
概要
临床特征.
15q重复综合征及其相关疾病(dup15q)是指母源性15号染色体15q11.2-q13.1的Prader-Willi/Angelman关键区域 (PWACR)表现为至少一个额外拷贝。而该额外拷贝大部分情况下通常由以下两种机制之一产生:
- 母源性15q11.2-q13.1等臂双着丝粒额外染色体–idic(15)–通常包含两个15q11.2-q13.1额外拷贝,导致15q11.2-q13.1四体(约80%的病例);
Dup15q的临床表现主要为肌张力衰退和运动迟缓、智力障碍、孤独症谱系障碍(ASD)及癫痫如婴儿痉挛症。较罕见的是,该病可能与精神病或不明原因的猝死有关。携带母源性idic(15)的患者其表型通常比仅发生间隙重复的受累的个体更严重。
诊断/检测.
母源性的PWACR(即15号染色体15q11.2-q13.1约5Mb大小的区域)至少检测到一个额外拷贝。
疾病诊疗.
对症治疗: 建议多学科小组对婴儿的运动和言语发育进行评估,并随后协助推荐适当的指导性方案。支持性护理方法包括:物理职能治疗、辅助沟通、行为疗法(如应用行为分析疗法)、行为表现的精神药物治疗及癫痫发作的标准管理。
监测: 定期的: 神经发育性和/或发育性/行为方面的评估,对癫痫的发作和/或癫痫类型变化进行监测。
避免的药物和情况: 癫痫的诱因(如睡眠剥夺、压力)及不按照药物治疗。
亲属风险性评估:考虑对先证者(已明确为母源性15q11.2-q13.1间隙重复高风险)的同胞进行遗传检测,以便及时对这些携带者进行多学科小组评估。
遗传咨询.
dup15q由以下两种情况导致:
产前检测或植入前遗传诊断利用染色体芯片 (CMA) 检测dup15q;然而, 即使有生育dup15q后代的妊娠风险,产前筛查结果也不能可靠地预测患者表型的严重性。
GeneReview 内容范围
诊断
目前15q重复综合征及其相关疾病还没有正式的诊断标准(在本GeneReview称为dup15q)。
提示性表现
具有以下表型的患者应怀疑为15q重复综合征及其相关疾病(dup15q):
- 婴幼儿时期轻微到重度的肌张力衰退及运动障碍
- 发育迟缓,主要表现为智力障碍(ID)和/或语言发育迟缓
- 孤独症谱系障碍(ASD)
- 癫痫, 尤其是婴儿痉挛
以下症状同样在dup15q中常见:
- 轻度到中度变形的特征包括朝天鼻、内眦赘皮以及向下倾斜的睑裂[Battaglia et al 1997, Wolpert et al 2000, Orrico et al 2009, Hogart et al 2010, Urraca et al 2013]
- 行为异常包括过度活跃、焦虑或情绪不稳[Battaglia et al 1997, Wolpert et al 2000, Piard et al 2010, Al Ageeli et al 2014]
建立诊断
15q重复综合征及其相关疾病(dup15q)的诊断建立在检测到母源性Prader-Willi/Angelman 关键区域 (PWACR)至少一个额外的拷贝,即在染色体 15q11.2-q13.1片段上约为5Mb大小的重复。
15q近端区域包括5个片段性重复或低拷贝重复区域(称为断裂点[BPs]),这些区域导致基因组的重排易感性增加[Hogart et al 2010]。这5个区域分别定义为BP1、BP2、BP3、BP4、BP5。而PWACR关键区域位于BP2和BP3之间 (图1) ,并总是包含在15q间隙重复或idic(15)中。PWACR是具有印记的:即该区域母源性的拷贝数增加会导致dup15q(本GeneReview的主题),而父源性拷贝数增加通常与更多可变且不同的神经发育表型有关(见遗传上相关疾病疾病) [Cook et al 1997, Urraca et al 2013]。
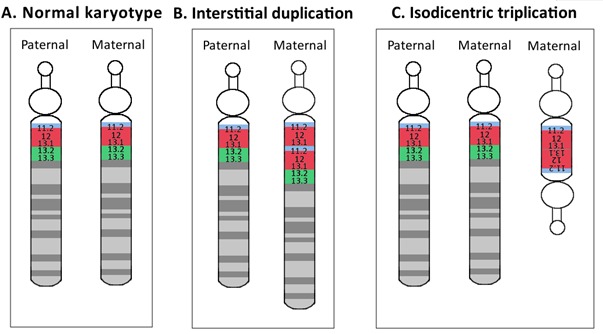
PWACR的额外拷贝通常由以下两种机制之一产生(图1):
- 母源性15q11.2-q13.1等臂双着丝粒额外染色体–idic(15)–通常包含两个15q11.2-q13.1额外拷贝,导致15q11.2-q13.1四体(约80%的病例 [Dup15q Alliance International Registry, 访问3-14-14])或
- 母源性15q11.2-q13.1间隙重复,通常在15号染色体上包含一个15q11.2-q13.1额外拷贝,导致15q11.2-q13.1三体(约20%病例[Dup15q Alliance International Registry, 访问3-14-14])
在本GeneReview中,dup15q指母源性15q11.2-q13.1区域至少发生一个额外拷贝,该区域覆盖PWACR,即基因组上23651570-28664979处(NCBI Build GRCh37/hg19, 详见此处)。重复片段的大小可能会有变化,目前最长为12Mb(详见此处),但都必须覆盖能导致dup15q的PWACR区域。
尽管有些关键基因(如, ATP10A, CYFIP1, MAGEL2, NECDIN, SNRPN, UBE3A, snoRNAs, 及一些编码GABAA受体的基因)均位于这4.5到12Mb大小的重复片段上,但并未发现有单个基因–当发生重复时–能导致dup15q(详见该区域相关基因的分子遗传学)。
基因组检测方法(检测DNA序列上拷贝数变异)包括染色体芯片分析(CMA)或靶向重复分析。注: (1) 母源性15q11.2-q13.1间隙重复通常不能利用常规的染色体G显带技术或其他传统的细胞遗传的显带技术鉴别;然而,母源性idic(15)及覆盖了PWACR的更大片段的间隙重复(>5 Mb)可通过细胞遗传学发现。(2) 有报道idic(15)嵌合体其有丝分裂期存在一定程度的不稳定性[Wang et al 2008],可能会影响表型以及用于诊断的基因组的检测方法的敏感性。
15q11.2-q13.1重复片段的遗传来源可以用以下方法检测:
- 在父母样本中验证15q11.2-q13.1间隙重复
表 1.
15q重复综合征及其相关疾病的基因组检测方法(dup15q)
| 重复 1 | ISCA ID 2 | 定位 3 | 检测方法 | 检测灵敏度 | |
|---|---|---|---|---|---|
| 先证者 | 家庭风险成员 | ||||
| 位于15q11.2-q13.1(覆盖PWACR)上的4.5至12Mb大小的重复 | ISCA-37404 4 或 ISCA-37478 5 | GRCh37/hg19 chr15: 21483759-32644465 | CMA 6, 7 | 100% | 100% |
| 靶向重复分析 8 | 不适用 9 | 100% | |||
2.基因组的变异的标准化临床注释及翻译来源于Clinical Genome Resource (ClinGen) project (原[ISCA]共识)。
3.15q11.2-q13.1复发性重复的最小片段大小由ClinGen定义。重复片段的大小可能随着微阵列的设计有略微的变化。请注意节段重复的断裂点,可能导致检测的基因组的片段与预期片段大小有所不同。该区域内更大或更小的缺失其患者的临床表型可能与15q11.2-q13.1复发性重复有所不同(详见遗传上相关的疾病)。
4.1类重复,约6Mb,从BP1到BP3
5.2类重复,约5Mb,从BP2到BP3
6.染色体微芯片(CMA)利用寡核苷酸微阵列或SNP分型微阵列。目前临床应用的CMA涵盖了15q11.2-q13.1区域。注意: 以往的寡核苷酸或BAC的CMA平台可能检测不到15q 重复及其相关疾病。
7.
大多数病例中为了确定重复是否为间隙串联重复或形成了另一额外染色体,FISH或细胞遗传的分析(如染色体G显带)检测也应进行作为CMA的参考。
8.靶向重复分析包括 FISH、定量PCR (qPCR)、多重连接探针扩增(MLPA)以及其他目标定量方法。
9.靶向重复分析不适用于已在15q11.2-q13.1区域有探针涵盖的CMA平台检测但未检测到15q11.2-q13.1复发性重复的个体。
亲属风险评估.FISH、qPCR或其他靶向重复定量分析方法可用来检测先证者家庭中15q11.2-q13.1高危风险的亲属。
临床特征
临床描述
15q重复综合征及其相关疾病(dup15q) 临床特征为肌张力衰退及运动迟缓、智力障碍、孤独症谱系障碍(ASD)、癫痫如婴儿痉挛症。该病的临床特征在母源性间隙重复和母源性等臂双着丝粒额外染色体idic(15)的患者中有很大不同(表2)。后者的表型往往比前者表型 受累的更为严重。然而,即使在同一类遗传机制中,剂量增加的个体,其表型严重性也会随之发生变化。一些表型如ASD,在母源性idic(15)患者或大于5Mb(覆盖PWACR关键区域)的间隙重复的个体中,更为常见[Hogart et al 2010]。
Table 2.
15q间隙重复和Idic(15): 临床特征比较
肌张力及运动技能.在dup15q新生儿和婴儿中,肌张力衰退与喂养困难和粗大运动发育迟缓有关[Depienne et al 2009, Hogart et al 2010, Urraca et al 2013]。
尽管儿童时期肌张力衰退会影响运动发育,但大部分儿童在两岁到三岁以后能独立行走(而母源性15q11.2-q13.1间隙重复的患儿更早就能行走)[Dennis et al 2006, Depienne et al 2009, Orrico et al 2009, Hogart et al 2010, Piard et al 2010, Al Ageeli et al 2014]。
走路外八字和共济失调也很常见[Bundey et al 1994]。发育迟缓及持久的损伤、粗大运动迟缓会影响适应性的生活技能。且需将15q重复综合征患儿与非综合性的ASD患儿区分开[DiStefano et al 2016]。
全身发育迟缓及智力障碍.早期的全身发育迟缓几乎很普遍。这可以在5岁以后被诊断为智力障碍。大部分15q重复综合征的儿童及青少年其智商在中度至重度障碍之间; 然而,也会有可变性,母源性15q11.2-q13.1间隙重复的个体其认知能力波动的范围更大。
语言发育尤其受到影响,普遍的发育情况在中度到严重迟缓不等[Grammatico et al 1994, Borgatti et al 2001, Hogart et al 2010]。有些患者表现出言语表达、代词颠倒和刻板的话语,而有些患者则可能缺乏功能性的语言[Battaglia et al 1997, Battaglia 2008]。
孤独症谱系障碍(ASD).大部分dup15q的儿童和青少年也符合ASD评估标准。与导致ASD的其他已知CNV相比,携带dup15q的个体患ASD的风险更大,比例约为42.6或更高[Malhotra & Sebat 2012, Moreno-De-Luca et al 2013]。ASD患者最主要的临床特征,尤其是社交障碍,可能会从儿童早期一直延续到晚期[Simon et al 2010]。
与非综合型ASD的患儿相比,dup15q/ASD的儿童表现出特定的不同的行为特征,如很保守的回应他人的微笑及面部表情–这些特征可能会影响对其行为干预[DiStefano et al 2016]。
癫痫.超过一半的dup15q个体患有癫痫,通常涉及到多种癫痫类型,如婴儿痉挛症、肌肉抽搐、强直痉挛、注意力不集中及病灶性癫痫[Conant et al 2014]。癫痫通常在6个月到9岁的之间开始发作[Battaglia 2008]。
dup15q是导致婴儿痉挛的最常见原因之一[Conant et al 2014]。dup15q引起的婴儿痉挛症通常会发展为Lennox Gastaut综合征及其他复杂的癫痫模式,并很难控制。多达40%的癫痫患者最初都是婴儿痉挛; 在这群个体中,约90%的人随后会发展为其他型癫痫。而dup15q患者可能仅仅表现出病灶性癫痫。dup15q
引起的顽固性癫痫可能会导致继发性副作用,如发育倒退。这种情况在超过一半的患者身上发生,他们经常出现不受控制的癫痫发作或癫痫状态[Battaglia et al 1997]。
在一项小型研究中,患有癫痫的儿童比未患癫痫的儿童具有更低的认知和适应性功能[DiStefano et al 2016]。
畸形特征.变形的畸形症状经常出现在dup15q患者中,如扁平的鼻梁及短翘鼻子、长人中、前倾的鼻孔、向下倾斜的眼睑、小颌畸形、低位耳、扁平枕、低额、高腭穹及厚嘴唇[Battaglia et al 1997, Borgatti et al 2001, Hogart et al 2010, Urraca et al 2013]。这些表型通常很微弱,可能在婴儿时期就被忽视。
精神错乱.尽管母源性idic(15)在精神病患者中已有报道[Bassett 2011, Ingason et al 2011, Costain et al 2013, Rees et al 2014], 但精神错乱在dup15q患者中并不是普遍的特征–这一发现可能反映了在认知功能低下及语言能力有限的个体中精神病的鉴别和诊断难度。例如,精神病在由于单亲二体性导致的Prader-Willi综合征中是一普遍的症状,同样也与母源性15q11.2-13.1间隙重复有关[Boer et al 2002, Vogels et al 2003, Bassett 2011]。这些个体相对dup15q患者来说,倾向于有更高的认知能力和语言能力。不同的是,由于dup15q更容易导致ASD表型,与情绪障碍有关的精神病可能被误诊为精神分裂症。
癫痫猝死症(SUDEP) 发生在一小部分dup15q患者中[Devinsky 2011, Wegiel et al 2012]。这种死亡通常在dup15q患者睡觉期间发生,大部分(但并非所有)在青少年及成年癫痫患者中发生。
SUDEP在其他一些涉及到神经发育疾病如严重的认知障碍和难治型癫痫中。其潜在的发病机制目前未可知。然而,现有的证据表明在大部分病例中,强直阵挛发作后伴随大脑功能的关闭及心跳呼吸的停止。SUDEP在9%的癫痫患者中有发生,但dup15q患者中SUDEP的发生率目前还不知道。
外显率
母源性idic(15)疾病的外显率是100%;表现度可能有差异。
母源性15q11.2-q13.1间隙重复尽管似乎外显率为完全外显,但有些个体也可能有一些轻微的表型或似乎不受影响,这也反映了可变的表现度而并非真正的外显率。
对于男女来说其外显率是一样的。
术语
以往用来描述15q重复综合征及其他相关疾病的术语:
患病率
在ASD患者中,dup15q是最常见的细胞遗传的异常。
- dup15q在人群中的患病率还不明确,但可能高达1:5000[Kirov et al 2014]。
- 在考虑为发育问题(发育迟缓、智力障碍或孤独症谱系障碍ASD)或多种先天的异常的临床CMA检测中,dup15q的患病率为1:508[Moreno-De-Luca et al 2013]。
- 在ASD队列研究中, dup15q的患病率为1:253-1:522 [Depienne et al 2009, Malhotra & Sebat 2012, Moreno-De-Luca et al 2013]。
- 在智力障碍的队列研究中,dup15q的患病率为1:584[Malhotra & Sebat 2012]。
遗传相关疾病
15q11.2-q13.1父源性间隙重复. 由于这个重复区域具有印记的作用,所以由父源性和母源性重复产生的表型不同:父源性间隙重复与更多变异的表型相关,包括睡眠问题,如深眠状态(异常或睡眠中出现异常行为)。临床发现,特别是自闭症表现,可有多达 50%受累的患者 [Urraca et al 2013]。由于母源性与父源性的表型特征有一定重叠,因此需进行父母源性的检测,以诊断先证者是dup15q综合征,还是父源性间隙重复。
Prader-Willi 综合征 (PWS) 是由于缺失, 单亲二体性, 或印记缺陷引起的,导致父源性15q11.2-q13.1区域丢失。尽管表型相似,PWS 与dup15q不同:PWS患者通常有特征性的面部特征、婴儿张力减退、性腺功能减退、轻度智力障碍、食欲过盛和强迫症行为 [Cassidy & Driscoll 2009]。与dup15q患者相比,PWS患者在语言和认知障碍方面更轻 (平均IQ:60-70s) [Cassidy & Driscoll 2009] 。
Angelman 综合征 (AS) 是由于 UBE3A 缺失, 单亲二体性, 印记缺陷或突变,导致母源性 UBE3A 等位基因的功能丧失。 AS不同于PWS和 dup15q不同,具有独特的面部特征,严重的智力障碍,严重语言表达障碍,癫痫发作,共济失调以及异常快乐或易兴奋的性格特点 [Dagli et al 2012]。
涉及 15q11.2 或 15q13.3 缺失–两者侧翼但不包括PWACR–具有不同神经行为的病理表型。
- 15q13.3缺失 (通常是 BP4至BP5) 与认知缺陷、ASD、癫痫、言语障碍以及包括精神分裂症、注意力问题、适应能力差和情绪障碍在内的行为/精神病相关 [Lowther et al 2015, Zhou et al 2016, Ziats et al 2016]。没有一致模式的变形的临床特征报道[Lowther et al 2015]。
- 15q11.2缺失 ( BP1至 BP2) 与言语延迟和认知缺陷相关,并且较少见的是癫痫,先天的心脏病和行为问题,包括注意力问题、极度活跃和ASD[Cox & Butler 2015, Vanlerberghe et al 2015]。没有一致模式的变形的临床特征报道 [Cox & Butler 2015, Vanlerberghe et al 2015]。
涉及 15q11.2 或 15q13.3 重复 但不包括 PWACR ,被认为涉及发育迟缓和自闭症 [Miller et al 2009, van Bon et al 2009, Burnside et al 2011], 但也有认为是意义不确定的变异[Kaminsky et al 2011, Chaste et al 2014]。
鉴别诊断
经典 Rett 综合征 是由 X 连锁基因MECP2 突变引起的神经发育障碍 [Chahrour & Zoghbi 2007]。 Rett 综合征主要见于女性,认为在大多数男性中是致命的。 经典Rett综合征的特征包括在出生后6至18个月正常发育,随后发育停滞,甚至技能快速倒退直到稳定。Rett综合征的一个标志性特征是用重复的、刻板的手部动作取代有目的的手部运动。其他特点还包括磨牙症(牙齿摩擦)、呼吸障碍、睡眠障碍、自闭症、癫痫发作,以及无端的哭泣或尖叫。
类似于15q重复综合征及其相关疾病(dup15q)临床表现包括运动和语言障碍,自闭症和癫痫发作。 然而,携带dup15q的个体 :
- 倾向于从婴儿早期延迟,在没有难治性癫痫的情况下很少出现精神运动性退化 ;
- 没有无目的的手部运动或Rett综合征的行为表型。
CDKL5 致病性变异 (OMIM), 是 X 连锁基因 , 发现 (1) 女性中有早发型严重癫痫发作,认知发育不良,但Rett综合征样特征少 [Archer et al 2006, Bahi-Buisson et al 2008] 和 (2) 男性表现为严重至极度的智力障碍和早发性难治性癫痫 [Elia et al 2008] 。
携带dup15q和 CDKL5致病性变异的个体可表现为严重的认知延迟、智力障碍和早发性癫痫; 然而,中至重度婴儿肌张力降低更是 dup15q的特征。
诊疗
初次诊断后的评估
为了确定被诊断为15q重复综合征及其相关疾病(dup15q)个体的疾病严重程度,建议使用以下评估 :
- 全面的系统回顾
- 体格检查
- 评估与张力低下相关的喂养困难
- 神经系统检查,包括癫痫程度和EEG基线评估
- 咨询临床遗传学专家和/或遗传咨询师
对症治疗
建议多学科团队从婴儿早期开始评估运动和语言发育,并随后协助推荐适当的教育计划。支持性护理可包括以下内容 :
- 专业物理治疗
- 替代和辅助沟通
- 行为疗法(例如应用行为分析疗法)
- 精神药物治疗行为症状
- 癫痫的标准治疗规范包括药物、迷走神经刺激剂和 / 或生酮饮食 [Conant et al 2014]。 抗癫痫药物(AED)治疗因癫痫类型和严重程度而疗效不一。 目前缺乏有关 dup15q的AED治疗前瞻性或随机对照数据。 参见 Conant et al [2014] 发表的由父母报道的各种药物和治疗的有效性。
癫痫处理对预防继发性并发症很重要,包括(最严重的)脑损伤、发育退化和发作期间的猝死(SUDEP) [Devinsky 2011]。
与癫痫有关的死亡有半数并非由于 SUDEP,而是其他原因,包括癫痫持续状态、溺水、跌倒和意外。 这些许多可以预防。 例如,癫痫持续状态可使用急救药物来预防,如直肠用地西泮或鼻内用咪达唑仑来预防。 证据表明,癫痫发作的及时发现和基本护理(例如,患者体位改为侧卧而不是俯卧位置)可有助于防止 SUDEP [Ryvlin et al 2013]。 然而,预防性治疗是已知唯一最好的控制癫痫的方法 [Ryvlin et al 2011]。 虽然很多监测器可以帮助发现 SUDEP(例如手腕和床垫监测器),但并不能防止 [Devinsky 2011]。
看护人. 有关癫痫儿童的父母或看护人的非医疗干预措施和应对策略的相关信息,请查阅 Epilepsy & My Child Toolkit (pdf)。
并发症的预防
需进行定期免疫接种的儿科护理。
监测
适当的监测如下:
- 定期进行神经发育和/或体格发育/行为评估
- 定期监测癫痫发作情况和/或癫痫发作类型的变化
应避免的药物/情况
应避免癫痫发作诱因(例如,睡眠不足、压力和不遵循药物治疗方案)。
在研治疗
目前还没有针对 dup15q的临床试验。 然而,正在进行的有关Angelman综合征和自闭症障碍(ASD)治疗的工作可能会为 dup15q未来的治疗提供信息。
检索 ClinicalTrials.gov 可获取大量疾病和环境的临床研究信息。
遗传咨询
遗传咨询是为个人和家庭提供关于遗传疾病的性质、遗传和影响等信息的过程,以帮助他们做出明智的诊疗和个人的决定。以下部分涉及遗传风险评估、家族史和 基因 检测来阐明家族成员的遗传状态。本部分的目的不是用来解决个人可能面对的所有个人、文化、伦理问题,或替代遗传学专家的咨询。 —ED.
家庭成员风险 – 母源性15q11.2-q13.1等臂双着丝粒额外染色体– Idic(15)
先证者父母
- 父母的检测不是常规。然而,有一例报道由母亲遗传的该区域的额外部分三体 [Michelson et al 2011],建议当母亲有临床症状(癫痫、ASD、精神分裂症或其他已报道症状)的,应及时考虑父母检测。
先证者同胞. 鉴于已报道所有 idic(15)受累的个体为新发,同胞的患病风险很低。 然而,由于母体胚系嵌合的可能性,推定其风险略高于一般人群。
先证者后代. Idic(15)个体的生育风险暂不明确。
其他家族成员. 鉴于迄今报道的所有idic(15)病例都是新发 ,其他家族成员的风险被认为是低的。
家庭成员风险 – 母源性15q间隙重复
先证者父母
先证者同胞
先证者后代. 15q间隙重复的患者其子代中每个孩子有 50% 的机会遗传该重复片段。
其他家族成员. 其他家族成员的风险取决于先证者父母的基因情况:如果父母之一有15q间隙重复 ,他或她的家族成员也可能有该重复。
相关遗传咨询问题
参见管理, 风险亲属评估了解有关评估风险亲属早期诊断和治疗的信息。
家庭生育计划
- 确定遗传风险和讨论产前检测的最佳时间应在怀孕前进行。 同样,高危家庭成员确定基因情况的检测,最好在怀孕前进行。
- 应当向可能生育 dup15q孩子风险的年轻人提供遗传咨询(包括讨论后代的潜在风险和生育选择)。
DNA 库 是储存 DNA (通常从血白细胞中提取)以备将来可能使用。因为将来实验技术以及我们对基因、等位基因变异和疾病的认识可能会提高,所以应考虑储存受累的个体的DNA。
产前检测
母源性 idic(15). 鉴于到目前为止,所有报道的 idic(15) 病例都是新发,因此推测未来怀孕的风险很低。 然而,夫妻可能希望考虑产前检测或植入前遗传诊断,由于父母胚系嵌合的可能,导致风险可能略大于一般人群。
母源性15q间隙重复
- 母亲携带父源性或遗传性15q间隙重复 ;
注意 : 不管妊娠是否确定 dup15q风险增加,产前检测结果无法准确预测表型的严重程度 (参见 Clinical Description) 。
对于产前检测,医学专家和家庭内部之间可能存在不同的看法,尤其在该检测的目的是作为终止妊娠而不是早期诊断时。尽管大多数中心会考虑将产前检测作为夫妇决定的选择,讨论这些问题是恰当的。
资源
为了患有该疾病的个人及其家庭的利益,GeneReviews 工作人员选择以下疾病特定的和/或伞形支持组织和/或登记处。 GeneReviews不为其它组织提供的信息负责。关于选择标准的信息,点击 这里。
- Dup15q AlliancePO Box 674Fayetteville NY 13066Phone: 855-DUP-15QAEmail: info@dup15q.org
- Unique: The Rare Chromosome Disorder Support GroupG1 The StablesStation Road WestOxted Surrey RH8 9EEUnited KingdomPhone: +44 (0) 1883 723356Email: info@rarechromo.org; rarechromo@aol.com
- Dup15q Alliance International RegistryPO Box 674Fayetteville NY 13066Email: coordinator@dup15qregistry.org
分子遗传学
分子遗传学和 OMIM 表格中的信息可能与 GeneReview 其它部分的信息不一致 : 表格可能包含更多的最新信息。 -ED.
表 A.
| 基因 | 染色体定位 | 蛋白质 | ClinVar |
|---|---|---|---|
| 不适用 | 15q11-.2-q13.1 | 不适用 | 不适用 |
分子遗传学发病机制
PWACR以外的拷贝通常由两种机制之一产生 ( 图 1 ) :
- 母源性15q11.2-q13.1等臂双着丝粒额外染色体 - idic(15) - 通常包含两个15q11.2-q13.1额外拷贝, 导致15q11.2-q13.1四体(约占~80% 的病例[Dup15q Alliance International Registry, 访问 3-14-14])或者
- 母源性15q11.2-q13.1间隙重复通常在15号染色体包含一个15q11.2-q13.1额外拷贝, 导致15q11.2-q13.1三体 (约占~20% 的病例 [Dup15q Alliance International Registry, 访问 3-14-14])
15q近端包括五个片段重复或低拷贝重复区域 (称为断点[BPs]), 这导致基因组的重排易感性增加 [Robinson et al 1993, Robinson et al 1998, Christian et al 1999]。这五个区域称为BP1到BP5。Prader-Willi/Angelman 的关键区域( PWACR ) 位于BP2和BP3之间 ( 图 1 ),并且总是包含在间隙重复或idic(15)中,从而导致dup15q。重复片段从BP1到BP3称为 I 类重复;那些仅限于BP2到BP3称为 II 类重复。 PWACR是具有印记的 : 母源性拷贝数增加导致dup15q,而父源性增加通常与更多可变且不同的神经发育表型相关 [Cook et al 1997, Urraca et al 2013]。
母源性15q11.2-q13.1等臂双着丝粒– 或 idic(15)–通常认为是在减数分裂期间由U型交换产生的一条双卫星染色体 。Idic(15)通常包括15pter-q13.1两个镜像拷贝( 15号短臂, 着丝粒, 和 15q11.2-q13.1) [Roberts et al 2003],有时也称为 inv dup(15)。对于不对称额外染色体,通常远端断点在真正等臂双着丝粒染色体两端的BP3 (近似位点 [hg19] 28800000) 和BP4以及BP5(近似位点 [hg19] 30700000 和 [hg19] 32500000) (Figure 2) [Hogart et al 2010]。Idic(15) 通常导致15q11.2-q13.1四体。
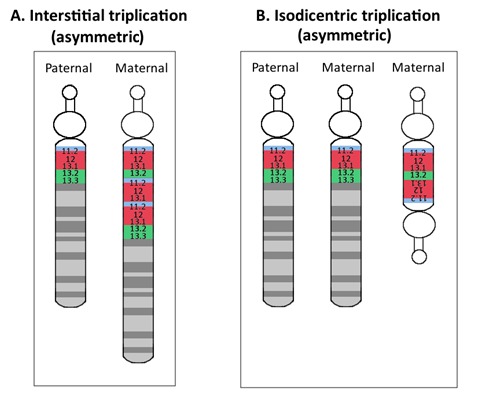
图2.
不对称的dup15q,如下所示:A.15q11.2-13.1间隙三体;和 B. 15q11.2-13.1等臂双着丝粒三体 。注意:浅蓝色表示BP1到BP2,红色表示BP2到BP3 (PWACR),绿色表示BP3到BP5。
通过两个不同断点区域 (即BP1到BP3) 之间的非等位同源重组(NAHR) ,导致间隙重复,dup15q的发生。母源性间隙重复的远端断点通常在BP3(近似位点 [hg19] 28812406), 而近端断点通常在BP1或BP2( 近似位点分别为 [hg19] 22964304 或 [hg19] 23966600 )。间隙重复通常导致15q11.2-q13.1三体。
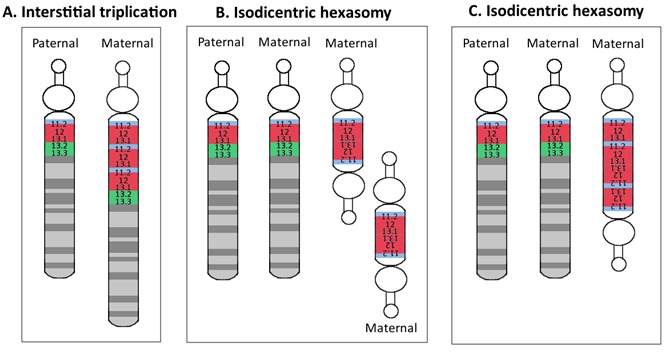
这些主要机制的变化包括以下:
- 间隙15q11.2-q13.1三体,导致15q11.2-q13.1四体。该表型倾向于比母源性15q11.2-q13.1间隙重复更严重,并且更像母源性15q11.2-q13.1等臂双着丝粒额外染色体 [Ungaro et al 2001, Hogart et al 2010] (图 3)。
- 不对称的等臂双着丝粒或间隙母源性三体,这通常导致15q11.2-q13.2四体和15q13.2-13.3三体。这些不对称的拷贝数变异在大约 10%-15%的等臂双着丝粒和间隙染色体中观察到[Dup15q Alliance International Registry, 访问 3-14-14] (图 2)。
该区域受关注的基因
- UBE3A, 与Angelman综合征密切相关的基因,被认为在dup15q中在导致智力障碍和自闭症特征起特殊作用[Glessner et al 2009, Greer et al 2010]. 在出生后神经元中,UBE3A 具有母源性特异性表达印记的,因此当个体具有母源性重复,其大脑具有高剂量表达。父源性15q11.2-q13.1重复与自闭症表型缺乏明确的一致性 [Cook et al 1997, Hogart et al 2010, Urraca et al 2013]。
- GABRB3, GABRA5, andGABRG3, 为编码 GABAA 受体亚基的基因,发现在dup15q中与癫痫发作关系密切 [Menold et al 2001, Samaco et al 2005, Hogart et al 2007]。在敲除这些基因的老鼠模型中,发展出包括癫痫发作的神经学问题 [DeLorey et al 1998, DeLorey et al 2008]。过度表达GABAA受体的转基因小鼠模型中癫痫发作迄今尚未表征 [Nakatani et al 2009]。GABRB3 由于该基因的单核苷酸多态性与自闭症相关 [Menold et al 2001],并且在 ASD 个体的脑组织样本中表达减少[Samaco et al 2005, Hogart et al 2007],GABRB3 可能与导致dup15q自闭症表型相关 [Conant et al 2014]。通过全基因组新发单碱基变异 研究,GABRB3 被认为在 15q11.2-q13.1 中唯一与 ASD 有密切相关性的基因 [Sanders et al 2015]。
- HERC2 是 E3泛素链接酶。具有双等位基因的HERC2 致病变异的个体,可能表现为智力障碍 [Puffenberger et al 2012] 或类似Angelman综合征样的严重神经发育障碍 [Harlalka et al 2013]。
参考文献
引用文献
- Al Ageeli E, Drunat S, Delanoë C, Perrin L, Baumann C, Capri Y, Fabre-Teste J, Aboura A, Dupont C, Auvin S, El Khattabi L, Chantereau D, Moncla A, Tabet AC, Verloes A. Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur J Med Genet. 2014;57:5-14. [PubMed: 24239951]
- Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, Kerr A, Bunyan D, Kerr B, Sweeney E, Davies SJ, Reardon W, Horn J, MacDermot KD, Smith RA, Magee A, Donaldson A, Crow Y, Hermon G, Miedzybrodzka Z, Cooper DN, Lazarou L, Butler R, Sampson J, Pilz DT, Laccone F, Clarke AJ. Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet. 2006;43:451-6. [PMC free article: PMC2564520] [PubMed: 16183801]
- Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, Cances C, Ville D, Afenjar A, Rio M, Héron D, N'guyen Morel MA, Arzimanoglou A, Philippe C, Jonveaux P, Chelly J, Bienvenu T. Key clinical features to identify girls with CDKL5 mutations. Brain. 2008;131:2647-61. [PubMed: 18790821]
- Bassett AS. Parental origin, DNA structure, and the schizophrenia spectrum. Am J Psychiatry. 2011;168:350-3. [PMC free article: PMC3276592] [PubMed: 21474594]
- Battaglia A. The inv dup (15) or idic(15) syndrome (Tetrasomy 15q). Orphanet J Rare Dis. 2008;3:30. [PMC free article: PMC2613132] [PubMed: 19019226]
- Battaglia A, Gurrieri F, Bertini E, Bellacosa A, Pomponi MG, Paravatou-Petsotas M, Mazza S, Neri G. The inv dup(15) syndrome: a clinically recognizable syndrome with altered behavior, mental retardation and epilepsy. Neurology. 1997;48:1081-6. [PubMed: 9109904]
- Battaglia A., Parrini B., Tancredi R. The behavioural phenotype of idic(15) syndrome. Am J Med Genet Part C Semin Med Genet. 2010;154C:448-55. [PubMed: 20981774]
- Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. 2002;359:135-6. [PubMed: 11809260]
- Borgatti R, Piccinelli P, Passoni D, Dalprà L, Miozzo M, Micheli R, Gagliardi C, Balottin U. Relationship between clinical and genetic features in "inverted duplicated chromosome 15" patients. Pediatr Neurol. 2001;24:111-6. [PubMed: 11275459]
- Bundey S, Hardy C, Vickers S, Kilpatrick MW, Corbett JA. Duplication of the 15q11-13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol. 1994;36:736-42. [PubMed: 8050626]
- Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, Gadi IK, Keitges E, Jaswaney VL, Papenhausen PR, Potluri VR, Risheg H, Rush B, Smith JL, Schwartz S, Tepperberg JH, Butler MG. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130:517-28. [PubMed: 21359847]
- Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17:3-13. [PMC free article: PMC2985966] [PubMed: 18781185]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422-37. [PubMed: 17988628]
- Chaste P, Sanders SJ, Mohan KN, Klei L, Song Y, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, Geschwind D, Grice DE, Ledbetter DH, Lord C, Mane SM, Martin DM, Morrow EM, Walsh CA, Sutcliffe JS, State MW, Martin CL, Devlin B, Beaudet AL, Cook EH Jr, Kim SJ. Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014;7:355-62. [PubMed: 24821083]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet. 1999;8:1025-37. [PubMed: 10332034]
- Conant KD, Finucane B, Cleary N, Martin A, Muss C, Delany M, Murphy EK, Rabe O, Luchsinger K, Spence SJ, Schanen C, Devinsky O, Cook EH, LaSalle J, Reiter LT, Thibert RL. A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia. 2014;55:396-402. [PubMed: 24502430]
- Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928-34. [PMC free article: PMC1712464] [PubMed: 9106540]
- Costain G, Lionel AC, Merico D, Forsythe P, Russell K, Lowther C, Yuen T, Husted J, Stavropoulos DJ, Speevak M, Chow EWC, Marshall CR, Scherer SW, Bassett AS. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum Mol Genet. 2013;22:4485-501. [PMC free article: PMC3889806] [PubMed: 23813976]
- Cox DM, Butler MG. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16:4068-82. [PMC free article: PMC4346944] [PubMed: 25689425]
- Dagli A, Buiting K, Williams CA. Molecular and Clinical Aspects of Angelman Syndrome. Mol Syndromol. 2012;2:100-12. [PMC free article: PMC3366701] [PubMed: 22670133]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505-14. [PubMed: 9763493]
- DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res. 2008;187:207-20. [PMC free article: PMC2684890] [PubMed: 17983671]
- Dennis NR, Veltman MW, Thompson R, Craig E, Bolton PF, Thomas NS. Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11-q13. Am J Med Genet A. 2006;140:434-41. [PubMed: 16470730]
- Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, Chaste P, Siffroi JP, Chantot-Bastaraud S, Benyahia B, Trouillard O, Nygren G, Kopp S, Johansson M, Rastam M, Burglen L, Leguern E, Verloes A, Leboyer M, Brice A, Gillberg C, Betancur C. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66:349-59. [PubMed: 19278672]
- Devinsky O. Sudden, unexpected death in epilepsy. N Engl J Med. 2011;365:1801-11. [PubMed: 22070477]
- DiStefano C, Gulsrud A, Huberty S, Kasari C, Cook E, Reiter L, Thibert R, Jeste SS. Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J Neurodev Disord. 2016;8:19. [PMC free article: PMC4858912] [PubMed: 27158270]
- Elia M, Falco M, Ferri R, Spalletta A, Bottitta M, Calabrese G, Carotenuto M, Musumeci SA, Lo Giudice M, Fichera M. CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy. Neurology. 2008;71:997-9. [PubMed: 18809835]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveal ubiquitin and neuronal genes. Nature. 2009;459:569-73. [PMC free article: PMC2925224] [PubMed: 19404257]
- Grammatico P, Di Rosa C, Roccella M, Falcolini M, Pelliccia A, Roccella F, Del Porto G. Inv dup(15): contribution to the clinical definition of phenotype. Clin Genet. 1994;46:233-7. [PubMed: 7820937]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J, Greenberg ME. The Angelman Syndrone protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704-16. [PMC free article: PMC2843143] [PubMed: 20211139]
- Harlalka GV, Baple EL, Cross H, Kühnle S, Cubillos-Rojas M, Matentzoglu K, Patton MA, Wagner K, Coblentz R, Ford DL, Mackay DJ, Chioza BA, Scheffner M, Rosa JL, Crosby AH. Mutation of HERC2 causes developmental delay with Angelman-like features. J Med Genet. 2013;50:65-73. [PubMed: 23243086]
- Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16:691-703. [PMC free article: PMC1934608] [PubMed: 17339270]
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38:181-91. [PMC free article: PMC2884398] [PubMed: 18840528]
- Ingason A, Kirov G, Giegling I, Hansen T, Isles AR, Jakobsen KD, Kristinsson KT, le Roux L, Gustafsson O, Craddock N, Möller HJ, McQuillin A, Muglia P, Cichon S, Rietschel M, Ophoff RA, Djurovic S, Andreassen OA, Pietiläinen OP, Peltonen L, Dempster E, Collier DA, St Clair D, Rasmussen HB, Glenthøj BY, Kiemeney LA, Franke B, Tosato S, Bonetto C, Saemundsen E, Hreidarsson SJ., GROUP Investigators. Nöthen MM, Gurling H, O'Donovan MC, Owen MJ, Sigurdsson E, Petursson H, Stefansson H, Rujescu D, Stefansson K, Werge T. Maternally derived microduplications at 15q11-q13: Implication of imprinted genes in psychotic illness. Am J Psychiatry. 2011;168:408-17. [PMC free article: PMC3428917] [PubMed: 21324950]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777-84. [PMC free article: PMC3661946] [PubMed: 21844811]
- Kirov G, Rees E, Walters JTR, Escott-Price V, Georgieva L, Richards AL, Chambert KD, Davies G, Legge SE, Moran JL, McCarroll SA, O’Donovan MC, Owen MJ. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2014;75:378-85. [PMC free article: PMC4229045] [PubMed: 23992924]
- Lowther C, Costain G, Stavropoulos DJ, Melvin R, Silversides CK, Andrade DM, So J, Faghfoury H, Lionel AC, Marshall CR, Scherer SW, Bassett AS. Delineating the 15q13.3 microdeletion phenotype: a case series and comprehensive review of the literature. Genet Med. 2015;17:149-57. [PMC free article: PMC4464824] [PubMed: 25077648]
- Malhotra D, Sebat J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223-41. [PMC free article: PMC3351385] [PubMed: 22424231]
- Mann SM, Wang NJ, Liu DH, Wang L, Schultz RA, Dorrani N, Sigman M, Schanen NC. Supernumerary tricentric derivative chromosome 15 in two boys with intractable epilepsy: another mechanismfor partial hexasomy. Hum Genet. 2004;115:104-11. [PubMed: 15141347]
- Menold MM, Shao Y, Wolpert CM, Donnelly SL, Raiford KL, Martin ER, Ravan SA, Abramson RK, Wright HH, Delong GR, Cuccaro ML, Pericak-Vance MA, Gilbert JR. Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J Neurogenet. 2001;15:245-59. [PubMed: 12092907]
- Michelson M, Eden A, Vinkler C, Leshinsky-Silver E, Kremer U, Lerman-Sagie T, Lev D. Familial partial trisomy 15q11-13 presenting as intractable epilepsy in the child and schizophrenia in the mother. Eur J Paediatr Neurol. 2011;15:230-3. [PubMed: 21145272]
- Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, Smith SE, Sobeih MM, Soul JS, Stoler J, Takeoka M, Tan WH, Thakuria J, Wolff R, Yusupov R, Gusella JF, Daly MJ, Wu BL. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242-8. [PMC free article: PMC4090085] [PubMed: 18805830]
- Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, State MW, Martin CL, Ledbetter DH. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18:1090-5. [PMC free article: PMC3720840] [PubMed: 23044707]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, Iwamoto K, Kato T, Okazawa M, Yamauchi K, Tanda K, Takao K, Miyakawa T, Bradley A, Takumi T. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–13 duplication seen in autism. Cell. 2009;137:1235-46. [PMC free article: PMC3710970] [PubMed: 19563756]
- Orrico A, Zollino M, Galli L, Buoni S, Marangi G, Sorrentino V. Late-onset Lennox-Gastaut syndrome in a patient with 15q11.2-q13.1 duplication. Am J Med Genet A. 2009;149A:1033-5. [PubMed: 19396834]
- Piard J, Philippe C, Marvier M, Beneteau C, Roth V, Valduga M, Béri M, Bonnet C, Grégoire MJ, Jonveaux P, Leheup B. Clinical and molecular characterization of a large family with an interstitial 15q11q13 duplication. Am J Med Genet A. 2010;152A:1933-41. [PubMed: 20635369]
- Puffenberger EG, Jinks RN, Wang H, Xin B, Fiorentini C, Sherman EA, Degrazio D, Shaw C, Sougnez C, Cibulskis K, Gabriel S, Kelley RI, Morton DH, Strauss KA. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat. 2012;33:1639-46. [PubMed: 23065719]
- Rees E, Walters JTR, Georgieva L, Isles AR, Chambert KD, Richards AL, Mahoney-Davies G, Legge SE, Moran JL, McCarroll SA, O’Donovan MC, Owen MJ, Kirov G. Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry. 2014;204:108-14. [PMC free article: PMC3909838] [PubMed: 24311552]
- Roberts SE, Maggouta F, Thomas NS, Jacobs PA, Crolla JA. Molecular and fluorescence in situ hybridization characterization of the breakpoints in 46 large supernumerary marker 15 chromosomes reveals an unexpected level of complexity. Am J Hum Genet. 2003;73:1061-72. [PMC free article: PMC1180486] [PubMed: 14560400]
- Robinson WP, Dutly F, Nicholls RD, Bernasconi F, Penaherrera M, Michaelis RC, Abeliovich D, Schinzel AA. The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet. 1998;35:130-6. [PMC free article: PMC1051217] [PubMed: 9580159]
- Robinson WP, Spiegel R, Schinzel AA. Deletion breakpoints associated with the Prader-Willi and Angelman syndromes (15q11-q13) are not sites of high homologous recombination. Hum Genet. 1993;91:181-4. [PubMed: 8462978]
- Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol. 2011;10:961-8. [PubMed: 21937278]
- Ryvlin P, Nashef L, Tomson T. Prevention of sudden unexpected death in epilepsy: a realistic goal? Epilepsia. 2013;54 Suppl 2:23-8. [PubMed: 23646967]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483-92. [PMC free article: PMC1224722] [PubMed: 15615769]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF III, Klei L, Mandell JD, Moreno-De-Luca D, Poultney CS, Robinson EB, Smith L, Solli-Nowlan T, Su MY, Teran NA, Walker MF, Werling DM, Beaudet AL, Cantor RM, Fombonne E, Geschwind DH, Grice DE, Lord C, Lowe JK, Mane SM, Martin DM, Morrow EM, Talkowski ME, Sutcliffe JS, Walsh CA, Yu TW., Autism Sequencing Consortium. Ledbetter DH, Martin CL, Cook EH, Buxbaum JD, Daly MJ, Devlin B, Roeder K, State MW. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215-33. [PMC free article: PMC4624267] [PubMed: 26402605]
- Simon EW, Haas-Givler B, Finucane B. A longitudinal follow-up study of autistic symptoms in children and adults with duplications of 15q11-13. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:463-7. [PubMed: 19548260]
- Ungaro P, Christian SL, Fantes JA, Mutirangura A, Black S, Reynolds J, Malcolm S, Dobyns WB, Ledbetter DH. Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J Med Genet. 2001;38:26-34. [PMC free article: PMC1734721] [PubMed: 11134237]
- Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, Schanen NC, Esmer C, Lamport D, Reiter LT. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6:268-79. [PMC free article: PMC3884762] [PubMed: 23495136]
- Urraca N, Davis L, Cook EH, Schanen NC, Reiter LT. A single-tube quantitative high-resolution melting curve method for parent-of-origin determination of 15q duplications. Genet Test Mol Biomarkers. 2010;14:571-6. [PMC free article: PMC3064527] [PubMed: 20642357]
- van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJ, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhé C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M, Vandeweyer G, Fryns JP, Janssens S, Foulds N, Reitano S, Smith K, Parkel S, Loeys B, Woods CG, Oostra A, Speleman F, Pereira AC, Kurg A, Willatt L, Knight SJ, Vermeesch JR, Romano C, Barber JC, Mortier G, Pérez-Jurado LA, Kooy F, Brunner HG, Eichler EE, Kleefstra T, de Vries BB. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511-23. [PMC free article: PMC3395372] [PubMed: 19372089]
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, Holder-Espinasse M, Delobel B, Duban B, Vallee L, Cuisset JM, Lemaitre MP, Vantyghem MC, Pigeyre M, Lanco-Dosen S, Plessis G, Gerard M, Decamp M, Mathieu M, Morin G, Jedraszak G, Bilan F, Gilbert-Dussardier B, Fauvert D, Roume J, Cormier-Daire V, Caumes R, Puechberty J, Genevieve D, Sarda P, Pinson L, Blanchet P, Lemeur N, Sheth F, Manouvrier-Hanu S, Andrieux J. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58:140-7. [PubMed: 25596525]
- Vogels A, Matthijs G, Legius E, Devriendt K, Fryns J. Chromosome 15 maternal uniparental disomy and psychosis in Prader-Willi syndrome. J Med Genet. 2003;40:72-73. [PMC free article: PMC1735257] [PubMed: 12525547]
- Wang NJ, Parokonny AS, Thatcher KN, Driscoll J, Malone BM, Dorrani N, Sigman M, LaSalle JM, Schanen NC. Multiple forms of atypical rearrangements generating supernumerary derivative chromosome 15. BMC Genet. 2008;9:2. [PMC free article: PMC2249594] [PubMed: 18177502]
- Wegiel J, Schanen NC, Cook EH, Sigman M, Brown WT, Kuchna I, Nowicki K, Wegiel J, Imaki H, Yong Ma S, Marchi E, Wierzba-Bobrowski T, Chauhan A, Chauhan V, Cohen IL, London E, Flory M, Lach B, Wisnewski T. Differences between the pattern of developmental abnormalities in autism associated with duplications 15q11.2-q13 and idiopathic autism. J Neuropathol Exp Neurol. 2012;71:382-97. [PMC free article: PMC3612833] [PubMed: 22487857]
- Wolpert CM, Menold MM, Bass MP, Qumsiyeh MB, Donnelly SL, Ravan SA, Vance JM, Gilbert JR, Abramson RK, Wright HH, Cuccaro ML, Pericak-Vance MA. Three probands with autistic disorder and isodicentric chromosome 15. Am J Med Genet. 2000;96:365-72. [PubMed: 10898916]
- Zhou D, Gochman P, Broadnax DD, Rapoport JL, Ahn K. 15q13.3 duplication in two patients with childhood-onset schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2016 Mar 10; [PMC free article: PMC5069586] [PubMed: 26968334]
- Ziats MN, Goin-Kochel RP, Berry LN, Ali M, Ge J, Guffey D, Rosenfeld JA, Bader P, Gambello MJ, Wolf V, Penney LS, Miller R, Lebel RR, Kane J, Bachman K, Troxell R, Clark G, Minard CG, Stankiewicz P, Beaudet A, Schaaf CP. The complex behavioral phenotype of 15q13.3 microdeletion syndrome. Genet Med. 2016 Mar 10; Epub ahead of print. [PubMed: 26963284]
- Zielinski C, Müller C, Smolen J. Use of plasmapheresis in therapy of systemic lupus erythematosus: a controlled study. Acta Med Austriaca. 1988;15:155-8. [PubMed: 3064527]
章节注释
致谢
The authors are indebted to the Dup15q Alliance for its efforts to advance research into dup15q. Many thanks go to Christa L Martin, PhD, Geisinger Health System, for technical assistance in the preparation of this review.
修订历史
- 2016年6月16日 (bp) 综述实时发布
- 2015年9月23日 (ll)首稿