关的社会技能及轻微的不良适应行为 [Wolters et al 2009]。患儿哭闹少见,但常常声音嘶哑,大多数患儿的发声减少。在2-3岁之间,可识别出患儿具有全面发育迟缓,语言的表达较接受能力更明显缺陷及适应不良行为[ Gropman et al 2006, Wolters et al 2009].。
在12-18月龄前,父母通常不会认识到存在的睡眠问题;他们经常描述自己的孩子为“爱笑”的“完美”孩子,很少哭闹并且“睡眠很好”。然而,睡眠质量测评提示早在9月龄时就已开始出现睡眠紊乱,情况从婴儿期至儿童期逐渐变差[ Duncan et al 2003, Gropman et al 2006]。
儿童/学龄期
体格症状。SMS的面容异常在儿童早期愈发明显(见 图 2, 图 3),同时伴有SMS行为 症状。眼科的常见问题,包括斜视,近视,虹膜异常,小角膜等,随年龄进行性加重。约60%的 受累患者在4岁后呈轻至重度的脊柱侧弯,最常见于胸骨中段,其他类型脊柱异常少见。67%患者手脚小,身材矮小(身高<第5百分位)。明显的扁平足、高拱形足、异常步态等常见,便秘常有报道。
50%SMS患者有高胆固醇血症 [Smith et al 2002]。
耳鼻喉科问题通常持续整个儿童期。中耳炎常见(≥3次/年),导致鼓膜置管术(85%),且有传导性耳聋的风险(65%),听觉过敏或对声音/频率过度敏感(78%) [Smith et al 2007]。喉部异常常见息肉,结节,水肿,部分声带麻痹。大部分SMS患者表现不伴声音亢进的腭咽闭合不全/功能不全。口腔感觉运动功能障碍的主要问题包括发音无力,不对称/运动受限,嘴唇密闭无力(64%),腭裂(64%),吐舌,流涎,鼻窦炎常报道需抗生素治疗。
耳鼻喉科的高发症状可解释患者的发声障碍(嘶哑)及语言表达延迟。通过适宜的干预及完全的语言交流项目包括手语交流,学龄前患者的语言发育通常会提高;发音问题会一直存在,但患者的语言强度可能会轻度提升,表现为语速增快及伴有鼻音及嘶哑的中等爆发音。听力障碍占 受累患者的2/3以上。
神经行为症状。儿童早期明显发育迟缓,大部分大龄儿童及成人有轻到中度的智力障碍。对顺序处理的认知能力相对较弱,短期记忆能力较差;长时记忆能力及知觉合闭(即将不完整的视觉刺激认为是完整的过程 即视觉不完全时对“整体中的部分”感知)能力相对较强。
SMS的行为 症状在儿童早期及学龄期明显,随着年龄加重,症状通常出现在三个时期:18-24月龄期,学龄期,青春期。18月龄时开始出现撞头行为。整个儿童期间可表现感统障碍。一种明显的感觉处理障碍可表现为神经性阈值失衡及对积极或消极状态的自我调节能力的波动 [Hildenbrand & Smith 2012],大部分SMS患者表现出伴或不伴多动症的注意力缺陷。
有研究通过SRS(社会反应量表)发现90%的SMS患者的评分位于自闭症区间(35% 轻/中度;55%重度),临床上符合普遍性发育障碍的诊断标准 [Laje et al 2010b]。SMS需与自闭症鉴别诊断,特别是那些表现为特殊或刻板行为、喂养困难、口腔运动功能障碍,白天多度嗜睡导致睡眠紊乱的SMS患者 [Smith & Gropman 2010]. 对自闭症的治疗干预可能对SMS患者有益。
适应不良行为是家庭及护理人员所面对的主要问题。适应不良行为包括经常性易怒,冲动,寻求注意(从成人),注意力缺陷,叛逆,攻击行为,自伤,及容忍困难。年龄及发育迟缓的程度与适应不良行为相关。睡眠紊乱的程度是适应不良行为的强指标 [Dykens & Smith 1998, Arron et al 2011, Sloneem et al 2011]。由于适应不良,许多患者真实的智力水平评估不准。
大部分SMS患者的自伤行为(SIB)见于2岁后 [Arron et al 2011, Sloneem et al 2011],常见行为包括自击(71%),自咬(77%),扯皮(65%) [Dykens & Smith 1998]。不同类型SIB的的发生率随年龄增加 [Finucane et al 2001],不同类型及范围的SIB与智力水平存在直接的相关性。剔甲癖 [Greenberg et al 1991]及插孔癖(将外物插入自身有孔部位 )是SMS中两种独特的自伤行为,在约25%-30% 受累患者中可见。在儿童晚期前,剔指甲一般不会成为严重的问题,从儿童早期开始一直到不合时宜的年龄持续出现吸手指或异物入口。
适应性不良行为反映出SMS患者在发育,尤其是在智力水平与情感成熟不同步的背景下对环境的复杂互动[ Finucane & Haas-Givler 2009]。随着年龄增长,SMS患者的智力水平与情感发育的差距逐渐加大,这种不一致现象导致了较大儿童及成人显著的行为及程式的挑战。
痉挛性上身挤压或“自我拥抱”行为是诊断该综合征的一种有效的症状[ Dykens et al 1997, Dykens & Smith 1998]。其他的刻板行为包括异物塞嘴或者将手插入嘴中(54%-69%),磨牙(54%),身体摇摆(43%),旋转物体(40%)。 由Dykens et al [1997] 发现的舔手指及翻页行为可能比之前报道的发生率低 [作者个人观察].
睡眠紊乱是护理人员需处理的一个主要问题[Foster et al 2010] 因为他们自己也可能睡眠不足。睡眠紊乱在儿童早期就是一个主要问题,对SMS患者的研究证实了患者深睡困难,频繁持续的夜间觉醒,白天过度嗜睡,随着年龄增长,午睡的次数及频率增加,夜间睡眠总时间减少。睡眠记录仪数据表明患者的快动眼睡眠期时间的减少超过一半 [Greenberg et al 1996, Potocki et al 2000b]。根据对婴儿(<1岁)至8岁的睡眠记录数据记录表明,与健康儿童对照人群相比,SMS患者的昼夜睡眠时间减少 [Gropman et al 2006].小于10岁的患儿很少表现出入睡困难,但在后半夜觉醒的次数增加[ Gropman et al 2007]。稍大年龄患儿及青少年表现出相对入睡困难。
自我伤害或在身体有孔部位插入异物(如阴道插入)会别误判为性虐待/虐待儿童
青春期
体格症状。 面部(图 3)外观逐渐成方形,伴有持续面中后缩,相对下颌前突,前额凸出,一字眉,浓眉(拳击状),面容粗糙。青春期发动时间正常,性早熟及性晚熟均可见。
神经行为症状。通常伴随青春期到来加重,睡眠紊乱仍然是主要问题。睡眠记录仪测试提示青春期的入睡比儿童早期更加困难[ Gropman et al 2007]。女性患者更易冲动 [Martin et al 2006],感情变化迅速,对恐惧的焦虑倾向逐渐加重成为青春期及成人期的主要问题。伴年龄升级的攻击性行为常见,某些女患者在月经期会发生癫痫 [Smith & Gropman 2010]。剔甲癖及插孔癖行为可能更加明显。异物插入耳朵在儿童及成人常见,其他身体有孔部位(鼻子,阴道,直肠)通常会到青少年期/成人期出现。
成人期
目前还缺乏足够的临床数据来预测SMS患者的平均寿命;已知SMS患者最大的年龄为88岁[Magenis, 个人通讯]。在去世前一个月,该患者仍跟往常一样,作为一名快乐的“SMS”患者,接受慢性鼻窦炎的治疗。在她去世前4天出现右侧脑卒中及左侧肢无力,无 病理解剖报告。
因没有涉及重要器官受损,SMS患者的预期寿命与其他一般认知障碍的人群相同。
体格症状。粗糙面容可见面中部连续性后缩及尖下巴导致的相对下颌前突,脊柱侧弯随年龄加重,身材矮小可能会或不会一直存在[ Smith et al 2004]。易怒,攻击行为和SIB可能会持续,但许多患者在成人期也可表现出相对”冷静“的行为。
基因型-表现型关联.
尚未见17p 缺失 的父母来源对 表型是否有影响的报道,提示印记 对SMS的临床表现影响不大。
目前报道的RAI1基因特异性致病突变的患者体型肥大,无矮小,不涉及器官系统 [Slager et al 2003, Bi et al 2004, Girirajan et al 2005]。携带RAI1致病突变的患者亦表现SMS的典型症状。在17p11.2区域是否存在修饰基因效应尚未可知。
发病率
预测的新生儿发病率为1:25,000[Greenberg et al 1991];实际发病率可能接近1:15,000[Smith et al 2005]。近5至10年由于细胞遗传学 技术的发展,大量的患者得到诊断。
该综合征在世界范围内不同人群中都有发现。
遗传相关(等位基因)的疾病
Persons with larger deletions extending distally to include PMP22 are also at risk for hereditary neuropathy with liability to pressure palsies (HNPP).
携带包含PMP22 基因片段缺失的患者会同时患有 遗传性压迫易感性周围神经病 (HNPP)的风险。
17p11.2 重复综合征 (Potocki-Lupski综合征) 患者发生 重排 的区域 与SMS缺失 区域一致,但与SMS患者的临床症状及行为不同。 [Potocki et al 2000a, Potocki et al 2007].
鉴别诊断
Smith-Magenis综合征(SMS)需要与其他包括发育迟缓,婴儿期肌张力减低,身材矮小,特殊面容,及行为 症状的综合征鉴别诊断。最常见的是以下综合征,下述综合征的鉴别诊断可应用 细胞遗传学 (FISH) 或 分子遗传学分析方法 :
- 22q11.2缺失综合征 (包括腭心面综合征, DiGeorge 综合征)
- Prader-Willi 综合征(PWS)
- 唐氏综合征(21三体;新生儿期)
- 2q23.1缺失综合征
- Kleefstra 综合征(9q34.3 缺失 或 EHMT1基因突变)
临床上,很多SMS患儿可能会被诊断为自闭症/自闭症谱系障碍(ASD)、注意力缺陷/多动症(ADHD),强迫症(OCD),及情感障碍等精神疾病患者。
患者出现语言发育延迟、适应不良行为或刻板行为的症状导致准确诊断更加困难。
SMS的延迟诊断常见。对先前染色体 分析阴性但怀疑为SMS的患者,重复aCGH或者特异性FISH检测可以确证。目前临床aCGH在对仅表现为“发育迟缓”的患儿的检测应用,提高了那些未被察觉的SMS 缺失 病例的检出率[作者经验]。
婴儿期肌张力低下,特殊面容(短头畸形,面中扁平,眼睑上斜), 先天性 心脏病的SMS婴儿经常被认为是唐氏综合征。携带提示性症状但不是21三体的患儿可通过使用针对SMS特异性探针 FISH确诊。
管理
初诊后评估
推荐以下评估手段来确定Smith-Magenis综合征( SMS)患者的疾病状况。
- 诊断时各系统的综合评估。
- 体格及神经学检查
- 如有泌尿系统感染史, 行肾脏超声检查以评估可能存在的肾脏/泌尿系统异常(~20%的SMS患者),包括泌尿系统评估。
- 超声心动图评估可能存在的心脏异常(<45%的SMS患者);根据心脏异常的严重程度制定随访计划。
- 脊柱X光评估可能存在的脊柱异常和脊柱侧凸(~60%) 。
- 血常规,免疫球蛋白定性测试,空腹血脂(高胆固醇血症评估),甲状腺功能测试
- 眼科评估,是否存在斜视,小角膜,虹膜异常,屈光不正。
- 语言发育的综合评估
- 热量摄入评估,胃食管反流的指征(GERD),吞咽能力,口腔运动技能评估。
- 耳鼻喉科评估耳朵,鼻子,喉咙问题,特别注意耳部畸形及腭畸形(腭裂,腭咽发育不全)
- 定期的听力评估,监测传导性/感觉神经性听力缺失。
- 多系统发育评估,包括运动,语言,社交,认知及职业技能。
- 早期的物理/职业理疗师治疗。
- 睡眠史需要特别注意,包括睡眠/觉醒规律及睡眠时的呼吸功能。睡眠日记有助于发现睡眠/觉醒的规律。睡眠呼吸障碍需要多导睡眠图(整夜的睡眠观测)的证据支持,以评估阻塞性睡眠呼吸暂停。
- 对有临床癫痫发作的病人,脑电图监测可指导抗癫痫药物的选择。对没有明显发作的癫痫患者,脑电图有助于评估对提升注意力/行为的治疗效果;行为及注意力的变化需要重新评估的亚临床事件。
- 通过神经系统影像(MRI或者CT扫描) 对癫痫及运动不对称等症状进行评估。
- 对缺
- 肾上腺功能的特异性筛查
- 对包含PMP22 基因的片段缺失患者外周神经功能的详细评估,是否存在 遗传性压迫易感性周围神经病 (HNPP)。
- 家庭支持及心理情感需求评估,为家庭干预提供方案
- 临床遗传咨询
症状治疗
以下为适宜的治疗方案:
- 进行儿科常规预防接种。
- 从幼儿期开始,参照儿童早期的干预项目,进行特殊教育及职业技能训练。
- 发音/语言发育、体格、职业、尤其是感统问题的治疗:
- 在儿童早期,发音/语言问题的干预应优先重视识别及治疗吞咽及喂养困难问题,以及对口腔感觉运动发育的优化治疗。
- 提高与吞咽及发音相关的技能需要设置以提高感觉输入,促进发音器官的运动,提高口腔运动耐力,降低过度敏感为目标的治疗
- 使用手语及完全的交流计划,使用计算机和平板电脑等设备作对发音/语言治疗的辅助工具,可提高交流技巧,并对行为产生积极影响。语言表达能力的提升与早期手语的 应用及语言学家的干预有关。
- 随着年龄增长,非典型感觉处理问题逐渐突出。了解SMS患者在感觉处理方式上的弱点及相对优势可有助于照顾他们的人更好的他们适应活动的需求,适应环境, 支持适当的社会交流活动。此外,SMS患者呈年龄增加而有更多的异常或非典型行为使早期干预及对照顾他们的人进行职业教育显得尤为重要。[ Hildenbrand & Smith 2012].
- 出现行为问题时立即实施包含家庭及学校在内的综合性行为支持计划,通常从小学开始。学校组织的1对1支持项目及与开展与SMS患者认知行为方式相匹配的课程可有效地解决SMS学生的需要。
- 智力障碍,重度行为困难,睡眠紊乱会对父母及同胞造成严重影响。父母往往处于沮丧及焦虑的状态,SMS患者家庭压力明显高于其他非特异性发育异常的患者家 庭。因此对家庭的服务,为他们提供可利用的资源是对SMS病人综合干预的组成部分。[ Hodapp et al 1998, Foster et al 2010]
- 使用精神类药物改善注意力缺陷及多动症。目前尚无有效的单一用药方案[ Laje et al 2010a]。根据一项对62个SMS患者精神类药物使用的队列研究综述表明,多重用药及系列用药的有效性不大。苯二氮类药物在"稍变坏"范围内的评分最低,提示该类药物的使用可能对SMS患者有害 [Laje et al 2010a]。
- 行为治疗包括着重于以个体指导, 结构化, 流程化为达到在校时最小化爆发性行为而设置的特殊教育。
- 睡眠紊乱的治疗管理。SMS患者的睡眠管理仍然是医生及父母面临的挑战。尚未报道有效的临床试验。
- 早先有关睡前使用褪黑激素的治疗效果令人鼓舞,褪黑激素的使用改善了睡眠且未见严重不良反应,但需保持低剂量(≤3 mg)。商店货架上的褪黑激素不受FDA的管制,因此剂量可能不准确。褪黑激素治疗实验尚没有对照研究。可考虑对睡眠紊乱的 受累 者进行4到6周褪黑激素的监测实验。
- 一项非对照研究报道,对9个SMS患者给予口服β-1肾上腺素拮抗剂(醋丁洛尔 10mg/kg),褪黑激素的白天峰值得到抑制且患者行为得到改善 [De Leersnyder et al 2001]。但该治疗没有回复夜间血浆褪黑激素的浓度[ De Leersnyder et al 2001]。
- 同一研究组 [De Leersnyder et al 2003] 另一项非对照研究联合白天给予醋丁洛尔晚上(晚8点)口服褪黑激素(6mg)方案,发现褪黑激素夜间的血浆浓度得以恢复且夜间觉醒现象消失,从而使睡眠时 间得到改善。父母报告用药后患者白天的行为及注意力都得到改善。β-1肾上腺素拮抗剂的禁忌症包括哮喘,肺病,某些心脏疾病,糖尿病。
- 在进行任何临床试验之前,必须考虑患儿的健康状况及基本的睡眠情况。
- 封闭式的床使睡眠时有所隔离
- 对 受累 患者临时护理及家庭心理支持,提供最优的生活环境。
- 高胆固醇血症监测(>50%的SMS患者);饮食或药物治疗。
- 使用正确的矫正镜片对眼科异常进行治疗。
- 使用鼓膜置管治疗复发性中耳炎
- 如有听力缺失进行听觉放大。
- 对癫痫的标准化管理。
- 对心脏和肾脏畸形、脊柱异常的标准化药物治疗。虽然对生长激素治疗已有报道, 但尚没有进行有对照的效果评价 [ Itoh et al 2004, Spadoni et al 2004]。
监测
推荐一年一次:
- 进行多学科(包括体格、职业,语言治疗评估及儿科评估)的发展评估。定期的神经发育/儿科行为学咨询是多学科评估的重要补充。
- 甲状腺功能, 包括游离74及TSH。
- 空腹血脂
- 隐蔽性尿路感染的尿常规
- 脊柱侧弯的监测
- 眼科评估
- 对中耳炎及其他耳窦异常的随访及管理。
- 对传导性或感觉神经性听力缺失进行一年一次的听力评估及根据临床需要进行
需避免的因子/情况
至少一例SMS少年女性病例报道,使用思达(阿托莫西丁)会导致患者攻击行为升级至送医的严重事件,该患者的睡眠模式发生显著改变。使用该类药物时应注意睡眠 参数及行为变化的监测。
家属风险评估
见遗传咨询中与家属风险评估相关的问题。
孕期管理
目前为止,未见SMS患者生育的报道。
在研的治疗方法
搜索ClinicalTrials.gov 获取疾病及异常相关临床试验的信息。注:该疾病可能还没有临床试验。
其他
药物干预应当考虑个体化情况,某些药物的使用可能会加重睡眠或行为困难,导致体重增加。
遗传咨询
遗 传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,使他们在充足的知识及信息帮助下做出基于足够背景知识,以及符合个 人情况的医学选择或个人决定。 以下段落涉及的是如何根据家庭成员的遗传状况、家族史、及遗传学检测结果对家庭成员进行遗传风险评估。这部分目的不是为了解决所有患者所可能面临的个人、 文化、或伦理问题,也不能代替遗传学专业远远的咨询工作- ED
家庭成员的患病风险
先证者的父母
- 复杂性 家族性染色体 重排的17p11.2缺失所导致的SMS罕见 , 但有报道 [Zori et al 1993, Yang et al 1997, Park et al 1998]. 因此,对所有诊断为SMS的患者,需对父母行染色体分析。
先证者的同胞
先证者的后代
- 未见SMS患者有受累后代的报道。
- 理论上,SMS患者后代的患病风险为50%
- 尚未对SMS的生育能力进行研究。
先证者其他家庭成员 其他家庭成员的患病风险取决于先证者父母的遗传状态。如果父母一方有 染色体 异常,则可对有风险的家庭成员进行染色体分析及 FISH检测。
遗传咨询相关问题
家庭计划
- 孕前是遗传风险值评估和产前检测可行性分析的最佳时间。
DNA样本库用来储备未来可能用到的DNA(主要从白细胞中提取)。建立受累 患者的DNA样本库是基于 未来检测方法的发展、人们对基因、等位基因变异以及疾病理解的深入方面的考虑。
产前检测
高风险孕妇。SMS通常由17p11.2 新发缺失 导致。实际上所有的SMS患者表现为单一的病例(即家庭中单一出现)。对罕见性复杂的 家族性染色体 重排, 可取有风险孕妇的绒毛穿刺(通常在10-12孕周)或羊水(通常在15-18孕周)细胞,通过联合常规 细胞遗传学 分析及 FISH对进行产前检测。注:产前诊断 时需包括FISH检测。
低风险孕妇. 已有报道孕妇在对因其他原因进行羊水产前检测时意外发现17p11.2缺失。至少在两例因常规筛查血清AFP(MSAFP)浓度低而执行羊水穿刺的孕妇中发现17p11.2缺失 [Fan & Farrell 1994; Thomas et al 2000, 个人发现]。在一个大的产前研究,从455,121例连续性产前 细胞遗传学研究中发现10例17p11.2缺失。
对已鉴定有致病性缺失或等位基因突变的家庭, 胚胎植入前遗传学诊断(PGD)是一种可选择的方式。
资源
GeneReviews的员工已经为患者选择了针对该疾病的患者支持组织和患者注册组织,目的是利于该疾病患者及患者家庭,GeneReviews不为其他组织提供的信息承担责任。获取选择标准的更多信息,请点击这里。
- Association of Smith-Magenis France (ASM France)FranceEmail: association@smithmagenis.com
- National Library of Medicine Genetics Home Reference
- Parents and Researchers Interested in Smith-Magenis Syndrome (PRISMS)21800 Town Center PlazaSuite 266A-633Sterling VA 20164Phone: 972-231-0035Fax: 972-499-1832Email: info@prisms.org
- Smith-Magenis Syndrome Foundation57 Allen RoadNorthants NN10 0DYUnited KingdomPhone: +44 01933 389951Email: info@smith-magenis.co.uk
- National Institutes of Health (NIH) SMS Research Registry and Tissue BankAnn C. M. Smith, MA, DSc (Hon)Phone: 301-435-5475Fax: 301-496-7184Email: acmsmith@mail.nih.gov
分子遗传学
在分子生物学和OMIM表格内的信息可能和GeneReview里其他地方的信息不一致:表格可能包含最新的信息-ED。
表 A.
Smith-Magenis 综合征: 基因及数据库
表 B.
Smith-Magenis 综合征的OMIM条目 ( OMIM中预览)
分子遗传病理学
Smith-Magenis 综合征 是一种 邻近基因缺失综合征. 约70%患者的缺失区间约为3.5 Mb [Potocki et al 2003, Vlangos et al 2003]。SMS 关键区域 定位于 17p11.2 ,长度小于 650 kb [Schoumans et al 2005, Vlangos et al 2005]. RAI1基因致病性突变也会导致SMS (见 表 3).
基因结构. 该 基因 有6个外显子。基因的详细信息见 表A, 基因。
致病性变异. 在具有SMS表型但未检测出17p11.2缺失的患者中鉴定出相关的RAI1基因显性致病变异 [Slager et al 2003, Bi et al 2004, Girirajan et al 2005, Truong et al 2010]. 见 表 3.
表3
选择性的RAI1 致病性突变
| Nucleotide Change 核苷酸改变 | Predicted Protein Change 蛋白质改变预测 1 | Reference Sequences 参考序列 |
|---|---|---|
| c.253_271del19 | p.Leu85CysfsTer55 | NM_030665.3 NP_109590.3 |
| c.1119delC | p.Gln374SerfsTer65 | |
| c.1449delC | p.Glu484LysfsTer35 | |
| c.2773_2801del29 | p.Val1925ArgfsTer9 | |
| c.2878C>T | p.Arg960Ter | |
| c.3103delC | p.Gln1035ArgfsTer29 | |
| c.3103dupC | p.Gln1035ProfsTer31 | |
| c.3801delC | p.Thr1268ProfsTer47 | |
| c.4649delC | p.Ser1550PhefsTer37 | |
| c.4685A>G | p.Gln1562Arg | |
| c.4933_4936del | p.Ala1645GlyfsTer35 | |
| c.5423G>A | p.Ser1808Asn | |
| c.5265delC | p.Arg1756GlyfsTer94 |
变异分类备注:列表中的变异由作者提供。GeneReviews 工作人员不会对变异的分类单独核实。
命名备注:GeneReviews 遵循人类基因组变异协会 (www.hgvs.org)的标准命规则。见命名解释的快速参考。
致病性突变参考来源: Slager et al [2003], Bi et al [2004], Girirajan et al [2005], Girirajan et al [2006], Truong et al [2010]
1.
对框移突变的命名包含了发生在框移位点(fs)的氨基酸的改变,及跟随有"Ter#"表示终止密码子(Ter)。一个新阅读框的计算是从由框移导致的第一位发生改变的氨基酸开始,到第一个终止密码子(Ter#)结束(见www.hgvs.org).
正常的 基因产物. 正常的维甲酸诱导蛋白1被认为具有转录调控功能[Bi et al 2004, Burns et al 2010, Carmona-Mora et al 2010]; 但需要更多的研究来全面评估该蛋白在细胞内的功能。
异常的 基因产物. RAI1致病性突变对基因/蛋白质功能的影响机制尚未明确。维甲酸诱导蛋白1单倍剂量不足被认为是导致该疾病表型的机制;即认为基因的 致病性突变 产生了无功能的蛋白质产物。
参考文献
Literature Cited
- Allanson JE, Greenberg F, Smith AC. The face of Smith-Magenis syndrome: a subjective and objective study. J Med Genet. 1999;36:394-7. [PMC free article: PMC1734375] [PubMed: 10353786]
- Arron K, Oliver C, Moss J, Berg K, Burbidge C. The prevalence and phenomenology of self-injurious and aggressive behaviour in genetic syndromes. J Intellect Disabil Res. 2011;55:109-20. [PubMed: 20977515]
- Bi W, Saifi GM, Shaw CJ, Walz K, Fonseca P, Wilson M, Potocki L, Lupski JR. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004;115:515-24. [PubMed: 15565467]
- Boone PM, Reiter RJ, Glaze DG, Tan DX, Lupski JR, Potocki L. Abnormal circadian rhythm of melatonin in Smith-Magenis syndrome patients with RAI1 point mutations. Am J Med Genet A. 2011;155A:2024-7. [PMC free article: PMC3140606] [PubMed: 21739587]
- Boudreau EA, Johnson KP, Jackman AR, Blancato J, Huizing M, Bendavid C, Jones M, Chandrasekharappa SC, Lewy AJ, Smith AC, Magenis RE. Review of disrupted sleep patterns in Smith-Magenis syndrome and normal melatonin secretion in a patient with an atypical interstitial 17p11.2 deletion. Am J Med Genet A. 2009;149A:1382-91. [PMC free article: PMC2760428] [PubMed: 19530184]
- Burns B, Schmidt K, Williams SR, Kim S, Girirajan S, Elsea SH. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum Mol Genet. 2010;19:4026-42. [PubMed: 20663924]
- Carmona-Mora P, Encina CA, Canales CP, Cao L, Molina J, Kairath P, Young JI, Walz K. Functional and cellular characterization of human Retinoic Acid Induced 1 (RAI1) mutations associated with Smith-Magenis Syndrome. BMC Mol Biol. 2010;11:63. [PMC free article: PMC2939504] [PubMed: 20738874]
- Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet. 1997;17:154-63. [PubMed: 9326934]
- Chik CL, Rollag MD, Duncan WC, Smith AC. Diagnostic utility of daytime salivary melatonin levels in Smith-Magenis syndrome. Am J Med Genet A. 2010;152A:96-101. [PMC free article: PMC2802065] [PubMed: 20034098]
- De Leersnyder H, Bresson JL, de Blois MC, Souberbielle JC, Mogenet A, Delhotal-Landes B, Salefranque F, Munnich A. Beta 1-adrenergic antagonists and melatonin reset the clock and restore sleep in a circadian disorder, Smith-Magenis syndrome. J Med Genet. 2003;40:74-8. [PMC free article: PMC1735264] [PubMed: 12525548]
- De Leersnyder H, De Blois MC, Claustrat B, Romana S, Albrecht U, Von Kleist-Retzow JC, Delobel B, Viot G, Lyonnet S, Vekemans M, Munnich A. Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome. J Pediatr. 2001;139:111-6. [PubMed: 11445803]
- Duncan WC, Gropman A, Morse R, Krasnewich D, Smith ACM. Good babies sleeping poorly: Insufficient sleep in infants with Smith-Magenis syndrome. Am J Hum Genet. 2003;73 suppl:A896.
- Dykens EM, Finucane BM, Gayley C. Brief report: cognitive and behavioral profiles in persons with Smith-Magenis syndrome. J Autism Dev Disord. 1997;27:203-11. [PubMed: 9105971]
- Dykens EM, Smith AC. Distinctiveness and correlates of maladaptive behaviour in children and adolescents with Smith-Magenis syndrome. J Intellect Disabil Res. 1998;42:481-9. [PubMed: 10030444]
- Edelman EA, Girirajan S, Finucane B, Patel PI, Lupski JR, Smith AC, Elsea SH. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: a meta-analysis of 105 cases. Clin Genet. 2007;71:540-50. [PubMed: 17539903]
- Fan YS, Farrell SA. Prenatal diagnosis of interstitial deletion of 17(p11.2p11.2) (Smith-Magenis syndrome) Am J Med Genet. 1994;49:253-4. [PubMed: 8116679]
- Finucane B, Dirrigl KH, Simon EW. Characterization of self-injurious behaviors in children and adults with Smith-Magenis syndrome. Am J Ment Retard. 2001;106:52-8. [PubMed: 11246713]
- Finucane B, Haas-Givler B. Smith-Magenis syndrome: Genetic basis and clinical implications. J Ment Health Res Intel Disab. 2009;2:134-48.
- Finucane BM, Konar D, Haas-Givler B, Kurtz MB, Scott CI Jr. The spasmodic upper-body squeeze: a characteristic behavior in Smith- Magenis syndrome. Dev Med Child Neurol. 1994;36:78-83. [PubMed: 8132119]
- Foster RH, Kozachek S, Stern M, Elsea SH. Caring for the caregivers: an investigation of factors related to well-being among parents caring for a child with Smith-Magenis syndrome. J Genet Couns. 2010;19:187-98. [PubMed: 20151318]
- Girirajan S, Elsas Ii LJ, Devriendt KH, Elsea SH. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J Med Genet. 2005;42:820-8. [PMC free article: PMC1735950] [PubMed: 15788730]
- Girirajan S, Vlangos CN, Szomju BB, Edelman E, Trevors CD, Dupuis L, Nezarati M, Bunyan DJ, Elsea SH. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006;8:417-27. [PubMed: 16845274]
- Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, Kondo I, Dobyns WB, Patel PI, Lupski JR. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet. 1991;49:1207-18. [PMC free article: PMC1686451] [PubMed: 1746552]
- Greenberg F, Lewis RA, Potocki L, Glaze D, Parke J, Killian J, Murphy MA, Williamson D, Brown F, Dutton R, McCluggage C, Friedman E, Sulek M, Lupski JR. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2) Am J Med Genet. 1996;62:247-54. [PubMed: 8882782]
- Gropman AL, Duncan WC, Smith AC. Neurologic and developmental features of the Smith-Magenis syndrome (del 17p11.2). Pediatr Neurol. 2006;34:337-50. [PubMed: 16647992]
- Gropman AL, Elsea S, Duncan WC Jr, Smith AC. New developments in Smith-Magenis syndrome (del 17p11.2). Curr Opin Neurol. 2007;20:125-34. [PubMed: 17351481]
- Hildenbrand HL, Smith AC. Analysis of the sensory profile in children with smith-magenis syndrome. Phys Occup Ther Pediatr. 2012;32:48-65. [PubMed: 21599572]
- Hodapp RM, Fidler DJ, Smith AC. Stress and coping in families of children with Smith-Magenis syndrome. J Intellect Disabil Res. 1998;42:331-40. [PubMed: 9828063]
- Itoh M, Hayashi M, Hasegawa T, Shimohira M, Kohyama J. Systemic growth hormone corrects sleep disturbance in Smith-Magenis syndrome. Brain Dev. 2004;26:484-6. [PubMed: 15351087]
- Laje G, Bernert R, Morse R, Pao M, Smith AC. Pharmacological treatment of disruptive behavior in Smith-Magenis syndrome. Am J Med Genet C Semin Med Genet. 2010a;154C:463-8. [PMC free article: PMC3022344] [PubMed: 20981776]
- Laje G, Morse R, Richter W, Ball J, Pao M, Smith AC. Autism spectrum features in Smith-Magenis syndrome. Am J Med Genet C Semin Med Genet. 2010b;154C:456-62. [PMC free article: PMC2967410] [PubMed: 20981775]
- Martin SC, Wolters PL, Smith AC. Adaptive and maladaptive behavior in children with Smith-Magenis Syndrome. J Autism Dev Disord. 2006;36:541-52. [PubMed: 16570214]
- Nováková M, Nevsímalová S, Príhodová I, Sládek M, Sumová A. Alteration of the circadian clock in children with Smith-Magenis syndrome. J Clin Endocrinol Metab. 2012;97:E312-8. [PubMed: 22162479]
- Park JP, Moeschler JB, Davies WS, Patel PI, Mohandas TK. Smith-Magenis syndrome resulting from a de novo direct insertion of proximal 17q into 17p11.2. Am J Med Genet. 1998;77:23-7. [PubMed: 9557889]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633-49. [PMC free article: PMC1852712] [PubMed: 17357070]
- Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000a;24:84-7. [PubMed: 10615134]
- Potocki L, Glaze D, Tan DX, Park SS, Kashork CD, Shaffer LG, Reiter RJ, Lupski JR. Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J Med Genet. 2000b;37:428-33. [PMC free article: PMC1734604] [PubMed: 10851253]
- Potocki L, Shaw CJ, Stankiewicz P, Lupski JR. Variability in clinical phenotype despite common chromosomal deletion in Smith-Magenis syndrome. Genet Med. 2003;5:430-4. [del(17)(p11.2p11.2)] [PubMed: 14614393]
- Qin NG, Huang B (2007) Prenatal diagnosis of 10 cases with Smith-Magenis Syndrome. Cytogenet Genome Res. 116:324 (A10).
- Sarimski K. Communicative competence and behavioural phenotype in children with Smith-Magenis syndrome. Genet Couns. 2004;15:347-55. [PubMed: 15517828]
- Schoumans J, Staaf J, Jonsson G, Rantala J, Zimmer KS, Borg A, Nordenskjold M, Anderlid BM. Detection and delineation of an unusual 17p11.2 deletion by array-CGH and refinement of the Smith-Magenis syndrome minimum deletion to approximately 650 kb. Eur J Med Genet. 2005;48:290-300. [PubMed: 16179224]
- Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003;33:466-8. [PubMed: 12652298]
- Sloneem J, Oliver C, Udwin O, Woodcock KA. Prevalence, phenomenology, aetiology and predictors of challenging behaviour in Smith-Magenis syndrome. J Intellect Disabil Res. 2011;55:138-51. [PubMed: 21199049]
- Smith AC, Bentley J, Zalewski C, Morse R, Introne W, Brewer C. Hyperacusis in persons with Smith Magenis syndrome: Expanding the SMS phenotype. Abstract 652. San Diego, CA: American Society of Human Genetics 57th Annual Meeting. 2007.
- Smith AC, Duncan WC. Smith-Magenis syndrome: a developmental disorder with circadian dysfunction. In: Butler MG, Meaney FJ, eds. Genetics of Developmental Disabilities. Boca Raton, LA: Taylor and Francis Group; 2005.
- Smith AC, Dykens E, Greenberg F. Behavioral phenotype of Smith-Magenis syndrome (del 17p11.2). Am J Med Genet. 1998a;81:179-85. [PubMed: 9613859]
- Smith AC, Dykens E, Greenberg F. Sleep disturbance in Smith-Magenis syndrome (del 17 p11.2). Am J Med Genet. 1998b;81:186-91. [PubMed: 9613860]
- Smith AC, Gropman AL, Bailey-Wilson JE, Goker-Alpan O, Elsea SH, Blancato J, Lupski JR, Potocki L. Hypercholesterolemia in children with Smith-Magenis syndrome: del (17) (p11.2p11.2). Genet Med. 2002;4:118-25. [PubMed: 12180145]
- Smith AC, Gropman AL. Smith-Magenis syndrome. In: Cassidy S & Allanson J, eds. Management of Genetic Syndromes. 3 ed. New York, NY: Wiley-Blackwell; 2010:739-67.
- Smith AC, Leonard AK, Gropman A, Krasnewich D. Growth assessment of Smith-Magenis syndrome. Abstract 145. Toronto, Ontario: American Society of Human Genetics 54th Annual Meeting; 2004.
- Smith AC, Magenis RE, Elsea SH. Overview of Smith-Magenis syndrome. J Assoc Genet Technol. 2005;31:163-7. [PubMed: 16354942]
- Smith AC, Pletcher BA, Spilka J, Blancato J, Meck J. First report of two siblings with SMS due to maternal mosaicism. Poster session 829/C. New Orleans, LA: American Society of Human Genetics 56th Annual Meeting; 2006.
- Spadoni E, Colapietro P, Bozzola M, Marseglia GL, Repossi L, Danesino C, Larizza L, Maraschio P. Smith-Magenis syndrome and growth hormone deficiency. Eur J Pediatr. 2004;163:353-8. [PubMed: 15138811]
- Thomas DG, Jacques SM, Flore LA, Feldman B, Evans MI, Qureshi F. Prenatal diagnosis of smith-magenis syndrome (del 17p11.2). Fetal Diagn Ther. 2000;15:335-7. [PubMed: 11111213]
- Tomona N, Smith AC, Guadagnini JP, Hart TC. Craniofacial and dental phenotype of Smith-Magenis syndrome. Am J Med Genet A. 2006;140:2556-61. [PubMed: 17001665]
- Truong HT, Dudding T, Blanchard CL, Elsea SH. Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature. BMC Med Genet. 2010;11:142. [PMC free article: PMC2964533] [PubMed: 20932317]
- Truong HT, Solaymani-Kohal S, Baker KR, Girirajan S, Williams SR, Vlangos CN, Smith AC, Bunyan DJ, Roffey PE, Blanchard CL, Elsea SH. Diagnosing Smith-Magenis syndrome and duplication 17p11.2 syndrome by RAI1 gene copy number variation using quantitative real-time PCR. Genet Test. 2008;12:67-73. [PubMed: 18373405]
- Vilboux T, Ciccone C, Blancato JK, Cox GF, Deshpande C, Introne WJ, Gahl WA, Smith AC, Huizing M. Molecular analysis of the retinoic acid induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One. 2011;6:e22861. [PMC free article: PMC3152558] [PubMed: 21857958]
- Vlangos CN, Wilson M, Blancato J, Smith AC, Elsea SH. Diagnostic FISH probes for del(17)(p11.2p11.2) associated with Smith-Magenis syndrome should contain the RAI1 gene. Am J Med Genet A. 2005;132A:278-82. [PubMed: 15690371]
- Vlangos CN, Yim DK, Elsea SH. Refinement of the Smith-Magenis syndrome critical region to approximately 950kb and assessment of 17p11.2 deletions. Are all deletions created equally? Mol Genet Metab. 2003;79:134-41. [PubMed: 12809645]
- Wolters PL, Gropman AL, Martin SC, Smith MR, Hildenbrand HL, Brewer CC, Smith ACM. Neurodevelopment of children under three years with Smith-Magenis syndrome. Pediatr Neurol. 2009;41:250-8. [PMC free article: PMC2785222] [PubMed: 19748044]
- Yang SP, Bidichandani SI, Figuera LE, Juyal RC, Saxon PJ, Baldini A, Patel PI. Molecular analysis of deletion (17)(p11.2p11.2) in a family segregating a 17p paracentric inversion: implications for carriers of paracentric inversions. Am J Hum Genet. 1997;60:1184-93. [PMC free article: PMC1712444] [PubMed: 9150166]
- Zori RT, Lupski JR, Heju Z, Greenberg F, Killian JM, Gray BA, Driscoll DJ, Patel PI, Zackowski JL. Clinical, cytogenetic, and molecular evidence for an infant with Smith- Magenis syndrome born from a mother having a mosaic 17p11.2p12 deletion. Am J Med Genet. 1993;47:504-11. [PubMed: 8256814]
Suggested Reading
- Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16:412-21. [PubMed: 18231123]
- Elsea SH, Williams SR. Smith-Magenis syndrome: haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathways. Expert Rev Mol Med. 2011;13:e14. [PubMed: 21545756]
- Williams SR, Girirajan S, Tegay D, Nowak NJ, Hatchwell E, Elsea SH. Array comparative genomic hybridization of 52 subjects with a Smith-Magenis-like phenotype: identification of dosage-sensitive loci also associated with schizophrenia, autism, and developmental delay. J Med Genet. 2010;47:223-9. [PubMed: 19752160]
Chapter Notes 章节注解
Author Notes 作者注解
The authors of the Smith-Magenis Syndrome GeneReview are members of the PRISMS Professional Advisory Board.
Author History 作者简介
Judith E Allanson, MD; Children’s Hospital of Eastern Ottawa (2001-2009)
Albert J Allen, MD, PhD; Eli Lilly Laboratories, Inc (2001-2005)
Kerry E Boyd, MD, FRCP(C) (2009-present)
Elisabeth Dykens, PhD; University of California Los Angeles (2001-2005)
Sarah H Elsea, PhD, FACMG (2001-present)
Brenda M Finucane, MS, CGC (2001-present)
Andrea Gropman, MD, FAAP, FACMG (2005-present)
Barbara Haas-Givler, MEd, BCBA (2005-present)
Kyle P Johnson, MD; Oregon Health and Science University (2004-2012)
Gonzalo Laje, MD (2012-present)
James R Lupski, MD, PhD, FAAP, FACMG, FAAAS; Baylor College of Medicine (2001-2012)
Ellen Magenis, MD, FAAP, FACMG (2001-present)
Lorraine Potocki, MD, FACMG (2001-present)
Ann CM Smith, MA, DSc (hon), CGC (2001-present)
Beth Solomon, MS; National Institutes of Health (2001-2012)
Revision History 修改历史
- 28 June 2012 (me) Comprehensive update posted live
- 7 January 2010 (me) Comprehensive update posted live
- 11 August 2006 (me) Comprehensive update posted to live Web site
- 26 August 2005 (cd) Revision: 序列分析 of RAI1 clinically available
- 15 March 2004 (me) Comprehensive update posted to live Web site
- 15 January 2002 (as) Author revisions
- 22 October 2001 (me) Review posted to live Web site
- 23 May 2001 (as) Original submission