
摘要
临床特征.
CLCN7相关性骨硬化症包括:婴儿恶性 CLCN7相关常染色体隐性遗传骨硬化症( infantile malignant CLCN7-related recessive osteopetrosis, ARO),中间型 常染体遗传骨硬化症(intermediate 常染色体的osteopetrosis, IAO),和 常染色体显性遗传骨硬化症II型( 常染色体显性遗传osteopetrosis type II, ADOII, Albers-Schönberg 病)。
- ARO婴儿期起病. 临床表现包括:骨折;生长发育迟缓;颅底骨硬化(有或无后鼻孔狭窄或脑积水)导致视神经受压、面部神经麻痹和听力下降;骨髓腔 缺如致严重贫血和血小板减少;牙 齿异常,牙瘤,下颌骨骨髓炎风险;低钙血症并强直性癫痫发作和继发性甲状旁腺功能亢进。未经治疗的ARO最长寿命为10岁。
- IAO儿童期起病. 临床表现包括:轻微创伤后骨折,偶 然发现典型的骨骼X线改变,轻度贫血,偶见继发于视神经受压的视力 障碍。IAO的预期寿命通常是正常的。
- ADOII通常在儿童晚期或青春期起病. 临床表现包括:骨折(发生在任何长骨和/椎后弓),脊柱侧凸,髋骨骨关节炎,下颌骨骨髓炎或感染性骨炎或别处的骨关节炎。颅神经受压很罕见。
诊断及检查.
具有ARO特征性的影像学改变(周身性骨 硬化,棒状长骨,颅底骨硬化,骨中骨外观)和ADOII特征性改变( 脊柱硬化呈“三明治椎骨”外观,骨中骨外观(主要在髂骨翼),股骨干骺端呈酒瓶征,轻度颅底硬化,长骨硬化的横带状影,诊断需考虑 CLCN7相关性骨硬化症)。 CLCN7致病性变异的鉴定可明确诊断。
管理.
对症处理:
- ARO. 补钙治疗低钙性抽搐;每个个体需要钙稳态管理;必要时输注红细胞及血小板;白细胞减少时应用抗生素;免疫球蛋白治疗低丙球蛋白血症;外科手术视神经减压;经验丰富的骨科医师治疗骨折;牙齿保健注意 牙齿萌出、关节僵硬、脓肿、囊肿及瘘管形成。
- ADOII. 矫形治疗骨折和关节炎时注意潜在的术后并发症(骨折延迟愈合或不愈合、感染); 近关节骨折可能需行关节置换术。
主要症状的预防:ARO可被造血干细胞移植(Hematopoietic stem cell transplantation, HSCT)治愈;然而,颅神经功能障碍通常是不可逆的,即使HSCT成功后,神经型ARO儿童也会发生进行性神经系统后遗症。
继发性并发症的预防:
- ARO. 在造血干细胞移植前后及过程中,限制钙和维生素D的摄入以预防高钙血症。
- ADOII. 良好的常规牙齿保健和口腔卫生有助于预防下颌骨骨髓炎。
监管:ARO. 全血细胞计数和眼科检查至少每年一次;HSCT后按每个在移植中心随访。
需要避免的药物及环境:ADOII. 高骨折风险的活动。
GeneReview范围
诊断
CLCN7相关性骨硬化症疾病谱包括:
- 常染色体显性遗传骨硬化症 II 型(autosomal dominant osteopetrosis type II,ADOII,Albers-Schönberg病).
提示性表现
具有骨骼硬化影像学表现的个体(可合并有低钙血症及导致抽搐、贫血、血小板减少症、视力损害和/或中枢神经系统受累), 需考虑诊断CLCN7相关性骨硬化症 (见 表1)。
表1.
CLCN7相关性骨硬化症各亚型的诊断性特征
| 表现 | CLCN7相关性骨硬化症亚型 | ||
|---|---|---|---|
| ARO 1 | IAO 2 | ADOII 3 | |
| 影像学改变 | 能确定诊断的 4 | 特征性的 5 | 特征性的 6 |
| 低钙血症 | 从严重到无 | 无 | 无 |
| 贫血 | 严重到中度 | 轻度到无 | 无 |
| 血小板减少症 | 严重到无 | 无 | 无 |
| 视力损害 | 常见 | 罕见 | 非常罕见 |
| 中枢神经系统受累 | 严重到无 | 无 | 无 |
| 症状出现的年龄 | 出生 | 2岁前 | 10岁前 |
确诊依据
先证者CLCN7相关性骨硬化症的诊断 依据:通过 分子遗传学检测鉴定 CLCN7的 双等位基因致病性变异或 杂合致病性变异(见 表3)。
CLCN7基因致病性变异导致的骨硬化症占所有骨硬化症比例的总结见 表2。
表 2.
CLCN7致病性变异导致的骨硬化症所占的比例
| 骨硬化症表型 | # CLCN7的致病性变异 | CLCN7致病变异所致骨硬化所占百分比 |
|---|---|---|
| 婴儿恶性 CLCN7相关 常染色体隐性遗传骨硬化症( 常染色体隐性遗传osteopetrosis, ARO) | 2 | 13% |
| 中间型 常染色体遗传骨硬化症(intermediate 常染色体的osteopetrosis,IAO) | 2 | 40% 1 |
| 1 | 60% 1 | |
| 常染色体显性遗传骨硬化症 II 型(autosomal dominant osteopetrosis type II,ADOII) | 1 | 75% 2 |
2.
Del Fattore et al [2006]发现 CLCN7致病性变异占ADOII患者的78%; Frattini et al [2003]发现 CLCN7致病性变异占ADOII患者的72%。在其他队列研究,比例可能更高[未发表的观察]。在某些案例中,可能是另一个基因的致病性变异导致ADOII表型 .
表 3.
用于 CLCN7相关性骨硬化症的分子遗传学检测
| 基因 1 | 检测方法 | 使用该方法检测到先证者致病变异 2的比例 |
|---|---|---|
| CLCN7 | 测序分析 3 | ~95% 4 |
| 基因靶向性 缺失/重复分析 5 | 未知 6, 7 |
临床特征
临床叙述
婴儿恶性 CLCN7相关常染色体隐性遗传骨硬化症(Autosomal Recessive Osteopetrosis, ARO)
ARO是系统性的、危及生命的疾病。如果不治疗,寿命大约是十年(尽管有生存更长时间的病例,但非常罕见)。ARO可能的临床表现有:
- 颅骨.在某些严重 受累儿童,1岁内出现大头畸形和前额突出。这并不一定与颅顶骨硬化是平行的 。颅底骨硬化通常导致后鼻孔狭窄。颅骨改变也可导致脑积水。
- 神经系统并发症
- 出生后不久即常出现视力障碍。大多数案例是由颅底硬化导致视神经受压引起的。
- 突出的大前囟常见,有时与脑积水有关,可能是由骨硬化导致脑血流和脑脊液(cerebrospinal fluid, CSF)循环梗阻引起。
- 面部神经麻痹引起的面瘫是一种不常见的临床表现。
- 低钙血症可导致癫痫发作。
- 神经型.如果出现血钙正常范围内的癫痫发作和发育迟缓,须考虑神经型。在这些案例中,神经系统的 表型类似 神经元蜡样质脂褐素质沉积病 [ Steward 2003]。在此非常严重的 受累儿 童亚型中,视网膜和中枢神经系统发生原发性退行性变。区分神经型的这些罕见的原发性神经系统表现(其不良预后)与更常见的颅底骨硬化引起的继发 性损伤是非常重要的。值得注意的是,特征性的病理性脑电图变化(非常频发的多灶性棘波和锐波),通常出现在临床症状和脑核磁共振发现神经退行性病变之前[A Schulz, 未发表结果]。神经型ARO的基础尚不完全清楚,但携带 CLCN7致病性变异的患者比 TCIRG1相关性ARO患者的神经系统并发症更常见且更严重[ Pangrazio et al 2010; A Schulz, 未发表结果],而所有 OSTM1相关性ARO个体均有中枢神经系统受累。这在 Clcn7和 Ostm1基因敲除老鼠的神经退行性变被反映[ Kasper et al 2005, Lange et al 2006]。Ostm1是ClC-7的一个β亚基,为其转运活性所需 [ Leisle et al 2011]。
- 耳的临床表现. 根据 Dozier et al [2005]的数据,78% ARO个体表现出不同程度的听力损失。乳突骨气腔形成不良和外耳道、咽鼓管和内听道狭窄经常导致中耳炎、传导性和感觉神经性听力下降以及面神经瘫痪 [ Dozier et al 2005]。
- 牙齿.ARO的口腔问题有牙齿萌出延迟、牙发育不全、牙齿畸形、牙釉质发育不全、牙釉质和牙本质矿化不足、牙瘤和严重的下颌骨髓炎。即使乳牙受损,在成功干细胞移植后恒牙可正常[ Jälevik et al 2002, Helfrich 2005, Luzzi et al 2006]。
- 低钙血症.低钙血症可导致强直性癫痫发作和继发性甲状旁腺功能亢进。
- 贫血和血小板减少症.骨髓腔缺如导致髓外造血、肝脾肿大、贫血和血小板减少症。血小板减少症相关的出血可非常严重,危及生命,尤其是在中枢神经系统。
- 免疫功能.免疫功能可受损。发生在疾病早期阶段的白细胞增多可变为白细胞减少。与常见的后鼻孔狭窄协同作用,免疫功能受损可能导致慢性鼻炎。据报道,ARO中粒细胞和单核细胞超氧化物生成缺陷 [ Wilson & Vellodi 2000]。
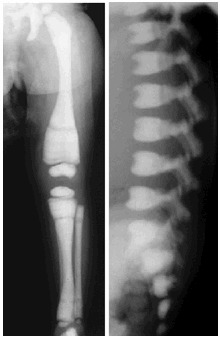
图 1.
ARO X线
中间型常染色体遗传骨硬化症(intermediate Autosomal Osteopetrosis, IAO)
IAO的特点是儿童期发病,病程较ARO轻。大多数案例的预期寿命正常。儿童可能因轻微外伤后骨折或因其它临床指征行X线检查发现特征性改变而就诊。血液系统的症状比ARO轻,而且通常仅限于贫血。尽管通常无中枢神经系统受累,但可出现继发于视神经受侵犯的 视力障碍[ Campos-Xavier et al 2003, Frattini et al 2003]。最近, TCIRG1隐性 亚效内含子的致病性变异在该病的中间型中被报告 [ Sobacchi et al 2014, Palagano et al 2015]。这些变异不仅导致异常 剪接,而且还允许产生有限数量的正常转录本,从而抑制通常与 TCIRG1致病性变异有关疾病的严重程度。事实上,三个拥有这样变异 纯合子的个体,到成年时是相对健康的,主要的问题是复发性非创伤性骨折 [ Palagano et al 2015]。
常染色体显性遗传骨硬化症 II 型(autosomal Dominant Osteopetrosis Type II, ADOII)
虽然ADOII有时被称为“良性骨硬化病”,多达60%-80%有ADOII影像学表现的个体经历过临床问题 (见 图 2)。
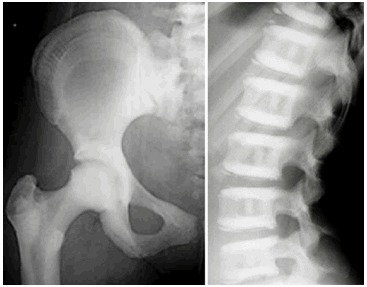
图 2.
ADOII X线转载至Bénichou et al [2000] ,经Elsevier许可
ADOII的临床和影像学表现通常在儿童后期或者青春期出现,尽管有更早发病的报道。以脊椎骨硬化为主,呈“三明治椎骨”外观,为ADOII的一个诊断标准。多数 受累的个体有“骨中骨”外观,主要在髂骨翼,也可出现在其它长骨。通常在长骨中观察到与主轴垂直的骨硬化横带状影。可见颅底骨密度增加[ Bénichou et al 2000, Cleiren et al 2001]。
临床表现有差异,甚至在同一家庭中[ Chu et al 2006]。在多数 受累个体有轻度ADOII表型的3个家庭中,疾病早发、贫血以及视神经受压导致的失明见于某些受累家庭成员。这种 表型曾被称为“中间型骨硬化病”,因为它与轻型的ARO有重叠。
影响骨骼的主要并发症:
- 骨折在最大规模的研究中,骨折发生在80%的 受累个体,平均每人有3次骨折[ Bénichou et al 2000]。一些个体有十次以上的骨折。最常受累的是股骨,但骨折也可发生在任何长骨和脊椎后弓,从而导致脊椎滑脱。
- 脊柱侧凸见于许多案例。
- 髋关节骨关节炎常见(27%),可能因软骨下骨张力过大所致。
- 骨髓炎下颌骨骨髓炎常与牙脓肿或龋齿有关[ Bénichou et al 2000]。其它部位的化脓性骨炎或骨关节炎也可能发生。
颅底骨硬化导致的颅神经受压罕见。听力和视力损失发生于不到5%的 受累个体。
外显率
基于人群和致病性变异研究,在ADOII家系中 外显率范围从60%到90% [ Bollerslev 1989, Bénichou et al 2000, Waguespack et al 2003]。
流行病学
ADOII的患病率估计高达1/20,000 [ Bénichou et al 2001]. 这种疾病在症状轻的案例中,可能漏诊。
ARO较不常见,患病率约1/250,000。在特定的地域更高的发病率被报道,最可能是由 建立者效应导致:在Chuvashiya每3500个新生儿中有1例,在俄罗斯的毛伊岛人群每14,000新生儿中有1例[ Bliznetz et al 2009];在中东患病率约为5.4/100,000[ Souraty et al 2007]。更高的患病率记录也见于哥斯达黎加[ Sobacchi et al 2001]和瑞典的 西博滕省[ Pangrazio et al 2013] 。
遗传学相关(等位基因)疾病
除了那些在本GeneReview中被讨论的已知与 CLCN7致病性变异相关的表型外,无其它相关表型。
鉴别诊断
常染色体隐性骨硬化症
- 继发于TCIRG1致病性变异的ARO( OMIM 259700). 超过50%的ARO 由 TCIRG1致病性变异导致。 TCIRG1致病性变异引起的ARO可与 CLCN7相关性疾病鉴别 [ Frattini et al 2000, Kornak et al 2000],基于后一种形式的骨硬化病有更高频率的神经发育延迟和癫痫发作。
- 破骨细胞数目减少型ARO. 破骨细胞数目减少型ARO的特点是:1岁以内发病,有典型的ARO临床表现。骨活检是可靠诊断的必备条件。然而, TNFSF11(OMIM 259710) 致病性变异引起轻微的T细胞缺陷和 TNFRSF11A(OMIM 612301) 致病性变异会导致低丙球蛋白血症,类似于一种常见的可变免疫缺陷病(common variable immune deficiency, CVID) [ Sobacchi et al 2007, Guerrini et al 2008]。排除 TNFSF11相关性ARO是至关重要的,因为造血干细胞移植在这 受累个体是不成功的。
- ARO合并肾小管酸中毒(renal tubular acidosis,RTA) (OMIM 259730). ARO合并RTA的发病通常晚于恶性婴儿发病形式的ARO,病情较轻 。除了周身性骨硬化外,大脑钙化为典型的表现,可能与智力障碍相关[ Jacquemin et al 1998]。该病致病性变异见于 CA2,该 基因编码碳酸酐酶2[ Bolt et al 2005]。
- 继发于OSTM1致病性变异的ARO (OMIM 259720). 大约有4%的ARO由 OSTM1的致病性变异导致。 OSTM1致病性变异导致一种非常严重的、伴有中枢神经系统受累的ARO形式 [ Pangrazio et al 2006]。也见OSTM1 位点缺失[ Ott et al 2013]。
- 继发于PLEKHM1致病性变异的ARO(OMIM 611497) . 到目前为止,有2例携带 PLEKHM1致病性变异个体被描述。 表型似乎非常轻,随着年龄的增长可逆转 [ Van Wesenbeeck et al 2007]。
- 继发于SNX10致病性变异的ARO(OMIM 615085). 大约有4%的ARO由 SNX10致病性变异导致;特别是,“Västerbottenian 骨硬化症”由该 基因致病性变异导致。这种形式的ARO严重程度似乎略低于 CLCN7相关性者。但是常见视力下降、贫血和骨质疏松,故可行 HSCT[ Aker et al 2012, Pangrazio et al 2013] 。
见 骨硬化症,常染色体隐性:OMIM表型系列,OMIM中与这种 表型相关的基因。
轻型 ARO
- 继发于TCIRG1内含子致病性变异的ARO(OMIM 259700). 轻型ARO可由 TCIRG1深部内含子或剪切位点变异导致 [ Sobacchi et al 2014, Palagano et al 2015]。这种类型非常类似于 CLCN7相关性 IAO或ADOII。
常染色体显性骨硬化症
- 常染色体显性骨硬化病I型(Autosomal dominant osteopetrosis type I, ADOI)(OMIM 607634) ADOI的骨硬化最明显的是颅骨,不会导致三明治样椎骨。这个疾病实体是否应该被称为骨内膜骨质增生或高骨密度症是有争议的,因为骨硬化病应保留给破骨细胞相关性疾病。ADOI与骨折率增高无关。其由 LRP5的致病性变异导致[ Van Wesenbeeck et al 2003]。
见 骨硬化症,常染色体隐性:OMIM表型系列,OMIM中与这种 表型相关的基因。
其它
- 致密成骨不全症(OMIM 265800). 受累个体身材矮小(成人身高< 150厘米)、前额突出、缝间骨、前囟持续开放、和末端趾骨溶解[ Vanhoenacker et al 2000]。骨骼广泛硬化且易骨折。一些病人的临床表现类似于IAO[ Pangrazio et al 2014]。 致密成骨不全症由 CTSK的致病性变异导致,该 基因编码组织蛋白酶K[ Gelb et al 1996],以 常染色体隐性模式遗传。
- SOST相关性硬化性骨发育不良,包括van Buchem病和骨硬化,特点是中至重度颅骨质增生导致的颅神经功能障碍、下颌骨增大和周身性骨硬化。骨硬化还包括并指、身材高大以及可因颅内压增高致命[ Hamersma et al 2003]。 SOST相关性硬化性骨发育不良以 常染色体隐性模式遗传。
- 常染色体显性 颅干骺端发育不良症( Autosomal dominant craniometaphyseal dysplasia, AD-CMD). CMD的临床标志是颅骨骨质增生导致 眼睛深陷 、鼻翼突起。面部神经麻痹常见,且比视神经受压更常见[ Braun et al 2001 ]。 股骨表现出建模缺陷,但无骨硬化。骨折易感性不增加。 由 ANKH 的致病性变异引起[ Nürnberg et al 2001 ]。
- 常染色体隐性颅干骺端发育不良症(Autosomal recessive craniometaphyseal dysplasia, AR-CMD)(OMIM 218400). 受累个体表现为典型的CMD特征(大头畸形、听力下降、颅骨质增生伴鼻翼突起、干骺端增宽),但颅顶增厚并不明显[ Iughetti et al 2000] 。由于 骨干骨硬化,这种疾病有时可类似轻型的骨硬化病。 由 GJA1 的致病性变异 引起 [ Hu et al 2013 ]。
- 白细胞粘附缺陷病III型(Leukocyte adhesion deficiency type III,LAD-III)(OMIM 612840). 受累个体表现为反复感染和出血倾向,且与血小板和白细胞的计数无关。因 FERMT3(编码蛋白fermitin家族同源体3)的致病性变异而致病。因为fermitin家族同源体3(也称为kindlin-3)信号为破骨细胞介导的骨吸收所必需,所以在一些LAD-III个体中可发现高骨密度[ Crazzolara et al 2015]。
管理
最近更新的诊断、治疗和随访指南在 网络上可找到。
初次诊断后评估
为确定一个诊断为婴儿恶性 CLCN7相关性 常染色体隐性遗传骨硬化症( 常染色体隐性遗传osteopetrosis, ARO)个体的疾病和需求程度,推荐以下评估:
- 全血细胞计数以评估白细胞增多或白细胞减少、血小板减少症及贫血伴网织红细胞计数降低
- 检查血液和尿液中钙离子浓度以评估低血钙症和继发性甲状旁腺功能亢进
- 腹部超声以评估肝脾肿大
- 颅脑MRI和/或CT以评估颅神经孔缩小、脑积水和神经型骨硬化症的脑部异常
- 眼科检查包括VEPs以评估视神经萎缩
- 耳鼻喉科检查以评估后鼻孔狭窄
- EEG检测与神经退化行性变相关的病理性变化;神经系统体格检查以评估疾病进展
- 向临床遗传学专家和/或遗传咨询师咨询
临床症状的治疗
由于疾病严重程度的差异,ARO和ADOII的治疗方案不同。IAO介于这两者之间,其预后多变。因此,治疗方案选择评估必须以个体为基础。
ARO
低血钙抽搐,作为疾病首发临床表现,发生于相当多的新生儿,应予补钙治疗。钙稳态的管理可能有困难,而建议有争议:尽管生理剂量的钙和维生素D已经用于治疗骨硬化病儿童的佝偻病,但限制钙和维生素D的摄入被用 来造血干细胞移植(hematopoietic stem cell transplantation, HSCT)后, 预防疾病进展和高钙危象。治疗需要考虑到 受累个体的特殊情况。
骨髓衰竭可能需要输注红细胞和血小板(辐照产品)。一些患病个体可发生白细胞减少和/或低丙球蛋白血症,抗生素和免疫球蛋白可作为一种预防或治疗方式。
新诊断的个体应尽快转到有本病行同种异体干细胞移植经验的儿科中心。
感觉和神经系统临床表现需要儿科、小儿神经科、眼科和心理医生的协作。视神经的外科减压术是一个艰难的手术,已在预防视力下降上取得一定的成功 [ Hwang et al 2000]。
骨折需要由经验丰富的骨外科医生或整形外科医师与儿科治疗医生协作来治疗和监督。
牙齿. 如不行HSCT,多数孩子不能活到恒牙萌发的年龄。尽管乳牙有缺陷,早期接受HSCT的儿童可能有正常的恒牙[ Jälevik et al 2002]。
某些案例,有缺陷的牙齿萌发、关节僵硬、脓肿、囊肿和瘘管的形成可能需要外科手术治疗。需要特别注意以预防下颌骨骨髓炎和牙槽骨的极度脆性[ Luzzi et al 2006]。
ADOII
骨折和关节炎常常需要矫形治疗。由于骨的脆性,术后并发症如骨折延迟愈合或不愈合和感染常见(50%)。近关节骨折可能需行全关节置换术[ Strickland & Berry 2005]。
主要症状的预防
ARO
造血干细胞移植(hematopoietic stem cell transplantation,HSCT).由于骨硬化病有缺陷的破骨细胞来源于造血细胞,故同种异体HSCT有效。大多数临床表现(骨硬化、骨髓衰竭和髓外造血)可通过HSCT预防或逆转。
早期移植可能预防由神经受压迫导致的继发性感觉神经障碍,但已经存在的损害是不能逆转的。
然而,发生在神经型ARO的原发性神经系统问题和视网膜变性不依赖于骨骼疾病,因此不能被造血干细胞移植改善或预防。由 CLCN7致病性变异导致的ARO而不发生神经系统并发症的患者已经被报道 [ Pangrazio et al 2010; A Schulz and U Kornak, 未发表结果]。
将神经型ARO患者排除在这种侵入性治疗之外非常重要,但有困难。另一方面,大多数那些没有原发性神经系统后遗症的患者,应该尽快行HSCT,以防止不可逆的继发性并发症,包括视力障碍。因此, 受累个体和造血干细胞移植治疗应在多学科评估疾病的严重程度和个体的预后因素之后,有经验的儿科中心进行。
欧洲免疫缺陷协会( the European Society of Immunodeficiencies ,ESID)和欧洲骨髓移植组( the European Group of Bone Marrow Transplantation,EBMT)的一项回顾性调查已经分析了ARO的HSCT结局[ Sobacchi et al 2013]。5年无病生存率: 全相合移植者约为88%、 无关相合移植者为80%、 单倍体相合移植为66%[ Sobacchi et al 2013] 。在最近发表的一份报告中,对193例来自不同移植中心以环磷酰胺为基础方案的患者,同胞HLA匹配移植和替代供体移植后5年存活率分别为62%和42%[ Orchard et al 2015]。最近报道的进一步改进的结果,来自3个大型移植中心以氟达拉滨为基础的预处理方案[ Natsheh et al 2016; Schulz and Moshous, 个人交流]。
注:因为ARO较常由 TCIRG1致病性变异导致而不是 CLCN7致病性变异,所以HSCTs主要在具有 TCIRG1而不是 CLCN7致病性变异的儿童中进行。然而,在 TCIRG1和 CLCN7致病性变异的个体间,治疗结果似乎没有显著性差异。[A Schulz et al, 未发表结果]。
造血干细胞移植后严重并发症的发生率高,特别是使用替代供体干细胞来源时。并发症包括排斥反应、延迟的造血重建、静脉闭塞性疾病、肺动脉高压和高钙危象 [ Steward et al 2004, Corbacioglu et al 2006, Shroff et al 2012]。
颅神经功能障碍(视神经萎缩导致的视力障碍)在大多数案例中是不可逆转的。在作者包含大约30个人的对列中,约三分之二的 受累个体在成功移植后有视力障碍 [A Schulz, 未发表结果]。
进行性神经系统后遗症、发育迟缓和反复癫痫发作发生在一些成功行HSCT的个体中[ Steward 2003]。除了视力障碍外,在作者的队列中,严重神经系统表现见于约10%个体。
其它. 保守治疗措施包括通过钙限制、骨化三醇、类固醇、甲状旁腺激素和干扰素刺激宿主的破骨细胞[ Kocher & Kasser 2003]。因为严重骨硬化病好结局良好的证据有限,而且因为其副作用严重(尤其是在婴儿中),这些仅在特殊情况下使用。
继发性并发症的预防
ARO. 在造血干细胞移植前后及过程中,推荐限制钙和维生素D的摄入以预防高钙血症。
应告知双亲和患者疾病可能的并发症,并给予相应的建议(如血小板减少症患者严重的中枢神经系统出血、病理性骨折)。由于疾病的异质性,建议应以个体为基础。
ADOII.良好的常规牙齿保健和口腔卫生可能有助于预防下颌骨骨髓炎。
监管
ARO
- 骨硬化病可能的临床表现和并发症需要反复研究,然而,对于研究的范围和频率没有公认的建议。所有ARO个体应该每年进行至少一次全血细胞计数和眼科检查。
ADOII. ADOII的骨骼表现不进展,因此无需特别的监管。
需要避免的药物/环境
ADOII
- 应该避免高骨折风险的活动。
- 矫形外科手术应该只在绝对需要的情况才执行,在处理硬化骨时,外科医生应该意识到潜在的并发症和困难。
研究中的治疗方法
搜索 ClinicalTrials.gov,可获取广泛疾病的临床研究和条件的信息。注:可能没有本病的临床试验。
遗传咨询
遗传咨询是向个体和家庭提供关于遗传性疾病的本质、遗传和影响的信息,以帮助他们至的医疗和个人决定。以下部分涉及遗传风险评估、家族史和遗传性检测,以阐明家系成员的遗传现状。本节不是为了解决患者可能面临的所有个人、文化、伦理问题或代替与遗传学专家的咨询—ED.
家庭成员风险— 常染色体隐性骨硬化症
先证者父母
先证者同胞
先证者后代
- 总的来说,只有成功接受HSCT(hematopoietic stem cell transplantation, HSCT),否则ARO个体不能生育。
家庭成员风险—常染色体显性骨硬化症
先证者父母
注:如果父母是第一个发生 致病性变异的个体,他/她可能有该变异的 体细胞嵌合体,可能轻度/最低程度 受累。
先证者同胞
先证者后代. 常染色体显性遗传 CLCN7相关性骨硬化症个体的每个孩子有50%机会获得 致病性变异。
其他家庭成员.其他家庭成员的风险取决于 先证者父母的状态。如果父母任一有 CLCN7致病性变异,他或她的家庭成员可能有风险。
相关遗传咨询问题
为早期诊断和治疗,见 高危亲属风险评估、管理,以获得评估高危亲属风险的信息。
貌似新发致病性变异家庭的注意事项。 当患常染色体显性遗传疾病的先证者父母双方均不具有先证者检测到的致病性变异或临床证据时,很可能为新发致病性变异。 然而,也可以探索非医学解释, 替代父本 或母本(例如辅助生殖)和未公开的收养关系。
计划生育
- 确定遗传风险、阐明 携带者状态的和讨论产前检测有效性的最佳时间是在怀孕之前。
DNA库是 DNA的储存库(通常从血液白细胞中提取),以备将来使用。因为检测方法和我们对基因、等位基因变异和疾病的理解可能会在将来得到提升,所以应考虑对受累个体的 DNA进行储存。
产前诊断和胚胎植入前遗传学诊断
一旦在 受累家系成员中被检测出 CLCN7致病性变异, 就可以对高风险孕妇进行产前检测和 胚胎植入前遗传学诊断。
关于使用产前检测,在医学专业人员和家庭内部可能存在认识的差异,特别是如果检测是为了终止妊娠而不是早期诊断。虽然大多数中心会考虑关于产前检测是父母做决定,但是这些问题的讨论是合适的。
信息来源
为了此病个体及其家庭的利益,GeneReviews员工挑选了以下疾病特定的和/或伞形支持组织和/或登记处。GeneReviews不为其他组织提供的信息负责。选择这些组织的标准,请点击获取详细信息。
- The OsteoPETrosis Society (OPETS)Phone:980-292-3921Email:osteopetrosispatient@gmail.com; janecastello.opets@gmail.com
- National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)1 AMS CircleBethesda MD 20892-3675Phone:877-226-4267 (toll-free); 301-565-2966 (TTY)Fax:301-718-6366Email:niamsinfo@mail.nih.gov
- National Library of Medicine Genetics Home Reference
- European Society for Immunodeficiencies (ESID) RegistryDr. Gerhard KindleUniversity Medical Center Freiburg Centre of Chronic ImmunodeficiencyEngesserstr. 479106 FreiburgGermanyPhone:49-761-270-34450Email:esid-registry@uniklinik-freiburg.de
- International Skeletal Dysplasia RegistryUCLA615 Charles E. Young DriveSouth Room 410Los Angeles CA 90095-7358Phone:310-825-8998Email:AZargaryan@mednet.ucla.edu
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能与GeneReview中其他部分的信息不一致;表格里可能包含更新的信息—ED.
表 A.
CLCN7-相关性骨硬化症: 基因和数据库
| Gene | Chromosome Locus | Protein | Locus Specific | HGMD |
|---|---|---|---|---|
| CLCN7 | 16p13 .3 | H(+)/Cl(-) 交换体 7 | CLCN7 数据库 | CLCN7 |
表 B.
分子遗传机制
ARO、IAO、ADOII是因破骨细胞功能障碍导致的疾病。破骨细胞是一个高度特化的细胞,具有独特的重吸收大量矿化骨组织的能力。像巨噬细胞一样, 破骨细胞是由单核造血前体细胞融合而形成的巨大多核细胞,随后在M-CSF和RANKL的作用下分化而成。
结合到骨组织表面后,形成一个密封区域,隔离吸收腔隙和细胞外环境。然后,大量的酸性囊泡与细胞质膜融合结合到骨表面形成褶皱缘。这种结构只存在于破骨细胞,且分泌大量的酸进入吸收腔隙,因此也被称为“细胞外溶酶体”[ Teitelbaum & Ross 2003]。溶解骨矿物质和酸性水解酶降解骨基质的最佳活性,特别是组织蛋白酶K,都需要低pH值。
到目前为止,大多数已明确遗传学病因的人类骨硬化病类型,是泌酸机制的缺陷所致。ClC-7氯通道位于溶酶体囊泡和褶皱缘上,充当 2Cl -/1H +交换体转运负电荷至吸收腔隙,其与通过褶皱缘上的V型H +-ATP酶从细胞外运输质子进入吸收腔隙同步[ Kornak et al 2001, Leisle et al 2011]。
TCIRG1基因编码此H +-ATP酶和由 CA2编码的碳酸酐酶2的一个重要亚基,在破骨细胞细胞质中产生必需的质子;因此,这些基因的缺陷也会导致骨硬化病。
CLCN7的致病性变异导致氯通道功能不同程度的丧失。大多病情严重的ARO案例中,H(+)/ Cl(-)交换转运体7(也称为ClC-7)缺失。正如基因敲除小鼠模型所阐述,蛋白质的完全缺失有致神经型ARO的高风险,表现为中枢神经系统和视网膜的变性 [ Kornak et al 2001, Steward 2003, Kasper et al 2005]。
不太严重的骨硬化病类型被认为是由致病突变引起蛋白质的不完全失活导致。 ADOII的致病性变异显然具有 显性负性效应。这些致病变异影响氯通道的功能已经得到证实,正如由 CLCN1显性致病变异引起的Thomsen型 先天性肌强直被描述的[ Jentsch et al 2005b]。 CLCN7的一些致病性变异被报导会改变蛋白质的表达和亚细胞定位。通过对局限于质膜的ClC-7突变体的电生理学测量证明,致ADOII的致病性变异不一定损害氯电流,但会导致通道的异常门控行为[ Leisle et al 2011]。
基因结构. 最相关的 CLCN7转录本 ( NM_001287.5, CCDS 32361.1)包含2418个碱基的 编码区,细分为25个外显子。 基因和蛋白质的详细摘要信息,见 表 A、 基因。
致病性变异. ARO的致病性变异有 无义变异(7.4%)、 剪接缺陷和插入缺失所致的移码变异(25.5%)、 框内缺失变异(0.01%)和 错义变异(66%)。这些致病性变异有大约 28.7%位于蛋白的C端CBS结构域。在一个ARO个体中发现一个 纯合的连续基因缺失[ Pangrazio et al 2012]。在ADOII中,致病性变异包括无义变异(2.5%)、移码变异(12.5%)、框内缺失变异(2.5%)和错义变异(82.5%)。这些致病性变异中,大约45%位于C端CBS结构域[Sobacchi, 未发表]。
表 4.
在该GeneReview中被讨论的 CLCN7变异
| 变异类型 | DNA核苷酸改变 | 预测的蛋白质改变 | 参考序列 |
|---|---|---|---|
| 良性 | c.1252G>A | p.Val418Met | NM_001287 .5 NP_001278 .1 |
| 致病性 | c.296A>G | p.Tyr99Cys | |
| c.643G>A | p.Gly215Arg | ||
| c.2299C>T | p.Arg767Trp |
关于变异分类的说明:表中所列的变异由作者提供,GeneReviews的员工没有独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会的标准命名惯例(varnomen .hgvs.org ). 有关命名的解释,见快速参考。
正常 基因产物.CLCN7基 因编码由805个氨基酸组成的H+/Cl-交换转运体7(ClC-7),它位于在大多数细胞的晚期核内体和溶酶体上。在破骨细胞内,蛋白质转移到褶皱缘。 像其它CLC通道一样,ClC-7包含两个C端CBS结构域,其被认为参与蛋白质的相互作用。这种相互作用可能促进功能通道二聚体的形成。CLC通道不是纯 氯离子通道而是起到氯离子/质子逆向转运作用 [ Jentsch et al 2005a]。OSTM1蛋白是功能性CLC通道的一个亚基,且影响它们的行为[ Lange et al 2006, Leisle et al 2011]。当蛋白复合物被驱动至细胞膜,即表现出慢门控和向外地整流氯电流[ Leisle et al 2011]。
异常 基因产物.在Clcn7 −/−小鼠破骨细胞中,ClC-7蛋白质完全丧失使破骨细胞失去重吸收活力;同样地,一些恶性婴幼儿骨硬化症个体有一个 无效等位基因[ Kornak et al 2001]。 错义变异的情况不太清楚。只有少数ClC-7突变体,已检测稳定性和亚细胞分布。检测的致病性错义变异显示不同程度的影响 Schulz et al 2010, Leisle et al 2011]。在ADOII相关的致病性变异(总共40个变异中的10个)的体外检测中,大多数通道的电生理学属性发生了改变 [ Leisle et al 2011]。有趣的是,其中一些变异显示正常电流振幅,甚至加速了门控。有人认为这些变化导致破骨细胞活动的可变衰减,引起临床病程的变异性[ Chu et al 2006, Del Fattore et al 2006]。
参考文献
引用文献
- Aker M, Rouvinski A, Hashavia S, Ta-Shma A, Shaag A, Zenvirt S, Israel S, Weintraub M, Taraboulos A, Bar-Shavit Z, Elpeleg O. An SNX10 mutation causes malignant osteopetrosis of infancy. J Med Genet.2012; 49:221–6.[ PubMed : 22499339]
- Bénichou O, Cleiren E, Gram J, Bollerslev J, de Vernejoul MC, Van Hul W. Mapping of autosomal dominant osteopetrosis type II (Albers-Schönberg disease) to chromosome 16p13.3. Am J Hum Genet.2001;2001; 69:647–54.[ PMC free article : PMC1235505] [ PubMed : 11468688]
- Bénichou OD, Laredo JD, de Vernejoul MC. Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestations in 42 patients. Bone.2000; 26:87–93.[ PubMed : 10617161]
- Bliznetz EA, Tverskaya SM, Zinchenko RA, Abrukova AV, Savaskina EN, Nikulin MV, Kirillov AG, Ginter EK, Polyakov AV. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: the unique splice site mutation in TCIRG1 gene spread by the founder effect. Eur J Hum Genet.2009; 17:664–72.[ PMC free article : PMC2986262] [ PubMed : 19172990]
- Bollerslev J. Autosomal dominant osteopetrosis: bone metabolism and epidemiological, clinical, and hormonal aspects. Endocr Rev.1989; 10:45–67.[ PubMed : 2666111]
- Bolt RJ, Wennink JM, Verbeke JI, Shah GN, Sly WS, Bökenkamp A. Carbonic anhydrase type II deficiency. Am J Kidney Dis.2005; 46(A50):e71–3.[ PubMed : 16265785]
- Braun HS, Nurnberg P, Tinschert S. Metaphyseal dysplasia: a new autosomal dominant type in a large German kindred. Am J Med Genet.2001; 101:74–7.[ PubMed : 11343343]
- Campeau PM, Lu JT, Sule G, Jiang MM, Bae Y, Madan S, Högler W, Shaw NJ, Mumm S, Gibbs RA, Whyte MP, Lee BH. Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum Mol Genet.2012; 21:4904–9.[ PMC free article : PMC3607481] [ PubMed : 22875837]
- Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V. Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum Genet.2003; 112:186–9.[ PubMed : 12522560]
- Chu K, Snyder R, Econs MJ. Disease status in autosomal dominant osteopetrosis type 2 is determined by osteoclastic properties. J Bone Miner Res.2006; 21:1089–97.[ PubMed : 16813529]
- Cleiren E, Bénichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, deVernejoul MC, Van Hul W. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet.2001; 10:2861–7.[ PubMed : 11741829]
- Corbacioglu S, Hönig M, Lahr G, Stöhr S, Berry G, Friedrich W, Schulz AS. Stem cell transplantation in children with infantile osteopetrosis is associated with a high incidence of VOD, which could be prevented with defibrotide. Bone Marrow Transplant.2006; 38:547–53.[ PubMed : 16953210]
- Crazzolara R, Maurer K, Schulze H, Zieger B, Zustin J, Schulz AS. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatr Blood Cancer.2015; 62:1677–9.[ PubMed : 25854317]
- Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M, Fortunati D, Ballanti P, Iacobini M, Luciani M, Devito R, Pinto R, Caniglia M, Lanino E, Messina C, Cesaro S, Letizia C, Bianchini G, Fryssira H, Grabowski P, Shaw N, Bishop N, Hughes D, Kapur RP, Datta HK, Taranta A, Fornari R, Migliaccio S, Teti A. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment. J Med Genet.2006; 43:315–25.[ PMC free article : PMC2563229] [ PubMed : 16118345]
- Dozier TS, Duncan IM, Klein AJ, Lambert PR, Key LL Jr. Otologic manifestations of malignant osteopetrosis. Otol Neurotol.2005; 26:762–6.[ PubMed : 16015181]
- Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet.2000; 25:343–6.[ PubMed : 10888887]
- Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM, Mihci E, Notarangelo LD, Ramenghi U, Teti A, Van Hove J, Vujic D, Young T, Albertini A, Orchard PJ, Vezzoni P, Villa A. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res.2003; 18:1740–7.[ PubMed : 14584882]
- Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science.1996; 273:1236–8.[ PubMed : 8703060]
- Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, Coxon FP, Helfrich MH, Crockett JC, Mellis D, Vellodi A, Tezcan I, Notarangelo J, Rogers MJ, Vezzoni P, Villa A, Frattini A. Humanosteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet.2008; 83:64–76.[ PMC free article : PMC2443850] [ PubMed : 18606301]
- Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet.2003; 63:192–7.[ PubMed : 12694228]
- Helfrich MH. Osteoclast diseases and dental abnormalities. Arch Oral Biol.2005; 50:115–22.[ PubMed : 15721137]
- Hu Y, Chen IP, de Almeida S, Tiziani V, Do Amaral CM, Gowrishankar K, Passos-Bueno MR, Reichenberger EJ. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS One.2013; 8:e73576.[ PMC free article : PMC3741164] [ PubMed : 23951358]
- Hwang JM, Kim IO, Wang KC. Complete visual recovery in osteopetrosis by early optic nerve decompression. Pediatr Neurosurg.2000; 33:328–32.[ PubMed : 11182645]
- Iughetti P, Alonso LG, Wilcox W, Alonso N, Passos-Bueno MR. Mapping of the autosomal recessive (AR) craniometaphyseal dysplasia locus to chromosome region 6q21-22 and confirmation of genetic heterogeneity for mild AR spondylocostal dysplasia. Am J Med Genet.2000; 95:482–91.[ PubMed : 11146471]
- Jacquemin C, Mullaney P, Svedberg E. Marble brain syndrome: osteopetrosis, renal acidosis and calcification of the brain. Neuroradiology.1998; 40:662–3.[ PubMed : 9833897]
- Jälevik B, Fasth A, Dahllöf G. Dental development after successful treatment of infantile osteopetrosis with bone marrow transplantation. Bone Marrow Transplant.2002; 29:537–40.[ PubMed : 11960278]
- Jentsch TJ, Maritzen T, Zdebik AA. Chloride channel diseases resulting from impaired transepithelial transport or vesicular function. J Clin Invest.2005a; 115:2039–46.[ PMC free article : PMC1180548] [ PubMed : 16075045]
- Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC. Annu Rev Physiol.2005b; 67:779–807.[ PubMed : 15709978]
- Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poet M, Steinfeld R, Schweizer M, Kornak U, Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J.2005; 24:1079–91.[ PMC free article : PMC554126] [ PubMed : 15706348]
- Kocher MS, Kasser JR. Osteopetrosis. Am J Orthop.2003; 32:222–8.[ PubMed : 12772872]
- Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell.2001; 104:205–15.[ PubMed : 11207362]
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet.2000; 9:2059–63.[ PubMed : 10942435]
- Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature.2006; 440:220–3.[ PubMed : 16525474]
- Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J.2011; 30:2140–52.[ PMC free article : PMC3117652] [ PubMed : 21527911]
- Luzzi V, Consoli G, Daryanani V, Santoro G, Sfasciotti GL, Polimeni A. Malignant infantile osteopetrosis: dental effects in paediatric patients. Case reports. Eur J Paediatr Dent.2006; 7:39–44.[ PubMed : 16646644]
- Natsheh J, Drozdinsky G, Simanovsky N, Lamdan R, Erlich O, Gorelik N, Or R, Weintraub M, Stepensky P. Improved outcomes of hematopoietic stem cell transplantation in patients with infantile malignant osteopetrosis using fludarabine-based conditioning. Pediatr Blood Cancer.2016; 63:535–40.[ PubMed : 26485304]
- Nürnberg P, Thiele H, Chandler D, Höhne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet.2001; 28:37–41.[ PubMed : 11326272]
- Ott C-E, Fischer B, Schröter P, Richter R, Gupta N, Verma N, Kabra M, Mundlos S, Rajab A, Neitzel H, Kornak U. Severe neuronopathic autosomal recessive osteopetrosis due to homozygous deletions affecting OSTM1. Bone.2013; 55:292–7.[ PubMed : 23685543]
- Orchard PJ, Fasth AL, Le Rademacher J, He W, Boelens JJ, Horwitz EM, Al-Seraihy A, Ayas M, Bonfim CM, Boulad F, Lund T, Buchbinder DK, Kapoor N, O'Brien TA, Perez MA, Veys PA, Eapen M. Hematopoietic stem cell transplantation for infantile osteopetrosis. Blood.2015; 126:270–6.[ PMC free article : PMC4497967] [ PubMed : 26012570]
- Palagano E, Blair HC, Pangrazio A, Tourkova I, Strina D, Angius A, Cuccuru G, Oppo M, Uva P, Van Hul W, Boudin E, Superti-Furga A, Faletra F, Nocerino A, Ferrari MC, Grappiolo G, Monari M, Montanelli A, Vezzoni P, Villa A, Sobacchi C. Buried in the Middle but Guilty: Intronic Mutations in the TCIRG1 Gene Cause Human Autosomal Recessive Osteopetrosis. J Bone Miner Res.2015; 30:1814–21.[ PubMed : 25829125]
- Pangrazio A, Fasth A, Sbardellati A, Orchard PJ, Kasow KA, Raza J, Albayrak C, Albayrak D, Vanakker OM, De Moerloose B, Vellodi A, Notarangelo LD, Schlack C, Strauss G, Kühl JS, Caldana E, Iacono NL, Susani L, Kornak U, Schulz A, Vezzoni P, Villa A, Sobacchi C. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J Bone Miner Res.2013; 28:1041–9.[ PubMed : 23280965]
- Pangrazio A, Frattini A, Valli R, Maserati E, Susani L, Vezzoni P, Villa A, Al-Herz W, Sobacchi C. A homozygous contiguous gene deletion in chromosome 16p13.3 leads to autosomal recessive osteopetrosis in a Jordanian patient. Calcif Tissue Int.2012; 91:250–4.[ PubMed : 22847576]
- Pangrazio A, Poliani PL, Megarbane A, Lefranc G, Lanino E, Di Rocco M, Rucci F, Lucchini F, Ravanini M, Facchetti F, Abinun M, Vezzoni P, Villa A, Frattini A. Mutations in OSTM1 (grey lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement. J Bone Miner Res.2006; 21:1098–105.[ PubMed : 16813530]
- Pangrazio A, Pusch M, Caldana E, Frattini A, Lanino E, Tamhankar PM, Phadke S, Lopez AG, Orchard P, Mihci E, Abinun M, Wright M, Vettenranta K, Bariae I, Melis D, Tezcan I, Baumann C, Locatelli F, Zecca M, Horwitz E, Mansour LS, Van Roij M, Vezzoni P, Villa A, Sobacchi C. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum Mutat.2010; 31:E1071–80.[ PubMed : 19953639]
- Pangrazio A, Puddu A, Oppo M, Valentini M, Zammataro L, Vellodi A, Gener B, Llano-Rivas I, Raza J, Atta I, Vezzoni P, Superti-Furga A, Villa A, Sobacchi C. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis. Bone.2014; 59:122–6.[ PMC free article : PMC3885796] [ PubMed : 24269275]
- Schulz P, Werner J, Stauber T, Henriksen K, Fendler K. The G215R mutation in the Cl-/H+-antiporter ClC-7 found in ADO II osteopetrosis does not abolish function but causes a severe trafficking defect. PLoS One.2010; 5:e12585.[ PMC free article : PMC2935355] [ PubMed : 20830208]
- Shroff R, Beringer O, Rao K, Hofbauer L, Schulz A. Denosumab for post-transplantation hypercalcemia in osteopetrosis. N Engl J Med.2012; 367:1766–7.[ PubMed : 23113501]
- Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M, Babul-Hirji R, Baric I, Canham N, Chitayat D, Dupuis-Girod S, Ellis I, Etzioni A, Fasth A, Fisher A, Gerritsen B, Gulino V, Horwitz E, Klamroth V, Lanino E, Mirolo M, Musio A, Matthijs G, Nonomaya S, Notarangelo LD, Ochs HD, Superti Furga A, Valiaho J, van Hove JL, Vihinen M, Vujic D, Vezzoni P, Villa A. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet.2001; 10:1767–73.[ PubMed : 11532986]
- Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, Del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P, Villa A Helfrich MH. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet.2007; 39:960–2.[ PubMed : 17632511]
- Sobacchi C, Pangrazio A, Lopez AG, Gomez DP, Caldana ME, Susani L, Vezzoni P, Villa A. As little as needed: the extraordinary case of a mild recessive osteopetrosis owing to a novel splicing hypomorphic mutation in the TCIRG1 gene. J Bone Miner Res.2014; 29:1646–50.[ PMC free article : PMC4258090] [ PubMed : 24535816]
- Souraty N, Noun P, Djambas-Khayat C, Chouery E, Pangrazio A, Villa A, Lefranc G, Frattini A, Mégarbané A. Molecular study of six families originating from the Middle-East and presenting with autosomal recessive osteopetrosis. Eur J Med Genet.2007; 50:188–99.[ PubMed : 17400532]
- Steward CG. Neurological aspects of osteopetrosis. Neuropathol Appl Neurobiol.2003; 29:87–97.[ PubMed : 12662317]
- Steward CG, Pellier I, Mahajan A, Ashworth MT, Stuart AG, Fasth A, Lang D, Fischer A, Friedrich W, Schulz AS. Severe pulmonary hypertension: a frequent complication of stem cell transplantation for malignant infantile osteopetrosis. Br J Haematol.2004; 124:63–71.[ PubMed : 14675409]
- Strickland JP, Berry DJ. Total joint arthroplasty in patients with osteopetrosis: a report of 5 cases and review of the literature. J Arthroplasty.2005; 20:815–20.[ PubMed : 16139724]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet.2003; 4:638–49.[ PubMed : 12897775]
- Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Bénichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, Warman ML, De Vernejoul MC, Bollerslev J, Van Hul W. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet.2003; 72:763–71.[ PMC free article : PMC1180253] [ PubMed : 12579474]
- Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, MacKay CA, Van Hul E, Timmermans JP, Vanhoenacker F, Jacobs R, Peruzzi B, Teti A, Helfrich MH, Rogers MJ, Villa A, Van Hul W. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest.2007; 117:919–30.[ PMC free article : PMC1838941] [ PubMed : 17404618]
- Vanhoenacker FM, De Beuckeleer LH, Van Hul W, Balemans W, Tan GJ, Hill SC, De Schepper AM. Sclerosing bone dysplasias: genetic and radioclinical features. Eur Radiol.2000; 10:1423–33.[ PubMed : 10997431]
- Waguespack SG, Koller DL, White KE, Fishburn T, Carn G, Buckwalter KA, Johnson M, Kocisko M, Evans WE, Foroud T, Econs MJ. Chloride channel 7 (ClCN7) gene mutations and autosomal dominant osteopetrosis, type II. J Bone Miner Res.2003; 18:1513–8.[ PubMed : 12929941]
- Wilson CJ, Vellodi A. Autosomal recessive osteopetrosis: diagnosis, management, and outcome. Arch Dis Child.2000; 83:449–52.[ PMC free article : PMC1718540] [ PubMed : 11040159]
推荐阅读
- de Vernejoul MC, Kornak U. Heritable sclerosing bone disorders: presentation and new molecular mechanisms. Ann N Y Acad Sci.2010; 1192:269–77.[ PubMed : 20392246]
- Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol.2013; 9:522–36.[ PubMed : 23877423]
- Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N Engl J Med.2004; 351:2839–49.[ PubMed : 15625335]
- Villa A, Guerrini MM, Cassani B, Pangrazio A, Sobacchi C. Infantile malignant, autosomal recessive osteopetrosis: the rich and the poor. Calcif Tissue Int.2009; 84:1–12.[ PubMed : 19082854]
章节注释
作者信息
该疾病的诊断、治疗及追踪随访指南在 网络上(pdf)及从作者邮箱Ansgar Schulz, MD at ansgar.schulz@uniklinik-ulm.de可查询到 。
作者履历
Marie-Christine de Vernejoul, MD, PhD; Hôpital Lariboisière (2007-2016)
Uwe Kornak, MD, PhD (2007-present)
Ansgar Schulz, MD (2007-present)
Cristina Sobacchi, MS (2016-present)
Anna Villa, MD, PhD (2016-present)
修订沿革
- 9 June 2016 (ha) 全面更新实时发布
- 20 June 2013 (me) 全面更新实时发布
- 14 October 2010 (me) 全面更新实时发布
- 12 February 2007 (me) 综述实时发布至网络
- 8 September 2006 (uk) 首稿