
摘要
临床特征
肥 厚性心肌病(Hypertrophic cardiomyopathy,HCM)普遍被认为是一种病因不明确的以左心室肥厚(left ventricular hypertrophy,LVH)为特征的心脏疾病。通常一些其他心血管疾病(例如长期高血压,主动脉狭窄造成的心脏压力负荷增大)或全身性疾病(例如法布 瑞氏症,淀粉样变性等沉积侵润性疾病)也能够导致左心室肥厚并心室腔扩张,但HCM患者并不伴随这些疾病同时也不具有心室腔扩张的特征。HCM的临床表现多变,从无症状LVH到进展性心力衰竭再到心源性猝死(sudden cardiac death,SCD),即使是在同一家系中,HCM的临床表现也可因人而异。HCM症状包括呼吸短促(尤其是在活动后)、胸痛、心悸、直立性低血压、晕厥前 兆以及晕厥。尽管HCM在各个年龄段皆可表现出LVH,但是更多见于青春期及青年患者。
诊断/临床检测
HCM的临床诊断普遍依赖于超声心动图或心脏核磁共振(MRI)两种非侵入性心脏影像检查,此外,具有特征性的心脏组织活检结果(心肌细胞排列紊乱与心脏纤维化)也可辅助诊断。对于具有家族史的患者,针对心脏肌小节组份相关基因的遗传检测有助于诊断遗传性HCM。
遗传咨询
位于心脏肌小节复合体相关基因上的致病变异导致的HCM通常呈现常染色体显性遗传模式。
临床管理
对症治疗: 一个对HCM诊断与治疗富有经验的内科医师有助于提高患者的生存与改善生活质量。治疗措施主要有药物治疗,室间隔削减治疗以及起搏器或除颤器的植入治疗。对于药物与器械治疗无效的晚期心衰患者,必要时可考虑心脏移植治疗。
并发症的预防: 对于合并持续性或阵发性心房颤动的HCM患者,应早期行抗凝治疗以降低血栓栓塞风险。另外依据病情可以给予抗生素预防感染性心内膜炎的产生。
监测随访: 评估患者的心源性猝死风险是临床管理中一项重要工作。对于无SCD风险的HCM患者,长时间定期的监测随访有助于评估患者SCD风险因素的变化并指导除颤器(implantable cardioverter defibrillator,ICD)的植入治疗决策。
生活/药物禁忌: 生活中应避免竞技性,耐久性训练活动、爆发性体育活动(如冲刺性短跑)、激烈的健身活动(如举重)、身体脱水(如使用利尿剂),并禁用可以降低心脏后负荷的药物(如血管紧张素转换酶抑制剂,血管紧张素受体阻滞剂以及其他血管扩张剂)。
患者亲属的风险评估: 一但在患者中检测出致病变异,则对家庭其他成员进行遗传筛查有助于针对变异携带者进行纵向发病风险评估。如果没有在患者中找到HCM致病变异,则推荐对患者的无症状一级亲属进行纵向发病风险评估。
孕产管理: HCM女性患者有经验的心脏科医生与熟悉高危产科的妇产科医生一同监管。
定义
本篇GeneReview将对肥厚性心肌病做一个全面的综述,以期帮助读者理解遗传检测在疾病的诊断与患者亲属的风险管理中的作用。
肥厚性心肌病(hypertrophic cardiomyopathy, HCM)的定义。HCM普遍被认为是一种病因不明确的以左心室肥厚(left ventricular hypertrophy,LVH)为特征的心脏疾病。通常一些其他心血管疾病(如长期高血压,主动脉狭窄造成的心脏压力负荷增大)或全身性疾病(如法布 瑞氏症,淀粉样变性等沉积侵润性疾病)同样能够引起左心室肥厚并心室腔扩张,但HCM患者并不伴随这些疾病同时也不具有心室腔扩张的特征。
HCM患者LVH的外显率与年龄高度相关。LVH多在患者青少年时出现,围绕青春期前后发病。尽管如此LVH也可以发生在其它年龄段,如成年后期 [Niimura et al 2002],婴幼儿及儿童时期。
诊断
HCM的临床诊断普遍依赖于超声心动图或心脏核磁共振(MRI)两种非侵入性的心脏影像检查,此外具有特征性的心脏组织活检结果(心肌细胞排列紊乱与心脏纤维化)也可辅助诊断。对于有家族史的患者,围绕心脏肌小节相关基因的遗传检测有助于诊断遗传性HCM.
患者心肌的不同部位均可发生不同程度的肥厚,肥厚可以是向心型,局限于某个室壁或者局限于左室心尖部,但是以室间隔的非对称性肥厚最为常见。
经胸超声心动图的观察要点主要有:
左室流出道梗阻相关的二尖瓣收缩期前向运动(Systolic anterior motion, SAM)与二尖瓣反流;
收缩期心室腔闭塞导致的左心室中部梗阻;
舒张功能受损,舒张期室壁活动受限。需要注意的是,在一些由肌小节组分相关基因致病变异导致的病例中,心脏超声可表现为左心室壁厚度正常,但心室舒张功能受损 [Nagueh et al 2001, Ho et al 2002],提示心肌舒张功能的异常可能是早于左心室肥厚出现的临床表型。
HCM的临床表现
HCM包含如无症状LVH,多种心律失常(心房颤动与恶性室性心律失常)及难治性心力衰竭等一系列临床表现,个体化差异很大,甚至在同一家系中,临床表现也会因人而异。常见临床症状有呼吸短促(尤其是活动后),胸痛,心悸,体位性低血压,晕厥前兆以及晕厥。
尽管HCM一开始被认为与高死亡率相关,但是现在人们倾向于HCM患者也可以具有较缓的病程,正常的生命预期以及可控的临床表现 [Maron et al 2003a, Maron et al 2003b, Elliott et al 2006]。
大约25%的HCM患者有静息状态下明显的心腔内梗阻,而大多数患者可发展为应激后(如心脏前负荷或后负荷骤减)流出道梗阻征象[Maron et al 2003c, Elliott et al 2006, Maron et al 2006]。梗阻程度与症状的严重程度或心源性猝死危险性之间并无明显的相关性。一些观察性的研究发现具有流出道梗阻的HCM患者通常具有较快的症状进展速度与较高的死亡的风险 [Maron et al 2003c, Sorajja et al 2009],但有时患者也可以长时间耐受高程度的梗阻。
HCM患者同时伴有心房颤动(atrial fibrillation, AF)的危险性也较高,而AF带来的血栓栓塞及症状恶化的风险也可加重此类HCM患者的病情。AF的发生率随年龄的增大而升高。
大约5%-10%的HCM患者进展为收缩功能受损的终末期心脏病,而一些患者则单纯表现为左心室扩张以及左心室肥厚。需要心脏移植治疗的终末期HCM患者的年死亡率约为11% [Harris et al 2006] 。
有重要的一小部分HCM患者有较高的风险发生SCD,这些SCD多由室性心动过速/心室颤动导致。
SCD 可能是本病的首发临床表现 [Maron et al 2000]。
在美国,HCM是导致竞技运动员发生SCD的最为重要的原因之一,现已被详细地描述 [Maron 2003]。
虽然青少年时期具有较高的SCD发生率,但其他年龄段同样伴随发生SCD的风险,而且此风险伴随患者终生。
SCD的风险因素将会在患者管理中进一步探讨。
HCM的流行病学
作为最常见的心血管单基因遗传病之一。HCM在普通人群中的发病率约为1:500,意味着在美国估计有60万人患有HCM。
HCM的鉴别诊断
其他也能够导致心肌肥厚,需要与HCM进行鉴别诊断的疾病包括:
获得性左心室肥厚 继发性病理性LVH可见 于心脏后负荷增大的疾病(如系统性高血压病,主动脉狭窄),并导致心脏舒张功能异常性心衰。生理性肥厚也就是因左心室容积增大而发生代偿性左心室壁增厚, 常见于运动量较大的运动员,这种心肌肥厚是机体生理性的适应机制所导致的,并不会引起病理性后果。通过有效降压治疗与脱离高强度运动训 练,两种继发性的心肌肥厚都可以在刺激因素消失之后恢复。
表现出左心室肥厚表型的综合征 (如LVH 相关的代谢性疾病或肌肉疾病)
PRKAG2是编码AMP激酶蛋白γ亚基的基因,PRKAG2的致病变异可引起充满糖原的空泡沉积病变,从而导致原因不明的心肌肥厚。本病多发心脏传导系统异常 [Konno et al 2010] 与室性早搏,可帮助与HCM鉴别。本病是常染色体显性遗传病。
Danon病,心脏表现为严重的LVH与频繁室性早搏,发病机制与编码溶酶体相关膜蛋白LAMP2基因的突变导致细胞内自噬泡堆积有关。致死性室性心律失常或合并终末期心力衰竭是导致患者死亡的普遍原因,心脏外临床表现主要集中在骨骼肌病变、神经系统病变及眼底病变(视网膜萎缩)。本病是X染色体连锁遗传病,通常是携带有杂合子突变的女性表现出心脏表型。
法布里病(Fabry disease), 本病是以α-半乳糖苷酶 (α-Gal)活性缺陷引起的全身各系统溶酶体进行性消耗为特征。典型的Fabry病男性患者仅有不到1%的α-Gal酶活性,青少年起病,表现为四肢周 期性剧烈疼痛(烧灼样疼痛),皮肤浅表血管损伤(血管角质瘤),少汗,特征性角膜混浊和蛋白尿,患者肾功能通常在起病后30-50年逐渐恶化为终末期肾病 (end-stage renal disease, ESRD),步入中年后逐渐出现心血管及脑血管病变。
相比之下,保留有1% 以上α-Gal酶活性的男性患者则表现出不同的心脏与肾脏表型。
心脏表型通常在患者16-18岁出现,以左心室肥厚,二尖瓣关闭不全或合并心肌病以及无ESRD性蛋白尿为临床特征。研究估计在成年男性特发LVH病例中,大约3%-4%由法布里病引起 [Sachdev et al 2002]。
本病为X染色体连锁遗传,但是在杂合突变女性携带者中,更低的α-Gal 酶活性可以导致明显的临床症状。LVH也可见于心肌淀粉样变性类疾病,由细胞内淀粉样蛋白累积造成,通常表现为限制性心肌病 [Shah et al 2006, Dubrey et al 2011]。淀粉样变性的类型(原发性,家族性或老年性)由细胞内淀粉样蛋白沉积的程度所决定,同时影响疾病的预后。
常见于儿童的LVH 在一个有关与孤立性或综合征性HCM儿童患者的注册研究中,Colan 等研究人员将患病儿童主要归为以下三类疾病因素[Colan et al 2007] (全文):(详见表1)
先天性代谢类疾病 II型糖原累积病(Glycogen storage disease type II, GSD II),庞贝氏症(Pompe disease)占总代谢疾病相关性心肌肥厚患儿的34% (25例/74例)。
先天畸形综合征 努南综合征占先天畸形综合征患儿的78%(60例/77例)。
神经肌肉接头疾病 弗里德赖希共济失调(Friedreich ataxia, FRDA)占总神经肌肉接头疾病相关心肌肥厚的88%(56例/64例)。
HCM的遗传特点
在本篇综述中,HCM被特指为一种排除后天刺激因素以及其他综合征型的先天原发性心肌肥厚性疾病。HCM的大多数致病突变集中在编码组成肌小节复合体多个基因中(在表1中列出)。大约50%-60%具有家族史的HCM患者或20%-30% 孤立性的HCM患者中可检测到至少一个编码肌小节组成蛋白的基因突变[Gersh et al 2011]。大约6%的患者携带一个以上的肌小节基因变异(同一基因不同变异位点或不同基因上多个杂合子位点变异),但是很少有患者同时携带一个以上的致病变异[Ingles et al 2013]。
至今已经鉴定出超过1500个致病变异。
评价策略
常见HCM患者遗传致病因素的检测流程见图 1。对HCM家系发病情况的准确评估依赖于多个家系成员的临床检查(心脏超声,心电图等)与基因检测结果的整合分析,因此对整个家庭成员进行遗传检测优于只对患者进行遗传检测。通过这种全面的整合分析才能确定整个家庭的疾病表型特征与基因型的遗传分离情况。
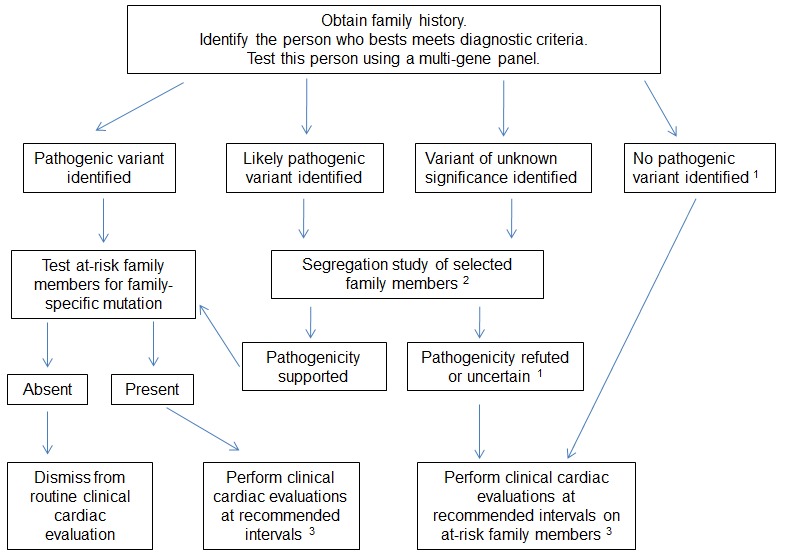
在进行遗传检测之前需要了解其对患者与家属带来的获益以及对HCM患者的局限性。
获益
遗传检测可以进一步确认HCM的临床诊断,并有助于与其他原因导致的心肌肥厚进行鉴别。
遗传检测可以增加鉴定出家庭成员的发病危险的可能性。
遗传检测可以为疾病发生机制提供线索。
局限性
检测结果具有不确定性。准确定性一个变异是否为致病变异,疾病相关变异或与疾病无关的变异是十分困难的。并且,对基因变异致病性的解读可能会随着时间而改变。
遗传检测不可能检测出所有致病变异。利用现有的筛查策略并不能在所有明确诊断的HCM患者中都找到致病变异。
遗传检测结果很难对HCM患者的管理造成直接的影响。
遗传检测策略
家族史
应 详细收集家族中3-4代亲属的病史信息,需要特别关注的病史如下:心衰,肥厚性心肌病,心脏移植,原因不明的猝死(特别是40岁以下的年轻人),心脏传导 系统疾病和/或心律失常,无法解释的卒中或血拴栓塞性疾病。注:在家族中,应该针对明确诊断为HCM或临床表现非常严重的患病亲属进行分子遗传学检测。
注意事项:(1)当未检出其他患病亲属时,应注意一些可变因素的影响,如选择了不合适的心脏检查方式,疾病外显率低,发病前已死亡和/或社会因素(如家庭的孤立、私下收养以及非亲生父母)。因此,这些情况下,不能够确定患有HCM的先证者是否为家族中的单发患者(simplex case)。(2)对家庭成员进行临床评估可以为家族史提供有价值的信息(如为之前没有HCM病史的家族成员做出诊断)。(3)家族史需要定期收集、更新。
多基因打包测序(Multi-Gene Panel)中候选基因的选择与检测可能带来的后果
靶向测序基因包(Multi-gene panels)中一般包含已知的HCM或其他心肌病相关的致病基因(详见表1)。需要注意的是,不同的实验室甚至在同一实验室不同时段,基因包中基因的种类与突变检出率也会不同。
不 同实验室通常拥有自己特有的变异分类体系,现在尚缺乏鉴定变异致病性的标准共识。『更新:2015年5月,美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布了关于遗传检测结果解读与遗传诊断指南(Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology)。本指南提供了一系列的标准与网络工具以帮助遗传检测实验室与遗传咨询提供者评价检出的变异的性质,并将其分为致病的 (Pathogenic),可疑致病的(Likely pathogenic),良性的(Benign),可疑良性的(Likely benign),意义不明的(Uncertain significance)。全文:http://www.nature.com/gim/journal/v17/n5/full/gim201530a.html』
变异结果一般分为以下几类:
阳性:实验室认为检测出的变异能够被足够数据支持其致病性(在患者中是致病的)。
意义不明的变异:并不能确定检测出的变异是致病的还是良性的,此时可能需要更多的信息去解读此变异对疾病的意义。
阴性:实验室没有鉴定出任何潜在的致病变异。重要的是,阴性结果没有诊断意义也不具有决定意义,并不能排除发病的遗传因素。在未来一旦有新的含有致病变异的基因被报道发现,则可以考虑重新检测阴性患者。
遗传检测实验室提供的变异分类信息为变异致病性的评价提供了参考基础,同时还要充分考虑个人的病史与先证者/家族病史信息。通常认为对家系成员进行遗传检测并分析遗传共分离情况可以对家族病史的评价做出有力的补充支撑(详见 具有发病风险的家庭成员的遗传检测)。
表1.
肥厚性心肌病的分子遗传学(按照发病频率从高到低排列)
| 这个基因中的突变占HCM患者的百分比 | 基因 1 | 蛋白 1 | OMIM号 1 |
|---|---|---|---|
| 40% | MYH7 | Myosin-7 | 160760 192600 |
| 40% | MYBPC3 | Myosin-binding protein C, cardiac type | 115197 600958 |
| 5% | TNNT2 | Troponin T, cardiac muscle | 115195 191045 |
| 5% | TNNI3 | Troponin I, cardiac muscle | 191044 613690 |
| 2% | TPM1 | Tropomyosin alpha-1 chain | 115196 191010 |
| 未知 | MYL2 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 160781 608758 |
| 1% | MYL3 | Myosin light chain 3 | 160790 608751 |
| 未知 | ACTC1 | Actin, alpha cardiac muscle 1 | 102540 612098 |
| 未知 | CSRP3 | Cysteine and glycine-rich protein 3 | 600824 612124 |
| 未知 | ACTN2 | Alpha-actinin-2 | 102573 |
| 未知 | MYH6 | Myosin-6 | 160710 613251 |
| 未知 | TCAP | Telethonin | 604488 |
| 未知 | TNNC1 | Troponin C, slow skeletal and cardiac muscles | 191040 613243 |
| 未知 | PLN | Cardiac phospholamban | 172405 613874 |
| 未知 | MYOZ2 | Myozenin-2 | 605602 613838 |
| 未知 | NEXN | Nexilin | 613121 613876 |
遗传检测的特征。 遗传检测手段的敏感性和特异性特征可以参考Clinical Utility Gene Card [Pinto et al 2011]。
具有发病风险的家庭成员的遗传检测
如果在一个家系成员中检测出明确致病变异,那么携带有杂合子变异的家庭成员有相当大的风险发生HCM,需要进行遗传检测。
如果家系中检测出变异的致病性是不确定的(可疑致病或意义不明),检测家庭中患病亲属后进行遗传分离分析有助于判断变异的性质。若观察到变异与家庭中HCM患者共分离的现象(而不与正常表型共分离)则可以进一步支持变异的致病性。
家庭中做遗传检测的成员数量是一个重要的参考指标,因为先证者的一级亲属有50%的概率遗传到变异。与此相对的,若有一个家族中有一个患者没有携带变异,那么这个变异不太可能被认为是致病变异。
对于具有发病风险的家庭成员,将基因检测与临床评估结果结合分析,能够提供更为全面的关于变异与疾病在家庭中的信息。
当遗传分离分析不支持变异的致病性时,此信息则需要反馈给遗传检测实验室。
如果在受检者中检测出的变异意义不明,那么对具有患病危险的健康家庭成员进行检测并不能为变异的解读提供有帮助的信息,也不能为这些家属的患病风险进行有效切实的预测。
如果受检者身上并未找到任何变异,则不需要(在当下)对具有患病风险的家属做进一步的遗传检测来明确其基因型状态。
心血管临床检查
依据指南[Gersh et al 2011]对家庭中具有患病风险成员进行心电图与超声心动图的心血管临床检查(具体参见监测随访中关于“具有HCM发病风险亲属”部分)。其中被检查者包含已知的致病变异携带者以及还未做遗传检测的风险成员。
依据临床检查诊断为HCM的家庭成员需要同时进行猝死风险分层评估(具体参见诊断后评估)[Gersh et al 2011]。
是否对不携带家族遗传变异的成员进行观察随访取决于支持此变异致病性的证据。
如果有充分证据支持被检出变异的致病性,则这些家庭成员毋须进行定期心血管临床检查除非出现临床表现。
如果缺乏足够的证据支持被检出变异的致病性,则这些家庭成员需要考虑进行定期心血管临床复检,并且一旦出现临床表现及时评估发病状态。
注意:随着人们对DNA变异有更多的了解,变异的分类可能会随之改变,从而潜在地影响对家庭及其成员的决策推荐。
遗传咨询
遗传咨询是向个人与家庭提供关于遗传病的疾病性质、遗传特征与影响等信息的过程,以帮助他们做出恰当的有利的医疗及个人决策。以下部分主要讨论遗传 危险因素的评估以及利用家族史和遗传检测结果解读家庭成员的遗传状况,并不能解决咨询对象可能面对的所有关于个人的、文化的或伦理的问题,也不是为了替代 专业人士的遗传咨询。 —ED.
遗传方式
多数引起肥厚性心肌病的突变均位于目前已知编码不同肌小节组分的基因座上,并且以常染色体显性方式遗传。遗传咨询和风险评估主要取决于患者HCM表型遗传的确定性与分子遗传检测的结果。
家庭成员的风险
先证者的父母
一些确诊为HCM的患者遗传自同样患病的父亲或母亲
此外HCM先证者也可能是由于新发(de novo)致病变异导致,尽管具体比例尚不确定,但估计约30%的病例是由新发突变引起[Morita et al 2008; H Rehm, personal communication]。
对于新发致病变异导致的先证者,推荐通过熟悉HCM的心脏病专家评估其亲本的心电图,超声心动图,体格检查等检查。在此评价策略下,可以确定之前因为不完全外显率和/或轻度临床表现而未被诊断的患病亲本。因此,只有进行了适当的诊断评估才能明确阴性家族史。
注意:尽管一些诊断为家族性HCM的先证者具有患病亲本,但家系中存在无症状或症状轻微的家庭成员,亲本提早于表型出现前的过早死亡或患病亲本症状延迟出现均可以降低家系中的疾病检出率,从而得到表面上阴性家族史。
先证者的亲属
先证者同胞亲属的发病风险依赖于其父母的遗传状态
如果先证者的亲本之一携带有致病变异,则同胞亲属遗传到等位基因的风险为50%,然而并不能同时预测临床症状的严重程度与发病年龄。
如果亲本中并不能找到先证者携带的致病变异,此时同胞亲属的风险是极低的。但是由于生殖细胞嵌合体的可能性,同胞亲属的发病风险依然高于正常人群。他们的遗传状态可以通过对存在发病风险的亲属进行遗传检测来明确。此外,不论家族性致病变异是否出现在先证者亲本中,具有临床表现的患病亲属都应该确保接受遗传检测。
当先证者父母无临床表现且没有找到遗传性致病变异时,仍然存在家族遗传疾病的可能性。此时先证者的亲属仍具有高于正常人群且无法预估的发病风险。
先证者的子女。 每一个家族性HCM患者的子女具有50%的可能性遗传到致病变异从而具有发病风险。但可能存在疾病表型不完全外显的情况并且较难预测其发病年龄与疾病严重程度。
其他家族成员. 先证者父母的遗传状态决定了其他家族成员的发病风险,若亲本之一携带致病变异,则他/她的家族成员处于发病风险之中。
遗传咨询相关事项
指南推荐建立三代以上的家族史以帮助识别有风险的家庭成员[Hershberger et al 2009]。具有风险的家庭成员应根据表 2所列的指南寻求临床评估,如果家系中存在已知致病变异,可考虑进行遗传检测。
由于HCM具有疾病表现多样以及外显性与年龄相关的特点,应定期对家族史进行更新。
确定遗传方式 已经发现单个患者中可以同时携带同一个肌小节蛋白基因上一个以上的致病变异(例如双重杂合),因此准确评估其他家庭成员的风险的关键是确定疾病的遗传模式 [Richard et al 2006, Girolami et al 2010]。
当在患病成员中检测到致病变异时,才有必要对处于风险中的无症状家属进行遗传检测。
对家庭中处于发病风险中的成年家属的遗传检测需要在接受了正式的遗传咨询之后进行。对于家庭中无症状亲属而言,检测并不能预测其发病年龄,疾病严重程度,症状类型与病情进展速度。而对于无明确疾病症状但有风险的家属,检测只是预测性质(而非诊断性质),起到发现那些需要长期监测HCM发病以及排除那些发病风险并不会增加的家庭人群的作用。
对具有风险的18岁以下年轻家属需要仔细平衡潜在的风险与获益。
潜在风险 对于遗传检测的核心争议在于它剥夺了当事人对这一信息知晓的选择权,从而导致受检者可能受到来自于家庭或社会中潜在的歧视,进而对其教育与就业造成影响。
潜在获益 早期检测出家族特异性的致病变异可以:(1)为未成年人管理提供帮助性信息,特别是已知发病早和/或恶化迅速的疾病;(2)允许对症状不明显但可被临床检测发现的HCM发作进行严格监控,还可以对猝死进行早期风险评估,尤其是对常参与竞技体育的无症状风险年轻人群,这是最重要的关注点。
家庭特异的致病变异的阴性检测结果将排除受检者发生HCM的可能性,因此亦可减少其不必要的临床监管。
注意:在家族管理中利用预测性遗传检测需要高度可信的证据表明先证者中的致病变异是家族患病的病因。
临床检测总是针对有症状的个体,一般不考虑年龄因素。有症状的儿童患者往往能够从早期基因诊断中获益。另见国家遗传咨询委员会(National Society of Genetic Counselors)的立场申明以及美国儿科学会(American Academy of Pediatrics)和美国医学遗传与基因组学会(American College of Medical Genetics and Genomics)的政策申明:儿童遗传检测和筛查的伦理与政策条款。
DNA标本库(DNA banking)是将以备将来使用的DNA(通常从血白细胞中提取)长期保存的策略。遗传检测手段与人们对基因、等位基因变异和疾病的认识随着时间不断的提高与改善,因此疾病患者的DNA应该考虑长期妥善保存。
产前检测与胚胎植入前遗传诊断
一旦在家庭里的患病成员中检测出致病变异,那么针对具有发病风险的孕妇进行产前检测或胚胎植入前遗传诊断是受检夫妇可以考虑的选择。
通常针对那些发病期在成年的疾病进行产前检测并不常见,原因是医学专家与家庭成员之间对产前检测持有不同的意见,特别是当产前检测用于终止妊娠而非早期诊断。尽管大多数检测中心在做决定时会将选择权交给受检父母,但双方对这些事项进行沟通讨论是十分必要的。
附加资源
以下经过GeneReviews工作人员筛选的信息来自于能够为患者及患者所在家庭提供帮助的保护支持组织和/或注册单位,GeneReviews 并不对除以下信息以外的提供方负责。点此查看筛选标准的相关信息。
- Children's Cardiomyopathy Foundation (CCF)PO Box 547Tenafly NJ 07670Phone: 866-808-2873 (toll-free)Fax: 201-227-7016Email: info@childrenscardiomyopathy.org
- Hypertrophic Cardiomyopathy Association (HCMA)328 Green Pond RoadPO Box 306Hibernia NJ 07842Phone: 973-983-7429Fax: 973-983-7870Email: support@4hcm.org
- My46 Trait Profile
- American Heart Association (AHA)7272 Greenville AvenueDallas TX 75231Phone: 800-242-8721 (toll-free)Email: review.personal.info@heart.org
- Cardiomyopathy UKChiltern CourtAsheridge RoadUnit 10Chesham Buckinghamshire HP5 2PXUnited KingdomPhone: 0800 018 1024 (UK only); 0800 018 1024 (UK only)Email: info@cardiomyopathy.org
患者管理
首次诊断后评估
一 部分重要的HCM患者具有较高的心源性猝死(sudden cardiac death ,SCD )发生风险,选择植入式心律转复除颤器(implantable cardioverter defibrillator,ICD)可以使这部分患者受益。进一步评估是否存在SCD相关风险预测因素使患者管理的常规部分。
SCD 危险因素包括:
室颤(VF),猝死/心跳骤停中止/恢复,或持续性室速(VT)的发作史
SCD家族史
左心室过度肥厚(>30mm)
运动反应性低血压
动态心电图检测显示非持续性VT
不明原因晕厥
危险因素信息来源于病史、家族史以及下列心血管临床检查,包括:
超声心动图用于测量LVH;
运动测试用于评价运动后血压变化;
动态心电图监测异位心室心律。
很难进行准确的风险评估,因为任何单一阳性预测值均相对较低(除了已经发生的心脏骤停或持续性VT以外)。虽然2个以上的危险因素与患者发生SCD风险增加有关 [Elliott et al 2000, Dimitrow et al 2010],但如果存在单个关键的危险因素,同样也可以考虑植入ICD预防猝死发生。相反,无任何危险因素会将患者归为低风险人群[Maron et al 2003b],尽管如此,这类人群仍然占SCD的3%-5%。
指南 [Garratt et al 2010, Gersh et al 2011]* 推荐ICD植入治疗用于:
目前植入ICD是预防SCD的唯一有效的治疗手段,但是也与累积发病相关。
对症治疗
有些医生对HCM的诊断与临床管理具有丰富的经验,通过他们的治疗可以提高生存率并改善生活质量。治疗手段包括药物治疗,侵入性室间隔削减治疗与起搏器或ICD植入治疗。心脏移植适用于那些对其他治疗无效的难治性晚期心力衰竭患者。
目前尚没有能够阻止、降低疾病进展或逆转临床表现的治疗手段。
用于缓解症状的药物治疗主要包含以下几类:
β受体阻滞剂
L型钙离子通道阻断剂
异丙吡胺(其负性肌力作用可以减少梗阻引起的生理反应)
抗心律失常药物,用于治疗心房颤动和/或室性心律失常。
注意:在出现梗阻表现的患者中,应避免使用直接血管扩张剂(例如ACE抑制剂、血管紧张素受体阻滞剂以及二氢吡啶类钙通道拮抗剂),因为这些药物可能加重患者的梗阻症状。
心脏舒张功能障碍是家族性HCM患者的常见的共同表现,它可明显导致非梗阻性劳力性呼吸困难与心脏容积负荷增加,通常是较难治疗的:
β-阻断剂和钙通道阻断剂可用于减慢心率并延长心室舒张期充盈时间。
利尿剂可以考虑用来缓解容积负荷增加引起的症状,但是需要警惕容量负荷减低导致的心输出量不足,尤其是在有梗阻的患者中。
当梗阻症状用药物治疗治疗无效时,可以考虑采用侵入性室间隔削减治疗来改善症状:
外科切除术(从室间隔切除一部分肌肉)很早就已开展并在很长一段时间里都被证明能够缓解或消除症状。
酒精性室间隔消融是最近逐步发展起来的基于导管的介入手段,定位于肥厚的室间隔部分,通过室间隔贯穿血管注入酒精诱导局部心肌梗死,从而起到缓解梗阻的作用。
对于心房颤动(AF),可能需要根据症状需要控制心率,利用药物或侵入性治疗控制节律[Gersh et al 2011]。因为合并AF的HCM患者具有较高的血栓栓塞风险,即使是阵发性房颤也建议抗凝治疗。
主要症状的预防
考虑到家族性HCM患者发生SCD的风险增加,应用ICD治疗是个重要的选择。ICD因为其在感知与终止VT和VF上的有效性被视为预防SCD的首选干预措施。
ICD作为一级预防的年使用率大约为2%-4%,作为二级预防的年使用率约为4%-11% [Maron et al 2007, O'Mahony et al 2012]。
在 涉及ICD的使用上有必要考虑到其可能伴随的并发症。ICD是安全的,但它并不是无害的,植入的年龄与治疗的时间长短都与累计发病率相关。每年约有5%的 HCM患者出现并发症。在那些接受了ICD作为一级预防治疗的HCM患者中,异常放电的发生率大约是正常除颤放电的两倍 [Maron et al 2007, Lin et al 2009, O'Mahony et al 2012]。
如果缺乏高度敏感性的患者特异性的预测因素,ICD的植入需要详尽细致的评估来决定,同时还需要患者完全的知情同意。
继发症状的预防
因为合并有AF的HCM患者发生血栓栓塞的风险也较高,因此强烈推荐持续性或阵发性房颤患者进行抗凝治疗。
具有梗阻表现的患者通常被认为有一定发生感染性心内膜炎的风险,既往指南建议对此人群进行抗生素预防感染。现有的指南已经将此修订,干预决策应该根据患者个体化情况而定 [Wilson et al 2007]。
临床监测
对那些并未符合ICD植入作为一级预防指征的HCM患者人群,应当每12-24个月重复评估发生SCD的危险因素(或一旦临床指标出现变化,立刻进行风险评估) [Gersh et al 2011]。
对有发生HCM风险的家属,已经提出了针对无临床表现但具有发病风险的家庭成员长期评价筛查指南(见表 2)。应该注意的是,以下筛查指南不仅适合已发现致病变异的亲属,也适合未检测出致病变异的HCM先证者的无症状一级亲属(成人以及儿童)。
因 为具有HCM诊断意义的表型的出现,如LVH,都与患者年龄相关,单个不明显临床表现不足以排除未来发展为HCM的可能性。具有诊断价值的临床表现通常不 会在婴儿/儿童早期出现,而常在青春期和成年早期出现,也可能在晚年出现。因此,对此类人群需要依据个体的年龄,家族史与医生判断进行一定频率的长期随 访。随访期间应关注任何症状的出现以及临床检查结果的变化。
表 2.
具有发病风险的健康家庭成员的临床筛查指南(包括体格检查,超声心动图,心电图)
| 年龄 | 筛查指南 |
|---|---|
| <12 岁 | 可选,除非具有下列表现之一:
|
| 12-18 岁 | 每12-18个月复检 |
| >18-21 岁 | 每≤5年复检或一出现症状立刻进行就医 如果家庭中有迟发性LVH或HCM相关并发症,则增加临床检测随访的频率 |
根据 Gersh et al [2011]修改
药品/生活禁忌
建议家族性HCM患者适度进行身体活动。已经出台了相关指南详细阐述了患者各种体育活动上的限制[Maron et al 2004, Gersh et al 2011]:
避免竞争性耐力训练和参与同竞技体育竞技强度相似的娱乐活动。
避免突发活动,如冲刺跑,以及高强度原地运动,如举重。
避免在极端环境条件下进行运动,并保持身体具有充足的水分
为了避免阻塞性生理恶化及症状加重,流出道梗阻的患者应当在饮酒、使用浴缸、蒸汽房、桑拿房时小心谨慎,还应避免以下活动:
脱水/血容量不足(因此应谨慎使用利尿剂)
减少心脏后负荷的药物(如:ACE抑制剂、血管紧张素受体阻滞剂与包括二氢吡啶类钙通道阻断剂在内的其他直接血管扩张剂)
勃起功能障碍药物(如:西地那非,他达拉非)
只有在已经试用了其他治疗方法后,才能考虑在确诊为HCM的儿童中谨慎使用兴奋类药物,并且需要在儿科心脏病专家的检测下使用兴奋类药物 [Vetter et al 2008]。
具有风险的家属的评估
如果在患病家庭成员中检测出致病变异,则需明确具有风险的家庭成员的遗传状态(见图1),这有助于对携带致病变异的亲属进行长期合理的随访评估(见监测随访,表2)。
生育管理
怀孕与分娩引起的血流动力学改变将增加家族性女性患者发生产科并发症的风险,特别是具有梗阻生理改变的患者。强烈建议通过对心血管医疗与高危产科都有经验的专家进行围产期护理。
正在进行的治疗研究
搜索ClinicalTrials.gov获取关于广泛疾病类别的临床研究信息。注意:不一定含有本病的临床研究。
References
已经发表的指南/共识
- Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA (2007) Epidemiology and case-specific outcomes in Hypertrophic Cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Available online. 2007. Accessed 2-8-17.
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 2-8-17. [PubMed: 23428972]
- Garratt CJ, Elliott P, Behr E, Camm AJ, Cowan C, Cruickshank S, Grace A, Griffith MJ, Jolly A, Lambiase P, McKeown P, O’Callagan P, Stuart G, Watkins H. Heart Rhythm UK position statement on clinical indications for implantable cardioverter defibrillators in adult patients with familial sudden cardiac death syndrome. Available online. 2010. Accessed 2-8-17. [PubMed: 20663787]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines. Available online. 2011. Accessed 2-8-17. [PubMed: 22068435]
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset disorders. Available online. 2012. Accessed 2-8-17.
参考文献
- Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA. Epidemiology and case-specific outcomes in Hypertrophic Cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115:773 - 81. [PubMed: 17261650]
- Dimitrow PP, Chojnowska L, Rudzinski T, Piotrowski W, Ziólkowska L, Wojtarowicz A, Wycisk A, Dabrowska-Kugacka A, Nowalany-Kozielska E, Sobkowicz B, Wróbel W, Aleszewicz-Baranowska J, Rynkiewicz A, Loboz-Grudzien K, Marchel M, Wysokinski A. Sudden death in hypertrophic cardiomyopathy: old risk factors re-assessed in a new model of maximalized follow-up. Eur Heart J. 2010;31:3084 - 93. [PubMed: 20843960]
- Dubrey SW, Hawkins PN, Falk RH. Amyloid Disease of the heart: assessment, diagnosis, and referral. Heart. 2011;97:75 - 84. [PubMed: 21148582]
- Elliott PM, Gimeno JR, Thaman R, Shah J, Ward D, Dickie S, Tome Esteban MT, McKenna WJ. Historical trends in reported survival rates in hypertrophic cardiomyopathy. Heart. 2006;92:785 - 91. [PMC free article: PMC1860645] [PubMed: 16216855]
- Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212 - 8. [PubMed: 11127463]
- Garratt CJ, Elliott P, Behr E, Camm AJ, Cowan C, Cruickshank S, Grace A, Griffith MJ, Jolly A, Lambiase P, McKeown P, O’Callagan P, Stuart G, Watkins H. Heart Rhythm UK position statement on clinical indications for implantable cardioverter defibrillators in adult patients with familial sudden cardiac death syndrome. Europace. 2010;12:1156 - 75. [PubMed: 20663787]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy:Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines. Circulation. 2011;124:2761 - 96. [PubMed: 22068435]
- Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444 - 53. [PubMed: 20359594]
- Harris KM, Spirito P, Maron MS, Zenovich AG, Formisano F, Lesser JR, Mackey-Bojack S, Manning WJ, Udelson JE, Maron BJ. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114:216 - 25. [PubMed: 16831987]
- Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA, et al. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009 Mar;15:83 - 97. [PubMed: 19254666]
- Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, Seidman CE, Solomon SD. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992 - 7. [PubMed: 12081993]
- Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, McCormack L, Winship I, McGaughran J, Atherton J, Semsarian C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15:972 - 7. [PubMed: 23598715]
- Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol. 2010;25:205 - 9. [PMC free article: PMC2932754] [PubMed: 20124998]
- Lin G, Nishimura RA, Gersh BJ, Phil D, Ommen SR, Ackerman MJ, Brady PA. Device complications and inappropriate implatable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709 - 14. [PubMed: 19282314]
- Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064 - 75. [PubMed: 12968091]
- Maron BJ, Casey SA, Hauser RG, Aeppli DM. Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol. 2003a;42:882 - 8. [PubMed: 12957437]
- Maron BJ, Chaitman BR, Ackerman MJ, Bayes de Luna A, Corrado D, Crosson JE, Deal BJ, Driscoll DJ, Estes NA 3rd, Araujo CGS, Liang DH, Mitten MJ, Myerburg MJ, Pelliccia A, Thompson PD, Towbin JA, Van Camp SP. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation. 2004;109:2807 - 16. [PubMed: 15184297]
- Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH 3rd, Spirito P, Ten Cate FJ, Wigle ED, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003b;42:1687 - 713. [PubMed: 14607462]
- Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of Hypertrophic Cardiomyopathy-related death: Revisited in a large non-referral-based patient population. Circulation. 2000;102:858 - 64. [PubMed: 10952953]
- Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, Epstein AE, Almquist AK, Daubert JP, Lawrenz T, Boriani G, Estes NA 3rd, Favale S, Piccininno M, Winters SL, Santini M, Betocchi S, Arribas F, Sherrid MV, Buja G, Semsarian C, Bruzzi P. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405 - 12. [PubMed: 17652294]
- Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, Cecchi F, Maron BJ. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003c;348:295 - 303. [PubMed: 12540642]
- Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, Nistri S, Cecchi S, Udelson JE, Maron BJ. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114:2232 - 9. [PubMed: 17088454]
- Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899 - 908. [PMC free article: PMC2752150] [PubMed: 18403758]
- Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128 - 30. [PMC free article: PMC2900859] [PubMed: 11447072]
- Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446 - 51. [PubMed: 11815426]
- O'Mahony C, Lambiase PD, Quarta G, Cardona M, Calcagnino M, Tsovolas K, Al-Shaikh S, Rahman SM, Arnous S, Jones S, McKenna W, Elliott P. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophic cardiomyopathy. Heart. 2012;98:116 - 25. [PubMed: 21757459]
- Pinto YM, Wilde AA, van Rijsingen IA, Christiaans I, Deprez RH, Elliott PM. Clinical utility gene card for: hypertrophic cardiomyopathy (type 1-14). Eur J Hum Genet. 2011;19(8) [PMC free article: PMC3172916] [PubMed: 21267010] [Cross Ref]
- Richard P, Villard E, Charron P, Isnard R. The genetic bases of cardiomyopathies. J Am Coll Cardiol. 2006;48:A79 - 89.
- Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407 - 11. [PubMed: 11914245]
- Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart. Arch Intern Med. 2006;166:1805 - 13. [PubMed: 17000935]
- Sorajja P, Nishimura RA, Gersh BJ, Dearani JA, Hodge DO, Wiste HJ, Ommen SR. Outcome of mildly symptomatic or asymptomatic obstructive hypertrophic cardiomyopathy: a long-term follow-up study. J Am Coll Cardiol. 2009;54:234 - 41. [PubMed: 19589436]
- Vetter VL, Elia J, Erickson C, Berger S, Blum N, Uzark K, Webb CL. Cardiovascular monitoring of children and adolescents with heart disease receiving stimulant drugs: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing. Circulation. 2008;117:2407 - 23. [PubMed: 18427125]
- Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT. Prevention of Infective Endocarditis. Guidelines from the American Heart Association. A Guideline From the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. J Am Dent Assoc. 2007;138:739 - 45. [PubMed: 17545263]
章注
作者注
布莱根妇女医院,心血管遗传中心(Brigham and Women’s Hospital Cardiovascular Genetics Center )
网址: www.brighamandwomens.org
修订历史
2014年1月16日 (me),本词条建立后重大更新
2011年5月17日 (cd),修订:ACTN2与CSRP3基因相关临床检测
2009年5月26日 (cd),修订:TNNC-1相关家族性肥厚性心肌病的序列分析与临床产前检测
2008年8月5日 (me),本词条建立后审查
2007年6月11日 (ac),首次提交