
概要
临床特征.
视网膜母细胞瘤(Rb) 是一种发生于儿童的发育中的视网膜的恶性肿瘤, 通常发生在5岁前。视网膜母细胞瘤从含有两个RB1基因拷贝都有癌症易感变体的细胞发育而来。视网膜母细胞瘤可以是单灶性或多灶性的。大约60%的患者有单侧视网膜母细胞瘤,他们的平均的确诊年龄是24个月。大约40%的患者有双侧视网膜母细胞瘤, 平均诊断年龄是15个月。遗传性视网膜母细胞瘤呈常染色体显性遗传,遗传性视网膜母细胞瘤患者也具有更高的发生非眼部肿瘤的风险。
诊断/检测.
视网膜母细胞瘤的诊断通常是通过间接的眼底镜检来确定。影像学检查可以用来辅助诊断和肿瘤分期。遗传性视网膜母细胞瘤的诊断需要确认一个患有视网膜母细胞瘤或者视网膜瘤,以及有视网膜母细胞瘤家族史的先证者,或者在 RB1中检测到胚系致病性变异。
管理.
临床表现的治疗: 尽早地诊断和治疗视网膜母细胞瘤和非眼部肿瘤可以降低发病率并增加寿命;对患者的护理最好是由包括眼科,儿科肿瘤学,病理学和放射肿瘤学在内的多学科专家团队提供。治疗选择取决于肿瘤分期, 肿瘤灶数 (单灶性, 单侧多灶性, 或双侧),眼内肿瘤的位置和体积,玻璃体种植的存在与否,有用视力的潜力,眼外受累的程度,以及可用资源。治疗选择包括眼球摘除、冷冻疗法、激光、全部或局部眼部化疗(包括动脉内化疗结合或继续激光或冷冻疗法)、使用巩膜斑块放疗,还有外束放疗作为最后的手段。
预防次级临床表现: 如果可能的话,对于遗传性视网膜母细胞瘤患者应避免放疗(包括X线、CT扫描,和外束放疗)以尽量降低发生第二种迟发癌症的终身风险。
监控: 已知有RB1胚系致病性变异的儿童: 每三到四周进行一次在麻醉状态下的眼科检查直到6个月大,然后减低检查频率直到3岁。能够合作的儿童的临床检查每三到六个月进行一次,七岁后每年检查一次,最终是每两年检查一次。患有单侧Rb的没有被检出杂合致病性变异的个体有低水平嵌合风险,应该进行常规的临床检查,包括临床超声。患有视网膜母细胞瘤的个体每隔一到两年应该进行一次视网膜检查和成像。为了检测视网膜母细胞瘤患者的第二种非眼部肿瘤,医生和家长应及时观察骨痛或肿块的表现,因为患肉瘤和其他肿瘤的风险很高;然而,目前有效的筛查方案还没有被定制。
避免接触有害物质/环境: 避免接触DNA损伤物质(辐射、烟草和紫外线)可以降低遗传性视网膜母细胞瘤幸存者患其他癌症的风险。
评估有风险的亲属: 对无症状高危儿童进行分子遗传学检测进行早期识别可以在那些没有遗传到致病性变异的风险家庭中减少昂贵的检查。
遗传咨询.
遗传学视网膜母细胞瘤以常染色体显性模式遗传。遗传性视网膜母细胞瘤的患者有杂合的新发突变或遗传的胚系RB1致病性变异。受累的个体的下一代有50%的概率遗传到致病性变异。如家系中RB1致病变异已知,可对高危孕妇进行产前诊断。
诊断
儿童和家庭受累的视网膜母细胞瘤的诊断和治疗指南已经出版 [Canadian Retinoblastoma Society 2009] (full text).
拟诊
有以下表现的个体应怀疑视网膜母细胞瘤:
- 白瞳症(白色瞳孔)
- 斜视
- 眼外观改变
- 视敏度降低
有以下表现和家族史的个体应怀疑遗传性视网膜母细胞瘤:
- 任何被诊断为视网膜母细胞瘤的个体,包括单侧(单灶和多灶)和双侧
- 有视网膜瘤的个体
- 有视网膜母细胞瘤家族史的个体
确诊
视网膜母细胞瘤的诊断的建立在先证者在眼科医生或验光师视网膜检查发现完全的瞳孔扩张。麻醉下的检查可完成对疾病诊断和程度的确定。眼球成像可以帮助诊断的建立。不要求有病理检查。注意:病理活检可能导致肿瘤传播到眼外组织,从而影响到个体的寿命。
遗传性视网膜的诊断建立在一个有视网膜母细胞瘤或视网膜瘤的先证者和视网膜母细胞瘤的家族史。大多数Rb患者没有家族史,因此通过分子遗传学检测 (见 Table 1)识别 杂合的 胚系 RB1 致病性变异 是有必要的。如果Rb在先证者中遗传,那么必须对相关风险家属进行早期诊断和筛查。
分子遗传学检测用于识别遗传学视网膜母细胞瘤的方法包括单基因检测和使用表型靶向检测。
单基因检测
- 患有双边的,单边且有家族史的或单边多灶性的Rb个体。 对外周血DNA进行序列分析和RB1基因靶向缺失/重复分析。注意:一些实验室可能提供对甲基化CpG二核苷酸致病性变异的靶向分析。
- 患有单边单灶性的或没有家族史的Rb个体
- 如果肿瘤组织不可获得,可对外周血DNA进行RB1序列分析和基因靶向缺失/重复分析
- 如果肿瘤组织可以获得,则对肿瘤DNA进行RB1序列分析和基因靶向缺失/重复分析。如果发现致病性变异,则进行外周血DNA检测看是否外周血DNA存在这些变异。如果未发现致病性变异,对RB1启动区CpG岛进行甲基化分析以识别由于RB1启动区甲基化导致的RB1表观遗传失活。如果启动子区未发现甲基化,则对肿瘤DNA检测是否存在MYCN扩增,约有1.5%的孤立的单边Rb个体,检测不到RB1致病性变异,其Rb是由MYCN扩增导致的。
A 表型靶向检测 包含对RB1的检测。然而,视网膜母细胞瘤没有发现位点异质性,数据分析的评估仅局限于RB1。
Table 1.
遗传学视网膜母细胞瘤的分子遗传学检测。
| 基因 1 | 检测方法 | 样本 | 该检测方法检出有生殖系致病性变异先证者比例 2 |
|---|---|---|---|
| RB1 | 序列分析 3 | 生殖系,肿瘤 | 70%-75% |
| 基因靶向缺失/重复分析 4 | 生殖系,肿瘤 | 8%-16% 5 | |
| 致病变异靶向分析 | 生殖系,肿瘤 | 25% 6 | |
| 甲基化分析 | 肿瘤 | 见脚注 7 | |
| MYCN | 基因靶向缺失/重复分析 4 | 肿瘤 | 见脚注 8 |
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
由于CpG-转换导致的提前终止的致病性变异占致病性变异的25%。 [Rushlow et al 2009].
- 7.
在散发性,单侧视网膜母细胞瘤的患者中,有10%-20%观察到RB1 启动子的超甲基化(使基因表达沉默) [Zeschnigk et al 2004]。在这些患者中,需要分析肿瘤DNA中的启动子甲基化状态,以鉴定引发肿瘤发展的两个非活性RB1 等位基因。
- 8.
约有1.5%的散发的单侧的Rb儿童在肿瘤组织检测中具有高水平MYCN扩增,但没有致病性变异导致RB1失活[Rushlow et al 2013]。
Table 2.
基于家族史和肿瘤表现的视网膜母细胞瘤先证者中存在胚系致病性变异的可能性。
| 家族史 | Rb表型 | 存在RB1胚系致病性变异的可能性 | ||
|---|---|---|---|---|
| 单侧的 | 双侧的 | |||
| 多灶性 | 单灶性 | |||
| 阳性 1 | + | 100% | ||
| + | 100% | |||
| + | 100% | |||
| 阴性 2 | + | 约100% 3 | ||
| + | 14%-95% | |||
| + | ~14% | |||
- 1.
阳性=超过1受累的家庭成员(视网膜母细胞瘤的10%)
- 2.
阴性=该家族只有一个受累的视网膜母细胞瘤患者(视网膜母细胞瘤的90%)
- 3.
RB1致病性变异是通过常规分子检测在90%-97%的双侧受累的单发病例中鉴定的;其余5%的可能具有易位,深内含子的剪接变异或低水平嵌合致病变体,它们可能存在胚系中,也可能不存在胚系中
注意:(1)Note: (1)如果在肿瘤细胞和非肿瘤细胞(结构DNA)中都未检出RB1 致病性变异 ,那么受累的患者有低概率有RB1 胚系致病性变异。(2)由于通常可以通过常规分子分析(例如测序)检测到低至20%的血液嵌合体,因此未能检测到结构DNA的致病性变异可以减少但无法完成消除患者具有胚系RB1致病性可能性。
临床特征
临床描述
视网膜母细胞瘤(Rb). 最常见的特征是白色瞳孔反射(白瞳症)。斜视是第二常见的症状,可能伴随白瞳症出现在或在其之前 [Abramson et al 2003]。异常的体征包括青光眼,眼眶蜂窝织炎,葡萄膜炎,前房积血或玻璃体出血。大多数受累的儿童在五岁前被诊断。大龄儿童的非典型表现更常见。
具有Rb的先证者通常存在以下临床特点中的一种:
- 阴性家族史和单侧Rb(60%的先证者)
- 阴性家族史和双侧Rb(30%的先证者)
- 阳性家族史和单侧或双侧Rb(约10%的先证者)。对于家族史阳性的且通过连续视网膜检查进行临床监测的个体,通常会在生命的第一个月发现肿瘤。
- 涉及染色体13q14区域缺失。所有被检索到的单病灶Rb病例中高达5%和所有被检索到的多灶性Rb病例中高达7.5%有染色体13q14区域缺失。这种染色体异常通常与发育迟缓和出生缺陷有关。 [Mitter et al 2011, Castéra et al 2013].
视网膜母细胞瘤是:
- 单侧的 如果Rb受累的只有一只眼睛。约60%的患者具有单侧Rb,诊断年龄平均是24个月。通常,在具有单侧Rb的患者中,肿瘤也是单灶的,即,肿瘤存在单一肿瘤。有些患者一只眼睛患有多灶性肿瘤(单侧多灶性Rb)。眼内传播可能与真实的多灶性肿瘤生长相似。在大多数无家族史的单侧Rb患者中,肿瘤较大,无法确定是否只存在单个肿瘤。
- 双侧的 如果Rb两只眼睛均受累的。约40%的患者具有双侧Rb,诊断年龄平均是15个月。在大多数患有双侧肿瘤的儿童中,在初诊时两只都有受累。在患有双侧Rb的个体中,双眼可能显示出多个肿瘤。一些初诊被诊断为单侧的Rb儿童后来在对侧未受累的眼睛中出现了肿瘤。
- 三侧的 双侧的(或极少单侧的)Rb和松果体肿瘤一起发生。
视网膜瘤和相关眼部病变。自发性生长停滞的良性视网膜肿瘤(称为视网膜瘤)可能出现在视网膜瘢痕内 [Dimaras et al 2008]。钙化的眼睛可能是由于Rb血管闭塞相关的自发消退所致 [Valverde et al 2002]。
松果体母细胞瘤 发生在大脑松果体的“视网膜样”组织中。松果体母细胞瘤或原始神经外胚层肿瘤与Rb并存,称为三侧Rb。松果体母细胞瘤很少见,通常是致命性的,不像眼睛的Rb,通常是可以治愈的。 [de Jong et al 2014]。
其他肿瘤。 其他特定眼外原发性肿瘤(统称第二原发肿瘤)的风险增加。大多数第二原发肿瘤是骨肉瘤,软组织瘤(主要是平滑肌肉瘤和横纹肌肉瘤)或黑色素瘤 [Kleinerman et al 2007, Marees et al 2008, Kleinerman et al 2012]。这些肿瘤通常在青春期或者成年期。接受外照射治疗的Rb患者中,第二原发肿瘤的发生率增加到50%以上 [Wong et al 1997]。未接受大剂量放疗的遗传性视网膜母细胞瘤幸存者终生罹患晚期癌症的风险很高 [Fletcher et al 2004, Kleinerman et al 2012, Dommering et al 2012b, Temming et al 2015]。
基因型-表型关联
在大多数患有遗传性视网膜母细胞瘤的家庭中,所有继承了胚系致病性变异的成员都在双眼中出现了多个肿瘤。然而,发现先证者(即家庭中第一个拥有Rb的人)只有单边的Rb并不罕见。这些家族中的大多数将RB1无效等位基因隔离,这些等位基因由移码或无义突变改变。除少数特殊情况外,RB1无效等位基因显示出几乎完整的外显率(>99%)[Lohmann et al 1996, Sippel et al 1998]。
少于10%的家庭表现出“低外显率”表型,表现力降低(即单侧Rb患病率增加)且不完全外显(即≤25%)。这种低外显率表型通常与RB1等位基因突变相关,主要是非移码或错义突变,特殊的剪接位点,外显子1的插入缺失变异,或者启动子区的致病性变异。
第三类家庭显示出不同的外部显着率,具体取决于致病性等位基因的父母起源(父母起源的影响) [Klutz et al 2002].
细胞遗传学上涉及13q14的缺失,也导致与RB1相同的染色体区域中其他基因的缺失,可能会导致发育延迟 [Castéra et al 2013] 和轻度至中度的面部畸形。由于13q14的大量缺失显示出基因表达能力降低,因此,相当大比例的具有此类缺失的个体仅表现出单侧Rb;这些孩子中有些根本没有肿瘤 [Mitter et al 2011]。MED4的连续丢失(位于RB1的中心)解释了具有大缺失(包括RB1和MED4)的个体的表达降低 [Dehainault et al 2014]。
外显率
见基因型-表型关联。
命名规则
视网膜母细胞瘤曾称视网膜胶质瘤。
发病率
Rb的发生率估计在1:15,000和1:20,000活产婴儿之间 [Moll et al 1997, Seregard et al 2004].
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,没有其他表型与RB1中的致病变异有关。
鉴别诊断
儿童期的几种眼疾可能临床与视网膜母细胞瘤相似:
- 零星的先天性疾病,包括持续性增生性原发性玻璃体病和Coats病 (OMIM 300216)
- 遗传性包括结节性硬化症,Norrie病,色素失禁症,家族性渗出性玻璃体视网膜病变(请参见 Autosomal Dominant Familial Exudative Vitreoretinopathy)和von Hippel-Lindau病。
- 犬弓形虫感染的眼病。
管理
已经制定了视网膜母细胞瘤(Rb)护理指南 [Canadian Retinoblastoma Society 2009] (full text).
初步诊断后的评估
为了确定诊断为Rb的个体的疾病程度和需求,建议进行以下评估:
- 在计划治疗之前,应确定眼内外的肿瘤范围。根据疾病的程度以及癌症扩散到眼睛外部的风险,为每只受累的眼睛分配一个分类。通过麻醉和超声或MRI的临床检查来评估肿瘤的范围,特别是关注肿瘤与视神经的关系。头部MRI也可用于评估松果体细胞瘤,表明三侧视网膜母细胞瘤。
- 对于具有眼外疾病危险因素的非常大的肿瘤,也可以在诊断时进行骨髓穿刺和脑脊液检查(CSF),或者在摘除眼睛的病理检查发现视神经浸润或有明显眼外扩散风险时进行检查。
- 如果Rb已散布到眼外,则需要评估癌症的阶段以确定对儿童的最适当护理。
- 在具有Rb家族史的孩子中,并且在孩子出现斜视或视力不佳的罕见情况下,视网膜肿瘤可能很小,并且在麻醉下通过临床检查可以发现。
- 需要咨询医学遗传学家和/或遗传咨询师。
对症治疗
治疗的目标是首先保存生命,然后是视觉。由于最佳治疗可能很复杂,因此,来自眼科,儿科肿瘤学,病理学和放射肿瘤学等各个领域的Rb治疗专家需要共同提供最佳护理。
除眼睛分类和肿瘤分期外,治疗的选择还取决于许多因素,包括肿瘤灶的数量(单灶,单侧,多灶或双侧),眼内肿瘤的位置和大小,是否存在玻璃体移植,有用视力的潜力,眼外扩散的范围和种类以及可用资源。
眼睛的治疗选择包括摘除眼球;冷冻疗法激光,全身或局部眼部化学疗法,包括动脉内化学疗法与激光或冷冻疗法联合或之后进行;使用巩膜外层斑块进行放射治疗;体外放射疗法作为最后治疗手段,一般不首选。
预防继发并发症
如果可能,应避免使用任何辐射(包括X射线,CT扫描和外部射线辐射),以最大程度地减少发生第二次晚期癌症的终生风险。只有在基本医疗保健中绝对必要时才应使用此类检测。
监视
有关风险患者或已经患Rb的人的医疗监视的更多信息,请参见 guidelines for retinoblastoma care.
初步诊断Rb之后的检测 成功治疗后,儿童需要频繁的随访检查以及早发现新出现的眼内肿瘤:
- 建议对已知患有RB1胚系致病性变异的儿童在麻醉下每三至四周进行一次眼科检查,直到六个月大,然后在三岁之前有所减少频率。每三到六个月对能够配合的儿童进行一次临床检查,直到七岁,然后每年一次,最终每两年一次。
- 对于未检测到杂合的 胚系 RB1 致病性变异的单侧Rb患者,有低水平嵌合的风险并且可能另一只眼睛也发展出肿瘤 [Rushlow et al 2009, Temming et al 2013]。这种风险很小,可以用常规的临床眼科检查代替麻醉下的检查,包括临床超声检查(一种简单的无创手术)
- 患有视网膜瘤的患者(与Rb相关的恶性视网膜前病变)每1至2年进行一次视网膜检查和成像,以尽早发现任何变化。
视网膜母细胞瘤个体中第二种非眼部肿瘤的检测。由于包括肉瘤,黑色素瘤和其他特定癌症在内的第二种癌症的高风险,因此建议立即检查任何体征或症状。正在对定期进行的全身MRI进行研究,以确定该技术何时将具有足够的特异性和敏感性,以筛查患有杂合的胚系RB1致病性变异的患者中的第二种肿瘤。
媒介/环境的避免
Fletcher et al [2004] 已经建议遗传性视网膜母细胞瘤幸存者的癌症风险可通过限制接触DNA破坏剂(放射疗法,烟草和紫外线)来降低。可以通过限制化学疗法的暴露量来降低这些人的肿瘤风险。
评估亲属风险
美国临床肿瘤医师学会将遗传性视网膜母细胞瘤确定为第1类疾病,即遗传综合征,遗传检测被认为是高危家庭成员标准管理的一部分 [American Society of Clinical Oncology 2003]。评估受累个体的明显无症状高危亲属是合适的,以便尽早发现那些将从经验丰富的眼科医生的眼科检查中受益的人,并尽早发现视网膜母细胞瘤。
评估可以包括:
- 如果已知该家族中的致病性变异,则可以进行分子遗传学检测,从而降低了那些没有遗传致病性变异的高风险家庭成员对昂贵筛查程序的需求 [Noorani et al 1996, Richter et al 2003];
- 如前所述,出生后立即由有治疗视网膜母细胞瘤经验的眼科医生进行眼科检查(参见监测,初步诊断后检测随后的Rb。年幼或不合作的孩子可能需要在麻醉下检查)。
参见遗传咨询,用于与出于遗传咨询目的的高危亲属评估相关风险。
正在研究中的疗法
检索 ClinicalTrials.gov 以获得有关各种疾病和状况的临床研究信息。注意:目前这种疾病的临床试验并不多。
遗传咨询
遗传咨询是一个给患者及家属提供关于遗传性疾病本质、遗传特性以及影响并帮助他们做出知情的医疗决定的过程。下列段落描述遗传风险的评估以及根据家族史和基因检测判断家族成员遗传状态。本段落描述不适用于解决患者实际面对的个人、文化或伦理问题,也不能代替专业的遗传咨询。—ED.
遗传模式
遗传性视网膜母细胞瘤遗传模式是常染色体显性遗传。
家庭成员的风险
先证者父母。
- 部分被诊断为遗传性视网膜母细胞瘤的个体都有一位受累的父(母)亲。
- 患有遗传性视网膜母细胞瘤的先证者,可能由于新发胚系RB1致病性变异。患有遗传性视网膜母细胞瘤且无视网膜母细胞瘤家族史的大多数患者是由于新发致病性变异。
- 如果在任一亲本的白细胞DNA中都无法检测到先证者中发现的致病性变异,则两种可能的原因是亲本中的胚系嵌合或先证者中的新发致病性变异。胚系嵌合的发生率约为6%。胚系嵌合可以通过敏感的方法检测,例如等位基因特异性PCR [Rushlow et al 2009] 或二代测序 [Chen et al 2014].
- 建议对具有明显新发致病性变异的先证者的父母进行分子遗传学检测。如果先证者中的RB1致病变异未知,则对先证者父母进行评估的建议包括由对视网膜母细胞瘤(Rb),视网膜瘤和与视网膜母细胞瘤相关的眼部病变由有丰富知识的眼科医生进行检查。
- 一些确诊为遗传性视网膜母细胞瘤的个体的家族史可能是阴性的,原因是家族成员未能识别该疾病(视网膜瘤),水平嵌合,或外显率降低。因此,除非已对先证者的父母进行了适当的临床评估和/或分子遗传学检测,否则无法确认为明确阴性的家族史。。约有10%的有杂合胚系RB1致病性的个体是单发的,先证者中未受影响的父母之一也有致病性变异。在大多数情况下,这是由于嵌合或杂合的“外显率降低”致病性变异,例如错义变体 [Rushlow et al 2009].
- 如果具有Rb的先证者由于嵌合RB1致病性变异而患有疾病,则父母没有致病性变异。
- 注意:如果父母是首次发生致病性变异的个体,则他/她可能具有该变异的体细胞嵌合,并且可能有单侧(较少)或没有视网膜母细胞瘤
先证者同胞
- 同胞患病风险取决于表型和父母遗传状态。
- 如果先证者的父母和先证者的双侧性Rb,则同胞的风险几乎为50%。在罕见的具有“家族性低外显率Rb”的家庭中,具有胚系致病性变异的同胞可降低发生肿瘤的风险。
- 当父母在临床上未受影响时,先证者同胞的风险似乎较低(即1%-2%;请参阅表3)。
- 父母临床上未受影响的先证者同胞仍然有遗传性视网膜母细胞瘤的风险增加,因为父母存在外显率降低的可能性。
- 如果在任一亲本的白细胞DNA中均无法检测到先证者中发现的RB1致病性变异,其同胞的患病风险低,但仍高于一般人群,由于理论上存在生殖系嵌合 胚系嵌合的可能性。因此,建议对每个同胞进行先证者中鉴定出的RB1致病性变异的检测。
- 如果先证者清楚地显示出非癌细胞(如白血球DNA)中有嵌合RB1肿瘤易感基因的变异,则认为致病性变异是由合子后引起的,并且父母双方均没有RB1胚系致病变异。同胞的风险没有增加,因此可以不要求对先证者中鉴定出的RB1致病变异进行同胞检测。
- 如果分子遗传学检测不可用或没有信息,则可以使用基于肿瘤表现(即单灶性或多灶性)和家族史的经验风险 (Table 3). 家族史为阴性的先证者同胞的患病风险低但不可忽略,可能反映出某一亲本中存在外显率降低的胚系RB1致病性变异或体细胞嵌合(包括胚系突变)。
- 如果亲本具有细胞遗传学上可检测到的平衡13号染色体易位或重排,则同胞具有增加遗传不平衡染色体重排的风险。
先证者后代。 遗传性视网膜母细胞瘤个体的每个孩子都有50%的机会遗传到RB1致病性变异。
- 如果先证者具有双侧Rb且无Rb家族史,则认为存在胚系RB1致癌变体,并且每个后代遗传致病性变异的风险为50%。如果先证者中发现了肿瘤易感基因的RB1变异,则可以在后代中进行预测性DNA检测。
- 如果先证者有单侧多灶性Rb且没有Rb家族史,则后代的再发风险降低 [Sippel et al 1998, Rushlow et al 2009]。
- 具有单侧单灶性疾病和阴性家族史的先证者的后代风险为6%,这反映了先证者可能患有嵌合的致病性变异或与较温和的表型表达相关的胚系RB1致病变异。在低外显率RB1等位基因患者中,具有“家族性低外显率Rb”的家庭发生肿瘤的风险低于高外显率RB1“无效”等位基因所观察到的风险的95%。
- 如果在先证者白细胞的DNA中未检测到而在肿瘤组织中检测到的RB1致病变体,则先证者存在大约1.2%的肿瘤组织识别出胚系嵌合致病性变异风险。先证者的后代遗传胚系致病性变异的风险为0.6% [Richter et al 2003]. 后代的分子遗传学检测必须鉴定先证者肿瘤中检测出的两种致病变异。
- 如果在肿瘤中鉴定出的一种致病变异是先证者白细胞的DNA嵌合的,则胚系受累的水平是不确定的。应检查所有后代的白细胞DNA中是否存在致病性变异。
Table 3.
当RB1胚系致病性变异体尚未明确时,先证者的同胞和后代中视网膜母细胞瘤发展的经验性风险
| 索引病例的肿瘤表型 | 家族史 | 索引病例同胞风险 | 索引病例后代风险 | ||
|---|---|---|---|---|---|
| 双侧性 | 单侧性 | ||||
| 多灶性 | 单灶性 | ||||
| X | 阴性 | 2% 1 | 50% | ||
| X | 阴性 | 1%-2% 1 | 6%-50% | ||
| X | 阴性 | ~1% | 6% | ||
| X | 阳性 | 可变的 2 | 可变的 2 | ||
| X | 阳性 | 50% | 50% | ||
- 1.
如果没有受影响的同胞 [Draper et al 1992]。
- 2.
在单侧Rb家庭中,外显率差异很大。
先证者的其他家庭成员
- 对其他家庭成员的风险取决于先证者父母的遗传状态。
- 如果父母是受累的,他或她的家人可能有风险。
相关遗传咨询问题
有关评估以早期诊断和治疗为目的的高危亲属的信息,请参阅管理,高危亲属评估。
预测性检测 有风险的无症状成人家庭成员,需要事先确定家族中的RB1致病性变异。
有明确致病性变异家庭的注意事项。 当具有遗传性视网膜母细胞瘤的先证者的父母均没有致病性变异或该疾病的临床证据时,RB1病原体变异可能是新发的。但是,可以探索的其他可能的非医学解释包括非生物学父亲或产妇(例如辅助生殖)或未公开收养。
遗传性癌症风险评估和咨询。 对于在有或没有分子遗传学检测的情况下通过癌症风险评估确定高危人群的医学,心理,伦理后果的详细说明,请参阅 Cancer Genetics Risk Assessment and Counseling – for health professionals (part of PDQ®, National Cancer Institute).
家庭计划
- 确定遗传风险和讨论是否可以进行产前检查的最佳时间是在怀孕之前。
- 适当地向受累的或有风险的年轻人提供遗传咨询(包括对后代和生殖选择的潜在风险的讨论)[Dommering et al 2012a]。
DNA银行 是DNA的存储,以备将来使用。考虑到检测手段以及我们对基因、等位基因变异及疾病的理解认识在未来将进一步提升,受累的患者可以保存基因。如果可能,应保存从白细胞提取的DNA和从肿瘤提取的DNA。
产前检测
如果有Rb家族史,则可以使用多种选择来优化高危妊娠的管理 [Canadian Retinoblastoma Society 2009]
- 如果RB1 致病性变异在一个受累的家族已被识别,高危孕妇产前测试怀孕的风险可以从提供的基因或定制产前检测致病性变异检测的临床实验室获得。
- 如果在胎儿中发现了RB1致病性变异,则可以使用超声检查来鉴定中等大小的眼内肿瘤。如果存在肿瘤,可以考虑早产以进行早期治疗 [Sahgal et al 2006]. 即使在产科超声检查中看不到肿瘤,也可能建议在妊娠36周时分娩,因为RB1致病性变异的婴儿中有30%会患有威胁视觉的微小肿瘤 [作者,未发表的数据].
- 如果尚未在受累的家庭成员中发现RB1致病性变异,则产前超声或MRI可能会在受影响的胎儿的眼睛中显示出中等大小的Rb。但是,这些测试不够灵敏,无法检测出小的Rb肿瘤。
在医学专家之间和家庭内部,在使用产前检查方面可能存在观点差异,特别是如果考虑将检查用于终止妊娠而不是早期诊断的时候。尽管大多数中心都将有关产前检查的决定视为父母的选择,但对这些问题的讨论是合适的。
胚胎植入前遗传学诊断 (PGD) 对于某些已经确定了RB1致病性变异的家庭可能是一个选择。
资源
工作人员已经筛选了以下专科疾病和患者帮扶组织,注册登记能使患者及其家庭获益。GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处 here.
- A Parent's Guide to Understanding RetinoblastomaAdobe Acrobat Reader requiredIRIS Medical
- Childhood Eye Cancer Trust (CHECT)The Royal London HospitalWhitechapel RoadLondon E1 1BBUnited KingdomPhone: +44 020 7377 5578Fax: +44 020 7377 0740Email: info@chect.org.uk
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- National Retinoblastoma Parents GroupPO Box 317Watertown MA 02471Phone: 800-562-6265Fax: 617-972-7444Email: napvi@perkins.pvt.k12.ma.us
- NCBI Genes and Disease
- World Eye Cancer Hope (WE C Hope)
- American Childhood Cancer Organization (ACCO)PO Box 498Kensington MD 20895-0498Phone: 800-366-2223 (toll-free); 301-962-3520Fax: 301-962-3521Email: staff@acco.org
- National Cancer Institute (NCI)6116 Executive BoulevardSuite 300Bethesda MD 20892-8322Phone: 800-4-CANCER
- National Federation of the Blind (NFB)200 East Wells Street(at Jernigan Place)Baltimore MD 21230Phone: 410-659-9314Fax: 410-685-5653Email: pmaurer@nfb.org
- eyeGENE - National Ophthalmic Disease Genotyping Network RegistryPhone: 301-435-3032Email: eyeGENEinfo@nei.nih.gov
分子遗传学检测
分子遗传学检测的信息和OMIM相关列表可能同其它GeneReview信息会有不同。(可能有更新原因)—ED.
分子遗传性致病机制
除极少数例外,肿瘤的发展始于没有正常RB1等位基因的细胞 [Rushlow et al 2013]. 参见 Figure 1.
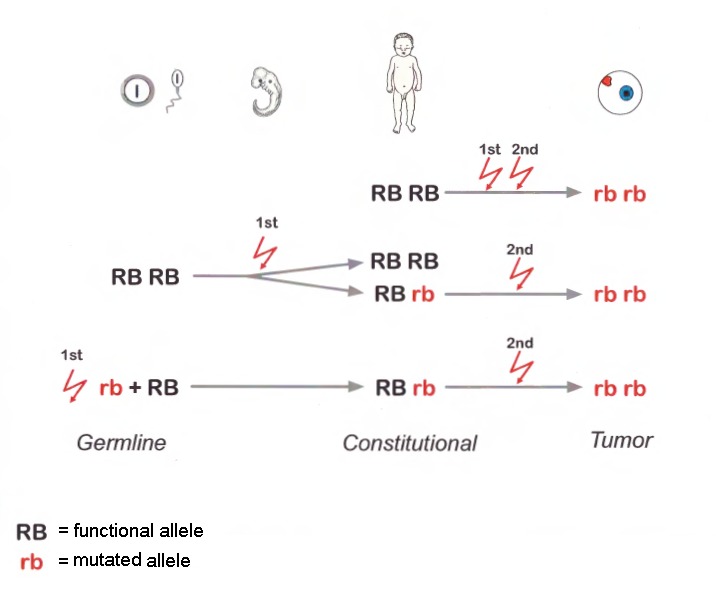
基因结构。 27个外显子转录病剪接成4.7-kbmRNA。没有迹象表明有其他功能可变剪接。常用的参考序列转率本为 NM_000321.2. 有关基因和蛋白质信息的详细摘要请参见 Table A, 基因。
良性等位基因变异。 已知在2.7-kb 开放阅读框架没有频繁的多态性位置,但在内含子的变异,有两个高度多态性微卫星 (Rb1.20, Rbi2),和一个小卫星 (RBD)。
致病性等位基因变异。 在视网膜母细胞瘤患者或肿瘤患者的白细胞DNA中已观察到2500多个核苷酸变体。超过1700已被记录 (参见 Table A, 特点位点). 多数RB1致病性变体通常通过单碱基取代,移码变体或剪接位点变体引起的框外外显子跳跃而导致过早终止密码子。已发现致病性变异散布在RB1启动子区的第1外显子至第25外显子中。在一个家庭中,在第27外显子中发现了可能的致病性变异 [Mitter et al 2009]。在CGA密码子或内含子12的剪接供体位点的甲基化CpG二核苷酸处观察到复发的致病变异。其他重要类型的致病性变异是复杂的重排和缺失 [Albrecht et al 2005, Rushlow et al 2009, Castéra et al 2013].
正常基因产物.RB1编码一种普遍表达的核蛋白,该蛋白参与细胞周期调节(从G1到S的转变)。在进入S期之前,RB蛋白被细胞周期蛋白依赖性激酶(cdk)系统的成员磷酸化。磷酸化后,口袋结构域的结合活性丧失,导致细胞蛋白释放。 综述请参见 Dick & Rubin [2013] 和 Dimaras et al [2015].
异常基因产物. RB1中的致病性变异导致失去细胞周期调节功能的蛋白质表达。在与低外显率视网膜母细胞瘤相关的致病变体产生的蛋白质中已观察到部分活性的保留 [Lohmann et al 1994, Bremner et al 1997, Otterson et al 1997].
参考文献
已发表的指南和共识
- American Society of Clinical Oncology. Policy statement update: genetic testing for cancer susceptibility. Available online; registration or institutional access required. 2010. Accessed 2-10-16.
- American Society of Clinical Oncology. Statement on genetic testing for cancer susceptibility. Available online. 2003. Accessed 2-10-16.
- Canadian Retinoblastoma Society. National Retinoblastoma Strategy Canadian Guidelines for Care: Stratégie thérapeutique du rétinoblastome guide clinique canadien. Available online. 2009. Accessed 2-10-16. [PubMed: 20237571]
文献引用
- Abramson DH, Beaverson K, Sangani P, Vora RA, Lee TC, Hochberg HM, Kirszrot J, Ranjithan M. Screening for retinoblastoma: presenting signs as prognosticators of patient and ocular survival. Pediatrics. 2003;112:1248鈥�55. [PubMed: 14654593]
- Albrecht P, Ansperger-Rescher B, Schüler A, Zeschnigk M, Gallie B, Lohmann DR. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum Mutat. 2005;26:437鈥�45. [PubMed: 16127685]
- American Society of Clinical Oncology. Statement on genetic testing for cancer susceptibility. Available online. 2003. Accessed 2-10-16.
- Bremner R, Du DC, Connolly-Wilson MJ, Bridge P, Ahmad KF, Mostachfi H, Rushlow D, Dunn JM, Gallie BL. Deletion of RB exons 24 and 25 causes low-penetrance retinoblastoma. Am J Hum Genet. 1997;61:556鈥�70. [PMC free article: PMC1715941] [PubMed: 9326321]
- Canadian Retinoblastoma Society. National Retinoblastoma Strategy Canadian Guidelines for Care: Stratégie thérapeutique du rétinoblastome guide clinique canadien. Can J Ophthalmol. 2009;44:S1鈥�88. [PubMed: 20237571]
- Castéra L, Dehainault C, Michaux D, Lumbroso-Le Rouic L, Aerts I, Doz F, Pelet A, Couturier J, Stoppa-Lyonnet D, Gauthier-Villars M, Houdayer C. Fine mapping of whole RB1 gene deletions in retinoblastoma patients confirms PCDH8 as a candidate gene for psychomotor delay. Eur J Hum Genet. 2013;21:460鈥�4. [PMC free article: PMC3598316] [PubMed: 22909775]
- Chen Z, Moran K, Richards-Yutz J, Toorens E, Gerhart D, Ganguly T, Shields CL, Ganguly A. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35:384鈥�91. [PMC free article: PMC4112364] [PubMed: 24282159]
- de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivela T, Moll AC. Trilateral retinoblastoma: a systematic review and meta-analysis. The lancet oncology. 2014;15:1157鈥�67. [PubMed: 25126964]
- Dehainault C, Garancher A, Castéra L, Cassoux N, Aerts I, Doz F, Desjardins L, Lumbroso L, Montes de Oca R, Almouzni G, Stoppa-Lyonnet D, Pouponnot C, Gauthier-Villars M, Houdayer C. The survival gene MED4 explains low penetrance retinoblastoma in patients with large RB1 deletion. Hum Mol Genet. 2014;23:5243鈥�50. [PubMed: 24858910]
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297鈥�306. [PMC free article: PMC4754300] [PubMed: 23594950]
- Dimaras H, Corson D, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, Gallie BL. Retinoblastoma. Nature Reviews Disease Primers. Available online. 2015. Accessed 2-10-16.
- Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, Marrano P, Corson TW, Eagle RC Jr, Squire JA, Gallie BL. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363鈥�72. [PubMed: 18211953]
- Dommering CJ, Garvelink MM, Moll AC, van Dijk J, Imhof SM, Meijers-Heijboer H, Henneman L. Reproductive behavior of individuals with increased risk of having a child with retinoblastoma. Clin Genet. 2012a;81:216鈥�23. [PubMed: 21954974]
- Dommering CJ, Marees T, van der Hout AH, Imhof SM, Meijers-Heijboer H, Ringens PJ, van Leeuwen FE, Moll AC. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2012b;11:225鈥�33. [PMC free article: PMC3365233] [PubMed: 22205104]
- Draper GJ, Sanders BM, Brownbill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br J Cancer. 1992;66:211鈥�9. [PMC free article: PMC1977909] [PubMed: 1637670]
- Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357鈥�63. [PubMed: 14996857]
- Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF Jr. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24鈥�31. [PubMed: 17202110]
- Kleinerman RA, Yu CL, Little MP, Li Y, Abramson D, Seddon J, Tucker MA. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol. 2012;30:950鈥�7. [PMC free article: PMC3341108] [PubMed: 22355046]
- Klutz M, Brockmann D, Lohmann DR. A parent-of-origin effect in two families with retinoblastoma is associated with a distinct splice mutation in the RB1 gene. Am J Hum Genet. 2002;71:174鈥�9. [PMC free article: PMC384976] [PubMed: 12016586]
- Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349鈥�54. [PubMed: 7927327]
- Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am J Hum Genet. 1996;58:940鈥�9. [PMC free article: PMC1914612] [PubMed: 8651278]
- Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771鈥�9. [PubMed: 19066271]
- Mitter D, Rushlow D, Nowak I, Ansperger-Rescher B, Gallie BL, Lohmann DR. Identification of a mutation in exon 27 of the RB1 gene associated with incomplete penetrance retinoblastoma. Fam Cancer. 2009;8:55鈥�8. [PubMed: 18509746]
- Mitter D, Ullmann R, Muradyan A, Klein-Hitpass L, Kanber D, Ounap K, Kaulisch M, Lohmann D. Genotype-phenotype correlations in patients with retinoblastoma and interstitial 13q deletions. Eur J Hum Genet. 2011;19:947鈥�58. [PMC free article: PMC3179359] [PubMed: 21505449]
- Moll AC, Kuik DJ, Bouter LM, Den Otter W, Bezemer PD, Koten JW, Imhof SM, Kuyt BP, Tan KE. Incidence and survival of retinoblastoma in The Netherlands: a register based study 1862-1995. Br J Ophthalmol. 1997;81:559鈥�62. [PMC free article: PMC1722238] [PubMed: 9290369]
- Noorani HZ, Khan HN, Gallie BL, Detsky AS. Cost comparison of molecular versus conventional screening of relatives at risk for retinoblastoma. Am J Hum Genet. 1996;59:301鈥�7. [PMC free article: PMC1914748] [PubMed: 8755916]
- Otterson GA, Chen WD, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci U S A. 1997;94:12036鈥�40. [PMC free article: PMC23695] [PubMed: 9342358]
- Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, Han L, Panton R, Branco P, Gallie B. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253鈥�69. [PMC free article: PMC379221] [PubMed: 12541220]
- Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, Gallie BL. Detection of mosaic RB1 mutations in families with retinoblastoma. Hum Mutat. 2009;30:842鈥�51. [PubMed: 19280657]
- Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, Prigoda-Lee NL, Spencer C, Dimaras H, Corson TW, Pang R, Massey C, Godbout R, Jiang Z, Zacksenhaus E, Paton K, Moll AC, Houdayer C, Raizis A, Halliday W, Lam WL, Boutros PC, Lohmann D, Dorsman JC, Gallie BL. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327鈥�34. [PubMed: 23498719]
- Sahgal A, Millar BA, Michaels H, Jaywant S, Chan HS, Heon E, Gallie B, Laperriere N. Focal stereotactic external beam radiotherapy as a vision-sparing method for the treatment of peripapillary and perimacular retinoblastoma: preliminary results. Clin Oncol (R Coll Radiol) 2006;18:628鈥�34. [PubMed: 17051954]
- Seregard S, Lundell G, Svedberg H, Kivelä T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: advantages of birth cohort analysis. Ophthalmology. 2004;111:1228鈥�32. [PubMed: 15177976]
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet. 1998;62:610鈥�9. [PMC free article: PMC1376960] [PubMed: 9497263]
- Temming P, Viehmann A, Arendt M, Eisele L, Spix C, Bornfeld N, Sauerwein W, Jöckel KH, Lohmann DR. Pediatric second primary malignancies after retinoblastoma treatment. Pediatr Blood Cancer. 2015;62:1799鈥�804. [PubMed: 25970657]
- Temming P, Viehmann A, Biewald E, Lohmann DR. Sporadic unilateral retinoblastoma or first sign of bilateral disease? Br J Ophthalmol. 2013;97:475鈥�80. [PubMed: 23355526]
- Valverde K, Pandya J, Heon E, Goh TS, Gallie BL, Chan HS. Retinoblastoma with central retinal artery thrombosis that mimics extraocular disease. Medical and pediatric oncology. 2002;38:277鈥�9. [PubMed: 11920797]
- Wong FL, Boice JD Jr, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF Jr, Li FP. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262鈥�7. [PubMed: 9333268]
- Zeschnigk M, Böhringer S, Price EA, Onadim Z, Masshöfer L, Lohmann DR. A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): analysis of the retinoblastoma locus. Nucleic Acids Res. 2004;32:e125. [PMC free article: PMC519124] [PubMed: 15353561]
章节注释
Author History
Norbert Bornfeld, MD; University of Essen (2000-2004)
Brenda L Gallie, MD (2004-present)
Bernhard Horsthemke, PhD; University of Essen (2000-2004)
Dietmar R Lohmann, MD (2000-present)
Eberhard Passarge, MD; University of Essen (2000-2004)
Revision History
- 19 November 2015 (me) Comprehensive update posted live
- 28 March 2013 (me) Comprehensive update posted live
- 10 June 2010 (me) Comprehensive update posted live
- 7 May 2007 (me) Comprehensive update posted to live Web site
- 21 January 2005 (dl) Revision: Risk to offspring of a 先证者
- 28 December 2004 (me) Comprehensive update posted to live Web site
- 21 January 2003 (me) Comprehensive update posted to live Web site
- 18 July 2000 (me) Review posted to live Web site
- 21 January 1999 (dl) Original submission