
概述
临床特点.
典型的德朗热综合征 (CdLS) 的特点是特殊面容、生长迟缓(宫内起病;一生中身高将小于第5百分位),多毛症和从细微的指骨异常到少指畸形等不同程度的上肢残缺。颅面特征包括连眉、高拱形眉毛、长睫毛、短鼻伴鼻孔前倾、牙齿小且有间隙和小头。IQ 范围30-102(均值:53)。许多个体表现出自闭和自残倾向。常见症状包括心脏间隔缺陷、胃肠道功能失调、听力丧失、近视和隐睾或生殖器发良不良。 表型 轻微的个体在生长、认知和四肢方面症状较轻,但通常具有CdLS的面容特征。
管理.
对症治疗: 积极管理胃食管反流,评估所有 受累的 个体的潜在胃肠旋转不良;如果反流严重,应考虑胃底折叠术。补充配方和/或胃造口管放置,以满足营养需求。 物理、职业和语言治疗,以优化精神运动发展和沟通技巧。对听力损失,心脏缺陷,癫痫发作,膀胱输尿管反流和隐睾症的进行标准治疗。
防止并发症: 进行血小板减少症和心脏病的手术前评估,麻醉期间仔细监控呼吸道情况;警惕恶性高热。.
监控: 每年进行 GI 评估,生长和精神运动发育监控;常规的眼、耳评估,以及心、肾异常的监控。
遗传咨询.
NIPBL-相关 CdLS, RAD21-相关 CdLS, 及 SMC3-相关 CdLS 为 常染色体显性遗传 ; HDAC8-相关 CdLS 及SMC1A-相关 CdLS 为 X-linked 遗传模式。大多数 受累的 个体携带NIPBL基因 de novo 杂合的 致病性变异 ; 少于1% 的 NIPBL-相关CdLS 患者有患病的父母。当父母无疾病临床表型,由于 胚系嵌合 的可能性,NIPBL-相关CdLS 先证者 的同胞患病风险估计为1.5%。HDAC8-相关 CdLS 及SMC1A-相关 CdLS的先证者,其同胞患病风险取决于先证者的母亲。对于已发现致病性变异的家庭,对高风险孕妇进行产前诊断是可能的。
诊断
诊断Cornelia de Lange综合征(CdLS) 的怀疑是基于临床发现,但是其临床诊断标准的共识尚未建立。
提示性发现
具有以下临床特征的个体应怀疑Cornelia de Lange综合征(CdLS) [Kline et al 2007a]:
颅面特征 (>95%)
- 经典型CdLS. 颅面特征容易识别,是诊断中最有用的部分 (见 Figure 1):
- 头部短小 (平均枕骨额叶周长 [OFC] <2nd百分位)
- 98%有连眉,高拱形眉 [Jackson et al 1993]
- 睫毛长而浓密
- 低耳位、耳后旋和/或多毛耳伴厚耳轮
- 扁或宽平鼻梁,上翘的鼻尖与前倾的鼻孔
- 人中长而平,上唇薄伴中线“水滴”样外观(在上唇的弧形下方的人中底部的上唇的顶部降低),嘴角下倾
- 30%有腭部高拱伴腭裂 [Sataloff et al 1990]
- 牙齿小而有间隙
- 80%有小颌畸形 [Jackson et al 1993]; 42%有下颌骨刺 [Braddock et al 1993]
- 短颈
- 轻型 CdLS. 一种较轻的 表型 一直被描述,其保留了许多特异面部特征但认知与肢体或结构性的异常较不严重,且更经常在SMC3 或 RAD21 基因 杂合的 致病性变异 或HDAC8 或 SMC1A基因 半合子的 致病性变异的患者中表现。 [Deardorff et al 2007, Selicorni et al 2007, Rohatgi et al 2010, Kaiser et al 2014, Minor et al 2014, Gil-Rodríguez et al 2015]. 轻型病例的临床诊断标准已被提出 [Ireland et al 1993, Jackson et al 1993, Selicorni et al 1993, Van Allen et al 1993, Allanson et al 1997] (see Figure 2).注: 这个较轻的 表型, 在临床上比经典或严重型CdLS更不引人注目,但可能代表了大多数的CdLS个体。 [Greenberg & Robinson 1989, Jackson et al 1993, Moeschler & Graham 1993, Selicorni et al 1993, Van Allen et al 1993, Allanson et al 1997, Deardorff et al 2007, Rohatgi et al 2010].
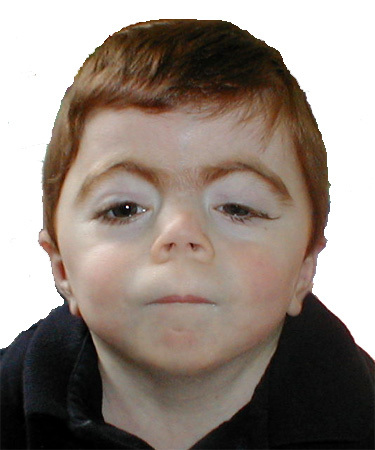
Figure 1.
经典 CdLS 颅面特征
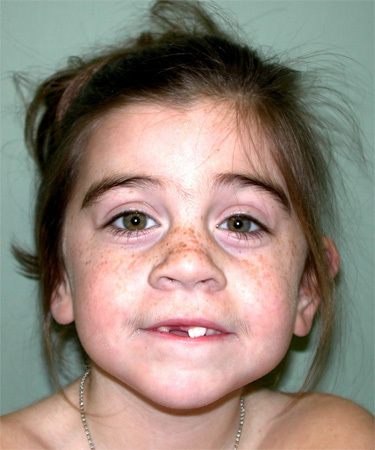
Figure 2.
SMC1A致病性变异的患者
生长迟滞 (>95%). 生长迟滞始于产前(尽管可能直到第9个月才引起注意)。终生身高和体重保持在小于第5个百分位 [Bruner & Hsia 1990, Kliewer et al 1993, Kline et al 1993a, Kousseff et al 1993, Boog et al 1999]. CdLS专用生长曲线表已制作 (www.cdlsusa.org).
此外,生长迟滞问题可能会叠加在胃食管反流继发的体质发育迟缓和其他喂养问题上。
智力障碍 (>95%)
- 经典型 CdLS. 重-极重度的普遍发育迟缓明显。
- 轻型 CdLS. 已发现较轻的 受累的 个体具有较高的机能和IQ(有一些在正常值范围)。CdLS 的IQ值总体处于30以下至102之间,平均IQ为53 [Kline et al 1993b, Saal et al 1993].
肢体异常 (>95%). 上肢主要受累,下肢相对较少。 肢体异常可以是对称的或不对称的。
- 经典型 CdLS. 从完全没有前臂的严重缺陷到各种形式的少指(缺失手指)的上肢缺陷发生在大约30%患者中。 在没有肢体缺陷的情况下,几乎所有个体都会出现小肢畸形(小手)、近端放置的拇指和第五指指侧曲畸形。 (see Figure 3).Radioulnar骨性关节炎很常见,可能导致肘关节屈曲挛缩。相对于上肢,下肢较少受累。双脚通常小,且超过80% 受累的 个体发生第2趾和中趾并趾。 [Jackson et al 1993].
- 轻型 CdLS. 放射检查发现第一掌骨短导致的拇指近端放置对于诊断可能有用。
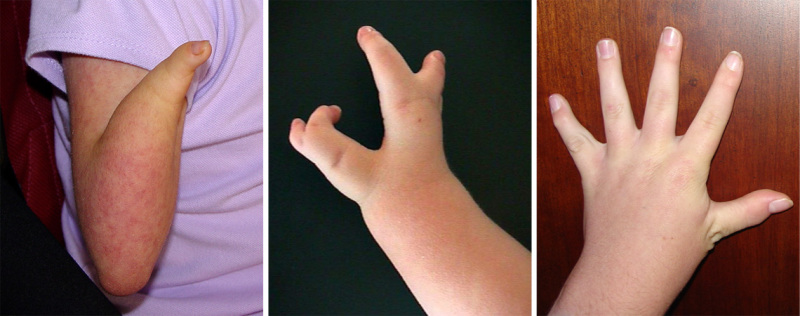
多毛 (>80%). 厚厚的头皮毛发延伸到颞部区域,有时涉及面部,耳朵,背部和手臂。
确诊
CdLS 的确诊通过在 先证者 发现上述临床特征和/或通过 分子遗传学检测 在NIPBL, RAD21, 或 SMC3基因发现 杂合的 致病性变异 或在 HDAC8,SMC1A 基因发现半合子致病性变异 (见 Table 1).
分子检测方法包括系列单个-基因检测, 表型靶向检测的一种应用,和更复杂的基因组的检测。
系列单个-基因 检测. 可考虑按顺序对NIPBL, SMC1A, HDAC8, SMC3, 和 RAD21 基因进行 分子遗传学检测 :
- 1.
应首先对 NIPBL 序列分析 . 若未发现 致病性变异 , 则下一步应考虑 NIPBL 基因-靶向 deletion/duplication analysis 。
- 2.
若未发现 NIPBL 致病性变异 且 受累的 个体具有较轻的CdLS身体特征,考虑 SMC1A 测序和 基因-靶向 deletion/duplication analysis
- 3.
若未发现 NIPBL 或 SMC1A 致病性变异 且高度怀疑为CdLS尤其是对表型较轻微的个体,考虑SMC3, RAD21, 和 HDAC8 序列分析 和 基因-靶向 deletion/duplication analysis。
NIPBL 体细胞 嵌合 已在一小部分个体中被报道过。可以考虑在收集外周血样品时获得口腔样本 [Huisman et al 2013]. 若高度怀疑为CdLS 且上述 分子遗传学检测 均为正常,可对NIPBL 嵌合体进行筛查。
注: 当CdLS诊断不明确,或 分子遗传学检测 未发现 致病性变异,考虑 细胞遗传的 检测或 染色体芯片 (CMA) ,因为已报道过少数个体具有包含NIPBL基因在内的 5p13缺失 [Taylor & Josifek 1981, Hulinsky et al 2005, Hayashi et al 2007]. 部分其他 染色体 异常的特征与CdLS有一部分相同之处 [DeScipio et al 2005, Rohatgi et al 2010].
也可以考虑进行包含NIPBL, SMC1A, HDAC8, SMC3, RAD21 和其它感兴趣基因的 表型靶向检测 (见 Differential Diagnosis) 。注:所包含基因以及多基因panel检测的 敏感性 随不同实验室、不同时期而有所不同。
当系列单个-基因 检测(和/或包括 NIPBL, SMC1A, HDAC8, SMC3, 和 RAD21基因的 表型靶向检测 )未能对具有CdLS特征的个体确诊时,更复杂的 基因组的 检测 (当可及时) 包括 外显子组测序, 基因组测序, 和线粒体测序应被考虑。点击 here,查看关于复杂基因组测序内容。
Table 1.
Cornelia de Lange综合征的分子遗传学检测
| 基因 1 | 该基因致病性变异所占CdLS患者比例 | 检测方法 |
|---|---|---|
| NIPBL | ~60% 2, 3 | 测序分析 4 |
| 靶向基因deletion/duplication analysis 5 | ||
| SMC1A | ~5% 6 | 测序分析 4 |
| 靶向基因 deletion/duplication analysis 5 | ||
| HDAC8 | ~4% | 测序分析 4 |
| 靶向基因 deletion/duplication analysis 5 | ||
| SMC3 | 1%-2% 7 | 测序分析 4 |
| 靶向基因 deletion/duplication analysis 5 | ||
| RAD21 | <1% | 测序分析 4 |
| 靶向基因 deletion/duplication analysis 5 | ||
| 未知 8 | NA | NA |
NA =不适用
- 1.
染色体 位点 和蛋白,见Table A. Genes and Databases 。该 基因检出的等位基因变异信息,见 Molecular Genetics
- 2.
- 3.
NIPBL 基因-靶向 deletion/duplication analysis 在~3%的NIPBL-相关CdLS中检出。 [Russo et al 2012].
- 4.
- 5.
基因-靶向 deletion/duplication analysis 可检测基因内缺失或重复。可采用的技术包括: quantitative PCR, 长片段PCR, 多重连接依赖探针扩增 (MLPA), 和针对检测单个外显子 缺失或重复设计的 基因-靶向微阵列。
6. Musio et al [2006], Borck et al [2007], Deardorff et al [2007]
- 7.
- 8.
50%CdLS个体未检出 NIPBL 致病性变异,提示可能存在 位点 异质性 [Borck et al 2004].
临床特征
临床描述
尽管CdLS的特征在70年前被正式确定,并且上临床上已有很好的描述 [Ptacek et al 1963, Motl & Opitz 1971, Jackson et al 1993], 但关于CdLS自然史直到最近才被研究 [Kline et al 2007a].
不具有以下症状的 受累的个体似乎有正常的寿命。见 Classic Cornelia de Lange Syndrome, 死亡原因.
大多数家族性 案例表明,表达性在家族中是相对一致的。
轻度和经典型CdLS个体面临的医疗问题是相似的; 然而,经典CdLS患者的认知障碍越大,其医学并发症的识别可能就越困难。
典型Cornelia de Lange综合征
生长. 多数经典型CdLS新生儿在产前就发生生长迟滞。对称的缓慢增长导致成比例矮小的身材在生后六个月后变得更加显。终生平均身高和体重低于第五百分位。 [Bruner & Hsia 1990, Kliewer et al 1993, Kline et al 1993b, Kousseff et al 1993, Boog et al 1999].
智力障碍. 据报道,多数经典型CdLS个体具人重度-极重度的智力障碍,IQ值在30-86之间(平均:53)。然而,有关CdLS儿童和智力残疾较轻的成年人的报道提出了更广泛的智力范围。 [Barr et al 1971, Kline et al 1993b, Moeschler & Graham 1993]. 在同时具有 NIPBL基因致病性的 错义 突变和 非移码 缺失的个体及一位具有 SMC1A 基因致病性错义突变的个体中,发现较轻度的智力障碍。 [Ajmone et al 2014].
神经精神.
许多个体表现出自闭症行为,包括自残倾向,他们可能会避免或拒绝社交互动和身体接触。
行为问题通常与无法沟通的挫折直接相关。
大约25%的儿童癫痫发作。
一些家长描述了温度不耐受和减少的疼痛感。
肢体受累. 25%的CdLS患者出现严重的上肢异常。
胃肠. 胃食管反流(GER)就几乎普遍存在的问题 [Bull et al 1993, Sommer 1993, Kline et al 2007b]. 新生儿期GER的诊断和治疗可以避免GER的其他并发症,包括食管炎,吸入性肺炎,化学性肺炎和烦躁不安。 (见 Management).
幽门狭窄是新生儿期持续呕吐的最常见原因,在4%患者中发现。
其他胃肠道异常包括肠旋转不良(2%)和 congenital diaphragmatic hernia 先天性膈疝(CDH) (1%)。CDH在出生前和出生后均已有确诊报道[Fryns 1987, Cunniff et al 1993, Jelsema et al 1993, Pankau & Jänig 1993, Marino et al 2002], 但可能有未被确诊的,尤其是在围产期死亡的婴儿中。
耳鼻喉科. 80%的CdLS患儿观察到感觉神经性听力丧失, 其中40%为严重 受累的 [Sataloff et al 1990]. (见 Deafness and Hereditary Hearing Loss Overview.)
眼科. 多达50% 受累的 个体确认有上睑下垂表现以及其它眼病,包括近视(60%)和眼球震颤(37%) [Levin et al 1990]. 其他眼科异常包括青光眼,鼻泪管狭窄,微角膜,散光,视神经萎缩,视神经缺损,斜视和突眼 [Nicholson & Goldberg 1966, Milot & Demay 1972, Folk et al 1981, Levin et al 1990].
泌尿生殖系统. 73%的CdLS男性患者有隐睾症,57%有发育不全(小)生殖器。已报道12%患者有肾脏异常,主要是膀胱输尿管反流。[Jackson et al 1993].
心血管. 大约25%CdLS个体有 先天的 心脏疾病 [Jackson et al 1993, Mehta & Ambalavanan 1997, Tsukahara et al 1998]. 最常见异常包括(降序排列):室间隔缺损,房间隔缺损,肺动脉狭窄,法洛四联症,左心发育不全综合征和二尖瓣主动脉瓣。
免疫. 已在几名患CdLS个体中描述了抗体缺乏症,表明需要对患有严重或复发性感染的 受累的 个体进行免疫筛查和免疫缺陷管理。[Jyonouchi et al 2013]. 最常报道的反复性感染包括慢性耳部感染,慢性病毒性呼吸道感染和肺炎。受损的T细胞功能可能与CdLS个体中观察到的抗体缺乏有关。
其他特征
- 在75%的CdLS患儿中,已经描述了一种特征性的低音啼哭,这种啼哭在婴儿期后期趋于消失,并且与更严重的病例有关[Jackson et al 1993].
- 据报道,少数儿童患有血小板减少症 [Froster & Gortner 1993, Fryns & Vinken 1994], 其中两人随后发生全血细胞减少症。
- 60%有大理石样皮肤。
- 50%有发育不良的乳头和脐。
- 通贯手和异常的皮纹模式被报道过 [Smith 1966, Opitz 1985].
在20名 受累的 女性的5名(25%)中观察到可引起腹痛的双角子宫[Oliver et al 2010, Kline et al 2015].
死亡原因.Beck & Fenger [1985] 研究了1917年至1982年间出生的48例CdLS患者的死亡率,并且在比较累积存活率时发现比一般人群的死亡率略有增加;在较年轻的年龄组中,这种增加更为显着。他们还报告了两位分别在54岁和61岁时死亡个体。
Beck & Fenger [1985] and Jackson et al [1993] 报道共有24例死于吸入性肺炎(9),复发性呼吸暂停(3),先天的 心脏病(3),肠扭转和肠梗阻(2),手术并发症(血小板减少和颅内出血)(1),脊柱手术后脑水肿和脑疝(1)。严重的支气管肺发育不良,纵隔炎,尿毒症,支气管哮喘,冠状动脉闭塞和肺栓塞是其他死亡原因。
Schrier et al [2011] 报道了295名已知死因的 受累的 个体。呼吸原因,包括误吸/反流和肺炎,是最常见的主要原因(31%),其次是胃肠道疾病,包括梗阻/肠扭转(19%)。先天性异常占死亡人数的15%,包括 先天的 膈疝和先天性心脏缺陷。获得性心脏病占死亡人数的3%。神经系统原因和事故各占8%,败血症占4%,癌症占2%,肾脏病占1.7%,其他原因占死亡人数的9%。
基因型-表型关联
尽管在具有典型CdLS特征的个体(包括面部特征和肢体异常)中可能具有 NIPBL基因致病性变异 , 但在表型轻微和严重的个体中均发现了NIPBL 基因致病变异。NIPBL 基因致病变异平均地分布于整个编码序列。NIBPL 错义 变异的个体通常症状更轻微。
SMC1A 或 SMC3 致病性变异的个体通常比NIPBL 致病性变异个体具有更少的结构性异常和不太严重的生长受限;然而,他们有显著的智力障碍,从中度到重度[Deardorff et al 2007]. SMC1A 或 SMC3 致病性变异个体的面部特征包括比 NIPBL 致病性变异 个体稍微更平而宽的眉毛以及更宽而长的鼻梁[Rohatgi et al 2010]. SMC3 致病性变异的个体特别经常具有轻微或没有连眉、更宽的鼻子和长而明显的人中。SMC3 致病性变异的个体(~56%)也观察到心脏畸形--而较少在SMC1A 致病性变异的个体中见到 [Gil-Rodríguez et al 2015].
RAD21 杂合的 致病性变异 个体通常不具有主要结构异常。杂合RAD21 致病性变异个体比典型CdLS患者具有更轻微的认知缺陷。这些个体通常表现为生长迟缓、轻微骨骼异常和与CdLS重叠的面部特征 [Deardorff et al 2012b].
具有HDAC8 半合子的 致病性变异 的男性个体具有与CdLS重叠的面部特征,但通常表现为延迟关闭前囟门、眼睑下垂、眼距宽、宽鼻、皮肤色素斑块、牙齿异常和开朗友好的个性。生长也趋向于不那么严重 受累的,相关产后生长迟缓和小头畸形的报道频率较低。在女性中,HDAC8 杂合的 致病性变异所导致的临床表现的严重性,很大程度上受X失活模式的影响[Kaiser et al 2014].
外显率
具有NIPBL 体细胞 杂合的 致病性变异 而未受累的个体从未被报道过;因此,外显率 似乎为100%.
类似地, 具有RAD21 杂合的 致病性变异 或男性SMC1A 半合子的 致病性变异, 外显率 似乎非常高。已经注意到 SMC1A 或 RAD21 致病性变异的女性患病严重程度的一些差异。[Musio et al 2006, Deardorff et al 2007, Minor et al 2014].
具有HDAC8 杂合的 致病性变异 的女性的临床表型深受X-失活模式影响 [Kaiser et al 2014]. 有建议称大脑中具有不利的X-失活模式的女性将表现更严重的表型.
尽管发现SMC3 致病性变异的个体相对较少, 其 外显率 似乎很高,然而各个不同致病性变异关联的表型存在差异。
命名法
Cornelia de Lange syndrome (CdLS) 由Vrolik于1849年第一次描述, 其报道了一个案例作为少指畸形的极端病例 [Oostra et al 1994]. Brachmann [1916] 详细介绍了一例对称性单指、肘前织带、矮小症、颈椎肋骨和多毛案例。
在1930年代, 一位名叫Cornelia de Lange的荷兰儿科医生描述了两名具有类似特征的不相关的女孩,并以她工作所在城市名为该症状的命名:typus degenerativus amstelodamensis [de Lange 1933, de Knecht-van Eekelen & Hennekam 1994]. 文献中一些例子将该疾病称为Brachmann-de Lange综合征; 然而,它更广泛地被称为Cornelia de Lange综合征,以纪念de Lange博士对理解该疾病所作出的贡献。
患病率
由于轻微表型可能未被识别出来,CdLS的患病率很难估计。已报道的患病率范围从1:100,000 [Pearce & Pitt 1967] 到高达1:10,000 [Opitz 1985]. 近期EUROCAT数据显示的经典型CdLS的预估患病率为1:50,000 [Barisic et al 2008]; 这个数据不太可能包含轻微的、更常见的 表型.
遗传相关(等位)疾病
除本 GeneReview 所讨论的表型外,未发现其它表型与HDAC8, NIPBL, RAD21, SMC1A, 或 SMC3 基因致病性变异相关。
鉴别诊断
只个证明了临床特征与CdLS重叠的疾病:
- 3q的部分 重复 . 与CdLS相同的特征包括发育迟缓,生长迟滞,低前发际线,突出的睫毛,凹鼻梁,前倾的鼻孔,长而显著的人中(保留中央管),小颌骨,根胫缩短的肢体和生殖器发育不全。 然而,部分重复3q的个体通常具有正常的出生体重,眉毛浓密,眼睛间隔宽,向上倾斜的睑裂,内眦赘皮,鼻子宽阔,嘴唇正常朱红色 [Fineman et al 1978, Fear & Briggs 1979, Annerén & Gustavson 1984, Tranebjaerg et al 1987].
- 染色体 2q31缺失. 该区域中包含HOXD簇的区域的缺失产生类似于在CdLS中看到的肢体减少缺陷,以及泌尿生殖系统和发育异常 [Del Campo et al 1999]。2q31 缺失 的个体不具有CdLS的特征面容。
- Fryns syndrome 特征是膈肌缺损(膈疝,腹脏突出,发育不全或发育不良);特征性面部外观(粗面,眼距宽,鼻梁宽,凹鼻,鼻尖宽,人中长,耳位低,耳形成不良,上唇,大嘴,小颌);远端指趾发育不全(指甲,终末指骨); 肺发育不全; 和相关的异常(羊水过多,角膜混浊和/或小眼病,口面劈裂,肾发育不良/肾皮质囊肿和/或涉及脑,心血管系统,胃肠系统,生殖器的畸形)。 超过新生儿期的生存率很少。有关出生后生长和精神运动发育的数据有限;然而,严重的发育迟缓和智力残疾很常见。Fryns综合征是 常染色体隐性遗传. 见 Congenital Diaphragmatic Hernia Overview.
- 胎儿酒精综合征 (FAS). FAS和CdLS共同的特征包括:胎儿宫内发育迟缓,生长迟滞,发育异常,小头畸形,新生儿面部多毛症,短眼睑裂,鼻孔前倾的短鼻,长而光滑的人中,上唇薄,以及心脏缺陷。 然而,FAS中的手和脚并不小,语言的 受累的 程度低于CdLS。 怀孕期间饮酒的历史可用于区分FAS和CdLS。
- CHOPS syndrome 特征是认知障碍和面容粗糙,心脏缺陷,肥胖,肺部受累,身材矮小和骨骼发育不良。它是由AFF4基因中的 杂合的 致病性 功能获得性 变异引起的。虽然CHOPS综合征和CdLS具有表型重叠,但具有CHOPS综合征的个体具有粗糙的面部特征,超重并且具有显著的肺部受累,这通常在CdLS中观察不到。[Izumi et al 2015].
- TAF1 变异与 X-linked 智力障碍综合征相关,其特征在于全面发育迟缓,全面性肌张力减退,可变的神经系统表现和 变形的 特征,包括长脸,突出的眶上脊,长的人中,前倾的鼻孔,尖下巴和突出的耳朵[O'Rawe et al 2015]. O'Rawe et al [2015] 报道的 受累的 个体通常共有的其他特征包括小头畸形和听力损失。
- Bohring-Opitz syndrome 特征为小头畸形,三头畸形,严重发育迟缓,生长迟滞,喂养困难,腭异常,特征性面部特征包括凹陷的鼻梁,睑缘裂,前倾的鼻孔,低位后旋耳和低发际线伴多毛症。 患有Bohring-Opitz综合征(BOS)的个体可以具有称为“BOS姿势”的特定肢体姿势,其被描述为肩部的外旋或内收,其中腕部和手指在掌指关节处弯曲。 可与BOS相关的其他特征包括心脏异常,胃肠道表现(例如,腹股沟疝,旋转不良),结构性脑异常和眼科症状。 癫痫发作可能存在于一些 受累的 个体中。BOS是由ASXL1中 杂合的 致病性变异 引起的 [Dangiolo et al 2015].
管理
评估以下初步诊断:
以下对于诊断为德朗热综合征(CdLS)患者的评估建议,是基于近期的指南 [Kline et al 2007b] (full text) 及作者的经验:
- 胃肠道评估(包括上GI系列,内窥镜检查,牛奶扫描和/或pH探针)以评估旋转不良和胃肠道反流,该症状如果未确诊或不治疗,可导致喂养不耐受,危及生命的复发性抽吸和肠扭转。
- 若CdLS生长曲线显示生长障碍,则由营养学专家评估。
- 进行上肢X射线评估骨性关节炎。必须谨慎进行物理治疗,以避免在存在桡尺骨骨赘时引起骨折。
- 进行多学科发展评估,以制定教育/治疗干预措施,重点在沟通技巧的发展。
- 听觉评估采用听觉脑干反应测试和耳声发射测试,以评估听力损失。
- 眼科评估,包括视力评估,扩张眼底检查,眼压测量以及泪管通畅和功能评估。
- 进行超声心动图筛查心脏缺陷。ASDs(心房间隔缺损)很常见并可能不被听诊所查出。
- 对所有 受累的 个体进行神经系统评估和EEG.
- 肾超声检查肾结构异常;若提示异常,则通过囊泡造影(VCUG)评估囊泡回流。
- 尿道下裂和/或隐睾症男性的泌尿学评估。
- 盆腔超声评估女性双角子宫;盆腔超声可以与肾脏超声检查同时进行。
- 若出现贫血、淤青、出血症状,进行全血细胞计数。
- 全血细胞计数,免疫谱(由免疫球蛋白组成;破伤风,白喉和肺炎球菌的抗体;B细胞组;和T细胞组)及如果存在复发性感染则考虑免疫评估。
- 咨询临床遗传学家。
对症治疗
以下是合适的:
- 如果GER(胃食管返流)严重,应考虑胃底折叠术,积极管理胃食管反流。
- 如果存在肠道旋转不良,应行手术矫正。
- 若生长障碍,补充配方食品和/或放置胃造口管,以满足营养需求。
- 如果肢体缺陷妨碍使用或移动,则行手臂/手的外科手术干预。
- 正在进行的物理、职业和语言治疗,以优化生长发育结果;若口头表达技巧未能满足表达需求,则采用替代交际方法(例如,手语,图片交换通信系统[PECS])辅助沟通。
- 对听力丧失行标准化治疗。
- 由于导管畸形,按摩疗法等对鼻泪管阻塞的积极治疗通常是不成功的;对屈光不正,斜视,青光眼和眼睑下垂行标准化治疗。
- 心脏缺陷行标准干预措施。
- 对癫痫行适当治疗。
- 对膀胱输尿管反流行抗生素预防和随诊。
- 若存在隐睾症,行睾丸固定术。
- 若存在双子宫,行标准化干预。
预防并发症
预防继发性并发症:
- 血小板减少症和心脏疾病的术前评估。
- CdLS患儿行镇静和/或手术,需在具备小气道护理经验的儿科麻醉医师机构进行。
- 在麻醉期间,注意恶性高热的风险 (见 Malignant Hyperthermia Susceptibility), 已在一小部分CdLS患儿报道。 [Papadimos & Marco 2003]
监控
以下是合适的:
- 每年一次的胃肠道评估包括生长监控。
- 每年一次由发育儿科医生评估发育进程和确认治疗干预手段和教育模式。
- 定期跟踪眼科和/或听力异常。
- 对现有心脏或肾脏异常的常规监测。
应避免的媒介/环境
尚无已知药剂会加剧CdLS的严重程度;然而,应该谨慎行事,以避免现有合并症的恶化,包括胃食管反流,自我伤害行为,异食癖,以及较少见的血小板减少症和免疫学特征。
评估亲属的风险
与 遗传咨询 目的的风险亲属评估相关的问题,见 Genetic Counseling
处于研究中的疗法
搜索 ClinicalTrials.gov 临床试验,以获得关于各种疾病和疾病的临床研究信息。注意:该种疾病可能没有临床试验。
遗传咨询
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
遗传模式
NIPBL-相关Cornelia de Lange syndrome (CdLS), RAD21-相关 CdLS, 以及SMC3-相关 CdLS 均为 常染色体显性遗传 模式。上述基因中的变异最常见为 de novo; 大约1% 的父母存在 胚系嵌合。
家族成员风险-常染色体显性遗传
先证者 父母
- 少于1%确诊为Cornelia de Lange综合征的个体有 受累的 父/母。
- CdLS 再发风险估计高达1.5% [Jackson et al 1993]. 在一组351个家族中,大约3.4%-5.4%父母被发现具有致病性变异 的 胚系嵌合 [Slavin et al 2012]. 然而,然而,Slavin等人的这项研究代表了一个高度偏向的样本,因为 家族性 病例被特别确定为连锁研究提供动力,以帮助识别突变导致CdLS的 基因(s)。因此,该数据可能具有偏差。
先证者 同胞
- 先证者 同胞的风险取决于先证者父母的遗传状态。
- 若 先证者 的 致病性变异 未能在父母的白细胞DNA中检出, 致病原因可能为父母一方 胚系嵌合 或先证者 de novo 致病性变异。在鉴定NIPBL致病变异作为CdLS的病因学之前,基于有不止一个 受累的 儿童的父母们未受影响,提出了胚系嵌合体假设。[Gillis et al 2004, Krantz et al 2004]. 随着 杂合的 NIPBL 致病性变异导致CdLS的发现,在一部分病例中确认了胚系嵌合体的存在 (见 先证者父母).
先证者 后代
- CdLS个体的每个后代有50%概率遗传到该 致病性变异。
家族成员风险-X连锁遗传
先证者父母
- 他母亲有一个遗传自上一代的 致病性变异 。
注: 不像通常的 X-linked 基因, SMC1A 基因在 X-chromosome inactivation 过程中并不失活;因此, 携带者 状态母亲可能表现出比其 受累的 儿子轻微的一些CdLS特征。然而,至今发现的SMC1A-相关的CdLS家族还太少以至于不足以充分评估这一模型。
先证者同胞
- 同胞的风险取决于母亲的 携带者 状态.
先证者后代
- 尽管许多典型CdLS个体不生育,而轻微的 受累的 个体可能拥有后代。
先证者的其他家族成员。先证者母系姨妈可能有携带者的风险,并且该姨妈的子代可能具有携带者风险或 受累的风险(取决于其性别)。
携带进检测
对有风险女性的携带者检测要求事先发现家族中的SMC1A 或 HDAC8 致病性变异 .
相关遗传咨询事宜
家庭计划
- 确定遗传风险和产前检测可用性的讨论的最佳时间是在孕前。
- 向 受累的 或有风险的年轻人提供遗传咨询(包括对后代的潜在风险和生殖选择的讨论)是适当的。
DNA 银行 是存储DNA(通常是从白细胞中提取)以备将来可能的使用。由于检测技术和我们对于基因、等位基因变异及疾病的认识可能在未来有所提升,应考虑对 受累的 个体进行DNA存储。
产前检测和胚胎植入前遗传学诊断
分子遗传学检测. 一旦在 受累的 家庭成员中发现了 致病性变异 ,就可以对增加风险的怀孕者进行 产前诊断 以及对CdLS进行 植入前遗传诊断。
超声检查. 对尚未发现 致病性变异 的家族,可以提供高分辨率超声检查以跟踪生长并评估CdLS中 受累的 肢体、心脏、膈肌、上腭和其他器官或结构。已报道的产前超声检查结果有:
- 生长迟滞,通常出现在妊娠中期
- 典型的子宫内CdLS胎儿面部轮廓包含:小颌畸形,一个突出的上唇和一个凹陷的鼻梁及有一些前倾的鼻孔 [Ranzini et al 1997, Boog et al 1999, Urban & Hartung 2001]
孕妇血清筛查. 如果胎儿有CdLS,母亲血清PAPP-A(妊娠相关血浆蛋白A)水平在孕早期和孕中期可能较低 [Westergaard et al 1983, Aitken et al 1999, Arbuzova et al 2003, Clark et al 2012].
资源
GeneReviews的工作人员选择了以下疾病特异性和/或伞状支持组织和/或登记处,以使该疾病患者及其家人受益。 GeneReviews不对其他组织提供的信息负责。有关选择标准的信息请点击 here.
- CdLS WorldCdLS World is an international "hub" for worldwide organizations and communities united by Cornelia de Lange Syndrome. Country-specific contact information is available on the CdLS website.
- Cornelia de Lange Syndrome Foundation, Inc.302 West Main Street#100Avon CT 06001Phone: 800-223-8355 (Toll-free Support Line); 860-676-8166Fax: 860-676-8337Email: info@cdlsusa.org
- My46 Trait Profile
分子遗传学
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. - ED.
Table A.
Cornelia de Lange Syndrome: 基因与数据库
Table B.
OMIM数据库关于Cornelia de Lange Syndrome的词条 (View All in OMIM)
| 122470 | CORNELIA DE LANGE SYNDROME 1; CDLS1 |
| 300040 | STRUCTURAL MAINTENANCE OF CHROMOSOMES 1A; SMC1A |
| 300269 | HISTONE DEACETYLASE 8; HDAC8 |
| 300590 | CORNELIA DE LANGE SYNDROME 2; CDLS2 |
| 300882 | CORNELIA DE LANGE SYNDROME 5; CDLS5 |
| 606062 | STRUCTURAL MAINTENANCE OF CHROMOSOMES 3; SMC3 |
| 606462 | RAD21, S. POMBE, HOMOLOG OF; RAD21 |
| 608667 | NIPPED-B-LIKE; NIPBL |
| 610759 | CORNELIA DE LANGE SYNDROME 3; CDLS3 |
| 614701 | CORNELIA DE LANGE SYNDROME 4; CDLS4 |
NIPBL
基因结构. NIPBL 转录本长约 9.5 kb,包含 47个外显子 (NM_133433.3). 该 基因 于2004年由两个独立研究组发现 [Krantz et al 2004, Tonkin et al 2004]. 关于基因和蛋白信息的详细描述,见 Table A, 基因.
致病性变异. 超过80个致病性变异已被报道;包括 错义 (~20%), 移码 (~40%), nonsense (~20%), 和剪接位点变异 (~15%) [Borck et al 2004, Gillis et al 2004, Krantz et al 2004, Tonkin et al 2004, Bhuiyan et al 2006, Yan et al 2006, Selicorni et al 2007]. 大多数已发现变异为 私有的; 然而, 在几个不相关的先证者中发现过相同的致病性变异 [Gillis et al 2004].
有研究者提出,较轻型CdLS 更可能是由 错义 突变导致,而较严重类型由截短突变导致 [Gillis et al 2004]. (关于更多信息, 见 Table A.)
正常的 基因产物. NIPBL (delangin or nipped-B-like) 蛋白包含 2,804 个氨基酸,是一种新型蛋白质,与果蝇nipped-b和酵母姐妹染色单体黏着蛋白2(scc2)具有同源性 [Krantz et al 2004, Tonkin et al 2004]. 具有正常功能的 NIPBL蛋白似乎在姐妹染色单体黏着力和调节远程增强子-启动子相互作用中起作用,很可能是通过 Dorsett [2004]文中描述的黏着复合物的相互作用。参考序列为 NP_597677.2.
异常的 基因产物. NIPBL 基因的致病性变异可能导致 单倍剂量不足 (移码, nonsense, 和可能的剪接位点变异) 或改变蛋白质 (错义 变异) ,该蛋白的功能和活性至今未明。NIPBL 蛋白的单倍剂量不足导致CdLS已在两篇关于整个基因缺失的CdLS婴儿的报道中被证明 [Taylor & Josifek 1981, Hulinsky et al 2005]. NIPBL 的改变可能对姐妹染色单体黏着力产生一些影响 [Kaur et al 2005]; 然而,CdLS的表现更可能是由于远程增强子-启动子相互作用被破坏,导致多个下游基因失调。
SMC1A
基因结构. SMC1A 转录本长约 9.7 kb,包含25个外显子。关于 基因 和蛋白信息的详细描述,见 Table A, 基因.
致病性变异. 至少29个SMC1A 基因致病性变异已被报道 (NM_006306.2) [Liu et al 2009]. 所有这些致病性变异都导致了 开放阅读框架的保留;包含错义 和 非移码 缺失 [Musio et al 2006, Borck et al 2007, Deardorff et al 2007, Liu et al 2009, Mannini et al 2010]. 这些研究中报道的 受累的 个体的表型表明 SMC1A 基因致病性变异导致轻型的CdLS,没有显著的肢体和脏器结构异常,但许多患者认知受累显著。
正常的 基因产物. SMC1A 蛋白包含1233个氨基酸,是酵母Smc1基因的人类同源体,是黏着复合物的核心组分,与Smc3形成异二聚体。黏着复合物(The cohesin complex)在姐妹染色单体的内聚作用(cohesion)中起关键作用,并在通过远程增强子-启动子相互作用调节基因表达中起作用 [Dorsett 2004]. SMC1A, 尽管位于 染色体 Xp11.22, 已被报道逃脱了 X-chromosome inactivation [Brown et al 1995], 其对表型 外显率 和 遗传咨询 有重要提示。
异常的 基因产物. 已报道的致病变异导致被改变但可能是完整的蛋白质 [Deardorff et al 2007, Liu et al 2009, Revenkova et al 2009]. 据报道SMC1A 基因可逃脱 X-chromosome inactivation [Brown et al 1995], 因此推测女性中的正常 等位基因 具有一定的保护作用。没有报道过完全破坏蛋白质的致病性变异 (e.g., nonsense, 移码) , 尽管男性中完成缺失 SMC1A蛋白可能是胚胎致死性的。然而,目前数据不足以推断出任何结论。SMC1A 基因致病性变异导致的表现可能是与 NIPBL基因相似的机制的结果:即,基因表达的改变 [Dorsett et al 2005].
SMC3
基因结构. SMC3 转录本长约4.1 kb,包含29个外显子 (NM_005445.3). 关于 基因 和蛋白信息的详细描述,见 Table A, 基因.
致病性变异. 至今, 有一个 致病性变异 (一个3-bp 缺失) 在一位轻型变异型CdLS个体被报道。症状包括:弓形眉毛,连眉,长睫毛,薄唇,小手和小脚,拇指近端放置,第五指弯曲,肘关节运动受限,多毛症,以及高鼻梁和高腭。他没有短头畸形,低发前发际线,前倾的鼻孔,长的人中,嘴角下倾,小颌畸形和听力损失。他受雇于一个受监督的职位。作者指出,有SMC3或SMC1A致病性变异的个体表现出非常轻微的面部畸形,没有四肢或手指缺失或减少,也没有其他主要结构异常 [Deardorff et al 2007].
正常的 基因产物. SMC3蛋白包含 1217个氨基酸 (NP_005436.1) ,是酵母Smc3基因的人类同源体,是黏着复合物的核心组分,与Smc1形成异二聚体。
异常的 基因产物. 预计对一个 受累的 个体SMC3 de novo 3-bp 缺失 的惟一报道导致被改变但可能是完整的蛋白质,该蛋白质表现类似于SMC1A中发现的 错义 突变体 [Deardorff et al 2007].
HDAC8
基因结构. HDAC8 最长的转录本 NM_018486.2 有11个外显子,长约243 kb。关于 基因 和蛋白信息的详细描述,见 Table A, 基因.
致病性变异.HDAC8 致病性变异在大约4%的典型CdLS特征或有 表型 重叠的个体中检出。[Deardorff et al 2012a, Kaiser et al 2014]. 已报道包括对HDAC8 基因造成干扰的染色体缺失和重复、 nonsense, 错义, 和 剪接位点 变异。Kaiser et al [2014] 报道了一组25名先证者,3名 受累的 家族成员, 和3名未受累母亲携带HDAC8基因致病性变异。在已报道的致病性变异中,23个为受累个体的 de novo突变,6个遗传自母亲(在4个家族中),7名个体的遗传模式未能确认。
HDAC8 基因致病性变异的个体具有与CdLS重叠的表型但不表现为典型的CdLS。通常,个体不存在主要结构异常,并且他们倾向于表现前囟的延迟闭合、眼间距宽和宽的鼻尖。 Kaiser et al [2014] 报道20/23 名女性HDAC8 基因 杂合的 致病性变异具有 X-chromosome inactivation 偏斜 (>95:5), 1名未显示X染色体失活偏斜,2名由于在检测位点存在 纯合性 多态性而无法提供信息。
正常的 基因产物. 组蛋白去乙酰化酶 8 蛋白 NP_060956.1 包含377个氨基酸,是脊椎动物SMC3脱乙酰酶,在细胞周期过程中的内聚循环中起作用。
异常的 基因产物. 组蛋白去乙酰化酶 8的活性缺失,导致SMC3乙酰化增加和内聚位点的占位减少,从而改变转录反应 [Deardorff et al 2012a].
RAD21
基因结构.RAD21 包含14个外显子(NM_006265.2) ,长3,604 bp. 关于 基因 和蛋白信息的详细描述,见 Table A, 基因.
致病性变异. 已发现少数个体具有RAD21 基因致病性变异 (错义, frameshift, 非移码 缺失) 和整个-基因 缺失[Deardorff et al 2012b, Minor et al 2014]. 这些个体的表现特征中与CdLS一致的包括:高拱型浓眉、连眉和矮小,然而没有认知延迟。两外不具有典型CdLS特征的先证者均发现新的 杂合的 de novo 错义 致病性变异. 两名个体也均有轻微的神经发育影响。Minor et al [2014] 描述了另两名RAD21致病性变异的先证者, 一个移码突变和一个遗传自母亲的外显子 13框内缺失,其表现为非典型的CdLS 表型 且与更轻微的发育延迟一致。
Table 2.
本篇讨论的RAD21 基因致病性变异
| DNA 序列改变 | 蛋白质改变 | 参考序列 |
|---|---|---|
| c.1127C>G | p.Pro376Arg | NM_006265 NP_006256 |
| c.1753T>C | p.Cys585Arg |
变异分类的注意事项: 表上所表变异为作者提供。GeneReviews 工作人员尚未独立验证变异的分类。
命名的注意事项: GeneReviews 遵循人类基因组变异学会的命名标准惯例(varnomen
- .hgvs.org ). 关于命名的解释,见 Quick Reference .
正常的 基因产物. RAD21 包含631个氨基酸,是黏着复合物的一个亚基。RAD21连接SMC1/SMC3异二聚体和STAG1亚基,以适当调节内聚作用及染色质结合和解离。[Deardorff et al 2012b].
异常的 基因产物. 功能研究表明,RAD21 缺失 会导致 单倍剂量不足 ,而 错义 变异则导致蛋白质结构改变或功能丧失 [Deardorff et al 2012b]. 之前由 Deardorff et al [2012b] 发现的p.Pro376Arg错义变异,通过增加STAG1和STAG2与RAD21的结合,导致RAD21-STAG活性改变,从而干扰内聚活性。该活性改变可能导致姐妹染色单体内聚的改变,尤其是更紧密结合的姐妹染色单体,这表明细胞周期从G2/M到G1延迟。Deardorff et al [2012b] 发现的第二个致病性变异 p.Cys585Arg, 预测为将改变Rad21-SMC1A相相互作用; 功能研究表明其功能丧失。Minor et al [2014] 描述的外显子 6 的移码 重复 导致了未成熟的终止子,可能成为 nonsense-介导的衰变的目标。
参考文献
文献引用
- Aitken DA, Ireland M, Berry E, Crossley JA, Macri JN, Burn J, Connor JM. Second-trimester pregnancy associated plasma protein-A levels are reduced in Cornelia de Lange syndrome pregnancies. Prenat Diagn. 1999;19:706 - 10. [PubMed: 10451512]
- Ajmone PF, Rigamonti C, Dall'Ara F, Monti F, Vizziello P, Milani D, Sellicorni A, Costantino A. Communication, cognitive development and behavior in children with Cornelia de Lange Syndrome (CdLS): preliminary results. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:223 - 9. [PubMed: 24706566]
- Allanson JE, Hennekam RC, Ireland M. De Lange syndrome: subjective and objective comparison of the classical and mild phenotypes. J Med Genet. 1997;34:645 - 50. [PMC free article: PMC1051026] [PubMed: 9279756]
- Annerén G, Gustavson KH. Partial trisomy 3q (3q25----qter) syndrome in two siblings. Acta Paediatr Scand. 1984;73:281 - 4. [PubMed: 6741531]
- Arbuzova S, Nikolenko M, Krantz D, Hallahan T, Macri J. Low first-trimester pregnancy-associated plasma protein-A and Cornelia de Lange syndrome. Prenat Diagn. 2003;23:864. [PubMed: 14558036]
- Barisic I, Tokic V, Loane M, Bianchi F, Calzolari E, Garne E, Wellesley D, Dolk H., EUROCAT Working Group. Descriptive epidemiology of Cornelia de Lange syndrome in Europe. Am J Med Genet A. 2008;146A:51 - 9. [PubMed: 18074387]
- Barr AN, Grabow JD, Matthews CG, Grosse FR, Motl ML, Opitz JM. Neurologic and psychometric findings in the Brachmann-De Lange syndrome. Neuropadiatrie. 1971;3:46 - 66. [PubMed: 5170967]
- Beck B, Fenger K. Mortality, pathological findings and causes of death in the de Lange syndrome. Acta Paediatr Scand. 1985;74:765 - 9. [PubMed: 4050424]
- Bhuiyan ZA, Klein M, Hammond P, van Haeringen A, Mannens MM, Van Berckelaer-Onnes I, Hennekam RC. Genotype-phenotype correlations of 39 patients with Cornelia De Lange syndrome: the Dutch experience. J Med Genet. 2006;43:568 - 75. [PMC free article: PMC2564552] [PubMed: 16236812]
- Boog G, Sagot F, Winer N, David A, Nomballais MF. Brachmann-de Lange syndrome: a cause of early symmetric fetal growth delay. Eur J Obstet Gynecol Reprod Biol. 1999;85:173 - 7. [PubMed: 10584631]
- Borck G, Redon R, Sanlaville D, Rio M, Prieur M, Lyonnet S, Vekemans M, Carter NP, Munnich A, Colleaux L, Cormier-Daire V. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet. 2004;41:e128. [PMC free article: PMC1735640] [PubMed: 15591270]
- Borck G, Zarhrate M, Bonnefont JP, Munnich A, Cormier-Daire V, Colleaux L. Incidence and clinical features of X-linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum Mutat. 2007;28:205 - 6. [PubMed: 17221863]
- Brachmann W (1916) Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbogen, sowie anderen Abnormalitaten (Zwerghaftigkeit, Halsrippen, Behaarung). Jahrbuch Kinderheilkd phys Erzieh 84:225.
- Braddock SR, Lachman RS, Stoppenhagen CC, Carey JC, Ireland M, Moeschler JB, Cunniff C, Graham JM Jr. Radiological features in Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1006 - 13. [PubMed: 8291513]
- Brown CJ, Miller AP, Carrel L, Rupert JL, Davies KE, Willard HF. The DXS423E gene in Xp11.21 escapes X chromosome inactivation. Hum Mol Genet. 1995;4:251 - 5. [PubMed: 7757075]
- Bruner JP, Hsia YE. Prenatal findings in Brachmann-de Lange syndrome. Obstet Gynecol. 1990;76:966 - 8. [PubMed: 1699187]
- Bull MJ, Fitzgerald JF, Heifetz SA, Brei TJ. Gastrointestinal abnormalities: a significant cause of feeding difficulties and failure to thrive in Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1029 - 34. [PubMed: 8291519]
- Clark DM, Sherer I, Deardorff MA, Byrne JL, Loomes KM, Nowaczyk MJ, Jackson LG, Krantz ID. Identification of a prenatal profile of Cornelia de Lange syndrome (CdLS): a review of 53 CdLS pregnancies. Am J Med Genet A. 2012;158A:1848 - 56. [PMC free article: PMC3402646] [PubMed: 22740382]
- Cunniff C, Curry CJ, Carey JC, Graham JM Jr, Williams CA, Stengel-Rutkowski S, Luttgen S, Meinecke P. Congenital diaphragmatic hernia in the Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1018 - 21. [PubMed: 8291515]
- Dangiolo SB, Wilson A, Jobanputra V, Anyane-Yeboa K. Bohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: Review of the most prevalent molecular and phenotypic features of the syndrome. Am J Med Genet A. 2015;167A:3161 - 6. [PubMed: 26364555]
- de Knecht-van Eekelen A, Hennekam RC. Historical study: Cornelia C. de Lange (1871-1950)--a pioneer in clinical genetics. Am J Med Genet. 1994;52:257 - 66. [PubMed: 7810555]
- de Lange C (1933) Sur un type nouveau de degeneration (typus Amstelodamensis) [On a new type of degeneration (type Amsterdam)]. Arch Med Enfants 36.
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012a;489:313 - 7. [PMC free article: PMC3443318] [PubMed: 22885700]
- Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil-Rodríguez C, Arnedo M, Loeys B, Kline AD, Wilson M, Lillquist K, Siu V, Ramos FJ, Musio A, Jackson LS, Dorsett D, Krantz ID. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485 - 94. [PMC free article: PMC1821101] [PubMed: 17273969]
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Mönnich M, Yan Y, Xu W, Gil-Rodríguez MC, Clark D, Hakonarson H, Halbach S, Michelis LD, Rampuria A, Rossier E, Spranger S, Van Maldergem L, Lynch SA, Gillessen-Kaesbach G, Lüdecke HJ, Ramsay RG, McKay MJ, Krantz ID, Xu H, Horsfield JA, Kaiser FJ. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. 2012b;90:1014 - 27. [PMC free article: PMC3370273] [PubMed: 22633399]
- Del Campo M, Jones MC, Veraksa AN, Curry CJ, Jones KL, Mascarello JT, Ali-Kahn-Catts Z, Drumheller T, McGinnis W. Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am J Hum Genet. 1999;65:104 - 10. [PMC free article: PMC1378080] [PubMed: 10364522]
- DeScipio C, Kaur M, Yaeger D, Innis JW, Spinner NB, Jackson LG, Krantz ID. Chromosome rearrangements in cornelia de Lange syndrome (CdLS): report of a der(3)t(3;12)(p25.3;p13.3) in two half sibs with features of CdLS and review of reported CdLS cases with chromosome rearrangements. Am J Med Genet A. 2005;137A:276 - 82. [PMC free article: PMC4896149] [PubMed: 16075459]
- Dorsett D. Adherin: key to the cohesin ring and Cornelia de Lange syndrome. Curr Biol. 2004;14:R834 - 6. [PubMed: 15458660]
- Dorsett D, Eissenberg JC, Misulovin Z, Martens A, Redding B, McKim K. Effects of sister chromatid cohesion proteins on cut gene expression during wing development in Drosophila. Development. 2005;132:4743 - 53. [PMC free article: PMC1635493] [PubMed: 16207752]
- Fear C, Briggs A. Familial partial trisomy of the long arm of chromosome 3 (3q). Arch Dis Child. 1979;54:135 - 8. [PMC free article: PMC1545362] [PubMed: 434890]
- Fineman RM, Hecht F, Ablow RC, Howard RO, Breg WR. Chromosome 3 duplication q/deletion p syndrome. Pediatrics. 1978;61:611 - 8. [PubMed: 662487]
- Folk JC, Genovese FN, Biglan AW. Coats' disease in a patient with Cornelia de Lange syndrome. Am J Ophthalmol. 1981;91:607 - 10. [PubMed: 7234942]
- Froster UG, Gortner L. Thrombocytopenia in the Brachmann-de Lange syndrome. Am J Med Genet. 1993;46:730 - 1. [PubMed: 8362921]
- Fryns JP. Posterolateral diaphragmatic hernia and Brachmann-de-Lange syndrome. Arch Fr Pediatr. 1987;44:474. [PubMed: 3619588]
- Fryns JP, Vinken L. Thrombocytopenia in the Brachmann-de Lange syndrome. Am J Med Genet. 1994;49:360. [PubMed: 8209903]
- Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, Kline AD, Li HH, Devoto M, Jackson LG, Krantz ID. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet. 2004;75:610 - 23. [PMC free article: PMC1182048] [PubMed: 15318302]
- Gil-Rodríguez MC, Deardorff MA, Ansari M, Tan CA, Parenti I, Baquero-Montoya C, Ousager LB, Puisac B, Hernández-Marcos M, Teresa-Rodrigo ME, Marcos-Alcalde I, Wesselink JJ, Lusa-Bernal S, Bijlsma EK, Braunholz D, Bueno-Martinez I, Clark D, Cooper NS, Curry CJ, Fisher R, Fryer A, Ganesh J, Gervasini C, Gillessen-Kaesbach G, Guo Y, Hakonarson H, Hopkin RJ, Kaur M, Keating BJ, Kibaek M, Kinning E, Kleefstra T, Kline AD, Kuchinskaya E, Larizza L, Li YR, Liu X, Mariani M, Picker JD, Pié Á, Pozojevic J, Queralt E, Richer J, Roeder E, Sinha A, Scott RH, So J, Wusik KA, Wilson L, Zhang J, Gómez-Puertas P, Casale CH, Ström L, Selicorni A, Ramos FJ, Jackson LG, Krantz ID, Das S, Hennekam RC, Kaiser FJ, FitzPatrick DR, Pié J. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange syndrome-overlapping phenotypes. Hum Mutat. 2015;36:454 - 62. [PubMed: 25655089]
- Greenberg F, Robinson LK. Mild Brachmann-de Lange syndrome: changes of phenotype with age. Am J Med Genet. 1989;32:90 - 2. [PubMed: 2705489]
- Hayashi S, Ono M, Makita Y, Imoto I, Mizutani S, Inazawa J. Fortuitous detection of a submicroscopic deletion at 1q25 in a girl with Cornelia-de Lange syndrome carrying t(5;13)(p13.1;q12.1) by array-based comparative genomic hybridization. Am J Med Genet A. 2007;143A:1191 - 7. [PubMed: 17497725]
- Huang WH, Porto M. Abnormal first-trimester fetal nuchal translucency and Cornelia de Lange syndrome. Obstet Gynecol. 2002;99:956 - 8. [PubMed: 11975974]
- Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet. 2013;50:339 - 44. [PubMed: 23505322]
- Hulinsky R, Byrne JL, Lowichik A, Viskochil DH. Fetus with interstitial del(5)(p13.1p14.2) diagnosed postnatally with Cornelia de Lange syndrome. Am J Med Genet A. 2005;137A:336 - 8. [PubMed: 16086407]
- Ireland M, Donnai D, Burn J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am J Med Genet. 1993;47:959 - 64. [PubMed: 8291539]
- Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, Zackai EH, Deardorff MA, Clark D, Allen JL, Dorsett D, Misulovin Z, Komata M, Bando M, Kaur M, Katou Y, Shirahige K, Krantz ID. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet. 2015;47:338 - 44. [PMC free article: PMC4380798] [PubMed: 25730767]
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. Am J Med Genet. 1993;47:940 - 6. [PubMed: 8291537]
- Jelsema RD, Isada NB, Kazzi NJ, Sargent K, Harrison MR, Johnson MP, Evans MI. Prenatal diagnosis of congenital diaphragmatic hernia not amenable to prenatal or neonatal repair: Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1022 - 3. [PubMed: 8291516]
- Jyonouchi S, Orange J, Sullivan KE, Krantz I, Deardorff M. Immunologic features of cornelia de lange syndrome. Pediatrics. 2013;132:e484鈥揺489. [PMC free article: PMC4074671] [PubMed: 23821697]
- Kaiser FJ, Ansari M, Braunholz D, Gil-Rodriguez MC, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, DiDonato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillen-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, Mohammed S, Moran E, Mortier GR, Moser JS, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Perez-Aytes A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheurle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pie J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014;23:2888 - 900. [PMC free article: PMC4014191] [PubMed: 24403048]
- Kaur M, DeScipio C, McCallum J, Yaeger D, Devoto M, Jackson LG, Spinner NB, Krantz ID. Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. Am J Med Genet A. 2005;138:27 - 31. [PMC free article: PMC2766539] [PubMed: 16100726]
- Kliewer MA, Kahler SG, Hertzberg BS, Bowie JD. Fetal biometry in the Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1035 - 41. [PubMed: 8291520]
- Kline AD, Barr M, Jackson LG. Growth manifestations in the Brachmann-de Lange syndrome. Am J Med Genet. 1993a;47:1042 - 9. [PubMed: 8291521]
- Kline AD, Calof AL, Lander AD, Gerton JL, Krantz ID, Dorsett D, Deardorff MA, Blagowidow N, Yokomori K, Shirahige K, Santos R, Woodman J, Megee PC, O'Connor JT, Egense A, Noon S, Belote M, Goodban MT, Hansen BD, Timmons JG, Musio A, Ishman SL, Bryan Y, Wu Y, Bettini LR, Mehta D, Zakuri M, Mills JA, Sirvastava S, Haaland RE. Clinical, developmental and molecular update on Cornelia de Lange syndrome and the cohesin complex: abstracts from the 2014 Scientific and Educational Symposium. Am J Med Genet. 2015;167:1179 - 92. [PubMed: 25899772]
- Kline AD, Grados M, Sponseller P, Levy HP, Blagowidow N, Schoedel C, Rampolla J, Clemens DK, Krantz I, Kimball A, Pichard C, Tuchman D. Natural history of aging in Cornelia de Lange syndrome. Am J Med Genet C Semin Med Genet. 2007a;145C:248 - 60. [PMC free article: PMC4902018] [PubMed: 17640042]
- Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR, Levin AV, Selicorni A. Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am J Med Genet A. 2007b;143A:1287 - 96. [PubMed: 17508425]
- Kline AD, Stanley C, Belevich J, Brodsky K, Barr M, Jackson LG. Developmental data on individuals with the Brachmann-de Lange syndrome. Am J Med Genet. 1993b;47:1053 - 8. [PubMed: 7507292]
- Kousseff BG, Thomson-Meares J, Newkirk P, Root AW. Physical growth in Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1050 - 2. [PubMed: 8291522]
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631 - 5. [PMC free article: PMC4902017] [PubMed: 15146186]
- Levin AV, Seidman DJ, Nelson LB, Jackson LG. Ophthalmologic findings in the Cornelia de Lange syndrome. J Pediatr Ophthalmol Strabismus. 1990;27:94 - 102. [PubMed: 2348318]
- Liu J, Feldman R, Zhang Z, Deardorff MA, Haverfield EV, Kaur M, Li JR, Clark D, Kline AD, Waggoner DJ, Das S, Jackson LG, Krantz ID. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum Mutat. 2009;30:1535 - 42. [PMC free article: PMC2783874] [PubMed: 19701948]
- Mannini L, Liu J, Krantz ID, Musio A. Spectrum and consequences of SMC1A mutations: the unexpected involvement of a core component of cohesin in human disease. Hum Mutat. 2010;31:5 - 10. [PMC free article: PMC2797832] [PubMed: 19842212]
- Marino T, Wheeler PG, Simpson LL, Craigo SD, Bianchi DW. Fetal diaphragmatic hernia and upper limb anomalies suggest Brachmann-de Lange syndrome. Prenat Diagn. 2002;22:144 - 7. [PubMed: 11857622]
- Mehta AV, Ambalavanan SK. Occurrence of congenital heart disease in children with Brachmann-de Lange syndrome. Am J Med Genet. 1997;71:434 - 5. [PubMed: 9286451]
- Milot J, Demay F. Ocular anomalies in de Lange syndrome. Am J Ophthalmol. 1972;74:394 - 9. [PubMed: 4626527]
- Minor A, Shinawi M, Hogue JS, Vineyard M, Hamlin DR, Tan C, Donato K, Wysinger L, Botes S, Das S, Del Gaudio D. Two novel RAD21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene. 2014;537:279 - 84. [PubMed: 24378232]
- Moeschler JB, Graham JM Jr. Mild Brachmann-de Lange syndrome. Phenotypic and developmental characteristics of mildly affected individuals. Am J Med Genet. 1993;47:969 - 76. [PubMed: 7507295]
- Motl ML, Opitz JM. Studies of malformation syndromes XXVA. Phenotypic and genetic studies of the Brachmann-de Lange Syndrome. Hum Hered. 1971;21:1 - 16. [PubMed: 5092710]
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006;38:528 - 30. [PubMed: 16604071]
- Nicholson DH, Goldberg MF. Ocular abnormalities in the de Lange syndrome. Arch Ophthalmol. 1966;76:214 - 20. [PubMed: 4957698]
- Oliver C, Bedeschi MF, Blagowidow N, Carrico CS, Cereda A, Fitzpatrick DR, Gervasini C, Griffith GM, Kline AD, Marchisio P, Moss J, Ramos FJ, Selicorni A, Tunnicliffe P, Wierzba J, Hennekam CM. Cornelia de Lange syndrome: extending the physical and psychological phenotype. Am J Med Genet. 2010;152A:1127 - 35. [PubMed: 20425817]
- Oostra RJ, Baljet B, Hennekam RC. Brachmann-de Lange syndrome "avant la lettre". Am J Med Genet. 1994;52:267 - 8. [PubMed: 7810556]
- Opitz JM. The Brachmann-de Lange syndrome. Am J Med Genet. 1985;22:89 - 102. [PubMed: 3901753]
- O'Rawe JA, Wu Y, Dörfel MJ, Rope AF, Au PY, Parboosingh JS, Moon S, Kousi M, Kosma K, Smith CS, Tzetis M, Schuette JL, Hufnagel RB, Prada CE, Martinez F, Orellana C, Crain J, Caro-Llopis A, Oltra S, Monfort S, Jiménez-Barrón LT, Swensen J, Ellingwood S, Smith R, Fang H, Ospina S, Stegmann S, Den Hollander N, Mittelman D, Highnam G, Robison R, Yang E, Faivre L, Roubertie A, Rivière JB, Monaghan KG, Wang K, Davis EE, Katsanis N, Kalscheuer VM, Wang EH, Metcalfe K, Kleefstra T, Innes AM, Kitsiou-Tzeli S, Rosello M, Keegan CE, Lyon GJ. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. 2015;97:922 - 32. [PMC free article: PMC4678794] [PubMed: 26637982]
- Pankau R, Jänig U. Diaphragmatic defect in Brachmann-de Lange syndrome: a further observation. Am J Med Genet. 1993;47:1024 - 5. [PubMed: 8291517]
- Papadimos TJ, Marco AP. Cornelia de Lange syndrome, hyperthermia and a difficult airway. Anaesthesia. 2003;58:924 - 5. [PubMed: 12911384]
- Pearce PM, Pitt DB. Six cases of de Lange's syndrome; parental consanguinity in two. Med J Aust. 1967;1:502 - 6. [PubMed: 6022911]
- Ptacek LJ, Opitz JM, Smith DW, Gerritsen T, Waisman HA. The cornelia de lange syndrome. J Pediatr. 1963;63:1000 - 20. [PubMed: 14071035]
- Ranzini AC, Day-Salvatore D, Farren-Chavez D, McLean DA, Greco R. Prenatal diagnosis of de Lange syndrome. J Ultrasound Med. 1997;16:755 - 8. [PubMed: 9360240]
- Revenkova E, Focarelli ML, Susani L, Paulis M, Bassi MT, Mannini L, Frattini A, Delia D, Krantz I, Vezzoni P, Jessberger R, Musio A. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum Mol Genet. 2009;18:418 - 27. [PMC free article: PMC2722190] [PubMed: 18996922]
- Rohatgi S, Clark D, Kline AD, Jackson LG, Pie J, Siu V, Ramos FJ, Krantz ID, Deardorff MA. Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. Am J Med Genet A. 2010;152A:1641 - 53. [PMC free article: PMC4133091] [PubMed: 20583156]
- Russo S, Masciadri M, Gervasini C, Azzollini J, Cereda A, Zampino G, Haas O, Scarano G, Di Rocco M, Finelli P, Tenconi R, Selicorni A, Larizza L. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur J Hum Genet. 2012;20:734 - 41. [PMC free article: PMC3376273] [PubMed: 22353942]
- Saal HM, Samango-Sprouse CA, Rodnan LA, Rosenbaum KN, Custer DA. Brachmann-de Lange syndrome with normal IQ. Am J Med Genet. 1993;47:995 - 8. [PubMed: 8291543]
- Sataloff RT, Spiegel JR, Hawkshaw M, Epstein JM, Jackson L. Cornelia de Lange syndrome. Otolaryngologic manifestations. Arch Otolaryngol Head Neck Surg. 1990;116:1044 - 6. [PubMed: 2383389]
- Schrier SA, Sherer I, Deardorff MA, Clark D, Audette L, Gillis L, Kline AD, Ernst L, Loomes K, Krantz ID, Jackson LG. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet A. 2011;155A:3007 - 24. [PMC free article: PMC3222915] [PubMed: 22069164]
- Sekimoto H, Osada H, Kimura H, Kamiyama M, Arai K, Sekiya S. Prenatal findings in Brachmann-de Lange syndrome. Arch Gynecol Obstet. 2000;263:182 - 4. [PubMed: 10834327]
- Selicorni A, Lalatta F, Livini E, Briscioli V, Piguzzi T, Bagozzi DC, Mastroiacovo P, Zampino G, Gaeta G, Pugliese A, et al. Variability of the Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:977 - 82. [PubMed: 8291540]
- Selicorni A, Russo S, Gervasini C, Castronovo P, Milani D, Cavalleri F, Bentivegna A, Masciadri M, Domi A, Divizia MT, Sforzini C, Tarantino E, Memo L, Scarano G, Larizza L. Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin Genet. 2007;72:98 - 108. [PubMed: 17661813]
- Slavin TP, Lazebnik N, Clark DM, Vengoechea J, Cohen L, Kaur M, Konczal L, Crowe CA, Corteville JE, Nowaczyk MJ, Byrne JL, Jackson LG, Krantz ID. Germline mosaicism in Cornelia de Lange syndrome. Am J Med Genet A. 2012;158A:1481 - 5. [PMC free article: PMC3356507] [PubMed: 22581668]
- Smith GF. A study of the dermatoglyphs in the de Lange syndrome. J Ment Defic Res. 1966;10:241 - 54. [PubMed: 5972765]
- Sommer A. Occurrence of the Sandifer complex in the Brachmann-de Lange syndrome. Am J Med Genet. 1993;47:1026 - 8. [PubMed: 8291518]
- Taylor MJ, Josifek K. Multiple congenital anomalies, thymic dysplasia, severe congenital heart disease, and oligosyndactyly with a deletion of the short arm of chromosome 5. Am J Med Genet. 1981;9:5 - 11. [PubMed: 6264787]
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636 - 41. [PubMed: 15146185]
- Tranebjaerg L, Baekmark UB, Dyhr-Nielsen M, Kreiborg S. Partial trisomy 3q syndrome inherited from familial t(3;9)(q26.1; p23). Clin Genet. 1987;32:137 - 43. [PubMed: 3652493]
- Tsukahara M, Okamoto N, Ohashi H, Kuwajima K, Kondo I, Sugie H, Nagai T, Naritomi K, Hasegawa T, Fukushima Y, Masuno M, Kuroki Y. Brachmann-de Lange syndrome and congenital heart disease. Am J Med Genet. 1998;75:441 - 2. [PubMed: 9482657]
- Urban M, Hartung J. Ultrasonographic and clinical appearance of a 22-week-old fetus with Brachmann-de Lange syndrome. Am J Med Genet. 2001;102:73 - 5. [PubMed: 11471176]
- Van Allen MI, Filippi G, Siegel-Bartelt J, Yong SL, McGillivray B, Zuker RM, Smith CR, Magee JF, Ritchie S, Toi A, et al. Clinical variability within Brachmann-de Lange syndrome: a proposed classification system. Am J Med Genet. 1993;47:947 - 58. [PubMed: 8291538]
- Westergaard JG, Chemnitz J, Teisner B, Poulsen HK, Ipsen L, Beck B, Grudzinskas JG. Pregnancy-associated plasma protein A: a possible marker in the classification and prenatal diagnosis of Cornelia de Lange syndrome. Prenat Diagn. 1983;3:225 - 32. [PubMed: 6194522]
- Yan J, Saifi GM, Wierzba TH, Withers M, Bien-Willner GA, Limon J, Stankiewicz P, Lupski JR, Wierzba J. Mutational and genotype-phenotype correlation analyses in 28 Polish patients with Cornelia de Lange syndrome. Am J Med Genet A. 2006;140:1531 - 41. [PubMed: 16770807]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, Tos T, Koparir A, Koparir E, Beck CR, Gu S, Aslan H, Yuregir OO, Al Rubeaan K, Alnaqeb D, Alshammari MJ, Bayram Y, Atik MM, Aydin H, Geckinli BB, Seven M, Ulucan H, Fenercioglu E, Ozen M, Jhangiani S, Muzny DM, Boerwinkle E, Tuysuz B, Alkuraya FS, Gibbs RA, Lupski JR. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015;125:636 - 51. [PMC free article: PMC4319410] [PubMed: 25574841]
推荐阅读
- Fitzpatrick DR, Kline AD. Cornelia de Lange syndrome. In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. 3 ed. New York, NY: Wiley-Liss; 2010:195-210. (A recent chapter by Drs. Fitzpatrick and Kline, Directors of the Cornelia de Lange Foundation of the UK and the United States, respectively).
章节说明
致谢
We would like to acknowledge the continued support of the families we follow with CdLS as well as the CdLS-USA Foundation.
作者
Dinah M Clark, MS; The Children's Hospital of Philadelphia (2005-2016)
Matthew A Deardorff, MD, PhD (2005-present)
Ian D Krantz, MD (2005-present)
Sarah E Noon, MS (2016-present)
修订记录
- 28 January 2016 (me) Comprehensive update posted live
- 27 October 2011 (me) Comprehensive update posted live
- 14 August 2006 (cd) Revision: SMC1L1 mutation scanning clinically available
- 18 May 2006 (cd) Revision: mutations in SMC1L1 identified in some individuals with CdLS
- 24 March 2006 (cd) Revision: prenatal testing clinically available
- 16 September 2005 (me) Review posted to live Web site
- 12 January 2005 (ik) Original submission