
概要
- 先天性心脏病(74%的个体都存在),特别是圆椎动脉干畸形(法洛四联症,主动脉弓离断,室间隔缺损,和永存动脉干)
- 腭异常(69%),特别是腭咽不全、粘膜下腭裂、悬雍垂裂和腭裂
- 特殊面容(在大多数北欧人种病人中呈现)
- 学习困难(70%-90%)
- 免疫缺陷(无论临床表现如何)(77%)
其他临床表型包括:
- 低钙血症(50%)
- 明显的喂养和吞咽问题;便秘伴或不伴胃肠道结构异常(肠道畸形,肛门闭锁,以及巨结肠疾病)
- 肾脏异常(31%)
- 听力丧失(包括传导性和感觉神经性)
- 喉气管食道异常
- 生长激素缺乏
- 自身免疫性疾病
- 癫痫发作(特发性或与低钙血症相关)
- 包括脊髓栓系在内的中枢神经系统异常
- 骨骼畸形(脊柱侧凸伴或不伴椎体异常,马蹄内翻足,多趾畸形,颅缝早闭)
- 眼部异常(斜视、角膜后胚胎环、视网膜血管扭曲、巩角膜和无眼畸形)
- 牙釉质发育不全
- 恶性肿瘤(罕见)
发育迟缓(尤其是语言出现的延迟)、智力障碍和学习困难(非语言学习障碍,语言智商明显高于行为智商)是常见的。
在大约20%的儿童中存在自闭症或自闭症谱系障碍,25%的成年人中存在精神疾病(尤其是精神分裂症);然而,注意力缺陷障碍、焦虑、持续言语和社会交往的困难也很常见。
22q11.2缺失综合征通过荧光原位杂交(FISH)、多重连接探针扩增(MLPA)技术或染色体微阵列(CMA)技术对22号染色体上的亚显微缺失进行诊断。目前,在临床发现的22q11.2缺失综合征患者中,只有不到5%的患者的常规细胞遗传学检测和FISH检测为正常结果;然而,这一数字可能会随着在DGCR(DiGeorge染色体关键区域)中存在不典型的或嵌合缺失而改变,这些病例不包含可由微阵列或MLPA技术而检测到的N25或TUPLE FISH探针覆盖的区域,。
管理。
对症处理:根据受累个体的特殊需求,采用多学科组合的方法(可能包括以下的任意组合:过敏学,听力学,心脏病学,心脏手术学,儿童发展心理学,牙科,内分泌学,ENT,喂养 学,胃肠病学,儿科,普通外科,免疫学,临床遗传学,神经病学,神经外科学,眼科学,骨科学,耳鼻喉科学,整形外科学,精神病学,肺科学,风湿病学,语言 学和泌尿科学)进行治疗。在喂养困难时可采用补充钙质,并转由内分泌科医生进行低钙血症和甲状腺监测;使用特殊策略(吃饭时调整勺子容量,治疗胃食管反流 和胃肠运动障碍);由于有语言发育迟缓的风险,在确诊后应立即开始进行教育干预和言语治疗/引进手语治疗。生长激素缺乏与针对普通人群的治疗方法相同。免 疫缺陷需要积极治疗感染;基本不需要预防性抗生素、静脉注射免疫球蛋白治疗或胸腺移植。早期诊断和干预精神疾病可以改善长期预后。并发症的预防:不推荐使用活疫苗对有淋巴细胞异常的婴儿进行免疫接种;在儿童期接种活疫苗前应重新评估儿童的免疫状况;可通过抗体水平检测来评估免疫接种的效果;推荐使用辐照过的血液制 品直到确认免疫系统正常;应在术前和术后测量血清钙离子浓度以避免低钙性癫痫发作;在进行涉及咽部的外科手术之前应对颈动脉进行评估;在腺 样体切除术前应考虑可能对语言功能造成的影响;在外科手术或运动项目中,考虑颈椎异常在颈部过度伸展时的影响;在进行咽部手术操作时应考虑术前和术后对睡眠的影响;在手术前应对血 小板体积和功能进行评估。
监测:在“一个个系统”的基础上按照需求进行随访,但包括对血清钙离子和甲状腺状态的常规重新评估;在接种活病毒疫苗前重新评估免疫状态;每年进行全血细胞计数和 分类;学龄前的眼科评估;评估鼻音语言质量;入学前进行听力评估;监测脊柱侧弯;常规牙科保健;定期进行说话、语言和发育评估以提供适当的补救。
有害因素/环境:碳酸饮料和酒精摄入可能会加剧低钙血症。咖啡因的摄入可能会导致焦虑或加剧焦虑。
GeneReview范围
| 22q11.2缺失综合征:包含的症状 |
|---|
|
同义词和别名请参见Nomenclature.
诊断
临床诊断
22q11.2缺失综合征(22q11.2DS)的疑似患者包括以下临床表现:
- 先天性心脏病(特别是圆锥缺陷)
- 腭异常(特别是腭咽发育不全[VPI])
- 低钙血症
- 免疫缺陷
- 学习困难(特别是非语言学习障碍,在语言智商[VIQ]和行为智商之间有超过10分的差距[PIQ]:VIQ >PIQ)
- 特殊面容
较少见的功能异常包括:
- 重度吞咽困难
- 生长激素缺乏
- 自身免疫性疾病(血小板减少症,青少年类风湿性关节炎,Grave’s 病,白癜风,中性粒细胞减少,溶血性贫血)
- 听力丧失(包括感觉神经性和传导性)
- 精神疾病
- 自闭症
其他有助于诊断的重要结构异常:
- 骨骼系统异常:手部轴前及轴后性多指,足部轴后性多趾,多余的肋骨,半椎体和颅缝早闭(冠状缝,人字缝)
- 泌尿生殖系统异常:肾缺如、肾积水、多囊/发育不良肾、复肾、马蹄肾、子宫缺如、尿道下裂、腹股沟疝、隐睾
- 喉气管食道异常:血管环、喉蹼、喉气管软化、舌下狭窄
- 眼科异常:视网膜血管扭曲,上睑下垂,角膜后胚胎环,眼组织缺损,白内障,无眼畸形,斜视
- 中枢神经系统异常:小脑萎缩,多小脑回,大脑外侧裂增宽,神经管缺陷,栓系脊髓,无诱因性癫痫发作,歪嘴哭面容
- 胃肠道异常:肛门前置/闭锁,食管闭锁,空肠闭锁,副脾疝,脐疝,膈疝,小肠旋转畸形,巨结肠疾病
- 耳前皮赘和/或凹陷
肿瘤:
- 肝母细胞瘤、肾细胞癌、肾母细胞瘤和神经母细胞瘤
检测
细胞遗传学分析:
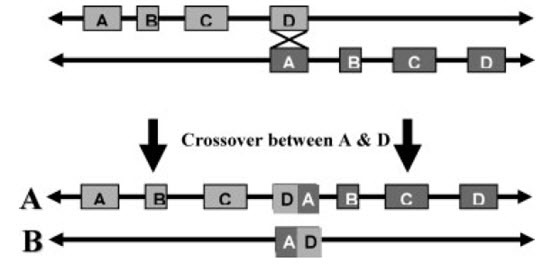
导致该综合征最常见的原因是染色体22q11.2区域上的一个300万碱基对(Mb)的缺失,这一区域由低拷贝数重复(LCRs)组成,该区域被划分为A-D区(请参阅分子遗传学部分)。该缺失是在精子发生或卵子发生时由非等位基因重组导致的(请参阅图1)。
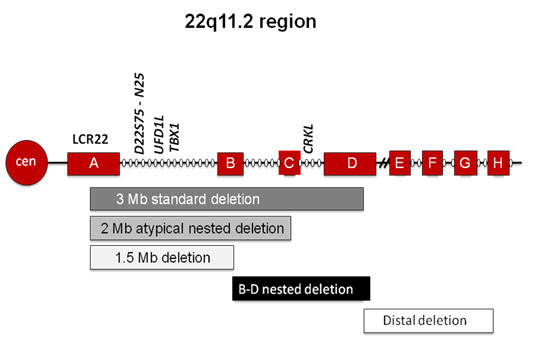
85%的患者存在从A延伸到D,包含TBX1基因的最常见的缺失(图2),TBX1基因是一个被认为与该综合征典型临床表型相关的基因,尤其是心脏异常。其余15%受影响的个体存在非典型的“嵌合”缺失。到目前为止,非典型性缺失包括A-B、B-D或C-D的缺失。这些非典型缺失可以采用染色体微阵列(CMA)或多重连接探针扩增(MLPA)等高分辨率的技术进行识别。在非典型的缺失中,34%不包含A-B区域。商业用的荧光原位杂交(FISH)探针N25和TUPLE,位于A-B区内,与TBX1基因相近;因此,不包含A-B的非典型缺失无法被商业用的FISH探针识别(图2)。
在临床上,有一小部分(<1%)的22q11.2DS个体存在涉及22q11.2区域的染色体重排,比如22号染色体和另一条染色体的易位。
分子遗传学检测
基因:在DiGeorge染色体区域(DGCR)中缺失基因是唯一已知与22q11.2DS相关的基因异常 [Driscoll et al 1992, Wilson et al 1992, Desmaze et al 1993, Driscoll et al 1993]。
临床检测
FISH:用 于22q11.2的FISH分析的两种探针是TUPLE1和N25。这两种探针进行FISH分析的检出率是等效的。然而,FISH检测中这两种探针 都不够敏感,无法检测22q11.2区域中较小片段的缺失(< 40kb)。现在大多数的缺失都是通过CMA或MLPA技术进行检测的,这两种方法都可以检测到FISH检测不到的较小片段缺失。然而,这些检测技术并不 能检测缺失区域内的基因的致病突变。
注:探针ARSA与染色体位点22q13.3杂交,也可用于检测,但仅用于对照。
重复/缺失分析是通过基因组DNA的序列分析鉴定不易检测到的缺失/重复。A-D区2.54Mb的常见缺失可通过任何检测缺失区域内基因组序列的拷贝数的分子检测方法检测到(见表1,脚注2)。全基因组或靶向检测方法均可采用(详细内容参见缺失区域分子遗传学部分):
注:是否能确定缺失片段的大小取决于22q11.2区域内探针的数量和分布。使用单个FISH探针(商业实验室的惯例)不能确定缺失片段大小。
表1
检测特性:检测敏感性和特异性请参阅 Clinical Utility Gene Card [Schwinger et al 2010]。
检测结果说明
临床特点
临床描述
一项针对250名22q11.2缺失综合征(DS)患者(48%为男性;52%为女性)的调查结果总结如下[McDonald-McGinn et al 1999]。根据一个大型多学科22q11缺失综合征中心对另外750名患者进行评估后的未发表数据,以下各表型所占的百分比仍然相同[作者2012年未公布的数据]。
心脏:在这一综合征中,有74%的受累个体存在先天性心脏缺陷,这是导致死亡的主要原因(在死亡个体中的比例>90% ) [McDonald-McGinn et al 2001]。最常见的异常是流出道的圆锥动脉干缺陷,如表2所示。值得注意的是,有一部分受影响的个体被发现有扩张的主动脉根部[John et al 2009]。目前尚不清楚受影响的个体的自然病史,但正在进行调查。
表2
222名22q11.2缺失综合征患者的心脏症状
| 心脏症状 | 占患者百分比 |
|---|---|
| 法洛四联症 (TOF) | 20% |
| 主动脉弓离断 (IAA) | 13% |
| 室中隔缺损 (VSD) | 14% |
| 永存动脉干 (TA) | 6% |
| 血管环 | 5.5% |
| 房中隔缺损 | 3.5% |
| 房间隔缺损;房间隔缺损 | 4% |
| 其他 1 | 10% |
| 正常 | 24% |
1.
左心房发育不良综合征;肺动脉瓣狭窄;右心室双出口;二叶主动脉瓣;动静脉管畸形;大血管移位;右肺动脉闭锁;主动脉根部扩张;异常锁骨下动脉
腭部:69%的22q11.2缺失患者有腭异常(表3), 最常见的是腭咽功能不全(VPI),可能是结构问题(短上颚),功能问题(下咽肌肌张力减退),或两者的结合。粘膜下腭裂和/或悬雍垂裂也相当普遍,而明 显的腭裂和唇腭裂较少见。经常有因心脏缺陷而被初诊为22q11.2缺失的儿童后来被发现存在未识别的有临床意义的腭咽功能不全[McDonald-McGinn et al 1997a]。值得注意的是,腭异常的报告发生率因多种因素而异,这些因素包括诊断技术,求诊断的意愿,患者进行评估的年龄,以及任何单一中心的固有测量偏差[Kirschner 2005]。大约17%的人没有腭部异常。
表3
22q11.2缺失综合征的腭部症状
| 腭部症状 | 占患者百分比 |
|---|---|
| 腭咽功能不全(VPI) | 27% |
| 粘膜下腭裂(SMCP) | 16% |
| 明显的腭裂 | 11% |
| 悬雍垂裂 | 5% |
| 唇裂/唇腭裂 1 | 2% |
| 婴儿腭咽功能不全 2 | 8% |
| 需随访/太年轻,无法充分评估 3 | 14% |
| 正常 | 17% |
1.
单边或双边
2.
“婴儿腭咽功能不全”或由病史诊断的隐蔽性粘膜下腭裂(鼻返流、中耳炎),在儿童年龄太小,不能提供言语证据以确诊时进行体格检查或 鼻内窥镜检查(在哭泣和吞咽过程中检测不完全闭合的情况)
3.
没有明显的异常,但儿童太小不能提供言语证据
喂养:大约36%的儿童有严重的进食困难,通常是严重的吞咽困 难,需要鼻胃管喂食和/或胃造口管放置。进食困难与心脏缺陷及腭部异常无关。对这些儿童的进一步评估常常发现鼻咽部返流较多,环状咽肌突出,异常的咽部闭 合和/或憩室。因此,许多儿童潜在的喂养问题是在咽食管区域,由第三和第四咽袋产生。
吞咽异常伴或不伴呼吸困难可能会被错误地归因于腭部或心脏异常,而不是归因于与蠕动异常及口咽和食管吞咽阶段异常。呼吸系统疾病或反复的肺部感染和反应性呼吸道疾病[Eicher et al 2000]应该被认为是一种可能的原因。便秘是大多数人的慢性特征。此外,结构异常如肛门闭锁、小肠扭转畸形、肠道不旋转、先天性膈疝、食道闭锁、气管食管瘘、巨结肠疾病、血管环引起的喂养困难都被报道过,可以导致严重的喂养和吞咽问题,以及在某些情况下可导致便秘[Digilio et al 1999; Kilic et al 2003; D McDonald-McGinn 2010, 未公布的数据]。
免疫功能:免疫缺陷是胸腺发育不全的结果。由于胸腺的作用是支持T细胞的成熟,受损T细胞的产生是主要缺陷。T细胞功能缺陷和抗体缺陷较不常见,为T细胞生产异常而继发的[Sullivan 2004]。
与没有缺失的对照组相比,22q11.2DS新生儿的胸腺谱系细胞数量明显减少;然而,随着时间的推移,T细胞的生产也有了改善。在一项研究中,儿童在T细胞生产中的严重缺陷在生命的第一年得到了改善[Sullivan et al 1999]。因此,在T细胞数量上有轻微下降的个体通常对病原体有正常的防御能力[Sullivan 2004]。
在60名受影响的6个月龄以上的儿童免疫功能研究中,77%被认为存在免疫缺陷,不论他们的临床表现如何。67%有T细胞生产受损,19%有T细胞功能受损,23%有体液缺陷,13%有IgA缺陷[Smith et al 1998, Sullivan et al 1998, Sullivan 2004]。
与22q11.2DS相关的其他表型如吸入性肺炎、腭功能障碍和胃食管反流都可能导致复发性感染,特别是在患有先天性心脏病的患者中。此外,吞咽困难可导致营养不良,进一步损害细胞免疫功能。因此,年龄较大的儿童和成人事实上继续存在感染,包括25% - 33%的复发性鼻窦炎或中耳炎,4% - 7%的复发性下呼吸道感染[Jawad et al 2001]。然而,尽管存在这些问题,很少有学龄儿童需要积极治疗他们的免疫缺陷[Sullivan 2004]。
在22q11.2缺失综合征患者中,免疫球蛋白水平通常是正常的,尽管可以注意到轻微的免疫球蛋白异常。低丙种球蛋白血症通常会在一岁之内得到控制,5岁以后高丙种球蛋白血症可能会发生。虽然大部分受影响的个体具有正常的抗体功能和抗体的作用,但也有一些有功能抗体缺陷。复发性的肺感染患者通常有免疫球蛋白异常,特别是对肺炎球菌多糖疫苗的抗体反应受损[Gennery et al 2002, Sullivan 2004]。
自体免疫疾病在22q11.2DS中是常见的。然而,它与严重的T细胞功能障碍并不相关,但可以导致一系列儿科疾病。自体免疫性细胞瘤和少年类风湿 关节炎(JRA)似乎是最常见的,可能比一般人群发病率高20-100倍。JRA通常是多关节的,可能很难治疗。自身免疫性甲状腺疾病和其他自身免疫性异 常也已被报道,很可能T细胞缺陷与其他诱发因素协同作用(如:主要组织相容性复合体)引起自身免疫性疾病。选择性IgA缺乏症可能发生在10%有缺失的个体中,而且在包括JRA在内的自身免疫性问题中尤为常见[Kawame et al 2001, Sullivan 2004]。
甲状旁腺功能:17%-60%的22q11.2DS患者有低钙 血症,在新生儿期通常是最严重的。钙稳态通常随着年龄的增长而正常化,尽管在儿童期晚期和/或青春期或怀孕期间低钙血症可能会复发。在某些情况下,接受持 续治疗的婴儿的低钙血症可能直到学龄时才能随着确诊22q11.2DS被诊断出来,而至少有一个无症状的成人在第四十岁左右时出现副甲状腺低能症[Kapadia et al 2008; M Eagen,个人沟通]。
颅面部:颅面部症状包括耳部异常、鼻畸形、闭眼眼睑、眼距过宽、唇腭裂、歪嘴哭面容、颅缝早闭[Gripp et al 1997, McDonald-McGinn et al 2001]。然而,这些症状以及其他面部特征,如长脸和颧骨扁平,都是可变的。事实上,一些人无法根据他们的面部特征提供诊断依据,尤其是非裔美国人[McDonald-McGinn et al 1996, McDonald-McGinn et al 2005]。
眼睛:对33位眼部异常患者的前瞻性评估显示,上眼睑悬垂 (41%)、上睑下垂(9%)、下眼睑悬垂(6%)、内眦皱褶(3%)和双行睫(从睑板腺上的睫毛异常生长)(3%)。其他发现包括角膜后胚胎环 (69%)、分离的角膜神经(3%)、巩角膜(3%)、深虹膜隐窝(10%)、视网膜血管迂曲(58%)、小视神经(7%)和倾斜的视盘(3%)。弱视为 13%,斜视为6%。在12% - 32%的对照组中观察到后角膜后胚胎环,在22q11.2DS患者中的发病率和Alagille综合征(89%)[Krantz et al 1997]几乎一致。散光、近视、远视的发生率与一般人群相同。少数人患有白内障和眼组织残缺[Forbes et al 2007];最近在一小部分人中发现了无眼畸形[作者,未发表的数据]。
耳鼻喉:耳部异常包括耳轮过度折叠;杯状且突起的小耳朵;耳前凹陷或皮赘,外耳道狭窄。鼻根突出,球型鼻尖,鼻部发育不全,鼻凹陷/鼻尖分叉较常见[Gripp et al 1997]。血管环导致的喘鸣、喉软骨软化病、喉蹼、喉部、喉闭锁、舌下狭窄等均可发生。慢性中耳炎和慢性鼻窦炎很常见。感觉神经性和传导性听力丧失均已有报道。也观察到气管食管瘘和食管闭锁的发生[M . Digilio 1999,未发表的数据]。
中枢神经系统:尽管大多数22q11.2DS患者在婴儿期有肌张力减退和学习障碍的历史[Moss et al 1995],但特定的神经症状并不常见。在一些研究中,有近50%的受影响的个体被报道存在小头畸型[Kobrynski & Sullivan 2007]。但根据作者的研究,129名患者中只有18%存在明显的中枢神经系统症状[作者,未发表的观察]。癫痫发作被认为在某些个体中最常见,但并非总是如此,与低血钙有关。在一项研究中,7%(27/383)的22q11.2DS患者存在无诱因发作 [Kao et al 2004]。
一些人存在歪嘴哭面容[Cayler 1969, Levin et al 1982, Silengo et al 1986, Sanklecha et al 1992, Giannotti et al 1994],这可能是进行诊断的独立线索。
小脑的共济失调和萎缩比较少见[Lynch et al 1995]。
其他CNS异常包括不明意义的多囊性脑白质病变和外侧裂周区发育不良[Bingham et al 1997],垂体发育不全和多小脑回(请参阅Polymicrogyria Overview)。
近期有研究利用功能性核磁共振扫描,结果显示与同样年龄和性别的对照组相比,后脑容量显著降低,左侧枕叶和顶叶区域的脑白质减少比额叶更多[Barnea-Goraly et al 2003, Bearden et al 2004, Bish et al 2004, Kates et al 2004]。大脑结构中的许多变化被认为与工作记忆、执行功能、视觉空间技能、语言和数学表现等领域的特定认知缺陷有关。
总的来说,中枢神经系统异常的种类非常多,与在一些Opitz G/BBB综合征患者中所见到的症状一致[Neri et al 1987, Guion-Almeida & Richieri-Costa 1992, MacDonald et al 1993]。
心理社会发展和认知功能:一般来说,22q11.2DS的儿童运动发育过程有延迟(开始走路的平均年龄是18个月),语言出现的延迟(很多在2-3岁时仍不会说话),还有大约20%有自闭症/自闭症谱系障碍[Fine et al 2005]。
具体地说,在一项对28名儿童进行标准化测试的研究中,精神发展是平均水平的占21%,轻度延迟的占32%,重度延迟的占46%;在运动发展中,8%是平均水平,13%是轻度延迟,79%重度延迟。
对一群12学龄前儿童使用WPPSI-R评估,全量表智商是78±11,平均作业智商是78±14,平均语言智商是82±15。在总语言中,16%是平均水平,44%是轻度延迟,40%是重度延迟[Solot et al 1998]。
在对一组80名学龄儿童进行的适龄韦氏智商测试中,平均IQ得分为76分;18%的人全量表智商得分在平均范围内,20%在低平均范围内,32%在边界范围内,30%为智力障碍。
年龄较大的有22q11.2DS患者通常有多领域的非典型的神经心理特征,其中最引人注目的是他们的语言智商比执行智商分数高得多。Moss et al [1995]观察到,在80名学龄儿童中,有66%语言智商和执行智商间存在平均差距,这与一般人群中罕见非语言学习障碍相一致[Wang et al 1998]。由于单凭全量表智商分数的高低并不能准确地反映22q11.2DS患者的能力,语言智商和执行智商分数需要分开考虑。此外,受累个 体在语言背诵学习和记忆、阅读理解和拼写方面表现出相对优势。在非语言处理、视觉空间技能、复杂的语言记忆、注意力、工作记忆、视觉空间记忆和数学等方面 存在缺陷。这一证据表明,语言比视觉记忆能力强,阅读能力也比数学能力强,这也支持了非语言学习障碍的存在,这种障碍需要特定的认知补救、行为管理和父母 咨询。精神疾病:在一些患者身上观察到的行为和性格包括去抑制和冲动,也包括害羞和回避[Swillen et al 1999]。除了自闭症和自闭症谱系障碍外,注意力缺陷、焦虑、执拗和社会交往困难也很常见[Swillen et al 1999, Niklasson et al 2001, Vorstman et al 2006]。包括精神分裂症、躁郁症、焦虑和抑郁在内的精神疾病的发病率在增加。这些精神疾病的流行和确切性质一直是调查的对象[Shprintzen et al 1992, Chow et al 1994, Bassett et al 1998, Yan et al 1998, Murphy et al 1999, Baker & Skuse 2005, Bassett et al 2005, Oskarsdóttir et al 2005a]。
有研究表明,60%的成年人患有精神疾病。最值得注意的是,在大约25%的个体中发现了精神分裂症,然而,焦虑和抑郁障碍也很常见[Bassett et al 2011]。行为差异可能在很小的时候就开始了;在10岁之前筛查有22q11.2DS精神疾病的儿童,可以为早期干预提供机会[Vorstman et al 2006]。
生长:大多数有22q11.2DS的成年人身高正常;然而,在 95名1到15岁的儿童中,有41%的儿童身高低于第五百分位数。其中有4人明显低于第五百分位;所有人的生长因子IGF1和IGFBP3浓度都较低。3 个人有生长激素缺乏症;3个人经MRI检测发现脑下垂体偏小;还有2人的人类生长激素疗法有效[Weinzimer et al 1998]。最近,已经开发了特定于22q11.2DS的生长图表[Habel et al 2012]。
自身免疫性疾病:多关节青少年类风湿性关节炎(JRA)在22q11.2DS儿童中发生的频率是一般人群的20倍。JRA的发病年龄从17个月到5岁不等。HLA类型对JRA的发展有很好的评估作用[Keenan et al 1997, Sullivan et al 1997]。 其他与22q11.2DS相关的自身免疫性疾病包括:特发性血小板减少症(ITP),甲亢(Grave’s病),甲状腺功能减退,白癜风,溶血性贫血,自 身免疫性嗜中性白血球减少,再生障碍性贫血和乳糜泻。在22q11.2的患者中,特发性血小板减少症的频率比一般人群高200倍[Sullivan et al 1997, Jawad et al 2001, Kawame et al 2001]。
肌肉骨骼系统:在108名骨骼异常患者中,6%有上肢异常,包括轴前性和轴后性多指趾畸形,15%有下肢异常包括轴后性多指趾畸形,马蹄内翻足,脚趾重叠弯曲,脚趾第2第3并趾[Ming et al 1997]。
在63名进行了胸片检查的患者中,19%有椎体异常,包括蝶骨、半椎和冠状裂隙异常;19%的人有肋骨异常,最常见的是多余肋或肋骨缺如。1.5%为低弹性肩胛骨[Ming et al 1997]。在79名进行前瞻性研究的患者中,50%存在严重的颈椎异常,其中有21%存在不伴有阻滞椎的C2-C3脊椎后融合,75%存在发育不良/异常的C1,59%存在C2畸形,13%存在伴有阻滞椎的C2-C3后融合[Ricchetti et al 2004]。 此外,有56%的颈椎异常患者存在屈曲位和伸展位的放射学不稳定性;33%的人在多于一个脊椎的水平面上增加运动;其中4例患儿后续通过颈椎CT扫描及/ 或MRI检查后发现了C2-C3颈椎前、后狭窄的节段运动。其中两名患者通过手术稳定;其中一名需要在脊髓压迫症状后进行紧急手术。最近,在1067名评 估的患者中,15%患有脊柱侧凸[D Colo, 个人交流]。
肾脏:一项使用肾超声检查了80例既往无泌尿科病史的22q11.2DS患者的前瞻性评估,在31%患者中发现肾或其他泌尿生殖系统异常[Wu et al 2002]。其中包括单肾、肾脏回声增强、多囊性发育不良肾/小肾脏、结石、膀胱壁增厚、马蹄肾、集合管系统重复、肾小管酸中毒、肾盂积水(5%)、遗尿。肾异常的高发生率与Devriendt et al [1996]报道的类似。此外,尿道下裂、睾丸未降[McDonald-McGinn et al 1995]和子宫缺如都已被报道[Sundaram et al 2007]。
最近,对859名22q11.2DS患者的记录进行了回顾,包括肾脏异常、单脐动脉、羊水过多和泌尿生殖道异常。在这些人中,有677人有泌尿生殖 系统的评估,其中45%的有异常表现,包括:脐疝,腹股沟疝,阴茎下弯,包茎,睾丸未降,和尿道下裂。有530人有肾脏数据,其中24.5%有异常,包括 肾盂积水和单侧肾发育不全[Amlie-Wolf et al 2012]。
其他:其他在22q11.2DS个人身上发现的症状包括:
- 肺分叶异常[McDonald-McGinn et al 1995]
- 恶性肿瘤包括肝母细胞瘤[Patrone et al 1990, Scattone et al 2003, McDonald-McGinn et al 2006],肾细胞瘤[Scattone et al 2003],肾母细胞瘤[McDonald-McGinn et al 2006],以及神经母细胞瘤。根据这些报告,由于肝母细胞瘤的人口发病率为1:10000,因此,22q11.2DS和肝母细胞瘤之间似乎很有可能存在因果关系。从理论上说,由于位于缺失区域内COMT基因产物的缺乏,导致肝母细胞瘤发病率的增加,COMT基因产物通常可以有效地解除环境致癌物的毒性,人们认为早期暴露于环境致癌物导致了恶性肿瘤发生[McDonald-McGinn et al 2006]。
基因型-表型相关性
该综合征在家庭间和内部,甚至在同卵双胞胎中的临床异质性很强,这使得基因型-表型的相关性很难确定[Driscoll et al 1995, McDonald-McGinn et al 2001]。有趣的是,在家族病例中,发育迟缓似乎更为明显;然而,这可能反映了社会经济因素,而不是遗传因素。
命名
现在认识到,22q11.2DS包含先前描述为先天性胸腺发育不全综合症的表型(DGS) [Kelley et al 1982],腭心面综合症(VCFS)[Scambler et al 1991, Driscoll et al 1992],圆锥动脉干-异常面容综合征(CTAF)[Matsuoka et al 1994],常染色体显性Opitz G/BBB综合症[McDonald-McGinn et al 1995, Fryburg et al 1996, Lacassie & Arriaza 1996]和Cayler心面综合征(歪嘴哭面容)[Giannotti et al 1994]。这些症状的临床描述源自一种确认偏倚。
DGS最初被描述为第三和第四咽囊发育缺陷,通常在新生儿中被发现,包括胸腺和甲状旁腺发育不全。后来,又增加了先天性心脏病,特别是心脏畸形。大多数DGS患者在新生儿期发现了主要的先天性心脏缺陷、低钙血症和免疫缺陷。
VCFS最初被描述为是腭部异常的组合,通常是腭咽功能不全(VPI),先天性心脏病(通常是室间隔缺损或法洛四联症),特殊面容,发育迟缓或学习困难。患有VCFS的儿童在进入学龄时,往往会被诊断为腭裂诊所或颅面部中心,并有明显的语言及学习障碍[Driscoll et al 1993, Wilson et al 1993, Wulfsberg et al 1996, McDonald-McGinn et al 1997b, Thomas & Graham 1997]。VCFS可被称为Shprintzen综合征。
常染色体显性OpitzG/BBB综合征也可被称为眼距过宽伴有食管异常和尿道下裂。
发病率
发病率估计在1/6395[Devriendt et al 1998]-1/4000[Wilson et al 1994]。考虑到22q11.2DS的临床异质性较大,其发生率可能比之前估计的要高得多。在瑞典的一项以人口为基础的研究中,平均每年发病率为14.1 / 10万活产[Oskarsdóttir et al 2004, Oskarsdóttir et al 2005a, Oskarsdóttir et al 2005b]。美国疾病控制中心(CDC)开展的一项以人口为基础的研究发现,白人、黑人和亚洲人的整体患病率约为1/6000,拉丁美洲裔人为1/3800[Botto et al 2003]。
在最近对两个大型参考实验室所提交样本的多个指征(如发育迟缓或多发自然流产)的回顾中,有30,153人接受了array CGH检测。其中,274人存在22q11.2缺失(约1/100)[McDonald-McGinn et al 2010a],这表明,尽管存在确认偏倚,目前的发病率仍然可能被低估了。此外,几个州目前正在进行的新生儿筛查工作将有助于更好地确定这些数据。
染色体微阵列技术广泛应用于检测较小的非典型性及“嵌合”缺失表明22q11.2DS的发病率甚至高于先前的估计。一份评估报告评估了904名受累个体,其中436人通过增强FISH或微阵列检测了缺失的大小。其中有63(15%)存在非典型缺失,包括28个A-B(44%),10个A-C(16%),17个B-D(27%)、2个C-D(3%)和 6个“其他”(10%)(非低拷贝数重复区侧翼序列)(见图2)。B-D和不太常见的C-D缺失不包含TBX1基因无法用标准FISH探针检测到。在这些非典型缺失的个体中,家族性缺失的发生率较高。因此许多成年人直到一个受影响儿童的缺失被检测到后才会被诊断出来。
遗传相关疾病
22q11.2DS与此GeneReview中描述的表型有关。其他表型与22q11.2缺失不相关。请参阅命名部分对之前由临床定义的综合征的讨论,这些症状现在都被认为是由22q11.2DS导致的。
鉴别诊断
所有与22q11.2DS相关的症状可以在独立的异常个体中被发现,也可以在正常人中被发现。
多达11%的人有明显的单纯腭裂,包括粘膜下裂,可能有22q11.2DS,这是最常见的与腭裂有关的遗传综合症。22q11.2DS也是先天性腭咽闭合不全最常见的遗传原因。
TBX1:TBX1基因位于22号染色体q11.2的A-B区域内,其致病突变可使不存在缺失的个体产生22q11.2染色体缺失综合征样临床症状,主要是先天性心脏异常[Gong et al 2001, Yagi et al 2003];其中至少有一种致病突变影响功能[Stoller & Epstein 2005]。目前尚不确定其他基因是否必须缺失,因为TBX1基因的致病突变并不能导致22q11.2缺失综合征的所有症状。现在已经报道了存在不包含TBX1基因的非典型嵌合缺失的受影响的个体(B-D缺失;图2),包括典型的圆椎动脉干心脏缺陷(如法洛四联症,主动脉弓离断和永存动脉干)的个体,因此可能存在下游的影响或CRKL基因的参与(请参阅图2)[McDonald-McGinn et al 2010a]。
此外,一些具有22q11.2缺失综合征症状的人,包括典型的心脏畸形,既没有明确的缺失,也没有TBX1基因的致病性突变。然而,其中许多人后来被发现有CHARGE综合症,这是由CHD7基因的致病突变引起的[Jyonouchi et al 2009]。
具有共享症状的疾病:
- Smith-Lemli-Opitz综合征(当出现多趾和腭裂时):Smith-Lemli-Opitz综合症是一种常染色体隐性遗传的先天性新陈代谢疾病,与血清7-脱氢胆甾醇(7-DHC)浓度或7-脱氢胆甾醇:胆固醇比率升高相关。由DHCR7基因的致突变导致。
- VATER联合征(当出现心脏疾病、椎体、肾、肢体异常时):VATER联合征是一种目前尚未确定的病因的排除性诊断。
- 眼- 耳-脊椎(Goldenhar)综合征(OAVS)(当出现耳异常、椎体缺损、心脏疾病、肾脏异常时):OAVS通常是散发病例。
FISH结果正常的疑有22q11.2DS患者可能存在(a)非典型的不包含22号染色体q11.2区域A-B区间的嵌合缺失,或(b)包含10p13-p14缺失的涉及其他染色体区域的染色体异常(尽管b被认为是一个独立实体)。
管理
初步诊断后的评估
针对22q11.2缺失综合征患者进行评估和治疗的临床实践指南已发表[Bassett et al 2011](全文)。
为了确定诊断为22q11.2DS患者的疾病程度和需求,建议进行以下评估(如果不作为诊断结果的一部分):
- 心脏医生对心脏进行基础评估,包括胸部X射线、心电图和超声心动图;如果怀疑有血管环的话,可能需要进行胸部MRI检查。
- 测定血清钙离子浓度和全段甲状旁腺激素水平,以评估甲状旁腺功能减退,并在异常时进行正式内分泌评估
- 促甲状腺激素水平
- 全血细胞计数与分类。低淋巴细胞绝对值计数需要对T细胞和B细胞亚群进行评估,并将其转诊给免疫学医生。
- 免疫评估可能包括流式细胞术、免疫球蛋白和T细胞功能
- 肾超声检查评估肾结构缺陷
- 可考虑进行眼科评估
- 听力学评估
- 胸部X射线可检查胸椎异常
- 在所有年龄超过4岁(颈椎会骨化得更好)的个体中进行颈椎影像学(六个部位:屈曲位、伸展位、正位、侧位、张口位、颅底位)检测
- 对可能影响食管和语言发育的腭部异常进行临床评估,
- 评估可能存在的喂养问题,包括严重的胃食管反流;吮吸/吞咽困难、提高喂养困难、添加结构性食物困难;呕吐和便秘
- 在一岁时进行的语言评估显示,几乎所有患有22q11.2缺失综合征的儿童的语言出现都有延迟,并将受益于早期干预策略。此外,这样的评估有助于诊断腭部畸形/ 腭咽功能不全。
必要时:
- 内分泌医生对身材矮小(身高低于第二百分位数)的儿童进行评估,以发现可能的生长激素缺乏
- 由心理学家或精神病医生对任何有焦虑、情绪障碍、行为异常或精神病的患者进行评估
- 血液科医生对有瘀伤或出血病史的患者进行评估
对症治疗
根据22q11.2缺失综合征患者的年龄和出现的问题,涉及卫生保健提供者在内的从以下专业进行的多学科评估往往是必要的:过敏学,听力学,心脏病学,心脏手术学,儿童发展心理学,牙科,内分泌学,ENT,喂养 学,胃肠病学,儿科,普通外科,免疫学,临床遗传学,神经病学,神经外科学,眼科学,骨科学,耳鼻喉科学,整形外科学,精神病学,肺科学,风湿病学,语言 学和泌尿科学。
低血清钙浓度需要以标准的方式补充钙质,在可能的情况下,转诊给一个内分泌医生/肾脏医生,因为长期钙补充会增加肾脏结石的风险。
喂养困难应由胃肠病学专家评估可能的结构异常,如小肠扭转畸形/肠道不旋转、巨结肠疾病和迟发性膈疝[McDonald-McGinn et al 2004]。
解决喂养困难的策略包括:吃饭时调整匙子容量;通过胃酸封闭、促运动物质、姿势疗法来治疗胃食管反流;通过药物治疗胃肠蠕动障碍,促进肠道疏通[Dinulos & Graf 1998, Eicher et al 2000]。
早期干预(职业疗法、物理疗法)和语言治疗/手语的引入应该在一岁或孩童时期开始,因为发生运动、认知、语言发育迟缓的风险较高。后续的教育和行为治疗通常是必要的。
如果存在生长激素缺乏,应与对一般人群的治疗方法相同。
除了积极治疗感染,免疫缺陷一般不需要特殊的干预。很少需要预防性抗生素、丙种球蛋白治疗或胸腺移植。
对精神疾病的早期诊断和早期干预可以改善精神分裂症,双相情感障碍[Clarke & O’Callaghan 2003]和其他疾病,如自闭症、焦虑、强迫症(OCD)和注意力缺陷多动障碍(ADHD/ADD)患者的长期预后[Vorstman et al 2006, Bassett et al 2011]。
并发症的预防
有淋巴细胞异常的婴儿不应该接种活疫苗(如口服脊髓灰质炎疫苗,麻疹、腮腺炎和风疹的混合疫苗)。在儿童时期接种活疫苗之前,他们的免疫状况应重新评估。
评估免疫接种后的抗体水平是必要的。
推荐使用辐照后血液制品,直到确认了免疫系统正常化。
在术前和术后均应测量血清钙离子的浓度,以避免因身体压力引起低钙性癫痫发作。
以下也应考虑:
- 在涉及到咽部的手术前进行颈动脉的评估
- 腺样体切除术前应考虑对语言的可能的影响
- 在外科手术和体育活动(如跌倒)导致颈部过度伸展时应进行颈椎异常的评估
- 执行咽部手术前和术后应研究睡眠状况
- 在手术前对血小板体积和功能进行评估
监测
受影响的个人需要根据“一个个系统”的基础来跟进:
- 对低钙血症的监测,包括婴儿时期每3到6个月,童年期每5年,之后每隔1到2年的钙离子水平监测
- 在术前和术后定期监测钙离子水平,并在怀孕期间定期监测
- 每年监测甲状腺情况
- 9-12月龄时重新评估免疫状态;在接种任何活病毒疫苗前都要重新进行免疫评估
- 每年进行全血细胞计数和分类
- 在一岁至五岁之间重复或根据症状的需要进行眼科检查,
- 语言出现后对鼻腔语音质量评估以筛查腭咽功能不全
- 在幼儿期和入学前重复听力学评估
- 定期监测脊柱侧弯的发展
- 常规的牙科保健,考虑对牙釉质发育不良/龋病风险增加的患者进行牙封闭处理
- 定期的发育评估使孩子受益,并可协助学校提供适当的补救措施。
临床遗传学家定期进行重新评估,可以为家庭提供新的发展和/或建议。
有害因素/环境
碳酸饮料和酒精摄入可能会加剧低钙血症。咖啡因的摄入可能会导致焦虑或加剧焦虑。
孕期管理
孕妇必须接受医学检查。应考虑到任何先决条件,包括先天性心脏病、脊柱侧凸和哮喘。额外的监测应该包括钙、甲状腺和血小板水平。此外,精神状态/行为改变时应立即请精神卫生保健医生进行评估。
一个高风险的胎儿应该通过胎儿超声心动图来评估以下异常情况:先天性心脏病;气道、上颚、吞咽功能和胃肠异常可能导致羊水过多(先天性膈疝、气管食管瘘、舌下狭窄、血管环、喉蹼、腭裂/唇腭裂);肾异常;骨骼异常如马蹄内翻足和颅骨关节病;以及脐疝和腹股沟疝。
在研的治疗方法
可通过搜索ClinicalTrials.gov获取各种疾病表型的临床试验的信息。注:该疾病可能没有临床试验。
遗传咨询
遗 传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识,以及符合个人情况的决定。 接着几个段落是涉及遗传风险评估, 根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的咨 询工作。-作者ED
遗传模式
22q11.2DS是一种连续的基因缺失综合征,其遗传方式为常染色体显性。
家庭成员风险
先证者的父母
- 10%的先证者缺失遗传自父母[McDonald-McGinn et al 2001]。
先证者的同胞。先证者同胞的风险取决于父母的遗传状态
- 如果父母也存在22q11.2DS,那么每个同胞的患病风险是50%。
遗传咨询相关问题
家庭计划
- 确定遗传风险的最佳时间以及产前检查有效性的讨论是怀孕之前。
DNA银行是储存DNA(通常是从白细胞中提取),以备将来使用。由于检测方法和我们对基因、基因突变和疾病的理解将在未来得到改善,因此应该考虑对受影响个体进行DNA储存。
产前诊断
高风险妊娠
- 分子遗传学检测:使用FISH,MLPA或微阵列检测技术的产前诊断可能是基于家族史或超声检查结果。羊膜穿刺术通常在大约妊娠15到18周进行,或在大约妊娠10到12周进行绒毛穿刺,可对获得的胎儿细胞进行分析。
- 超声评估:通过高分辨率超声检查腭部和其他相关异常,通过超声心动图检查心脏异常,可以在妊娠期18到22周评估患病风险。
注:孕周的计算是从上一个正常月经周期的第一天开始计算或通过超声测量。
低风险妊娠:对于一些根据家族史不能明确是否有患22 q11.2DS风险的妊娠, 在常规超声检查中发现先天性心脏病和/或腭裂/或唇腭裂可能可以明确诊断,尤其是圆椎动脉干心脏异常,如主动脉弓离断,永存动脉干,法 洛四联症和室中隔缺损。其他可在产前检测到的与22q11.2DS相关的结构异常包括:先天性膈疝、脐疝或腹股沟疝、气管食管瘘/食管闭锁/喉闭 锁、多趾畸形、颅骨关节病、多小脑回和肾脏异常。此外,羊水过多也经常出现。从胎儿细胞中获取并制备的染色体可以用微阵列技术,MLPA或FISH进行分析。 在妊娠晚期确诊22q11.2DS对围产期管理非常有帮助。
胚胎植入前遗传学诊断(PGD)可作为一些已经确认存在致病突变的家庭的选择。
资源
为了该病患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对该病的,或者包括其他GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击这里获取详细信息。
- 22q11 GroupPO Box 1302Milton Keynes MK13 0LZUnited KingdomPhone: +44 1908 320 852Email: 22q11@melcom.cix.co.uk
- International 22q11.2 Deletion Syndrome Foundation, Inc.P.O. Box 2269Cinnaminson NJ 08077Phone: 877-739-1849 (toll-free)Email: info@22q.org
- International DiGeorge/VCF Support Networkc/o Family Voices of New York46 1/2 Clinton AvenueCortland NY 13045Phone: 607-753-1621 (day); 607-753-1250 (eve)Fax: 607-758-7420
- Max Appeal15 Meridian AvenueStourbridge West Midlands DY8 1049United KingdomPhone: 0800 389 1049 toll freeEmail: info@maxappeal.org.uk
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- Chromosome 22 Centralc/o Murney Rinholm7108 Partinwood DriveFuquay-Varina NC 27526Phone: 919-567-8167Email: usinfo@c22c.org
- Chromosome Disorder Outreach (CDO)PO Box 724Boca Raton FL 33429-0724Phone: 561-395-4252 (Family Helpline)Email: info@chromodisorder.org
- European Society for Immunodeficiencies (ESID) RegistryDr. Gerhard KindleUniversity Medical Center Freiburg Centre of Chronic ImmunodeficiencyEngesserstr. 479106 FreiburgGermanyPhone: 49-761-270-34450Email: esid-registry@uniklinik-freiburg.de
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其他部分的信息不一致:表格里的信息可能更加新-ED
表A
22q11.2缺失综合征:基因和数据库
| 关键区域 | 基因 | 染色体位置 | 蛋白 | 位点特异性 | HGMD |
|---|---|---|---|---|---|
| DGCR | 不适用 | 22q11 .2 | 不适用 | ||
| TBX1 | 22q11 .21 | T-box transcription factor TBX1 | TBX1 database | TBX1 |
Table B. 表B
22q11.2缺失综合征OMIM 词条(在OMIM数据库中查看全部信息)
DiGeorge染色体区域:许多基因已经匹配到22q11.2的DIGEORGE综合征关键区域(请参阅表4[pdf])。
缺失:超过85%的22q11.2缺失的个体有一段大约2.54Mb的区域重叠;其余的要么包含不同的缺失端点,要么是复发性的,非典型地嵌在较大的典型缺失区域(TDR)内比较短的片段缺失(请参阅图2) [Levy et al 1995, Kurahashi et al 1996, O’Donnell et al 1997, McQuade et al 1999]。在一个具有典型的VCFS / DGS表型的个体中发现存在一个典型缺失区域范围内20 kb的小缺失 [Yamagishi et al 1999]。这个较小的缺失影响了UFD1L和CDC45L基因。在一些受影响的个体中,由于缺失从远端开始并延伸至端粒,因此这些个体缺失的部分没有覆盖到典型缺失区域。22q11.2缺失端点附近重复序列块的位置,在导致典型和非典型缺失事件中起着至关重要的作用。
一小部分个体的缺失由缺失了22号染色体短臂末端→q11区域的非平衡易位导致。(如欲了解更多信息,请参阅表A,位点特异性)
正常的基因产物:其中一些基因产物已被识别并被进一步鉴定。请参阅表4以获取基因及其相关基因产物列表。
非正常的基因产物:未知
参考文献
引用文献
- Amlie-Wolf L, McDonald-McGinn DM, Valverde K, Devoto M, Mennuti M, Dickinson K, Bailey A, Lande R, Emanuel BS, Zackai EH. Renal anomalies, polyhydramnios, and single umbilical artery as prenatal clues to the diagnosis of 22q11.2 deletion syndrome. Lake Buena Vista, FL: Eighth Biennial International 22q11.2DS Conference. 2012.
- Baker KD, Skuse DH. Adolescents and young adults with 22q11 deletion syndrome: psychopathology in an at-risk group. Br J Psychiatry. 2005;186:115–20. [PubMed: 15684233]
- Barnea-Goraly N, Menon V, Krasnow B, Ko A, Reiss A, Eliez S. Investigation of white matter structure in velocardiofacial syndrome: a diffusion tensor imaging study. Am J Psychiatry. 2003;160:1863–9. [PubMed: 14514502]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307–13. [PMC free article: PMC3127862] [PubMed: 16208694]
- Bassett AS, Hodgkinson K, Chow EW, Correia S, Scutt LE, Weksberg R. 22q11 deletion syndrome in adults with schizophrenia. Am J Med Genet. 1998;81:328–37. [PMC free article: PMC3173497] [PubMed: 9674980]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J. International 22q11.2 Deletion Syndrome Consortium. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011 Aug;159(2):332–9.e1. [PMC free article: PMC3197829] [PubMed: 21570089]
- Bearden CE, van Erp TG, Monterosso JR, Simon TJ, Glahn DC, Saleh PA, Hill NM, McDonald-McGinn DM, Zackai E, Emanuel BS, Cannon TD. Regional brain abnormalities in 22q11.2 deletion syndrome: association with cognitive abilities and behavioral symptoms. Neurocase. 2004;10:198–206. [PubMed: 15788257]
- Bingham PM, Zimmerman RA, McDonald-McGinn D, Driscoll D, Emanuel BS, Zackai E. Enlarged Sylvian fissures in infants with interstitial deletion of chromosome 22q11. Am J Med Genet. 1997;74:538–43. [PubMed: 9342208]
- Bish JP, Nguyen V, Ding L, Ferrante S, Simon TJ. Thalamic reductions in children with chromosome 22q11.2 deletion syndrome. Neuroreport. 2004;15:1413–5. [PubMed: 15194864]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–7. [PubMed: 12837874]
- Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ, McDonald-McGinn D, Zackai EH, Driscoll DA. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum Mol Genet. 1995;4:763–6. [PubMed: 7633430]
- Cayler GG. Cardiofacial syndrome. Congenital heart disease and facial weakness, a hitherto unrecognized association. Arch Dis Child. 1969;44:69–75. [PMC free article: PMC2020193] [PubMed: 5765991]
- Chow EW, Bassett AS, Weksberg R. Velo-cardio-facial syndrome and psychotic disorders: implications for psychiatric genetics. Am J Med Genet. 1994;54:107–12. [PMC free article: PMC3142271] [PubMed: 8074160]
- Clarke M, O’Callaghan E. Is earlier better? At the beginning of schizophrenia: timing and opportunities for early intervention. Psychiatr Clin North Am. 2003;26:65–83. [PubMed: 12683260]
- Desmaze C, Scambler P, Prieur M, Halford S, Sidi D, Le Deist F, Aurias A. Routine diagnosis of DiGeorge syndrome by fluorescent in situ hybridization. Hum Genet. 1993;90:663–5. [PubMed: 8444474]
- Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35:789–90. [letter] [PMC free article: PMC1051442] [PubMed: 9733045]
- Devriendt K, Swillen A, Fryns JP, Proesmans W, Gewillig M. Renal and urological tract malformations caused by a 22q11 deletion. J Med Genet. 1996;33:349. [letter] [PMC free article: PMC1050591] [PubMed: 8730297]
- Digilio MC, Marino B, Bagolan P, Giannotti A, Dallapiccola B. Microdeletion 22q11 and oesophageal atresia. J Med Genet. 1999;36:137–9. [PMC free article: PMC1734297] [PubMed: 10051013]
- Dinulos MB, Graf WD. DiGeorge syndrome and velocardiofacial syndrome. In: Gilman S, Goldstein GW, Waxman SG, eds. Neurobase. San Diego, CA: Arbor Press; 1998.
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–33. [PMC free article: PMC1682598] [PubMed: 1349199]
- Driscoll DA, Randall P, McDonald-McGinn DM, et al. Are 22q11 chromosomal deletions a major cause of isolated cleft palate? 52nd Annual Meeting. Tampa, FL: American Cleft Palate-Craniofacial Association; 1995.
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813–7. [PMC free article: PMC1016560] [PubMed: 8230155]
- Eicher PS, McDonald-Mcginn DM, Fox CA, Driscoll DA, Emanuel BS, Zackai EH. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J Pediatr. 2000;137:158–64. [PubMed: 10931405]
- Fine SE, Weissman A, Gerdes M, Pinto-Martin J, Zackai EH, McDonald-McGinn DM, Emanuel BS. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord. 2005;35:461–70. [PMC free article: PMC2814423] [PubMed: 16134031]
- Forbes BJ, Binenbaum G, Edmond JC, DeLarato N, McDonald-McGinn DM, Zackai EH. Ocular findings in the chromosome 22q11.2 deletion syndrome. J AAPOS. 2007;11(2):179–82. [PubMed: 17140829]
- Fryburg JS, Lin KY, Golden WL. Chromosome 22q11.2 deletion in a boy with Opitz (G/BBB) syndrome. Am J Med Genet. 1996;62:274–5. [PubMed: 8882786]
- Gennery AR, Barge D, O’Sullivan JJ, Flood TJ, Abinun M, Cant AJ. Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child. 2002 Jun;86:422–5. [PMC free article: PMC1763000] [PubMed: 12023174]
- Giannotti A, Digilio MC, Marino B, Mingarelli R, Dallapiccola B. Cayler cardiofacial syndrome and del 22q11: part of the CATCH22 phenotype. Am J Med Genet. 1994;53:303. [letter] [PubMed: 7856669]
- Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Budarf ML. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. [PMC free article: PMC1734783] [PubMed: 11748311]
- Gripp KW, McDonald-McGinn DM, Driscoll DA, Reed LA, Emanuel BS, Zackai EH. Nasal dimple as part of the 22q11.2 deletion syndrome. Am J Med Genet. 1997;69:290–2. [PubMed: 9096759]
- Guion-Almeida ML, Richieri-Costa A. CNS midline anomalies in the Opitz G/BBB syndrome: report on 12 Brazilian patients. Am J Med Genet. 1992;43:918–28. [PubMed: 1415340]
- Habel A, McGinn MJ 2nd, Zackai EH, Unanue N, McDonald-McGinn DM. Syndrome-specific growth charts for 22q11.2 deletion syndrome in Caucasian children. Am J Med Genet A. 2012 Nov;158A:2665–71. [PubMed: 22711268]
- Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. 2001;139:715–23. [PubMed: 11713452]
- John AS, McDonald-McGinn DM, Zackai EH, Goldmuntz E. Aortic root dilation in patients with 22q11.2 deletion syndrome. Am J Med Genet A. 2009;149A:939–42. [PMC free article: PMC4080309] [PubMed: 19353635]
- Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:e871–7. [PMC free article: PMC4098848] [PubMed: 19403480]
- Kao A, Mariani J, McDonald-McGinn DM, Maisenbacher MK, Brooks-Kayal AR, Zackai EH, Lynch DR. Increased prevalence of unprovoked seizures in patients with a 22q11.2 deletion. Am J Med Genet A. 2004;129A:29–34. [PubMed: 15266612]
- Kapadia CR, Kim YE, McDonald-McGinn DM, Zackai EH, Katz L. Parathyroid hormone reserve in 22q11.2 deletion syndrome. Genet Med. 2008;10:224–8. [PubMed: 18344713]
- Kates WR, Burnette CP, Bessette BA, Folley BS, Strunge L, Jabs EW, Pearlson GD. Frontal and caudate alterations in velocardiofacial syndrome (deletion at chromosome 22q11.2). J Child Neurol. 2004;19:337–42. [PubMed: 15224707]
- Kawame H, Adachi M, Tachibana K, Kurosawa K, Ito F, Gleason MM, Weinzimer S, Levitt-Katz L, Sullivan K, McDonald-McGinn DM. Graves’ disease in patients with 22q11.2 deletion. J Pediatr. 2001;139:892–5. [PubMed: 11743521]
- Keenan GF, Sullivan KE, McDonald-McGinn DM, Zackai EH. Arthritis associated with deletion of 22q11.2: more common than previously suspected. Am J Med Genet. 1997;71:488. [letter; comment] [PubMed: 9286462]
- Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr. 1982;101:197–200. [PubMed: 7097410]
- Kilic SS, Gurpinar A, Yakut T, Egeli U, Dogruyol H. Esophageal atresia and tracheo-esophageal fistula in a patient with Digeorge syndrome. J Pediatr Surg. 2003 Aug;38:E21–3. [PubMed: 12891520]
- Kirschner RE. Palatal anomalies and velopharyngeal dysfunction associated with velo-cardio-facial syndrome. In: Murphy KC, Scambler PJ. Velo-Cardio-Facial Syndrome: A Model for Understanding Microdeletion Disorders. New York, NY: Cambridge University Press; 2005:83-104.
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–52. [PubMed: 17950858]
- Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34:152–7. [PMC free article: PMC1050871] [PubMed: 9039994]
- Kurahashi H, Nakayama T, Osugi Y, Tsuda E, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Deletion mapping of 22q11 in CATCH22 syndrome: identification of a second critical region. Am J Hum Genet. 1996;58:1377–81. [PMC free article: PMC1915078] [PubMed: 8651317]
- Lacassie Y, Arriaza MI. Letter to the editor. Opitz GBBB syndrome and the 22q11 deletion syndrome. Am J Med Genet. 1996;62:318. [PubMed: 8882795]
- Levin SE, Silverman NH, Milner S. Hypoplasia or absence of the depressor anguli oris muscle and congenital abnormalities, with special reference to the cardiofacial syndrome. S Afr Med J. 1982;61:227–31. [PubMed: 7058445]
- Levy A, Demczuk S, Aurias A, Depetris D, Mattei MG, Philip N. Interstitial 22q11 microdeletion excluding the ADU breakpoint in a patient with DiGeorge syndrome. Hum Mol Genet. 1995;4:2417–9. [PubMed: 8634722]
- Lynch DR, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Whitaker LA, Fischbeck KH. Cerebellar atrophy in a patient with velocardiofacial syndrome. J Med Genet. 1995;32:561–3. [see comments] [PMC free article: PMC1050553] [PubMed: 7562973]
- MacDonald MR, Schaefer GB, Olney AH, Tamayo M, Frias JL. Brain magnetic resonance imaging findings in the Opitz G/BBB syndrome: extension of the spectrum of midline brain anomalies. Am J Med Genet. 1993;46:706–11. [PubMed: 8362914]
- Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, Ikeda K, Nishibatake M, Ando M, Momma K. Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11.2. Am J Med Genet. 1994;53:285–9. [PubMed: 7856665]
- McDonald-McGinn D, Kohut A, Saitta S, Laskin R, Hacker A, Bale S, Aradhav S, Rosenfeld J, Shaffer L, Sullivan K, Emanuel B, Zackai E. Chromosome 22q11.2 “B-D” Deletion – New Diagnosis or More of the Same? Platform Presentation. Washington, DC: American Society of Human Genetics Meeting; 2010a.
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. Autosomal dominant "Opitz" GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995 Oct 23;59:103–13. [PubMed: 8849001]
- McDonald-McGinn DM, Emanuel BS, Zackai EH. Autosomal dominant "Opitz" GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1996;64:525–6. [PubMed: 8862635]
- McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Mlynarski E, Lynch DR, Yan AC, Bilaniuk LT, Sullivan KE, Warren ST, Emanuel BS, Vermeesch JR, Zackai EH, Jerome-Majewska LA. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet. 2013;50:80–90. [PMC free article: PMC3585484] [PubMed: 23231787]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, Larossa D, Emanuel BS, Zackai EH. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed: 10191425]
- McDonald-McGinn DM, Kohut T, Zackai EH. Deletion 22q11.2 (velo-cardio-facial syndrome/DiGeorge syndrome). In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. 3 ed. Hoboken, NJ: Wiley-Blackwell; 2010b:263-84.
- McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Wang P, Solot C, Schultz P, Lynch D, Bingham P, Keenan G, Weinzimer S, Ming JE, Driscoll D, Clark BJ 3rd, Markowitz R, Cohen A, Moshang T, Pasquariello P, Randall P, Emanuel BS, Zackai EH. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997a;1:99–108. [PubMed: 10464633]
- McDonald-McGinn DM, Maisenbacher M, Ciprero K, Hoffman T, Catanzaro J, Armeli C, Goldmuntz E, Gleason M, Gillis L, Mascarenhas M, Porter B, Hedricks H, Nance M, Adzick A, Drummond D, Pasquariello P, Driscoll D, Emanuel B, Zackai EH. Warning: Hear Hoofbeats - Think Zebras in the 22q11.2 Deletion! Toronto, Ontario: American Society of Human Genetics 54th Annual Meeting; 2004.
- McDonald-McGinn DM, Minugh-Purvis N, Kirschner RE, Jawad A, Tonnesen MK, Catanzaro JR, Goldmuntz E, Driscoll D, Larossa D, Emanuel BS, Zackai EH. The 22q11.2 deletion in African-American patients: an underdiagnosed population? Am J Med Genet A. 2005;134:242–6. [PMC free article: PMC2810968] [PubMed: 15754359]
- McDonald-McGinn DM, Reilly A, Wallgren-Pettersson C, Hoyme HE, Yang SP, Adam MP, Zackai EH, Sullivan KE. Malignancy in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Am J Med Genet A. 2006;140:906–9. [PubMed: 16532473]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–9. [PubMed: 11339373]
- McDonald-McGinn DM, Zackai EH. Genetic counseling for the 22q11.2 deletion. Dev Disabil Res Rev. 2008;14:69–74. [PubMed: 18636638]
- McDonald-McGinn DM, Zackai EH, Low D. What’s in a name? The 22q11.2 deletion. Am J Med Genet. 1997b;72:247–9. [PubMed: 9382154]
- McQuade L, Christodoulou J, Budarf M, Sachdev R, Wilson M, Emanuel B, Colley A. Patient with a 22q11.2 deletion with no overlap of the minimal DiGeorge syndrome critical region (MDGCR). Am J Med Genet. 1999;86:27–33. [PubMed: 10440825]
- Ming JE, McDonald-McGinn DM, Megerian TE, Driscoll DA, Elias ER, Russell BM, Irons M, Emanuel BS, Markowitz RI, Zackai EH. Skeletal anomalies and deformities in patients with deletions of 22q11. Am J Med Genet. 1997;72:210–5. [PubMed: 9382145]
- Moss E, Wang PP, McDonald-McGinn DM, et al. Characteristic cognitive profile in patients with a 22q11 deletion: verbal IQ exceeds nonverbal IQ. Am J Hum Genet. 1995;57:A42.
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–5. [PubMed: 10530637]
- Neri G, Genuardi M, Natoli G, Costa P, Maggioni G. A girl with G syndrome and agenesis of the corpus callosum. Am J Med Genet. 1987;28:287–91. [PubMed: 3425610]
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Neuropsychiatric disorders in the 22q11 deletion syndrome. Genet Med. 2001;3:79–84. [PubMed: 11339385]
- O’Donnell H, McKeown C, Gould C, Morrow B, Scambler P. Detection of an atypical 22q11 deletion that has no overlap with the DiGeorge syndrome critical region. Am J Hum Genet. 1997;60:1544. [letter] [PMC free article: PMC1716117] [PubMed: 9199579]
- Oskarsdóttir S, Belfrage M, Sandstedt E, Viggedal G, Uvebrant P. Disabilities and cognition in children and adolescents with 22q11 deletion syndrome. Dev Med Child Neurol. 2005a;47:177–84. [PubMed: 15739722]
- Oskarsdóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005b;164:146–53. [PubMed: 15565286]
- Oskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. 2004;89:148–51. [PMC free article: PMC1719787] [PubMed: 14736631]
- Patrone PM, Chatten J, Weinberg P. Neuroblastoma and DiGeorge anomaly. Pediatr Pathol. 1990;10:425–30. [PubMed: 2161524]
- Ricchetti ET, States L, Hosalkar HS, Tamai J, Maisenbacher M, McDonald-McGinn DM, Zackai EH, Drummond DS. Radiographic study of the upper cervical spine in the 22q11.2 deletion syndrome. J Bone Joint Surg Am. 2004;86-A:1751–60. [PubMed: 15292424]
- Sanklecha M, Kher A, Bharucha BA. Asymmetric crying facies: the cardiofacial syndrome. J Postgrad Med. 1992 Jul-Sep;38(3):147–8,150. [PubMed: 1303422]
- Scattone A, Caruso G, Marzullo A, Piscitelli D, Gentile M, Bonadonna L, Balducci G, Digilio MC, Jenkner A, Camassei FD, Boldrini R, Nazzaro P, Pollice L, Serio G. Neoplastic disease and deletion 22q11.2: a multicentric study and report of two cases. Pediatr Pathol Mol Med. 2003;22:323–41. [PubMed: 14692228]
- Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, Williamson R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–6. [PubMed: 2045103]
- Schwinger E, Devriendt K, Rauch A, Philip N. Clinical utility gene card for: DiGeorge syndrome, velocardiofacial syndrome, Shprintzen syndrome, chromosome 22q11.2 deletion syndrome (22q11.2, TBX1). Eur J Hum Genet. 2010 Sep;18(9) [PMC free article: PMC2987430] [PubMed: 20125192]
- Shprintzen RJ, Goldberg R, Golding-Kushner KJ, Marion RW. Late-onset psychosis in the velo-cardio-facial syndrome. Am J Med Genet. 1992;42:141–2. [letter] [PubMed: 1308357]
- Silengo MC, Bell GL, Biagioli M, Guala A, Bianco R, Strandoni P, De Sario PN, Franceschini P. Asymmetric crying facies with microcephaly and mental retardation. An autosomal dominant syndrome with variable expressivity. Clin Genet. 1986;30:481–4. [PubMed: 3815881]
- Smith CA, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH, Sullivan KE. Increased prevalence of immunoglobulin A deficiency in patients with the chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1998;5:415–7. [PMC free article: PMC104536] [PubMed: 9606003]
- Solot C, Gerdes M, McDonald-McGinn D, et al. Developmental profiles in a pre-school population with 22q11 deletion. Baltimore, MD: American Cleft Palate-Craniofacial Association 55th Annual Meeting and Conference Symposium; 1998.
- Stoller JZ, Epstein JA. Identification of a novel nuclear localization signal in Tbx1 that is deleted in DiGeorge syndrome patients harboring the 1223delC mutation. Hum Mol Genet. 2005;14:885–92. [PubMed: 15703190]
- Sullivan KE. The clinical, immunological, and molecular spectrum of chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Curr Opin Allergy Clin Immunol. 2004;4:505–12. [PubMed: 15640691]
- Sullivan KE, Jawad AF, Randall P, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin Immunol Immunopathol. 1998;86:141–6. [PubMed: 9473376]
- Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF. Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1999;6:906–11. [PMC free article: PMC95796] [PubMed: 10548584]
- Sullivan KE, McDonald-McGinn DM, Driscoll DA, Zmijewski CM, Ellabban AS, Reed L, Emanuel BS, Zackai EH, Athreya BH, Keenan G. Juvenile rheumatoid arthritis-like polyarthritis in chromosome 22q11.2 deletion syndrome (DiGeorge anomalad/velocardiofacial syndrome/conotruncal anomaly face syndrome). Arthritis Rheum. 1997;40:430–6. [PubMed: 9082929]
- Sundaram UT, McDonald-McGinn D, Huff D, Emanuel B, Zackai E, Driscoll D, Bodurtha J. Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am J Med Genet A. 2007;143A:2016–8. [PMC free article: PMC2810967] [PubMed: 17676598]
- Swillen A, Vandeputte L, Cracco J, Maes B, Ghesquière P, Devriendt K, Fryns JP. Neuropsychological, learning and psychosocial profile of primary school aged children with the velo-cardio-facial syndrome (22q11 deletion): evidence for a nonverbal learning disability? Child Neuropsychol. 1999;5:230–41. [PubMed: 10925707]
- Thomas JA, Graham JM Jr. Chromosomes 22q11 deletion syndrome: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1997;36:253–66. [PubMed: 9152551]
- Vorstman JA, Morcus ME, Duijff SN, Klaassen PW, Heineman-de Boer JA, Beemer FA, Swaab H, Kahn RS, van Engeland H. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry. 2006 Sep;45:1104–13. [PubMed: 16926618]
- Wang PP, Solot C, Moss EM, Gerdes M, McDonald-McGinn DM, Driscoll DA, Emanuel BS, Zackai EH. Developmental presentation of 22q11.2 deletion (DiGeorge/velocardiofacial syndrome). J Dev Behav Pediatr. 1998;19:342–5. [PubMed: 9809264]
- Weinzimer SA, McDonald-McGinn DM, Driscoll DA, Emanuel BS, Zackai EH, Moshang T Jr. Growth hormone deficiency in patients with 22q11.2 deletion: expanding the phenotype. Pediatrics. 1998;101:929–32. [PubMed: 9565428]
- Wilson DI, Burn J, Scambler P, Goodship J. DiGeorge syndrome: part of CATCH 22. J Med Genet. 1993;30:852–6. [PMC free article: PMC1016569] [PubMed: 8230162]
- Wilson DI, Cross IE, Goodship JA, Brown J, Scambler PJ, Bain HH, Taylor JF, Walsh K, Bankier A, Burn J, Wolstenholme J. A prospective cytogenetic study of 36 cases of DiGeorge syndrome. Am J Hum Genet. 1992;51:957–63. [PMC free article: PMC1682842] [PubMed: 1415264]
- Wilson DI, Cross IE, Wren C, et al. Minimum prevalence of chromosome 22q11 deletions. Am J Hum Genet. 1994;55:A169.
- Wu HY, Rusnack SL, Bellah RD, Plachter N, McDonald-McGinn DM, Zackai EH, Canning DA. Genitourinary malformations in chromosome 22q11.2 deletion. J Urol. 2002;168:2564–5. [PubMed: 12441983]
- Wulfsberg EA, Leana-Cox J, Neri G. What’s in a name? Chromosome 22q abnormalities and the DiGeorge, velocardiofacial, and conotruncal anomalies face syndromes. Am J Med Genet. 1996;65:317–9. [see comments] [PubMed: 8923942]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–73. [PubMed: 14585638]
- Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science. 1999;283:1158–61. [PubMed: 10024240]
- Yan W, Jacobsen LK, Krasnewich DM, Guan XY, Lenane MC, Paul SP, Dalwadi HN, Zhang H, Long RT, Kumra S, Martin BM, Scambler PJ, Trent JM, Sidransky E, Ginns EI, Rapoport JL. Chromosome 22q11.2 interstitial deletions among childhood-onset schizophrenics and "multidimensionally impaired". Am J Med Genet. 1998;81:41–3. [PubMed: 9514586]
推荐阅读
- Alkalay AA, Guo T, Montagna C, Digilio MC, Dallapiccola B, Marino B, Morrow B. Genetic dosage compensation in a family with velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome. Am J Med Genet A. 2011;155A:548–54. [PMC free article: PMC4081864] [PubMed: 21337693]
- Bassett AS, Chow EW, Husted J, Hodgkinson KA, Oechslin E, Harris L, Silversides C. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet. 2009;46:324–30. [PMC free article: PMC3188306] [PubMed: 19246480]
- De Smedt B, Swillen A, Verschaffel L, Ghesquiere P. Mathematical learning disabilities in children with 22q11.2 deletion syndrome: a review. Dev Disabil Res Rev. 2009;15:4–10. [PubMed: 19213009]
- Fung WLA, McEvilly R, Fong J, Silversides C, Chow E, Bassett AS. Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. Am J Psychiatry. 2010;167:998. [PMC free article: PMC4516404] [PubMed: 20693476]
- Green T, Gothelf D, Glaser B, Debbane M, Frisch A, Kotler M, Weizman A, Eliez S. Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry. 2009;48:1060–8. [PubMed: 19797984]
- Kiehl TR, Chow EW, Mikulis DJ, George SR, Bassett AS. Neuropathologic features in adults with 22q11.2 deletion syndrome. Cereb Cortex. 2009;19:153–64. [PubMed: 18483005]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. [PMC free article: PMC2869000] [PubMed: 20466091]
- Swaby JA, Silversides CK, Bekeschus SC, Piran S, Oechslin EN, Chow EW, Bassett AS. Complex congenital heart disease in unaffected relatives of adults with 22q11.2 deletion syndrome. Am J Cardiol. 2011;107:466–71. [PMC free article: PMC3188300] [PubMed: 21257016]
- Toriello HV, Goldenberg P. Evidence-based medicine and practice guidelines: application to genetics. Am J Med Genet C Semin Med Genet. 2009;151C:235–40. [PubMed: 19621463]
- Van Aken K, Swillen A, Beirinckx M, Janssens L, Caevenberghs K, Smits-Engelsman B. Prospective control abilities during visuo-manual tracking in children with 22q11.2 Deletion syndrome compared to age- and IQ-matched controls. Res Dev Disabil. 2010a;31:634–41. [PubMed: 20181458]
- Van Aken K, Swillen A, Beirinckx M, Janssens L, Caevenberghs K, Smits-Engelsman B. Kinematic movement strategies in primary school children with 22q11.2 Deletion syndrome compared to age- and IQ-matched controls during visuo-manual tracking. Res Dev Disabil. 2010b;31:768–76. [PubMed: 20181457]
本章节的注解
更新历史
- 28 February 2013 系统性更新发布到公开网页上
- 16 December 2005 系统性更新发布到公开网页上
- 23 July 2003 系统性更新发布到公开网页上
- 23 September 1999内容发布到公开网页上
- 7 August 1998 (dmm) 最早稿件