
简介
临床特征 肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的特征是:
- 基底神经节中锰积累导致的运动障碍
- 全血锰浓度通常超过2000 nmol / L(正常值:<320 nmol / L)
- 红细胞增多症
- 肝肿大伴可变性肝纤维化/肝硬化
神经系统学发现可表现为:在儿童期(2-15岁)为四肢肌张力障碍,导致特征性的高步态(“鸡走步态”),构音障碍,细微震颤和运动迟缓,或有时为痉挛性截瘫。或在成年期因对L-多巴的治疗无反应而出现帕金森综合症(步态蹒跚,僵硬,运动迟缓,记忆力减退和单调语音)。肝功能衰竭,肝硬化的继发性并发症和神经系统疾病会缩短预期寿命。
诊断/测试 临床和脑部MRI的特征性发现,锰全血浓度升高和红细胞增多症提示诊断。通过在SLC30A10中鉴定双等位基因的致病变异可以证实这一点。
管理 表现的治疗:定期静脉滴注乙二胺四乙酸二钠钙的螯合疗法可改善血锰水平和神经系统发现,并导致肝脏疾病消失的迹象。此外,补充口服铁疗法(尽管血清铁水平正常)可以降低血锰水平并解决红细胞增多症。终末期肝病患者应考虑进行肝移植,尽管尚未针对这种疾病进行过尝试。
预防主要症状:螯合疗法和补充铁剂可预防受累的无症状同胞的主要疾病表现。
预防继发性并发症:尽早开始理疗和骨科治疗旨在预防挛缩并保持下肢活动。
可以尝试使用抗痉挛药物(包括巴氯芬和肉毒杆菌毒素)和左旋多巴对症治疗。吞咽评估和定期饮食评估可确保营养。为了防止吸入性肺炎,可能需要胃饲管和/或气管切开术。仔细监视可减少螯合疗法和/或铁补充剂引起并发症的可能性
避免接触的物质/情况:锰含量高的食物:丁香;藏红花;坚果青口贝;黑巧克力;还有南瓜,芝麻和葵花籽
有风险的亲属的评估:由于螯合疗法和铁的补充可以预防受累的 无症状个体的原发疾病表现,因此建议通过分子遗传学检测评估先证者的高危同胞(如果家族致病变异是已知的)或通过定期监测全血锰浓度和血红蛋白。
遗传咨询 肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病以常染色体隐性遗传的方式遗传。 受累的个体的每个同胞都有25%的机会受累,有50%的机会成为无症状的 携带者,有25%的机会不受影响也不是携带者。如果已知该家族中的SLC30A10致病性变异,则可以对有风险的家庭成员进行携带者测试,并针对高危妊娠进行产前诊断。
诊断
提示性发现
具有典型临床,脑MRI和实验室检查结果的个体应怀疑肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病。
临床发现。 存在早发和晚发的形式:
- 童年发作形式(2至15岁之间)。 通常为四肢肌张力障碍(导致特征性的高步态 (“鸡步走步态”),构音障碍,微震和运动迟缓[Tuschl et al 2012, Quadri et al 2015]或偶尔出现痉挛性截瘫[Gospe et al 2000]。
- 成人发病形式 对左旋多巴治疗无反应的帕金森病(步态僵硬,运动迟缓,记忆力减退和单调语音)[Quadri et al 2012]。
脑 MRI. 见 Figure 1.
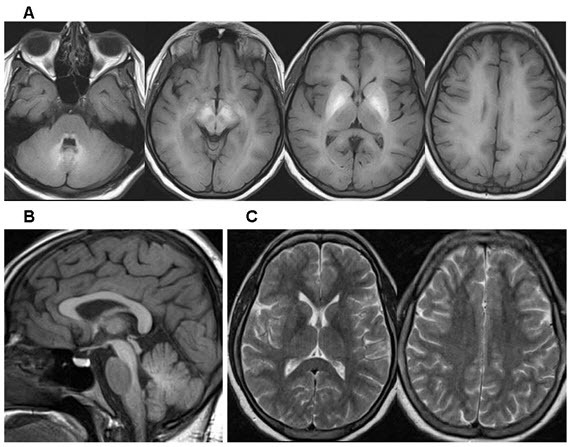
Figure 1.
受影响的个体的代表性脑MRI A.跨轴T1加权图像。 注意所有白质的异常高信号以及双侧壳壳和苍白球的异常明显信号。
- T1加权图像显示了基底神经节的特征性高信号包括苍白球的,壳核 ,尾状核,丘脑下和齿状核,并有丘脑和腹桥。 当疾病广泛时,可能会出现白质和垂体前叶受累。
- T2加权图像显示了相应的低强度变化。 但是,这些变化通常不太明显,因此可能被报告为正常。
- 注意:锰血药浓度的正常化(请参阅Management)改善了脑部MRI的发现[Quadri et al 2012, Stamelou et al 2012, Tuschl et al 2012].
实验室检查结果 高锰血症:
- 所有受累的患者的全血锰浓度均升高。 患病个体的平均值大于2000 nmol / L(正常值:<320 nmol / L)。
- 相反,获得性高锰血症中的血锰浓度通常低于2000 nmol / L。
伴随表现
- 红细胞增多症 锰诱导编码促红细胞生成素的基因的表达 [Ebert & Bunn 1999]。 具有特征的是,受累的人是多红细胞的。 文献中报道的血红蛋白浓度范围为15.9至22.5 g / dL(平均:18.6 g / dL)。 已经发现一些研究的个体的促红细胞生成素水平升高[Gospe et al 2000, Quadri et al 2012, Tuschl et al 2012].
- 铁减少的标记 锰和铁竞争相同的血清结合蛋白(转铁蛋白)和膜转运蛋白(二价金属转运蛋白1)。 因此,受累的个体显示出较低的血清铁蛋白浓度和血清铁水平,而总铁结合能力却有所提高[Quadri et al 2012, Tuschl et al 2012].
- 慢性肝病 肝脏受累的严重程度可能有所不同,并非本病的致病因素。 但是,如果存在,则肝脏受累应进一步提示诊断:
- 迄今报道的大多数受累的患者都有肝脏受累的证据,包括肝肿大,转氨酶升高(丙氨酸转氨酶[ALT],天冬氨酸转氨酶[AST])和未结合的高胆红素血症。
- 肝脏超声检查或MRI可确认肝肿大和肝硬化的特征。
- 六个受累的患者的肝活检/验尸检查的病理特征包括纤维化,脂肪变性和小结节性肝硬化。
注意:一个患有肝肿大和小结节性肝硬化的个体没有肝功能障碍的实验室证据 [Gospe et al 2000, Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012, Lechpammer et al 2014]. - 肝锰含量很高。 罗丹宁染色证实了锰在肝细胞中的沉积。 铜和锌的含量也可能随着肝酶水平的轻度升高而受累的 [Gospe et al 2000, Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012, Lechpammer et al 2014]。
建立诊断
在先证者中通过 分子遗传学检测鉴定SLC30A10的 双等位基因的致病变异,建立了肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的诊断[Quadri et al 2012, Tuschl et al 2012](请参阅Table 1)。
分子遗传测试方法可以包括单基因测试, multigene panel的使用以及更全面的基因组的测试:
- 单基因测试。如果仅发现一个或没有 致病性变异 ,则首先进行SLC30A10的序列分析,然后进行基因靶向的deletion/duplication analysis 。
- 也可以考虑包括SLC30A10和其他目的基因的multigene panel (请参阅Differential Diagnosis)。注意:(1)套餐中包含的基因 和每个基因所用测试的诊断敏感性因实验室而异,并可能随时间而变化。 (2)一些套餐可能包含与本《基因综述》中讨论的病症无关的基因;因此,临床医生需要确定哪个套餐最有可能以最合理的成本确定该病的遗传原因,同时减少无法解释潜在表型的意义不确定性基因和致病性变异基因 。 (3)在某些实验室中,面板选项可能包括定制的实验室设计套餐和/或定制的以表型为重点的 外显子组分析,其中包括临床医生指定的基因。 (4)套餐中使用的方法可能包括 序列分析,deletion/duplication analysis和/或其他非基于序列的测试。
有关多基因套餐的介绍,请单击 here。有关订购基因检测的临床医生的更多详细信息,请参见 here。
- 可以考虑进行更全面的基因组的测试(如果有),包括外显子组测序和 基因组测序。这样的测试可以提供或暗示先前未考虑的诊断(例如,导致相似临床表现的不同基因或基因的突变)。有关全面基因组测试的介绍,请单击here。可以在here找到有关订购基因组测试的临床医生的更多详细信息。
Table 1
用于肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的分子遗传学检测
| 基因 1 | 测试方法 | 具有致病变异的先证者所占比例 2 使用测试 |
|---|---|---|
| SLC30A10 | Sequence analysis 3 | 14/15 4 |
| Gene-targeted deletion/duplication analysis 5 | 1/15 6 |
- 1.
查染色体 位点 和蛋白质,见Table A. Genes and Databases.
- 2.
有关该 基因中检测到的等位基因变异的信息见Molecular Genetics .
- 3.
- 4.
来自14个家庭的24个个体(13个具有儿童期发病方式,1个具有成人发病方式)[Quadri et al 2012, Tuschl et al 2012, Avelino et al 2014, Quadri et al 2015, Mukhtiar et al 2016]。 在14个家庭的受累的个体中进行的序列分析确定了纯合性SLC30A10致病变异。
- 5.
基因靶向的 deletion/duplication analysis基因内的缺失或重复。 可以使用的方法包括: quantitative PCR,远程PCR,多重连接依赖性探针扩增(MLPA)和目标基因的微阵列,可检测单外显子缺失或重复。
- 6.
来自一个家庭的四个受累的同胞对于一个涉及外显子1和2的大型基因组的SLC30A10缺失是纯合性的[Tuschl et al 2012].
临床特征
临床表现
神经学发现
- 童年起病 在儿童期发作的肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病中, 受累的患者在2至15岁之间就有神经系统表现。许多人十几岁就坐轮椅了。
儿童期发作形式的神经系统症状和体征主要为锥体外系表现,包括肌张力障碍,构音障碍和僵硬。四肢肌张力障碍表现为行走困难,步 态迈步高(“公鸡步态”),肌张力障碍和伸肌痉挛疼痛。精细运动障碍会导致书写和绘画方面的问题,并且无法执行手的快速交替(运 动异常)。舌头肌张力障碍可导致构音障碍 [Quadri et al 2012, Tuschl et al 2012, Quadri et al 2015].
一位受累的患者表现了孤立的皮质脊髓束受累。发现痉挛性轻瘫的典型神经系统症状(例如,痉挛,反射亢进,伸肌足底反应)[Gospe et al 2000].
- 成年发病 Quadri et al [2012]报告了两个兄弟,分别在47岁和57岁时出现进行性步态障碍和运动迟缓。神经系统检查显示出帕金森氏症的特征,包括表情呆板,单调语音,轻度僵硬,整体运动迟缓,步态宽阔及姿势不稳,没有震颤,肌张力障碍或小脑或锥体束障碍的迹象。用左旋多巴和多巴胺激动剂治疗不能改善神经系统的表现。
在两名受累的患者中描述了感觉运动轴突性多发性神经病,具有迟发性神经系统症状[Quadri et al 2012].
高锰血症:所有受累的患者的全血锰浓度均升高。
- 由于数据有限,高锰血症的起病尚不清楚。
- 据记录,受累的儿童的全血锰浓度升高,年龄可早至五岁 。
- 但是,鉴于临床表现可以在出生头两年出现,因此预计高锰血症会在临床表现发作之前或同时发生。
- 必须排除因环境过度暴露(包括肠胃外营养)和晚期肝病患者获得性肝脑变性引起的高锰血症。请参阅 Differential Diagnosis.
- Quadri et al [2012]报道了一名受累的患者,其血液中锰的浓度仅一次微增。因此,单次测量锰浓度可能会产生误导,并且在临床怀疑强烈的情况下,建议重复测量。
注意:杂合子(即一种SLC30A10致病性变异的携带者)的血锰浓度在正常范围内或略有升高。 Gospe et al [2000]报告了杂合的亲本中的高血锰水平,临界水平为380 nmol / L,Tuschl et al [2012]报告了3个杂合亲本中的水平在380至649 nmol / L之间(正常:<320 nmol / L)。
红细胞增多症 迄今报告的所有受累的患者均在诊断时患有红细胞增多症。红细胞增多症可以在神经系统表现发作之前发生,因此,受影响的个体在接受正确的诊断之前通常会进行多次静脉切开术[Quadri et al 2012, Tuschl et al 2012]。从3岁开始就在受影响的儿童中描述了红细胞增多症。由于数据不足,不能排除较早出现的红细胞增多症。直到成年后期才出现神经系统症状的个体早在三十岁就患有红细胞增多症。在使用螯合疗法或铁治疗后,红细胞增多症可以消退。有证据表明,一名患者的红细胞增多症在疾病晚期阶段无需治疗即可治愈[Lechpammer et al 2014].
肝病 肝受累的范围从轻度的肝肿大到成年早期的肝衰竭[Tuschl et al 2012]。但是,已经报道了仅表现为肌张力障碍的纯神经系统表型[Quadri et al 2012].
在大多数受累的患者中,转氨酶轻度升高 [Quadri et al 2012, Tuschl et al 2012]。迄今为止,三名18至46岁受影响的人死于肝硬化并发症。作者所知的大多数受影响个体仍处于青少年或成年早期,因此没有长期随访数据。
即使在同一个家庭中也存在明显的表型变异:Quadri et al [2012]报道的两兄弟现年六十多岁,患有肌张力障碍而严重受累,但并未表现出肝脏受累。两人一生都具有正常的肝功能和肝超声检查。然而,尽管患病的妹妹神经系统受累最少,但她在三十岁左右发展为肝硬化,死于肝衰竭,享年46岁。
智力 在所有受累的人中看来都是正常的。 Quadri et al [2012]描述了一个出现认知和行为问题的患者,这些被认为与酒精有关。虽然已知环境锰暴露会引起认知和精神障碍(所谓的“锰疯狂”),包括情绪不稳,幻觉和强迫行为[Racette et al 2012],但在肌张力障碍/帕金森病,高锰血症红细胞增多症和慢性肝病患者中尚未观察到。
异食癖 几名受累的患者在童年早期有异食癖[Brna et al 2011; Brna,未发布的数据]。
肤色较深 一些受累的患者被描述为具有紫色或深色皮肤变色,其程度使得父母能够在临床症状出现之前区分患病和未患病的孩子[作者,未发表的数据]。
病理 对SLC30A10缺乏症患者进行的尸检研究表明,基底节的黄灰色斑纹与严重的神经元丢失,星形细胞增多,髓磷脂丢失,海绵体病和若丹宁阳性沉积有关,特别是在苍白球,而其他基底节受到影响程度较小。观察到了白质的胶质变性和皮质脊髓束的轴突损失[Lechpammer et al 2014].
基因型-表型相关
在十个与儿童或成人发病有关的不相关家庭中,已有十四个纯合性SLC30A10致病变异和一个纯合多外显子缺失 的报道[Quadri et al 2012, Tuschl et al 2012]。由于已知病例的数量很少,因此无法建立牢固的 基因型-表型相关性。然而,有趣的是,伴随成人成年帕金森病出现的同胞中 致病性变异接近SLC30A10的第四个(最后一个)外显子的末尾。该病原体变体((p.Gln412ArgfsTer26; c.1235delA)预计会引起移码,从而引入过早的终止密码子和缺少最后49个氨基酸的蛋白质的翻译[Quadri et al 2012]。这种特定的等位基因可能产生具有残留功能的蛋白质。
患病率
这种锰代谢的先天性错误直到最近才被发现。全世界共有来自十个家庭的28名受累的患者 [Quadri et al 2012, Tuschl et al 2012, Avelino et al 2014, Quadri et al 2015, Mukhtiar et al 2016]。患病率尚未确定。
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,没有其他表型与病原体SLC30A10相关。 但是,由SLC30A10致病性变异产生的完整表型谱可能还是未知的。
鉴别诊断
肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的鉴别诊断包括以下内容。
SLC39A14 deficiency (SLC39A14缺陷早发性帕金森病-肌张力障碍)。锰转运蛋白缺陷是由于锰摄入肝脏受损所致。由于脑锰沉积,受影响的个体还表现出高锰血症和迅速进展的儿童期帕金森氏肌张力障碍。脑部MRI表现与肌张力障碍/帕金森症,高锰血症,红细胞增多症和慢性肝病相同。显着特征是缺乏肝锰沉积而导致的肝脏疾病和红细胞增多症,可以通过肝脏MRI进行评估 [Tuschl et al 2016].
获得性高锰血症 过度暴露于锰会导致神经毒性,并导致“锰中毒”-锥体外系运动异常(肌张力障碍/帕金森综合症)的独特综合征,以及由于锰积累而导致的脑T1加权MR图像上基底神经节的高强度信号[Racette et al 2012].
- 在采矿和焊接行业环境暴露的工人中,吸入含锰的粉尘或烟雾,摄入被污染的饮用水的个人以及使用静脉内被高锰酸钾污染的美卡西酮的吸毒者 [Stepens et al 2008, Bouchard et al 2011, Racette et al 2012].
- 总肠胃外营养与锰毒性有关,因为绕过了肠道中锰吸收和随后肝排泄的控制机制[Chalela et al 2011].
- 在患有晚期肝硬化或门体系统分流的人中观察到获得性肝脑变性,其中锰的胆汁排泄受损会导致基底神经节中的锰蓄积,从而导致衰弱运动障碍[Meissner & Tison 2011].
Wilson disease威尔逊病
Parkinson disease 及其鉴别诊断包括:
- 非典型退行性帕金森综合症(多系统萎缩,进行性核上性麻痹)
- 血管帕金森病
- 药物诱发的帕金森症
- Huntington disease
- DRPLA(齿龈-前路尿道萎缩)
- 青少年帕金森病,包括parkin type of early-onset parkinsonism
肌张力障碍的遗传形式包括:
- DYT1 early-onset isolated dystonia DYT1早发性肌张力障碍(DYT-TOR1A)
- 多巴反应性肌张力障碍(DRD),包括多巴胺合成/转运疾病和影响四氢生物蝶呤合成/回收的疾病(请参阅 GTP Cyclohydrolase 1-Deficient Dopa-Responsive DystoniaGTP环水解酶1缺陷型多巴反应性肌张力障碍),
- 其他形式的原发性肌张力障碍和肌张力障碍加障碍(请参见Dystonia Overview)
与肌张力障碍有关的神经退行性疾病,包括:
- Pantothenate kinase-associated neurodegeneration 泛酸激酶相关神经变性(PKAN),一种具有脑铁蓄积的神经变性(NBIA)
- 由PANK2,PLA2G6,C19orf12,FA2H,ATP13A2,WDR45,COASY,FTL,CP和DCAF17中的致病性变异引起的其他形式的NBIA(请参阅NBIA Overview)。
- Niemann-Pick disease type C 尼曼-匹克病(C型)(与肝病相关)
- 有机酸血症(即戊二酸,甲基丙二酸,丙酸,3-羟基异丁酰-CoA水解酶缺乏症)
- Mitochondrial respiratory chain disorders线粒体呼吸链疾病
- 丙酮酸脱氢酶复合体缺乏症
- Gaucher disease高雪氏病
脑瘫 家族性和后天性痉挛性截瘫(请参见 Hereditary Spastic Paraplegia Overview)
处理
初步诊断后的评估
为了确定诊断为肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的个体的疾病程度和需求,建议进行以下评估:
- 对肌张力障碍,帕金森病和痉挛症进行神经系统检查,包括评估步行和言语
- 脑部核磁共振
- 肝评估,包括肝功能检查,肝超声检查和肝活检(如果有)。建议咨询肝脏科医生。
- 建立全血锰水平
- 物理治疗,职业治疗和/或言语治疗的评估
- 吞咽和营养状况评估
- 咨询临床遗传学家和/或遗传咨询师
表现治疗
依地酸钙钠螯合疗法 定期静脉滴注乙二胺四乙酸二钠钙的螯合疗法可以稳定血锰水平,改善神经系统症状,并制止肝病进展[Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012].
短期 个体对依地酸二钠钙的反应是通过一次为期五天的疗程,每天两次, 剂量为20 mg / kg /剂量(由250 mL的0.9%氯化钠组成,在1小时内静脉内给药)。每天测量血浆锰浓度和24小时尿锰水平。其他监测包括:血清中的电解质,钙,磷酸盐和镁的浓度;肾和肝功能;全血细胞计数;以及锌,铜和硒等微量金属的血清浓度。
注意:为避免血钙过低,需要在至少一小时内缓慢使用乙二胺四乙酸钙钠输液。如果钙水平(校正白蛋白浓度)低,则应在更长的时间范围内(即> 3小时)进行输注。
长期 如果螯合疗法在短期内被证明是有效的,则有望每月使用五天的依地酸二钠钙疗程(静脉内20 mg / kg /剂量2x /天)降低血锰水平并使血红蛋白浓度和铁指数正常化[Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012]。螯合疗法应终生持续。在治疗期间,每两个月进行一次监测,包括:电解质,钙,磷酸盐,镁的血清浓度;肾和肝功能;全血细胞计数;以及锌,铜和硒等微量金属的血清浓度(请参见Prevention of Secondary Complications)。
铁疗法 铁是肠锰吸收的竞争性抑制剂。因此,口服补铁(尽管血清铁水平正常)可以降低血锰水平并解决红细胞增多症[Tuschl et al 2008, Tuschl et al 2012]。人们认为,在受累的患者中发现高血清转铁蛋白水平可降低锰中毒的风险。
注意:在接受铁补充剂的患者中,需要经常(每隔约3个月)监测铁指数(请参阅Prevention of Secondary Complications)
肝移植 患有晚期肝病的个体应考虑进行肝移植。但是,尚未针对患有这种疾病的个体进行尝试,因此没有可用的数据。
其他
肌张力障碍可导致身体畸形和疼痛。应提供物理治疗,职业治疗和/或言语治疗。尝试使用抗痉挛药物和左旋多巴的对症治疗取得了有限的成功。
预防主要表现
螯合疗法和补铁可以预防无症状的受累的同胞的原发疾病表现(请参见Treatment of Manifestations)。
预防继发并发症
尽早开始理疗和骨科治疗的目的是预防挛缩并保持下肢活动。根据需要,应为个人提供适应性辅助工具(例如,步态异常的助行器或轮椅)和辅助通讯设备。
尝试用抗痉挛药物对症治疗,包括巴氯芬和肉毒杆菌毒素以及左旋多巴,但收效甚微。
吞咽评估和定期饮食评估可确保营养。一旦不再维持足够的口服饮食,应考虑胃造口管放置。为了防止吸入性肺炎,可能需要胃饲管和/或气管切开术。
仔细监测可以减少螯合疗法和/或铁补充剂引起并发症的可能性。
监测
每隔三个月就要密切监测肝功能和疾病指标,例如血红蛋白,铁指数和全血锰。
应向神经科医生和肝病医生进行常规随访,以对MRI脑,临床表现(例如肝功能恶化)和肝脏超声检查进行重复评估并监测治疗情况。
依地酸二钠钙螯合疗法的不良反应包括低血钙,肾毒性,微量金属和维生素缺乏症以及血小板减少症和白细胞减少症 [Lamas et al 2012].
在基线和此后每月评估全血细胞计数和肾功能,包括尿液分析。监测可以稳定剂量延长至每隔一个月一次。此外,还需要监测以下内容:痕量金属水平,包括锰,锌,铜和硒;肝功能;电解质钙,镁和磷酸盐的浓度;和铁状态。根据需要提供微量金属补充剂。
在以下情况下,可能需要中止治疗:
- 白细胞计数<3.5x10^9/L
- 中性粒细胞<2.0x10^9/L
- 血小板<150x10^9/L
- > 2+蛋白尿> 1次时(且无感染迹象)
以上临界值基于D-青霉胺治疗的指导原则[Chakravarty et al 2008]。由于乙二胺四乙酸钙钠的螯合治疗可以预防SLC39A14相关早发性帕金森病-肌张力障碍的死亡率和发病率,因此较低的临界值可能是可以接受的。需要仔细权衡临床治疗的益处与每个受累的患者的不良反应。
补铁的毒性 为了避免铁毒性,需要定期监测血清铁和总铁结合能力。如果血清铁超过总铁结合能力的80%,应停止或减少铁的补充。
避免的药物/情况
应当避免锰含量很高的食物(丁香,藏红花,坚果,贻贝,黑巧克力以及南瓜,芝麻和葵花籽)。
评估处于危险中亲戚
评估先证者的明显无症状同胞是适当的,以便尽早发现那些将从迅速采取治疗措施和预防措施中受益的同胞。螯合疗法和补铁可以潜在地预防无症状个体的原发疾病表现。
评估包括:
- 分子遗传学测试是否已知该家族中的SLC30A10致病变异;
- 如果尚不清楚该家族的致病变异,则应定期监测全血锰浓度和血红蛋白。
有关与遗传咨询目的相关的高危亲属测试的问题,请参阅Genetic Counseling。
孕期管理
对于受累的胎儿,不建议进行产前治疗,因为这种疾病不会在儿童早期出现。
对于受累的母亲,没有有关妊娠管理的数据或信息。
正在调查的疗法
搜索美国的ClinicalTrials.gov和欧洲的www.ClinicalTrialsRegister.eu,以获取有关多种疾病和状况的临床研究信息。注意:可能没有针对该疾病的临床试验。
遗传咨询
遗传咨询是为个人和家庭提供有关遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分介绍了遗传风险评估以及家族史和遗传测试的使用,以阐明家族成员的遗传状况。 本部分的目的不是要解决个人可能面临的所有个人,文化或伦理问题,也不能替代遗传专家的咨询。 —编者。
遗传模式
肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病以 常染色体隐性遗传的方式遗传。
家庭成员的风险
先证者的父母
先证者的同胞
先证者的后代
其他家庭成员。先证者’父母的每个同胞都有被SLC30A10 致病性变异携带者的风险。
携带者(杂合子)检测
对高危亲属进行携带者测试需要事先鉴定该家族中的SLC30A10致病变异。
相关的遗传咨询问题
有关评估以早期诊断和治疗为目的的高风险亲戚的信息,请参阅《管理,Evaluation of Relatives at Risk》。
家庭计划
- 确定遗传风险,阐明携带者身份以及讨论是否可以进行 prenatal testing的最佳时间是在怀孕之前。
- 适当地向受累的,携带者或有携带者风险的年轻人提供遗传咨询(包括对后代和生殖选择的潜在风险的讨论)。
DNA库是DNA(通常从白细胞中提取)的存储,以备将来使用。由于测试方法和我们对基因,等位基因变异和疾病的理解将来可能会改善,因此应考虑受累的患者的DNA储藏。
产前检查和植入前遗传学诊断
一旦在一个受累的家庭成员中发现了SLC30A10的致病变异,就可以对妊娠风险增加进行prenatal testing ,并进行肌张力障碍/帕金森综合症,高锰血症,红细胞增多症和慢性肝病的植入前遗传诊断。
资源
GeneReviews工作人员选择了以下特定疾病和/或综合保护组织和/或注册表,以保护患有这种疾病的个人及其家人。 GeneReviews对其他组织提供的信息概不负责。 有关选择标准的信息,请单击 here.
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- American Liver Foundation75 Maiden LaneSuite 603New York NY 10038Phone: 800-465-4837 (Toll-free HelpLine); 212-668-1000Fax: 212-483-8179Email: info@liverfoundation.org
- Dystonia Medical Research FoundationOne East Wacker DriveSuite 1730Chicago IL 60601-1905Phone: 800-377-3978 (toll-free); 312-755-0198Fax: 312-803-0138Email: dystonia@dystonia-foundation.org
分子遗传
分子遗传学和OMIM表中的信息可能不同于GeneReview中的其他信息:表中可能包含最新信息。 —编者。
Table A
肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病:基因和数据库
| 基因 | 染色体位点 | 蛋白 | 基因数据库 | HGMD | ClinVar |
|---|---|---|---|---|---|
| SLC30A10 | 1q41 | Zinc transporter 10 | SLC30A10 @ LOVD | SLC30A10 | SLC30A10 |
Table B
肌张力障碍/帕金森病,高锰血症,红细胞增多症和慢性肝病的OMIM条目(View All in OMIM)
基因结构 SLC30A10具有四个外显子。 有关基因和蛋白质信息的详细摘要,请参见 Table A,基因。
良性变异 在SLC30A10中已报道了二十种正常变异。
致病变异 在已经鉴定出受累的患者的所有15个家庭中,均记录了纯合的致病变异(见 Table 2)。 这些包括 错义和 nonsense 的变异,以及涉及至多两个SLC30A10外显子的单碱基和较大的缺失。 据预测,致病性变异要么(1)由于移码和过早的终止密码子或大 缺失而导致蛋白被截断,要么(2)影响蛋白的进化高度保守区域。 因此,这些序列改变对蛋白质功能具有有害作用 [Quadri et al 2012, Tuschl et al 2012].
Table 2MIM条目
选定的SLC30A10致病变异
| 起病 | DNA核苷酸改变 | 预测的蛋白质变化 | 参考序列 |
|---|---|---|---|
| 儿童1-5 | g.chr11:218.057.426_218.158.564del101139 | GRCh38 NM NP | |
| c.266T>C | p.Leu89Pro | ||
| c.292_402del | p.Val98_Phe134del | ||
| c.314_322del | p.Ala105_Pro107del | ||
| c.460C>T | p.Gln154Ter | ||
| c.492del | p.Gly165AlafsTer27 | ||
| c.496_553del | p.Ala166GlnfsTer7 | ||
| c.500T>C | p.Phe167Ser | ||
| c.507delG | p.Pro170LeufsTer22 | ||
| c.585del | p.Thr196ProfsTer17 | ||
| c.765_767del | p.Val256del | ||
| c.922C>T | p.Gln308Ter | ||
| c.1006C>T | p.His336Tyr | ||
| c.1046T>C | p.Leu349Pro | ||
| 成人1 | c.1235delA | p.Gln412ArgfsTer26 |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews员工尚未独立验证变异的分类。
关于术语的注释:GeneReviews遵循人类基因组变异学会 (varnomen
- .hgvs.org )的标准命名约定。 有关命名法的说明,请参见Quick Reference。- 1.
- 2.
- 3.
- 4.
- 5.
正常基因产物 SLC30A10是阳离子扩散促进剂(CDF)系列的SLC30溶质载体亚家族的成员。人SLC30A10是485个氨基酸的蛋白质 [Tuschl et al 2012]。该蛋白是在肝和脑中表达的跨膜锰转运蛋白,可促进锰在细胞表面的外排 [Leyva-Illades et al 2014]. Quadri et al [2012] 显示,SLC30A10的表达及其编码蛋白的水平在体外受到细胞外锰水平的严格控制。暴露于高锰浓度会导致SLC30A10 mRNA和蛋白质表达显着增加。
异常基因产物. 受累的患者中描述的异常基因产物.SLC30A10致病变异(Table 2)对蛋白质功能具有有害作用。虽然在锰敏感酵母细胞中表达的野生型SLC30A10可以在高锰浓度下挽救生长,但具有错义和nonsense 序列变化的SLC30A10无法恢复对锰的抗性 [Tuschl et al 2012]。此外,SLC30A10致病性变异导致受影响个体的肝脏和脑组织免疫反应性丧失,并且无法转运至细胞表面[Quadri et al 2012, Lechpammer et al 2014, Leyva-Illades et al 2014]。在人类中,SLC30A10功能受损会导致锰在肝和脑中积累 [Gospe et al 2000, Lechpammer et al 2014].
参考文献
引用文献
- Avelino MA, Fusão EF, Pedroso JL, Arita JH, Ribeiro RT, Pinho RS, Tuschl K, Barsottini OG, Masruha MR. Inherited manganism: the "cock-walk" gait and typical neuroimaging features. J Neurol Sci. 2014;341:150 - 2. [PubMed: 24746291]
- Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur MÈ, Bouffard T, Limoges E, Bellinger DC, Mergler D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect. 2011;119:138 - 43. [PMC free article: PMC3018493] [PubMed: 20855239]
- Brna P, Gordon K, Dooley JM, Price V. Manganese toxicity in a child with iron deficiency and polycythemia. J Child Neurol. 2011;26:891 - 4. [PubMed: 21596707]
- Chakravarty K, McDonald H, Pullar T, Taggart A, Chalmers R, Oliver S, Mooney J, Somerville M, Bosworth A, Kennedy T. BSR/BHPR guideline for disease-modifying anti-rheumatic drug (DMARD) therapy in consultation with the British Association of Dermatologists. Rheumatology. 2008;47:924 - 5. [PubMed: 16940305]
- Chalela JA, Bonillha L, Neyens R, Hays A. Manganese encephalopathy: an under-recognized condition in the intensive care unit. Neurocrit Care. 2011;14:456 - 8. [PubMed: 21174173]
- Ebert BL, Bunn HF. Regulation of the erythropoietin gene. Blood. 1999;94:1864 - 77. [PubMed: 10477715]
- Gospe SM Jr, Caruso RD, Clegg MS, Keen CL, Pimstone NR, Ducore JM, Gettner SS, Kreutzer RA. Paraparesis, hypermanganesaemia, and polycythaemia: a novel presentation of cirrhosis. Arch Dis Child. 2000;83:439 - 42. [PMC free article: PMC1718535] [PubMed: 11040156]
- Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Drisko JA, Lee KL. Design of the Trial to Assess Chelation Therapy (TACT). Am Heart J. 2012;163:7 - 12. [PMC free article: PMC3243954] [PubMed: 22172430]
- Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr. Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75:608 - 12. [PubMed: 24599576]
- Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci. 2014;34:14079 - 95. [PMC free article: PMC4198546] [PubMed: 25319704]
- Meissner W, Tison F. Acquired hepatocerebral degeneration. Handb Clin Neurol. 2011;100:193 - 7. [PubMed: 21496578]
- Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with dystonia, polycythemia and cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain Dev. 2016;38:862 - 5. [PubMed: 27117033]
- Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467 - 77. [PMC free article: PMC3309204] [PubMed: 22341971]
- Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, Kori I, Hattiholi V, Jain P, Aneja S, Kumar A, Gulati P, Goel M, Talukdar B, Bonifati V. Manganese transport disorder: novel SLC30A10 mutations and early phenotypes. Mov Disord. 2015;30:996 - 1001. [PubMed: 25778823]
- Racette BA, Aschner M, Guilarte TR, Dydak U, Criswell SR, Zheng W. Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology. 2012;33:881 - 6. [PMC free article: PMC3350837] [PubMed: 22202748]
- Stamelou M, Tuschl K, Chong WK, Burroughs AK, Mills PB, Bhatia KP, Clayton PT. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord. 2012;27:1317 - 22. [PMC free article: PMC3664426] [PubMed: 22926781]
- Stepens A, Logina I, Liguts V, Aldins P, Eksteina I, Platkājis A, Mārtinsone I, Tērauds E, Rozentāle B, Donaghy M. A Parkinsonian syndrome in methcathinone users and the role of manganese. N Engl J Med. 2008;358:1009 - 17. [PubMed: 18322282]
- Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Kling Chong WK, Mills PB. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90:457 - 66. [PMC free article: PMC3309187] [PubMed: 22341972]
- Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, Clayton PT. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia-A new metabolic disorder. J Inherit Metab Dis. 2008;31:151 - 63. [PubMed: 18392750]
- Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. [PMC free article: PMC4894980] [PubMed: 27231142]
本章节注解
更新历史
- 9 February 2017 (ha) 全面更新实时发布
- 11 September 2014 (me) 全面更新实时发布
- 30 August 2012 (me) 实时发布
- 1 June 2012 (kt) 初稿提交