
摘要
临床表现.PWS的典型症状为在婴儿早期严重的肌无力和进食困难,如不严格控制饮食,在接下来的婴儿晚期或者幼童时期会发生进食过量和逐次展开的病态肥胖症。患者运动和语言发展较为迟缓。所有患者都伴有一定程度的认知缺损。经典的动作表型(发脾气、执拗、操纵行为以及强迫性精神特征)较为普遍。生殖官能不良在男性和女性中均有发现,表现为为生殖器发育不全、青春期的发育不完整以及发生在大多数患者中的不育。身材矮小症普遍(如果不用生长激素治疗);特殊的面部特征如斜视和脊柱侧弯常常出现。
诊断/测验.
公认的诊断方法较为准确,但通过DNA甲基化测验发现15号染色体的PW关键区域(PWCR)出现不正常的亲本特异性印记是诊断的主要依据;这个测试决定了此区域是否仅遗传于母亲(即遗传于父亲的区域缺失)从而可以鉴别出超过99%的受累个体。DNA甲基化检测对于个体确诊PWS来说较为重要,尤其是那些没有典型症状和过于幼小而没有出现足够特征来进行临床诊断的个体。
处理
症状的处理方法:在幼儿期,使用特制的乳头或者肠管饲法来确保营养适量;物理疗法或许可以改善肌肉强度;对于隐睾症可以考虑激素或者外科疗法。在儿童期,根据儿童的身高、体重和BMI严格控制每日的食物摄入,以确保在获取足够营养的同时限制过度的体重增长(控制BMI Z分数<2或者更好),除此之外鼓励体育运动。使用生长激素替代疗法控制激素水平,促进瘦体质和运动性,减少脂肪量。按照正常人群标准评估和治疗睡眠障碍。应制定教育计划,如果需要可以使用言语疗法。通过强力的限制处理体态问题;对于大部分青少年和成人患者来说血清素再摄取抑制剂很有帮助。在青春期使用性激素替换法来产生完整的第二性征。N-乙酰半胱氨酸或者托吡或许可以帮助减少皮肤贴片。在许多儿童中,莫达非尼很成功的治疗了日间嗜睡。在成人期,为患有PWS的个体建立的group home通过规范行为和管理体重或许可以预防病态肥胖症,生长激素或许可以帮助维持肌肉体积。
二级并发症的预防:控制体重以预防2型糖尿病;补充钙和维生素D来防止骨质疏松症;若已发生骨质疏松症,考虑用二磷酸盐治疗。
监督:婴儿应用部分屏蔽纠正斜视;对身高、体重和BMI的常规检测来保证运动和饮食计划的适当性;每年进行甲状腺机能减退测试。
其他:目前未知可用药物帮助控制饮食过度,尽管有几种药物正在研究中。
遗传咨询
PWS的导致是由于父系遗传的15号染色体上的位于PWS/Angelman综合征(AS)区域(即15q11.2-q13)的基因印记缺失表达,这种缺失由若干遗传机制造成(父系遗传缺失,母系15号单亲二倍体并极少的伴有印记缺失)。患病儿童的亲属的患此病的风险取决于遗传机制对于父系遗传的15q11.2-q13区域的缺失表达的影响结果。若受累儿童有缺失或者是单亲二倍体,则亲属的得病风险通常小于1%,若受累儿童有印记缺失则风险将提高到50%,若出现了亲代染色体易位则风险将提高到25%。对于已查明遗传机制并在妊娠时已知患病风险将提高时可以做出生前检查。
诊断
PWS的普适诊断标准于1993年发展[Holm et al 1993]被证明较为准确[Gunay-Aygun et al 2001]并继续在临床上发挥作用。然而,诊断的证明需要分子遗传学检测,这在此检测方法刚出现时使用并不广泛。
提示性发现
当个体有如下特征性临床所见或者有在细胞遗传的/FISH/染色体芯片上的发现时,应该怀疑有PWS。
提示需要进行诊断测试的临床所见应该基于对PWS诊断标准在分子层面上被确认[Gunay-Aygun et al 2001]的个体诊断标准的分析。并且还需要根据年龄组改变。下面所出现的所有标明年龄的发现物足以证明PWS的DNA甲基化(见 Establishing the Diagnosis):
出生至两岁
- 肌无力 与 弱吸吮能力(新生儿期)
两岁至六岁
- 肌无力 与 弱吸吮能力史
- 整体发育迟缓
六岁至十二岁
- 肌无力和弱吸吮能力史(肌无力通常保留)
- 整体发育迟缓
- 若无控制出现进食过量伴随向心性肥胖
十三岁至成年期
- 认知损害,通常是轻度智力残疾
- 若无控制出现进食过量伴随向心性肥胖
- 下丘脑引起的生殖官能不良和/或典型的行为问题
细胞遗传的/FISH/染色体芯片发现。大约70%的患有PWS的个体15号染色体的一个缺失包括15q11.2-q13带,这个缺失可以被高分辨率染色体研究和荧光原位杂交技术(FISH)或者染色体芯片检测。
注:典型的缺失是两种情况之一:从远端的断裂点(BP3)到近端的两个断裂点之一(BP1或BP2)。临床的FISH 检测会发现这两种缺失但无法区分它们。另外的非经典的缺失和罕见的缺失在缺失案例中发生大约8% [Kim et al 2012]。
大约1%的受累个体有可检测的染色体重排(如易位或倒位),结果是15q11.2-q13带缺失。
少于1%的个体具有安定的染色体重排破坏包括15q11.2-q13,并且可以被染色体分析或者FISH检出。
确定诊断
PWS诊断建立于一个DNA甲基化后证明15号染色体上的Prader-Willi关键区域(PWCR)有非正常的亲本特异性印记的先证者,这个区域在15号染色体上证明了只有母本印记(即缺少只有父本留下的基因)。
造成PWS的主要三种分子学机制包括父本缺失、母本15号染色体单亲二倍体(UPD)和印记缺失(ID)。DNA甲基化是唯一的技术,他可以诊断出由三种遗传学机制导致的PWS以及在删除案例中区别PWS和 Angelman syndrome (AS) [Glenn et al 1996, Kubota et al 1996, Glenn et al 1997]。
见图表一中的分子遗传学检测摘要,用以定义先证者中的潜在遗传学机制。
主要的分子机制被更精细的通过遗传病因学分类成分子级别Ia-IIIb,这对于遗传咨询来说很重要(见图表三)。
见表一可得到全面测试方法来建立DNA甲基化符合PWS的个体的遗传机制。
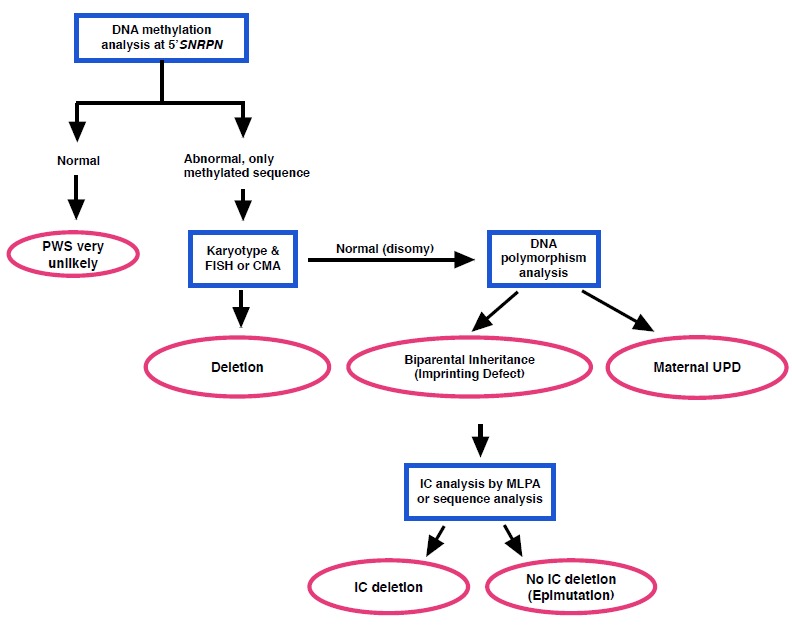
表一.
全面测试方法来建立PWS的分子级别。 FISH = fluorescence in situ hybridization CMA = chromosomal microarray UPD = uniparental disomy IC = imprinting center MLPA = multiplex ligation probe amplification
注:DNS甲基化测验符合PWS满足于临床诊断但不满足于遗传咨询,遗传咨询需要潜在遗传机制的确认(图表一)。见遗传咨询。
Table 1.
用在PWS中的测验
表一. 全面检测方法来建立PWS的分子类型。 FISH = fluorescence in situ hybridization CMA = chromosomal microarray UPD = uniparental disomy IC = imprinting center MLPA = multiplex ligation probe amplification 注:DNS甲基化测验符合PWS满足于临床诊断但不满足于遗传咨询,遗传咨询需要潜在遗传机制的确认(图表一)。见遗传咨询。 Table 1. 用在PWS中的测验
UPD = 单亲二体性 ID = 印记 缺失 IC = 印记 中心 1. 见分子遗传学更多细节。. 2. 可用于诊断,但不可以区分遗传学机制;可以用Southern blot 或者 methylation-specific PCR做。 3. 甲基化特异性多重连接依赖探针扩增(MS-MLPA): 4.FISH一般与核型结合做。利用特异性探针(比如SNRPN)将信息限制在AS/PWS区域。FISH不会问询整个AS/PWS区域同时会错过小的缺失。它不会给出余下染色体的信息而且不会区分正常、UPD和ID。 5. 染色体芯片(CMA)相比于FISH有略微高的检测频率同时可以根据缺失的大小提供具体的信息。同时,它也会根据基因组剩余物中的缺失和倍增提供信息。CMA比核型和FISH更加精确;它可以发现SNORD116微量缺失(见表格2和分子遗传发病机制)。 6. 与CMA相似;并且,使用小核苷酸多聚物(SNP)可以发现纯合型强直症案例的UPD。 7. 不是首选检测,在用DNA甲基化分析诊断PWS后进行FISH或者CMA都可以分析证明二体性。 8. DNA测序在IDs中有特殊作用,用来从表面突变中区分出IC。它被限制于PWS IC最小的缺失重叠区域(SRO)[Ohta et al 1999]中的小于4.3kb的区域。对映区域大约25,196,494 到25,200,794 bp[UCSC Genome Browser, hg19]。 | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
临床症状
临床描述
围产期。胎儿大小通常在正常范围内。出生前的肌无力通常表现为胎动减少,在分娩时胎位异常,辅助分娩和剖宫手术的需要率上升。出生体重和体重指数(BMI)平均比正常人小15% [Miller et al 2011]。
肌无力。幼儿肌无力几乎是一种普遍现象,造成运动减少和困倦伴随着自然苏醒减少,哭泣很弱和弱反射包括弱吸吮。肌无力是起源和神经肌肉研究包括肌肉活检的中心,在结束诊断目的后通常为正常或表现为废弃的无特异性标识。
弱吸吮,瞌睡和胃口不良导致在婴儿早期生存失败,在一段不确定的时间内通常需要肠管饲法或使用特制乳头,通常要几周到几个月。在儿童可以用杯子喝水或者吃流食时,就可以进行大致正常的进食了。
肌无力会经过时间好转。成年人仍有轻微的肌无力伴随肌肉体积和质量下降。
发育迟缓。患有PWS的儿童90%-100%有运动发展的迟缓,达到相同成就通常在正常人两倍的年龄时(例,在12个月时会坐,在24个月时会走)。语言能力同样典型的延迟。在学龄前时智力缺陷通常较明显。测试显示大部分患有PWS的人在轻度智力缺陷的范围内(平均IQ:60s到70s),大约40%的人有轻度失能或者低于正常的智力,大约20%的人有中度失能。不看测量的IQ,大部分患有PWS的儿童有多重严重的学习障碍以及在智力方面弱的学校表现[Whittington et al 2004a]。尽管一小部分受累人群有严重的语言发展缺陷,对大部分个体来说言语能力是相对强度。根据作者的经验,一小部分PWS患者能够参加并完成大学。
性腺功能减退症。在两种性别中都有出现性腺功能减退症并且表现为生殖器发育不全,
不完全青春期发育,大多数不育。生殖器发育不全在出生和终生内都很明显。
- 男性。阴茎可能会较小,大多数发育不全的特征为阴囊较小,缺少褶皱,浅着色。80%-90%的男性有单边或者双边隐睾。
- 女性。生殖器发育不全通常被忽略,大阴唇,阴唇和阴蒂通常自出生起较小。
性功能不全通常与血液内促性腺素类浓度较低有关,会引起青春期发育的不完整,延迟有时会混乱。早熟的肾上腺皮质功能初现约在15%-20%的人群中出现。不育已成为规律,即使有少数女性生育的例子被报道 [Akefeldt et al 1999; Schulze et al 2001;Vats & Cassidy, unpublished data]。即使PWS中的性功能不全长时间被认为是下丘脑的原因,最近的研究揭示了下丘脑和主要性腺缺乏的结合 [Eldar-Geva et al 2009, Hirsch et al 2009, Eldar-Geva et al 2010, Gross-Tsur et al 2012],这个结论基于低促性腺素症的缺乏和在一些受累两个性别的个体中的不正常的低抑制素B水平。
在一项84位患有PWS的个体(一半男性,一半女性)年龄在2-35岁的研究中,下列情况被确认:
- 男性。隐睾100%、小睾丸76%、阴囊发育不全69%
- 女性。小阴唇和/或阴蒂发育不全76%、原发闭经56%、大于15岁的自然初潮(大部分出血)44%
- 两种性别。过早的阴毛初现14%、性早熟3.6%(1男,2女)
食欲和肥胖。从目前来看在PWS(如,伴随在饮食过度后的发育停滞导致肥胖)中只有两种不同的营养阶段,一个多中心研究[Miller et al 2011]发现在各个营养阶段的转换更加复杂,在获得PWS的个体的正常发展过程中有七种不同的营养阶段(表2)。
Table 2.
PWS中的营养阶段
临床描述 围产期。胎儿大小通常在正常范围内。出生前的肌无力通常表现为胎动减少,在分娩时胎位异常,辅助分娩和剖宫手术的需要率上升。出生体重和体重指数(BMI)平均比正常人小15% [Miller et al 2011]。 肌无力。幼儿肌无力几乎是一种普遍现象,造成运动减少和困倦伴随着自然苏醒减少,哭泣很弱和弱反射包括弱吸吮。肌无力是起源和神经肌肉研究包括肌肉活检的中心,在结束诊断目的后通常为正常或表现为废弃的无特异性标识。 弱吸吮,瞌睡和胃口不良导致在婴儿早期生存失败,在一段不确定的时间内通常需要肠管饲法或使用特制乳头,通常要几周到几个月。在儿童可以用杯子喝水或者吃流食时,就可以进行大致正常的进食了。 肌无力会经过时间好转。成年人仍有轻微的肌无力伴随肌肉体积和质量下降。 发育迟缓。患有PWS的儿童90%-100%有运动发展的迟缓,达到相同成就通常在正常人两倍的年龄时(例,在12个月时会坐,在24个月时会走)。语言能力同样典型的延迟。在学龄前时智力缺陷通常较明显。测试显示大部分患有PWS的人在轻度智力缺陷的范围内(平均IQ:60s到70s),大约40%的人有轻度失能或者低于正常的智力,大约20%的人有中度失能。不看测量的IQ,大部分患有PWS的儿童有多重严重的学习障碍以及在智力方面弱的学校表现[Whittington et al 2004a]。尽管一小部分受累人群有严重的语言发展缺陷,对大部分个体来说言语能力是相对强度。根据作者的经验,一小部分PWS患者能够参加并完成大学。 性腺功能减退症。在两种性别中都有出现性腺功能减退症并且表现为生殖器发育不全, 不完全青春期发育,大多数不育。生殖器发育不全在出生和终生内都很明显。
性功能不全通常与血液内促性腺素类浓度较低有关,会引起青春期发育的不完整,延迟有时会混乱。早熟的肾上腺皮质功能初现约在15%-20%的人群中出现。通常不孕,即使有少数女性生育的例子被报道 [Akefeldt et al 1999; Schulze et al 2001;Vats & Cassidy, unpublished data]。即使PWS中的性功能不全长时间被认为是下丘脑的原因,最近的研究揭示了下丘脑和主要性腺缺乏的结合 [Eldar-Geva et al 2009, Hirsch et al 2009, Eldar-Geva et al 2010, Gross-Tsur et al 2012],这个结论基于低促性腺素症的缺乏和在一些受累两个性别的个体中的不正常的低抑制素B水平。 在一项84位患有PWS的个体(一半男性,一半女性)年龄在2-35岁的研究中,下列情况被确认:
食欲和肥胖。从目前来看在PWS(如,伴随在饮食过度后的发育停滞导致肥胖)中只有两种不同的营养阶段,一个多中心研究[Miller et al 2011]发现在各个营养阶段的转换更加复杂,在获得PWS的个体的正常发展过程中有七种不同的营养阶段(表2)。 Table 2. PWS中的营养阶段
PWS中发生的的摄食过度被认为是下丘脑的不正常导致缺少饱腹感。觅食行为,储存或者寻找食物,偷窃食物或者为了食物偷窃金钱很常见。在多数情况下,胃排空延迟,呕吐很罕见。这些行为以及整体热量需求的减少导致了肥胖。与未受累的个体比较后者出现的原因是运动量减少和瘦体质(主要是肌肉)减少导致的静息时能量消耗的减少。在两性中PWS导致的肥胖主要是中心性的(腹部、臀部、和大腿),有趣的是,那种程度的肥胖者比同等程度的正常肥胖者有更少的内脏脂肪。过度肥胖和它的并发症是发病率和死亡率的主要原因(见发病率和死亡率)。 几个独立组显示患有PWS的摄食过度的大龄儿童和成年人在餐前和餐后格瑞林水平显著上升 [Cummings et al 2002, DelParigi et al 2002, Haqq et al 2003b]。格瑞林是一种有力的循环性开胃激素,主要在胃内产生。循环性格瑞林水平在禁食后升高在摄食后降低。胃口诱导效应通过位于下丘脑的胃口调节通路发生。格瑞林水平在无PWS的肥胖者中较瘦人更低,并且随着年龄降低。 [Scerif et al 2011] 一项小研究发现9位无过量摄食的患有PWS的儿童(17-60个月)与8个控制匹配BMI、年龄和性别的正常儿童的格瑞林水平相似[Erdie-Lalena et al 2006]。作为对比,在两个更大更新的研究组发现患有PWS的儿童和青少年的格瑞林水平在任何年龄都显著高于与之匹配的对照组 [Feigerlová et al 2008, Kweh et al 2015]。事实上,最高的格瑞林水平在最幼小的孩子中被发现。因此,在这两项大的研究中hyperghrelinemia发生的比PWS中的肥胖和胃口增长的发展要早一段时间。此外,现在有几组发现了药理学上将PWS的格瑞林水平降低到正常,利用短效或者长效因子不影响摄食过量者的体重、胃口或者进食行为 [Haqq et al 2003a, Tan et al 2004, De Waele et al 2008]。目前没有持续确定的激素异常来解释摄食过量,PWS中摄食过量的代谢联系尚未确定。 内分泌学方面。高达25%的患有PWS的成年人(尤其是有明显肥胖的)有2型糖尿病 [Butler et al 2002]且平均发病年龄是20岁。在近15年内,对父母的早期诊断和教育、生长激素疗法的使用、以及PWS的群体家庭的出现频率增高造成有PWS的个体病态肥胖(结果之一,在二型糖尿病中)的发展减少。 中枢性甲状腺功能减退,有正常的促甲状腺激素水平和低甲状腺素水平,在PWS患者中有高达25%的记录,平均诊断和治疗年龄位两年 [Miller et al 2008, Diene et al 2010]。 在一项研究中的夜间甲吡酮试验发现60%的有PWS的儿童有肾上腺功能减退(CAI),提示只可能是造成这个人群中突然死亡的高发生率的原因 [de Lind van Wijngaarden et al 2008]。已知对有初始肾上腺功能减退的个体引入GH方法会加速肾上腺危象经促染皮质醇的周围代谢,这也许可以解释在开始GH治疗时猝死的发生和PWS中的CAI的关系 [Scaroni et al 2008]。然而,后续的实验发现了对低或高剂量的二十四肽促皮质[素]试验和胰岛素耐量试验的正常皮质醇反应 [Nyunt et al 2010, Farholt et al 2011];因此,CAI对于有PWS的个体来说是否是个真问题目前仍然不确定,同时在内分泌学家之间还未统一是应该在所有PWS个体间做CAI评定还是仅在有与肾上腺功能减退的症状相同的个体间进行。 有很多睡眠异常情况被记录包括REM(快眼动相)潜伏期的减少,改变的睡眠结构,氧去饱和以及中心和阻塞性呼吸暂停[Festen et al 2006, Priano et al 2006]。初级下丘脑功能障碍被认为是 和 的原因,研究发现脑脊髓液中低水平的阿里新和睡眠觉醒系统的一种神经以及脑桥脚被盖核中的乙酰胆碱神经元水平的降低 [Dauvilliers et al 2003, Nevsimalova et al 2005, Bruni et al 2010, Hayashi et al 2011]。 行为障碍:其中一个典型特点是,在70%-90%PWS人群中儿童会在早期呈现出脾气变得易怒,固执,出现极强的控制欲,并伴有强迫症,且难以改变自己的生活习惯。
精神问题与行为障碍是降低青春期与成年期生活质量的罪魁祸首。 发育障碍:在缺乏生长激素的替代品的情况下,如果在童年时期没有表现出身材矮小的特点,那么往往在10到20岁之间会出现身材矮小的情况。因为(缺少生长激素)患病者会出现全面发育迟缓,导致未接受治疗的男性平均身高为155cm而女性为148cm。 在10岁之前,患者手脚生长速度会极其缓慢,往往低于百分之5,以致于成年女性平均足尺寸为20.3cm男性为22.3cm。受影响的婴儿和儿童在未接受生长激素治疗情况下的生长图表已公布,并且接受生长激素治疗的pws儿童的生长图表也已经被制备。 300多名受影响儿童通过至少15项研究数据,记录了PWS情况下GH分泌的减少。 同时患有PWS的成年人也会出现GH缺乏症。[Burman et al 2001] [Grugni et al 2006, Höybye 2007]. 面部畸形:特征性的面部病症( 额头狭窄,杏仁状睑裂,鼻梁狭窄,薄上唇红,嘴角下弯)在出生时可能会或可能不会明显,但随着时间的推移会缓慢加剧。 由于缺失单拷贝的基因OCA2,患者经常发现毛发,虹膜和皮肤的色素减退。 视觉障碍:患者有60%-70%概率会患有斜视。 骨骼畸形:患者有10%-20%的概率会出现髋关节发育不良,因为骨质减少与骨质疏松的患病率增高,所以患者更易骨折。 [West & Ballock 2004, Shim et al 2010]. 脊柱侧弯的患病率为40%-80%,发病年龄与严重重程度各不相同。 其他症状:以下病症患病概率增加
发病率与死亡率:pws的死亡率高于其智力残疾的对照组,而死亡率高要因素是因为pws带来的肥胖等并发症。[Einfeld et al 2006].据患病人口研究,年死亡率大约为3%,而随着对于患病者的妥善管理,同一批次人的死亡率降低至1.5%。 [Butler et al 2002] [Whittington et al 2015]. 有两名多方面研究的PWS逝者被报道,接下来召开了对64名死亡患者的案例讨论与病历回顾,发现呼吸道疾病和其他发热性疾病是儿童死亡的最常见原因,而肥胖相关的心血管疾病和胃病或睡眠呼吸暂停导致的死亡在成人中最常见。 [Schrander-Stumpel et al 2004, Stevenson et al 2004] [Tauber et al 2008]其他发病的原因还包括糖尿病,血栓性静脉炎和皮肤问题(例如,慢性水肿,皮肤采摘感染) 研究表明,少数人因患有呼吸道或胃肠道感染,导致意外死亡; 罕见的,其中三人有小肾上腺,同时报告表明60%的人患有中心肾上腺皮质功能不全(CAI)这可能是意外性突然死亡的原因。 其他研究未能进一步探明PWS中CAI的高发病率的原因。[Stevenson et al 2004] [de Lind van Wijngaarden et al 2008] [Farholt et al 2011, Grugni et al 2013] 研究表明,许多PWS患者出现急性胃扩张和坏死,特别是在那些以前肥胖现在瘦弱的人进行暴饮暴食之后, 它可能是致命的并且可能因为高痛阈而无法识别。 [Stevenson et al 2007a] 研究表明,PWS患者中约有8%的人死于窒息,尤其是当他们在进食热狗的时候 [Stevenson et al 2007b]. 因为GH治疗开始几个月出现患者死亡,有关GH可能导致死亡的考量已经提出。[Eiholzer 2005, Sacco & Di Giorgio 2005]. 报告的死亡人员主要集中在已有呼吸道或心脏疾病的肥胖人群中,有上气道阻塞和未矫正的扁桃体和腺样体肥大的病史。在一家制药公司的数据库中,675名接受GH治疗的儿童中有5名突然死于呼吸系统疾病。在另一项调查中,受影响的GH患者的死亡率没有差异。一项关于英国某地区PWS自然史的研究发现,没有GH治疗,PWS患者的总体死亡率高达每年3%。因此,GH给药与意外死亡的关系仍不清楚。所以,建议在GH治疗开始前进行患者睡眠观察,并在GH治疗开始后再跟进4到8周,以确保GH治疗没有引起或恶化睡眠呼吸紊乱。对48名接受治疗的儿童进行的长期研究表明,GH治疗的益处大大超过了风险。[Craig et al 2006] [Nagai et al 2005] [Whittington et al 2001] [Miller et al 2006b][Carrel et al 2010]. 神经研究:在一项研究中,所有20名被评估为PWS的个体都患有21名对照个体与16名未患有PWS的早发性病态肥胖患者中未发现的脑部异常,pws个体都有脑室肿大;50%个体的顶叶 - 枕叶脑组织体积减少; 60%患有Sylvan裂隙多发性纤维; 65%的岛屿闭合不完整。在另一项研究中,发现了一些PWS患者的白质病变。 来自另一组的91名PWS患者的脑部MRI研究显示,垂体高度相较于普通人降低了49%,神经放射学异常区域占67%。 这些研究发现的异常对于人体的影响尚不清楚。[Miller et al 2007] [Miller et al 2006a][Iughetti et al 2008] 基因型-表型相关性 没有任何已知表型特征特异的与导致PWS的三种主要分子机制中的任何一种相关联,然而,却观察到两个最大分子类别(缺失和UPD)之间某些表型的频率与危害严重性的一些统计学上的差异。 UPD
基因缺失
图二:染色体区域15q11.2-q13的遗传和表达图谱总结Prader-Willi综合征(PWS)区域(以蓝色显示)具有五个仅有父本(PWS区域)表达的编码多肽的独特拷贝基因(MKRN3,MAGEL2) ,NECDIN和SNURF-SNRPN) (more...) 外显率: 完全外显 命名法 不再使用术语HHHO(性腺机能减退,肌张力减退,低血压,肥胖)。 这种情况有时被称为Willi-Prader综合征或Prader-Labhart-Willi综合征。 发病率: 在许多人群中,PWS的估计患病率为1:10,000至1:30,000。 | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|

遗传相关疾病
Angelman综合征(AS)是因为缺少源于母亲的PWS / AS区域引起的。两年后,它在临床上症状与PWS不同。
母亲遗传的PWS / AS区域如果重复则会导致智力残疾,癫痫发作和自闭症。 详见15q重复综合症和相关疾病。[Boyar et al 2001].
鉴别诊断:
许多疾病症状与PWS表现症状相似。
颅咽管瘤及其治疗结束后表现与PWS的十分的相似。特别是在颅咽管瘤发生在早期时,对下丘脑的损伤导致大多数与PWS相同的特征出现。如果不确定是何种疾病,病史调查与DNA甲基化分析将区分颅咽管瘤和PWS。
嗜食型矮小是一种与心理社会压力相关的后天性疾病,症状包括生长激素功能不全,饮食过多和轻度学习障碍[Gilmour et al 2001]。如果不确定是何种疾病,病史调查与DNA甲基化分析应该将这种疾病与PWS区分开来。[Gilmour et al 2001]
婴儿期的低血压症也见于以下情况:
- 新生儿败血症
- 中枢神经系统性抑郁症
- 先天性肌强直性营养不良1型,以出生时肌张力减退和严重全身无力为特征,常伴有呼吸功能不全和早期死亡; 智力残疾也十分常见。它是由DMPK中CTG三核苷酸重复的扩增引起的。
- 几种肌病和神经病,包括一些脊髓性肌萎缩症(SMA)[Miller et al 1999, Richer et al 2001]。 在这些情况下,可能出现呼吸能力弱的症状,这是PWS中很少见到的病症。 通常需要进行分子遗传学测试,EMG / NCV以及肌肉活检以区分这些病症。 [Miller et al 1999, Richer et al 2001]
- Angelman综合征(AS),其特征是重度发育迟缓与智力残疾,严重的言语障碍,步态共济失调和肢体震颤,以及具有特立独行不合时宜的兴奋行为,包括阵发性大笑,微笑和兴奋性行为。出生后小头畸形和癫痫发作也很常见。AS由母系遗传的UBE3A等位基因的基因表达缺陷或者表达却无功能所引起,15号染色体的DNA甲基化分析可诊断75%-80%的AS患者。在婴儿期,肌张力减退可能是AS的唯一病症。患有AS的个体没有PWS患者的特征性吸吮问题,性腺机能减退和异常面部外观。
- 脆性X智力低下综合征,其特征是男性患者有中度智力残疾,女性患者有轻度智力残疾。男性可能出现的典型的外观症状(巨头畸形,长脸,下颌前突与前额突出,巨耳畸形)结缔组织中出现(关节松弛)和大睾丸(后期)。 常见行为异常,有时出现自闭症谱系症状。脆性X智力低下综合征的诊断依赖于检测FMR1的改变,包括三联体重复的扩增和异常的基因甲基化。在婴儿期,肌张力减退可能是唯一的表现。 受影响的个体缺乏PWS患者的特征性吸吮问题,性腺机能减退和异常面部外观。
在以下疾病中可以看到发育迟缓,智力残疾和可能存在的性腺机能减退的肥胖症
- Angelman综合征(AS): 由父母遗传的UPD 15引起的AS患者,通常具有随年龄升高而增长的BMI。 在一项大型研究中,超重超过70%,肥胖超过40%。 [Lossie et al 2001].
- 脆性X综合症。已发现具有“Prader-Willi样”表型的子集,包括饮食过多和肥胖[de Vries et al 1993].
- 14号染色体的母系单亲二体型,包括产前发育迟缓,哺育困难,身材矮小和性早熟。 [Cox et al 2004, Hosoki et al 2009]
- 奥尔布赖特遗传性骨营养不良症(OMIM 103580),也出现身材矮小,并具有不同的特征性面部外观(圆脸)但没有肌张力减退。 通过测量Gs受体偶联蛋白可以进行特异性测试。
- Bardet-Beidl综合征(BBS),其特征为锥体营养不良,躯干肥胖,多指畸形,认知障碍,男性低促性腺激素性性腺功能减退,女性泌尿生殖系统畸形和肾功能障碍。 患有BBS的个体具有与PWS不同的特征面部表型。虽然不到10%的个体遗传可能很复杂但是遗传通常是常染色体隐性遗传。
- 科恩综合征,其特点是在婴儿期和儿童期不能正常发育; 青少年时期的躯干肥胖; 早发性肌张力减退和发育迟缓; 在出生后第一年易发生小头畸形; 中度到深度的精神反应迟缓; 进行性视网膜脉络膜营养不良和高度近视; 许多患有中性粒细胞减少症,有些患有反复感染和口疮性溃疡; 性格积极; 关节过度伸展 ; 有和PWS不同的特征性面部特征。 Cohen综合征是由于VPS13B的致病变异引起的。 遗传通常是常染色体隐性遗传。
- Borjeson-Forssman-Lehmann综合征(OMIM 301900),见于男性,其特征是严重的认知缺陷,癫痫,性腺机能衰退,代谢减退,明显肥胖,婴儿肌张力减弱和生长障碍,身材矮小。 它可以通过智力障碍的严重程度,是否存在眼球震颤,突出的眉脊,眼睑下垂和深陷眼睛的特征性面部外观来辨别。 PHF6中的变异是致病的。由X染色体遗传。患有此病症的杂合女性有已经扭曲失活的X染色体或存在PHG6的基因组缺失。
- Alstrom综合征,以锥体营养不良,早发性肥胖,进行性感音神经听力障碍,扩张型心肌病(> 60%),与黑棘皮病相关的胰岛素抵抗综合征,2型糖尿病和发育迟缓(~50%)为特征。 其他内分泌异常可包括甲状腺机能减退和男性低促性腺激素性性腺机能减退。 在十几岁的女性中出现以逼尿肌 - 尿道协同失调为特征的不同严重程度的泌尿系统疾病。 通常在晚期还会发现严重的肾脏疾病。70%-80%的北欧血统个体中发现ALMS1,并且在全世界约40%的个体患有此病。
具有相似表型的细胞遗传学疾病包括:
- 已经在具有6q16.2(包括SIM1)的间质缺失的个体中鉴定出综合征性肥胖的“PWS样表型 。据报道,这种缺失已经在以前综合征性肥胖症患者中已经重复发现五次以上。 此外,已经在患有严重肥胖的患者中发现了单独的SIM1中的致病变异。[Bonaglia et al 2008] [Desch et al 2015][Bonnefond et al 2013, Ramachandrappa et al 2013].
- 一些报道将Prader-Willi表型与1p36缺失相关联; 相关发现包括肌张力减退,发育迟缓,肥胖,饮食过多和行为障碍问题 [Tsuyusaki et al 2010, Stagi et al 2014].
- 多个报告描述了16p11.2的缺失,包括SHB2B1,其涉及瘦蛋白和胰岛素信号传导[Maillard et al 2015].
- 具有PWS样表型的个体的其他细胞遗传学异常的报告包括dupXq27.2-ter和del10q26 [Lukusa & Fryns 2000, Ben-Abdallah-Bouhjar et al 2012, Rocha & Paiva 2014].
管理:
对Prader-Willi综合征(PWS)患者的管理取决于年龄,应包括解决PWS的症状和后期指导。如果可能,建议进行团队管理。已经出版的几种管理方法[Goldstone et al 2008, Cassidy & Driscoll 2009, Cassidy & McCandless 2010, McCandless 2011, Cassidy et al 2012].
初步诊断后的评估
为确定被诊断患有PWS的个体的疾病和治疗需求程度,建议进行以下评估:
- 咨询临床遗传学家或遗传咨询师。
- 内分泌相关咨询
- 营养相关咨询
- 评估新生儿和幼儿是否有哺育问题和生长是否正常
- 年龄,身高,体重,头围和体重指数(BMI)等指标都源于且适用于PWS患者的生长图表 [Butler et al 2011, Butler et al 2015]
- 儿童甲状腺功能减退症的评估,特别是评估那些长期未能茁壮成长的儿童,在没有增加食物摄入的情况下体重增加过多的儿童,以及尽管生长激素治疗但生长线性较差的儿童
- 任何年龄段,都可进行研究睡眠的低阈值呼吸状态评估。这些检查在开始生长激素治疗(GHT)之前和开始GHT后四至八周尤其有意义,同时可以评估尤其是在肥胖个体中的扁桃体和腺样体的大小。
- 对婴儿的发育和儿童的智力发育与言语功能进行评估。
- 评估两年后存在的行为障碍和强迫症系症状,以及青少年和成人的相关精神病。如果评估记录显示出这存在这些问题,则需要转介进行更详细的评估。
- 对所有年龄段男性评估是否存在隐睾症。
- 如果存在斜视,则转诊进行眼科评估,并在一年内诊断时再次评估视力
- 对任意年龄段,临床评估是否有脊柱侧凸,如果有出现可能,则进一步进行X射线评估。
注意:非常肥胖的人在临床上无法充分评估脊柱侧凸必须进行X射线来确定诊断。
对症治疗
建议采用团队管理方法[ Goldstone等2008,Cassidy&McCandless 2010 ]。 [Goldstone et al 2008, Cassidy & McCandless 2010].
喂养,饮食过量和肥胖。Feeding, hyperphagia, and obesity. 在生命的头几周到几个月可能需要特殊的喂养方法,包括特殊的乳头或强饲喂养,以确保足够的营养并避免发育停滞。被诊断患有PWS的个体通常不需要G管,因为喂养会随着时间而改善。
当营养期2a(通常在18至36个月之间)体重百分比开始增加时,应制定一个均衡、低热量饮食、定期运动和密切监督以尽量减少偷吃食物的计划,以预防肥胖(即BMI Z评分<2)及其后果。如果肥胖随时存在,则相同的计划是适当的。通常需要咨询营养师和密切跟进,并且一旦孩子能够打开冰箱和橱柜,需要锁住厨房,冰箱和/或橱柜。 婴儿和PWS儿童患者的热量需求通常为推荐每日摄取量(RDA)的60%-80%。在计划每日食物摄入量时,应考虑PWS成年患者的能量需求,这些需求很少超过1200-1400 Kcal /天。 营养师评估了对维生素和矿物质摄入量的充分性以及适当补充的处方后表明,钙和维生素D尤其需要补充。
没有药物可以帮助控制饮食过多,但一些针对于此的临床试验正在进行(参见治疗调查)。
PWS患者不推荐胃旁路治疗,因为它似乎不能纠正缺乏饱腹感并且不能防止暴饮暴食。 此外,并发症发生率很高[ Scheimann等2012 ]。 [Scheimann et al 2012].
生长激素治疗使身高恢复正常,增加瘦体重,减少脂肪量,增加活动能力,这有利于控制体重。幼儿的剂量建议通常与患有单一的生长激素缺乏症(即~1 mg / m 2 /天)的剂量相似,但剂量必须随着孩子的成长而改变。治疗可以在婴儿期或有诊断结论时开始。成人的生长激素剂量是儿童推荐剂量的20%-25%。监测生长速度、头围和血清IGF-1对于避免过度治疗非常重要。
生长激素疗法的对照试验通过成年期证实从婴儿期显著益处[ Carrel等2010,Sode-Carlsen等2010,Wolfgram等2013 ]。[Carrel et al 2010, Sode-Carlsen et al 2010, Wolfgram et al 2013].
- 已经报道了基于对照试验的受治疗婴儿的语言和认知技能的提高[ Myers等2007 ]以及成人的心智速度和灵活性以及运动表现的改善[ Höybye等2005 ]。 [Myers et al 2007] [Höybye et al 2005]
- 在一家制药公司的数据库中记录的328名儿童的一到两年的生长激素治疗结果的回顾中表明,他们的生长速度有所提高,特别是在青春期前的儿童,但他们的BMI没有变化[ Craig等2006 ]。[Craig et al 2006].
- 相对于未有不良副作用增加的39名未治疗的个体来说,21名长期治疗的个体表现出显著更高的成年身高。[ Angulo等2007 ]。 [Angulo et al 2007].
- 一些关于PWS患者的生长激素治疗的更好的认识已经提出 [Osório 2012,Siemensma 2012 ],但仍有更多的工作和更长的研究需要完成。[Osório 2012, Siemensma 2012],
- 虽然最初担心生长激素治疗会导致PWS患者脊柱侧凸,但后来的研究显示,与未接受治疗的患者相比,治疗组的脊柱侧凸频率或严重程度没有差异[ Nagai等2006,Angulo等2007 ]。 [Nagai et al 2006, Angulo et al 2007].
用于治疗口干的产品可以解决唾液分泌减少的问题,包括特殊牙膏,凝胶剂,漱口水和口香糖。
治疗,教育和行为管理。对三岁以下儿童进行早期干预,特别是物理治疗,可以提高肌肉力量,并促进实现发育的重要阶段。 在老年人中,每日肌肉训练增加身体活动性和瘦体重[ Schlumpf等2006 ]。[Schlumpf et al 2006].
在儿童中启动适当的教育计划:
- 开始语言治疗,以解决婴儿期和儿童期的语言延迟和发音不清晰。
- 在学龄期间,通常需要进行特殊教育,无论是在包容环境中还是在独立的教室环境中。个人助理有助于确保出勤率。社会技能培训小组是有益的。
应通过行为管理程序解决行为障碍,包括牢固的限制设置。 虽然没有药物可以有效控制所有PWS患者的行为,但血清素再吸收抑制剂已经帮助了受影响青少年和成人的最大比例,特别是那些有强迫性症状的患者[ Brice 2000,Dykens&Shah 2003 ]。 [Brice 2000, Dykens & Shah 2003].
据报道,精神病人对选择性血清素再吸收抑制剂反应良好,但对情绪稳定剂没有反应[ Soni等2007 ]。 [Soni et al 2007]. 此药物对PWS精神病患者治疗的有效性没有精细深入的研究[ Ho&Dimitropoulos 2010 ]。 [Ho & Dimitropoulos 2010].
性腺功能低下。 甚至直到青春期,隐睾症都可以自发消退,但通常需要借助激素和手术方法;但保持生育能力不是问题。标准治疗是合适的。应该考虑对患有隐睾症的婴儿进行人绒毛膜促性腺激素治疗,因为它可以改善阴囊的大小并改善手术效果[ McCandless 2011 ; Angulo&Miller,未发表的数据]。[McCandless 2011; Angulo & Miller, unpublished data].
性激素的替代产生了足够的第二性征,但由于睾酮替代在男性行为问题中的可能作用和雌激素替代在中风风险中的作用以及与女性月经有关的卫生问题,因此存在争议。每天使用睾酮贴片或凝胶可以通过提供比每月注射缓释环戊丙酸睾酮更均匀的血液水平来避免行为问题的恶化。此外,在非PWS患者成人群体中显示,与凝胶和贴剂相比,环戊丙酸睾酮注射与心血管事件,住院和死亡的风险的相关性更高[ Layton等2015 ]。 [Layton et al 2015].
在决定激素替代时应考虑骨质疏松症。最近关于四名PWS女性患者生育能力的报告提出了对生育控制的需求问题[ Akefeldt等1999 ; Schulze等2001 ; Vats&Cassidy,未发表的数据]。 [Akefeldt et al 1999; Schulze et al 2001; Vats & Cassidy, unpublished data].
睡眠问题。儿童和成人的睡眠不安应该促进睡眠研究,因为可能有治疗方法。治疗取决于病因,并且可能包括扁桃体切除术和腺样体切除术和/或CPAP或BiPAP,像在一般人群一样。
在患有PWS的个体中经常看到过多的白天嗜睡(与睡眠呼吸暂停的程度无关)。莫达非尼已被证明是一种治疗此状况的安全有效的方法[ De Cock et al 2011 ]。 [De Cock et al 2011].
皮肤采摘。Skin picking. 一项研究表明,在一些人中采用托吡酯治疗皮肤采摘减少[ Shapira等2004 ]; 据其他临床医生报道,有趣的是大约一半患有皮肤病的PWS患者从低剂量(每日25-50毫克)托吡酯中获益。 [Shapira et al 2004]
最近一项针对35名PWS患者(年龄5-39岁)使用450-1200 mg /天N-乙酰半胱氨酸的研究发现,其在减少或消除皮肤采摘方面取得了很大成功[ Miller&Angulo 2014 ]。 [Miller & Angulo 2014].
其他
- • 斜视的管理与任何婴儿一样。
- • 脊柱侧凸,髋关节发育不良和肥胖并发症的管理与一般人群一样。
成年。. 对于患有PWS的成年人,行为和体重管理最成功的生活状况是专门为PWS患者指定的团体住所,其中饮食和获取食物受到严格限制,并且锻炼包括在日常活动中。受影响的个体通常需要有庇护的就业环境。environment.
监护,意愿,信托和倡导的问题应在青春期之前进行调查。
预防主要表现
如果制定治疗表现中描述的饮食,锻炼和监督计划,可以预防肥胖。 早期诊断允许临床医生开始关于PWS的自然史,特别是营养阶段的预期指导,告知家庭肥胖的风险以及监测体重增加的需要并于18-36个月左右开始限制热量摄入。
如果在年轻时开始,生长激素治疗以及良好的饮食控制可能会延缓肥胖和脂肪量的高比例。它还可以防止典型面部外观的发展并改善运动里程碑。
预防继发性并发症
在没有肥胖的情况下很少发生糖尿病。
补充钙和维生素D可能是有益的,因为低热量饮食通常在乳制品中较低,并且大多数年龄较大的PWS儿童患者和PWS成人患者已记录到骨质疏松症。
如果发生骨质疏松症,可考虑用二膦酸盐治疗。
虽然没有正式的研究,但PWS患者往往对各种药物都非常敏感。所以建议从较低剂量开始使用。
监控
美国儿科学会(AAP)的健康监督指南已经出版[ McCandless 2011 ](全文)。[McCandless 2011] (full text).
确保运动计划和饮食的适当性,包括维生素和矿物质摄入量的充足性以及监测身高,体重和BMI(以kg为单位的体重/身高(m 2)):
- • 婴儿期的每个月;
- • 十岁之前每六个月;
- • 此后至少每年一次。
隐睾症可以在睾丸固定术后复发; 因此,应监测睾丸位置。
在具有显著肥胖或显著的体重快速增加的任何人中通过标准方法(例如,获得糖基化血红蛋白浓度和/或葡萄糖耐量试验)评估糖尿病的存在。
每年测试甲状腺功能减退症,包括游离T4和TSH水平。
获得任何睡眠障碍的历史; 如果有,请进行睡眠研究。
在临床上监测脊柱侧凸的发展,或者在肥胖的情况下,至少每年进行一次放射照相监测。
通过DEXA进行骨密度测定,以在成年期每两年评估可能发生的骨质疏松症。
至少每年获得行为和精神障碍的病史。
风险亲属的评估
有关为遗传咨询目的检测有风险亲属的问题,请参阅遗传咨询。
正在调查的疗法
在过去的几年里,制药行业对测试PWS主要表现的治疗方法的产生了极大的兴趣 - 特别是饮食过多,肥胖和行为问题。 有关这些研究的详细说明,请单击此处(pdf)。
在ClinicalTrials.gov中搜索去获取有关各种疾病和病症的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。本节不是为了解决个人可能面临的所有个人,文化或道德问题,也不是为了替代与遗传专业人士的咨询。
遗传方式
Prader-Willi综合征(PWS)是由几种遗传机制之一的染色体 15q11.2-q13 的父本衍生的PWS / AS区域缺乏表达引起的。
对家庭成员的风险
先证者的父母
- • 先证者的父母不受影响。
- • 父母基因检测的建议取决于先证者的PWS的遗传机制(参见先证者的兄弟和表3)。(see Sibs of a proband and Table 3).
- • 注意:父亲中的种系镶嵌现象很少见,但在15q11.2缺失[ Fernández-Novoa等2001 ]和IC缺失[ Buiting et al 2003,Wey et al 2005 ]中观察到了此现象。[Fernández-Novoa et al 2001] [Buiting et al 2003, Wey et al 2005]. 还观察到母体 15号染色体的周期性减数分裂不分离[ Harpey等1998]。 [Harpey et al 1998].
先证者的同胞
PWS 先证者的同胞的风险取决于先证者和分子类中PWS的遗传机制(总结于表3)。 绝大多数家庭的复发风险不到1%。然而,某些病因有高达50%的复发风险,并且具有几乎100%风险的情景(即,具有15/15 罗伯逊易位的母亲)虽然不太可能,但理论上是可能的。
用于复发风险 评估。 如果DNA 甲基化模式仅是母系遗传的特征,则应根据遗传咨询目的确定潜在的遗传机制(缺失,UPD或ID)和特定的遗传病因(分子类别)(见图1和表3)。
对15q11.2-q13 缺失的核型和FISH分析开始通常是最有效的。I 同时细胞遗传学研究允许检测涉及近端15q 的易位或其他异常。随着在临床遗传学中越来越多地使用染色体微阵列(CMA),阵列(特别是SNP阵列)可以取代FISH分析以鉴定PWS中的缺失。 但每种技术都有其优点。
- • CMA将精确识别缺失大小,预计这对于未来的基因型 - 表型相关性将变得越来越重要[ Kim et al 2012]。 [Kim et al 2012]. 此外,在非删除情况下,SNP阵列将识别高百分比的UPD。
- • 然而,CMA不会检测到涉及近端15q 的罕见染色体重排(易位和倒位); 这些可通过同时进行染色体核型和FISH分析检测到,并且在复发风险测定中很重要。
- • 为了遗传咨询的目的,建议在先证者中进行染色体分析,以从涉及15q11.2区域的平衡或不平衡染色体重排中辨别出间质性从头缺失。
- • 如果患有PWS的个体具有比典型的更严重的表型,需要辨别是否存在更大的缺失或在基因组中的其他地方存在额外的染色体异常时,则还将指示CMA 。
如果未检测到缺失或其他染色体异常,则进行DNA 多态性研究(需要来自父母和先证者的血液)。I越来越多的临床遗传学家正在使用CMA结合SNP(即oligo-SNP阵列),通过显示仅限于染色体15s的长(> 13.5 Mb)连续的纯合子段,可以检测大约75%的UPD [ Papenhausen等2011 ; Butler&Driscoll,未发表的数据]。 [Papenhausen et al 2011; Butler & Driscoll, unpublished data]. 在这些情况下,DNA多态性分析可能不是诊断UPD所必需的。
如果未检测到UPD,则会转至专门的实验室进行印迹中心(IC)的微缺失分析。
表3。
基于遗传机制的PWS 先证者的同胞的风险
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。本节不是为了解决个人可能面临的所有个人,文化或道德问题,也不是为了替代与遗传专业人士的咨询。 遗传方式 Prader-Willi综合征(PWS)是由几种遗传机制之一的染色体 15q11.2-q13 的父本衍生的PWS / AS区域缺乏表达引起的。 对家庭成员的风险 先证者的父母
先证者的同胞 PWS 先证者的同胞的风险取决于先证者和分子类中PWS的遗传机制(总结于表3)。 绝大多数家庭的复发风险不到1%。然而,某些病因有高达50%的复发风险,并且具有几乎100%风险的情景(即,具有15/15 罗伯逊易位的母亲)虽然不太可能,但理论上是可能的。 用于再发风险 评估。 如果DNA 甲基化模式仅是母系遗传的特征,则应根据遗传咨询目的确定潜在的遗传机制(缺失,UPD或ID)和特定的遗传病因(分子类别)(见图1和表3)。 对15q11.2-q13 缺失的核型和FISH分析开始通常是最有效的。I 同时细胞遗传学研究允许检测涉及近端15q 的易位或其他异常。随着在临床遗传学中越来越多地使用染色体微阵列(CMA),阵列(特别是SNP阵列)可以取代FISH分析以鉴定PWS中的缺失。 但每种技术都有其优点。
如果未检测到缺失或其他染色体异常,则进行DNA 多态性研究(需要来自父母和先证者的血液)。I越来越多的临床遗传学家正在使用CMA结合SNP(即oligo-SNP阵列),通过显示仅限于染色体15s的长(> 13.5 Mb)连续的纯合子段,可以检测大约75%的UPD [ Papenhausen等2011 ; Butler&Driscoll,未发表的数据]。 [Papenhausen et al 2011; Butler & Driscoll, unpublished data]. 在这些情况下,DNA多态性分析可能不是诊断UPD所必需的。 如果未检测到UPD,则会转至专门的实验室进行印迹中心(IC)的微缺失分析。 表3。 基于遗传机制的PWS 先证者的同胞的风险
UPD = 单亲二体 ID = 印迹缺陷 IC = 印记中心 1、术语基于Jiang等[1999] Ia: 具有缺失的个体的父亲应该进行染色体和FISH分析以确定它们是否具有染色体重排。 对于具有典型(1型或2型)大型从头合成时有5至6-Mb缺失之一的先证者,同胞的风险小于1%。据报道,这种大型缺失的生殖腺嵌合现象很少见[ Kokkonen&Leisti 2000 ]。[Kokkonen & Leisti 2000]. Ⅰb:如果在先证者中发现了染色体重排,那么同胞和其他家庭成员面临的风险取决于重排是父系遗传或新发 [ Cassidy等2012 ]。 [Cassidy et al 2012]. IIa: 母系15 UPD是典型的同胞的新发与复发风险到小于1%的情况,除非任一亲本存在一个罗伯逊易位。因此,先证者中指出了染色体分析。 如果这不能识别染色体异常,应该给孩子的父亲提供染色体分析,以确保他没有罗伯逊易位。推测母亲没有罗伯逊易位,因为先证者中的两条母体15号染色体是正常的。 然而,理论上可能父亲的罗伯逊易位涉及15号染色体导致其在减数分裂I期异常分离,并导致一个15号染色体无效的精子。拯救二体与单体性结合,将导致胚胎具有母体15 UPD。 IIb:15 UPD 患者应进行染色体分析,以确保他们没有母系遗传的罗伯逊易位,理论上罗伯逊易位会增加家族的复发风险。在极少数情况下,UPD是由于罗伯逊易位的错误分离和随后的三体性拯救造成的。经验数据表明,大多数病例的复发风险也不到1%,尽管理论风险要高得多。 如果母亲有15/15 罗伯逊易位,三体性拯救将导致PWS。如果父亲有15/15 罗伯逊易位,单体拯救将导致PWS。虽然大多数15/15 罗伯逊易位的父母可能会自发流产而不是生出患有PWS的孩子,但有些人可能通过上述机制生下一个患有PWS的活产儿。 极少数情况下,一个小标记染色体也存在于母亲15 UPD的先证者中 [ Liehr等2005 ]。[Liehr et al 2005]. 在这些情况下,检查父母的核型是很重要的,因为看起来这些小标记染色体可能会增加不分离和UPD的风险[ Kotzot 2002 ]。[Kotzot 2002]. IIIa: 由印迹缺陷(ID)引起的患有PWS的个体应该由经验丰富的实验室检测IC 缺失。大约有15%的有ID的人会根据IC中的微缺失进行识别。在这些个体的大约一半中,IC缺失是家族性的,并且家族性再发风险是50%。 因此,具有IC缺失的儿童的父亲应在PWS最小缺失重叠区域(SRO)中进行DNA 甲基化和剂量分析(或序列分析)以确定它们是否携带IC缺失。 IIIb: 大多数(约85%)具有ID的患者具有新发表观遗传致病变异体,对于该组,同胞的复发风险小于1%。 先证者的后代
其他家庭成员 如果先证者或父母被诊断为染色体重排(如易位和倒置),携带者的亲属需接受遗传咨询和选择进行基因检测。 相关遗传咨询事宜 生育规划
DNA储存 是保存DNA的方式(典型的方式是从白细胞中提取),以备将来所用。因为我们的检测方式和我们对基因、等位基因的多样性以及疾病的理解将有可能得到进一步的发展,所以应当考虑保存受累患者的基因。 产前检测 家庭中已有患PWS的小孩(小孩患病的基因机制已查明) 需注意关于PWS的产前诊断是可行的,依赖分子机制、FISH、CMA、DNA甲基化分析、MS-MLPA和UPD的DNA多态性研究都适于检测,需要注意仅仅在DNA甲基化分析(如MS-MLPA)5' SNRPN位点会发现印记缺失[Kubota et al 1996, Glenn et al 2000, Ramsden et al 2010].。虽然可以对所有三种分子类别的PWS进行产前检测,但其可操作性可能有限。 通过DNA甲基化分析提供PWS产前诊断的实验室通常仅接受羊水细胞(与绒毛膜绒毛相比)进行分析,但是目前已知来自胎盘的组织通常表现为低甲基化 [Driscoll & Migeon 1990, Glenn et al 2000]。 [Kubota et al 1996, Glenn et al 2000, Ramsden et al 2010] [Driscoll & Migeon 1990, Glenn et al 2000].
对于无PWS家族史的妊娠。 在以下情况可能会出现PWS患者
最近利用胎儿无细胞DNA的进行非侵入性产前检测(NIPT)已经可用于检测主要的三体性病症以及常见的微缺失(包括PWS和AS)状况 [Norton et al 2012]。 如果在NIPT中进行CMA,它可以发现15q11.2缺失,但它不能区分AS缺失和PWS缺失,并且它不能得到UPD和ID的产生机制。[Norton et al 2012] 胚胎植入前遗传学诊断(PGD)可能适用于已确定为IC缺失的家庭。 PGD也可用于检测家族性易位以排除UPD。 | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
资源
GeneReviews的工作人员选择了以下疾病特异性和/或伞式支持组织和/或注册机构,以帮助患有此疾病的个人及其家人。 GeneReviews不对其他组织提供的信息负责。有关入选标准的信息,请单击此处here。
- 国际Prader-Willi综合症组织(IPWSO)地址:c/o BIRD Europe Foundation OnlusVia Bartolomeo Bizio 1Costozza 1-36023Italy电话: +39 0444 555557传真: +39 0444 555557邮件: info@ipwso.org
- My46 Trait Profile
- Prader-Willi Syndrome Association地址:8588 Potter Park DriveSuite 500Sarasota FL 34238电话: 800-926-4797 (toll-free); 941-312-0400传真: 941-312-0142邮件: national@pwsusa.org; pwsausa@pwsausa.org
- Foundation for Prader-Willi Research (FPWR)电话: 888-322-5487传真: 888-559-4105邮件: info@fpwr.org
- Prader-Willi Syndrome Association (UK)地址:125a London RoadDerby DE1 2QQUnited Kingdom电话:01332 365676传真: 01332 360401邮件: admin@pwsa.co.uk
分子遗传学
Molecular Genetics和OMIM表格中的信息可能与GeneReview中的信息有所不同:表格中可能包含更多最新的信息。-ED。
Table B.
Prader-Willi综合症的OMIM入口 (View All in OMIM)
| 137142 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, ALPHA-5; GABRA5 |
| 137192 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, BETA-3; GABRB3 |
| 176270 | PRADER-WILLI SYNDROME; PWS |
| 182279 | SMALL NUCLEAR RIBONUCLEOPROTEIN POLYPEPTIDE N; SNRPN |
| 600161 | PRADER-WILLI/ANGELMAN REGION RNA 1; PWAR1 |
| 600233 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, GAMMA-3; GABRG3 |
| 601491 | IMPRINTED IN PRADER-WILLI SYNDROME; IPW |
| 601623 | UBIQUITIN-PROTEIN LIGASE E3A; UBE3A |
| 602117 | NECDIN; NDN |
| 603856 | MAKORIN 3; MKRN3 |
| 603857 | MKRN3 ANTISENSE RNA; MKRN3AS |
| 605283 | MAGE-LIKE 2; MAGEL2 |
| 605436 | SMALL NUCLEOLAR RNA, C/D BOX, 116-1; SNORD116-1 |
| 605837 | HECT DOMAIN AND RCC1-LIKE DOMAIN 2; HERC2 |
| 605855 | ATPase, CLASS V, TYPE 10A; ATP10A |
| 609837 | SMALL NUCLEOLAR RNA, C/D BOX, 115-1; SNORD115-1 |
| 610922 | NUCLEAR PORE ASSOCIATED PROTEIN 1; NPAP1 |
| 611215 | PRADER-WILLI REGION NONCODING RNA 1; PWRN1 |
| 611409 | OCA2 GENE |
分子遗传发病机制
PWS区域定位于15号染色体(15q11.2-q13)近端长臂上的5至6-Mb基因组的区域(Figure 2),它位于较小的2.5 Mb差异印记区域内。PWS是一种连续的基因疾病:迄今为止的研究表明,完整的疾病表型是由于几种基因的表达缺失所致。这也是基因组印迹的一个例子,因为15q11.2-q13区域中相关基因的表达取决于亲本来源。 [Glenn et al 1997, Bittel et al 2006]。 [Glenn et al 1997, Bittel et al 2006].
导致PWS的基因组和表观遗传的变化都指向正常父源染色体15q11.2-q13区域基因表达缺失。缺乏这些基因的父系遗传拷贝,或未能表达它们,会导致受影响个体中这些基因的完全缺失表达,因为母系遗传的基因已被表观遗传因子编程沉默 [Glenn et al 1997, Cassidy & Driscoll 2009]。相反,在同一区域中母系遗传优先表达基因UBE3A的表达缺失会通过几种不同的机制导致Angelman综合症[Lossie et al 2001, Williams et al 2010]。 [Glenn et al 1997, Cassidy & Driscoll 2009] [Lossie et al 2001, Williams et al 2010].
15q11.2-q13区域可被三个常见的分段重复[Amos-Landgraf et al 1999] 的缺失位点 [Christian et al 1999]大致分为四个区域。(见 Figure 2 和 Table 4)
- 在两个近端断点(BP1和BP2)之间的近端非印迹区域包含四个双亲均表达的基因,NIPA1, NIPA2, CYF1P1, and GCP5A [Chai et al 2003] [Chai et al 2003]
- “PWS仅父源表达区域”包含五个多肽编码基因(MKRN3, MAGEL2, NECDIN,和双顺反子NURF-SNRPN);NPAP1(过去称为C15orf2)(一种无内含子基因,在睾丸中双等位基因表达,但仅在脑中由父源等位基因表达)一组C / D box 小核仁RNA基因(snoRNAs)和几个反义转录本(包括UBE3A的反义转录本)
- “Anglman综合症区域”包含母源优先表达基因UBE3A 和 ATP10A
- 远端非印迹区域,包含一个含三个GABA受体基因簇,眼皮肤白化病2型(OCA2)基因,HERC2和常见远端断点(BP3)
Table 4.
PWS现有缺失位点
| 缺失位点 | ISCA ID 1 | 定位2 |
|---|---|---|
| 15q11-q13, class 1 (type 1) | ISCA-37404 | GRCh37/hg19 chr15: 22,876,632-28,557,186 |
| 15q11-q13, class 2 (type 2) | ISCA-37478 | GRCh37/hg19 chr15: 23,758,390-28,557,186 |
- 1.
基因组变异标准化临床注释和解释,来自临床基因组资源(ClinGen)项目(以前称为国际细胞基因组数据标准(ISCA)联盟)
- 2.
基因组坐标代表与ClinGen指定的PWS复发性微缺失相关的最小缺失大小。根据不同测试实验室使用的阵列设计,缺失坐标可能略有不同,并且由于在断点附近存在节段重复,从这些基因组位置计算的微缺失的大小可能与预期的微缺失大小不同。
PWS区域的中心是SNURF-SNRPN,编码两种蛋白质的双顺反子基因。 SNURF-SNRPN的5'末端的CpG岛,包括启动子,外显子1和内含子1,是差异甲基化的:未甲基化的父系遗传等位基因被表达,甲基化的母系遗传等位基因被抑制 [Glenn et al 1996]。对于父本PWS印迹中心(IC),CpG岛和外显子1在4.3kb的最小缺失重叠区域(SRO)内[Ohta et al 1999]。位于该基因座的六个snoRNA基因受SNURF-SNRPN的表达调节。 UBE3A反义转录本也来自SNURF-SNRPN的转录,目前认为其可抑制父本UBE3A [Cavaillé et al 2000, Chamberlain & Brannan 2001, Runte et al 2001]。
缺失机制 大多数PWS是由父源等位基因上的间质15q11.2-q13微缺失引起的[Ledbetter et al 1981, Butler & Palmer 1983, Glenn et al 1997]。65%-75%的PWS是由于缺失引起的,绝大多数缺失具有两个常见近端断点中的一个(BP1或BP2)和一个常见远端断点(BP3)(见 Figure 2 和 Table 4) [Christian et al 1995, Amos-Landgraf et al 1999]。这些复发性常见间质缺失约为5-6 Mb,由共同断裂点(BP1,BP2和BP3)两侧的多个串联重复序列介导。些低拷贝250-400-kb重复序列在减数分裂过程中经历非同源配对,导致缺失(导致PWS或AS取决于亲本来源)、重复(来自父本和母本)、染色体三体和倒置重复(15) [Robinson et al 1998, Amos-Landgraf et al 1999, Boyar et al 2001, Maggouta et al 2003, Depienne et al 2009]。约8%的病例具有由多种病因引起的独特或非典型大小的缺失(不属于1型和2型),原因包括包括不平衡的易位[Kim et al 2012],导致的缺失比通常PWS中的缺失更大或更小,可能引起的临床特征也不同。
在母源基因发生DNA甲基化模式的PWS患者中可以鉴定出SNRPN(包括推测的印迹控制元件)启动子区域和近端上游区域的小缺失,但是对于既没有父源基因PWS/AS区域的大的缺失也不是母系UPD的患者,这种模式被认为为IC微缺失引起的ID。
其他具有双亲遗传的个体,但仅有母本的DNA甲基化模式,同时在IC的SRO中没有可检测到的异常,这些人被认为是通过表观突变的ID。 区域内的目的基因,以下基因都位于PWS/AS区中:
- SNURF-SNRPN是编码两种不同蛋白质的复杂双顺反子基因,外显子4-10编码SmN蛋白,SmN蛋白是参与mRNA剪接的剪接体蛋白 [Glenn et al 1996]。外显子1-3编码SNURF,产生一条未知功能的多肽链[Gray et al 1999]。它同时还作为定位在着丝粒上六个 snoRNA基因的宿主基因,这六个基因通过SNURF-SNRPN的表达来调控。
- IPW 是位于SNORD116-1和SNORD115-1之间的长非编码RNA。它过去曾被认为没有功能,但最近已被证明它是14号染色体上 DLK1-DIO3印迹区的调节因子,它也可能参与调节基因组中其他非15号染色体的印迹区域[Stelzer et al 2014]。在下述SNORD116 缺失的4名患者中IPW也缺失。
- PAR1, PAR4, PAR5, 和 PAR7是未知转录
- OCA2 (过去称为 P)编码酪氨酸酶阳性白化病;它的缺失与1/3PWS患者的色素减退相关。
- GABRB3,GABRA5, 和 GABRG3, 都是GABA受体亚基基因。
- UBE3A (过去称为 E6AP)与AS有关
- ATP10A,母源表达基因,位于AS的最常见缺失间隔中。
- HERC2在常见缺失位点中多次重复。
- NECDIN (NDN)编码DNA结合蛋白。Ndn敲除小鼠模型表明,NDN介导神经突向外生长所必需的细胞内过程,并且导致necdin的损失对轴突生长造成影响 [Lee et al 2005],也有报道小鼠Ndn敲除模型具有与PWS个体类似的缺陷,并表明necdin是神经系统早期发育中的抗细胞凋亡或存活因子[Andrieu et al 2006]。
- MAGEL2, 靠近NDN基因座的无内含子基因,仅由父本等位基因转录,主要在脑中表达PWS 最近还发现了四个个体在MAGEL2的父本等位基因中具有截短变体,具有与PWS重叠的特征[Schaaf et al 2013]。临床上所有四个人都患有自闭症谱系症,智力残疾,体重过度增加和/或饮食过多以及缺乏饱腹感。目前认为“这些患者在表型上与经典PWS患者不同”,并且将该病症从“Prader-Willi样综合征”改名为“Schaff-Yang综合征” [Fountain & Schaaf 2015; CP Schaaf, personal communication]。Magel2 也认为与小鼠昼夜节律有关 [Kozlov et al 2007] 最近有研究报道,Magel2缺失小鼠在弓状下丘脑神经元中的瘦素敏感性发生显着下降[Pravdivyi et al 2015]。
- MKRN3 (Markorin 3, ZNF127)是一个只在父源染色体表达的锌指蛋白,最近有研究报道,MKRN3父源遗传性功能丧失性突变是导致人类家族性性早熟的原因之一 [Abreu et al 2013, Macedo et al 2014]。
- NPAP1 是一个无内含子基因,在成年人睾丸中双等位基因表达,但在胎儿的脑中单等位基因表达。
- PWRN1, 要在睾丸中表达,在前列腺,心脏,肾,肝,肺,骨骼肌,气管,脊髓和胎儿脑中也有少量表达; 目前发现在胎儿大脑中为单等位基因表达。
- SNORD116 (snoRNA HBII-85). 两条证据表明 snoRNA SNORD116 簇,包含29个基因拷贝,是导致PWS特征的原因。为了保持SNURF-SNRPN表达而造成的平衡异位和将SNORD116簇从其启动子分离的着丝粒基因会导致PWS。最近,四名有PWS特征的患者中被报导有SNORD116簇上的微缺失[Sahoo et al 2008, de Smith et al 2009, Buiting 2010, Duker et al 2010, Bieth et al 2015]然而,其中三名患者[Sahoo et al 2008, de Smith et al 2009, Duker et al 2010]有不符合经典PWS的特征,包括儿童身材高大,头围大,缺乏“PWS面部形态”,手部特征不典型。snoRNAs可能通过选择性剪接参与mRNA的修饰,每个snoRNA基因可能有多个靶点,然而至今并没有在SNORD116发现靶点,而在家族性AS的情况下,相关基因簇SNORD115的关键作用被排除在外[Runte et al 2005]。 SNORD116 (snoRNA HBII-85).
- 一些其他的印迹基因和未知功能的转录也已被发现。
区域内的印迹基因 PWS区域中的几个基因 (SNURF-SNRPN, MKRN3, NDN, MAGEL2,NPAP1, PWRN1, SNORD116, IPW, and SNORD115)受基因组印记的影响,因此可以解释PWS表型仅在父源PWS区域缺失时存在。DNA甲基化参与了基因组印迹的过程,以证实甲基化存在于PWS区域中的一些基因中[Glenn et al 1996, Glenn et al 1997, MacDonald & Wevrick 1997]。在一些有母源DNA甲基化模式,但既没有父源PWS/AS区域大的缺失也不是母源UPD的PWS患者中,在SNRPN的上游检测到非常小的假定印迹控制元件上的缺失 [Saitoh et al 1997, Ohta et al 1999]。其他个体表现出散发的印迹突变,即表观突变 [Buiting et al 1998, Buiting et al 2003]。
References
Published Guidelines/Consensus Statements
- Deal CL, Tony M, Höybye C, Allen DB, Tauber M, Christiansen JS; 2011 Growth hormone in Prader-Willi Syndrome Clinical Care Guidelines Workshop Participants. Growth Hormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J Clin Endocrinol Metab. 2013;98:E1072-87. Available online. 2013. Accessed 7-6-16. [PMC free article: PMC3789886] [PubMed: 23543664]
- Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 93:4183-97. Available online. 2008. Accessed 7-6-16. [PubMed: 18697869]
- McCandless SE, Committee on Genetics. Clinical report -- health supervision for children with Prader-Willi Syndrome. Pediatrics. 127:195-204. Available online. 2011. Accessed 7-6-16. [PubMed: 21187304]
- Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 11:11-70. Available online. 2010. Accessed 7-6-16. [PMC free article: PMC2877670] [PubMed: 20459762]
Literature Cited
- Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, Cukier P, Thompson IR, Navarro VM, Gagliardi PC, Rodrigues T, Kochi C, Longui CA, Beckers D, de Zegher F, Montenegro LR, Mendonca BB, Carroll RS, Hirschhorn JN, Latronico AC, Kaiser UB. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. 2013;368:2467 - 75. [PMC free article: PMC3808195] [PubMed: 23738509]
- Akefeldt A, Tornhage CJ, Gillberg C. A woman with Prader-Willi syndrome gives birth to a healthy baby girl. Dev Med Child Neurol. 1999;41:789 - 90. [letter] [PubMed: 10576646]
- Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65:370 - 86. [PMC free article: PMC1377936] [PubMed: 10417280]
- Andrieu D, Meziane H, Marly F, Angelats C, Fernandez PA, Muscatelli F. Sensory defects in Necdin deficient mice result from a loss of sensory neurons correlated within an increase of developmental programmed cell death. BMC Dev Biol. 2006;6:56. [PMC free article: PMC1687209] [PubMed: 17116257]
- Angulo MA, Castro-Magana M, Lamerson M, Arguello R, Accacha S, Khan A. Final adult height in children with Prader-Willi syndrome with and without human growth hormone treatment. Am J Med Genet A. 2007;143A:1456 - 61. [PubMed: 17567883]
- Ben-Abdallah-Bouhjar I, Hannachi H, Labalme A, Gmidène A, Mougou S, Soyah N, Gribaa M, Sanlaville D, Elghezal H, Saad A. Chromosomal microarray analysis of functional Xq27-qter disomy and deletion 3p26.3 in a boy with Prader-Willi like features and hypotonia. Eur J Med Genet. 2012;55:461 - 5. [PubMed: 22683462]
- Bieth E, Eddiry S, Gaston V, Lorenzini F, Buffet A, Conte Auriol F, Molinas C, Cailley D, Rooryck C, Arveiler B, Cavaillé J, Salles JP, Tauber M. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet. 2015;23:252 - 5. [PMC free article: PMC4297892] [PubMed: 24916642]
- Bischof JM, Stewart CL, Wevrick R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum Mol Genet. 2007;16:2713 - 9. [PubMed: 17728320]
- Bittel DC, Kibiryeva N, Butler MG. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics. 2006;118:e1276 - 83. [PMC free article: PMC5453799] [PubMed: 16982806]
- Boer H, Holland A, Whittington J, Butler J, Webb T, Clarke D. Psychotic illness in people with Prader-Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. 2002;359:135 - 6. [PubMed: 11809260]
- Bonaglia MC, Ciccone R, Gimelli G, Gimelli S, Marelli S, Verheij J, Giorda R, Grasso R, Borgatti R, Pagone F, Rodrigueq L, Martinez-Frias ML, van Ravenswaaij C, Zuffardi O. Detailed phenotype-genotype study in five patients with chromosome 6q16 deletion: narrowing the critical region for Prader-Willi-like phenotype. Eur J Hum Genet. 2008;16:1443 - 9. [PubMed: 18648397]
- Bonnefond A, Raimondo A, Stutzmann F, Ghoussaini M, Ramachandrappa S, Bersten DC, Durand E, Vatin V, Balkau B, Lantieri O, Raverdy V, Pattou F, Van Hul W, Van Gaal L, Peet DJ, Weill J, Miller JL, Horber F, Goldstone AP, Driscoll DJ, Bruning JB, Meyre D, Whitelaw ML, Froguel P. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J Clin Invest. 2013;123:3037 - 41. [PMC free article: PMC3696559] [PubMed: 23778136]
- Boyar FZ, Whitney MM, Lossie AC, Gray BA, Keller KL, Stalker HJ, Zori RT, Geffken G, Mutch J, Edge PJ, Voeller KS, Williams CA, Driscoll DJ. A family with a grand-maternally derived interstitial duplication of proximal 15q. Clin Genet. 2001;60:421 - 30. [PubMed: 11846734]
- Brice JA. Behavorial and psychotropic interventions in persons with Prader-Willi syndrome. Endocrinologist. 2000;10:27S - 30S.
- Bruni O, Verrillo E, Novelli L, Ferri R. Prader-Willi syndrome: sorting out the relationships between obesity, hypersomnia, and sleep apnea. Curr Opin Pulm Med. 2010;16:568 - 73. [PubMed: 20814307]
- Buiting K, Cassidy SB, Driscoll DJ, Gillessen-Kaesbach G, Kanber D, Tauber M, Schwinger E, Horsthemke B. Clinical utility gene card for: Prader-Willi Syndrome. Eur J Hum Genet. 2014;22(9) [PMC free article: PMC4135421] [PubMed: 24736734]
- Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365 - 76. [PubMed: 20803659]
- Buiting K, Dittrich B, Gross S, Lich C, Färber C, Buchholz T, Smith E, Reis A, Bürger J, Nöthen MM, Barth-Witte U, Janssen B, Abeliovich D, Lerer I, van den Ouweland AM, Halley DJ, Schrander-Stumpel C, Smeets H, Meinecke P, Malcolm S, Gardner A, Lalande M, Nicholls RD, Friend K, Schulze A, Matthijs G, Kokkonen H, Hilbert P, Van Maldergem L, Glover G, Carbonell P, Willems P, Gillessen-Kaesbach G, Horsthemke B. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet. 1998;63:170 - 80. [PMC free article: PMC1377255] [PubMed: 9634532]
- Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72:571 - 7. [PMC free article: PMC1180233] [PubMed: 12545427]
- Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev. 2001;22:787 - 99. [PubMed: 11739333]
- Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44:248 - 55. [PubMed: 11995893]
- Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. 2004;113:565 - 73. [PubMed: 14993551]
- Butler MG, Lee J, Manzardo AM, Gold JA, Miller JL, Kimonis V, Driscoll DJ. Growth charts for non-growth hormone treated Prader-Wili syndrome. Pediatrics. 2015;135:e126 - 35. [PMC free article: PMC4279067] [PubMed: 25489013]
- Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. 1983;1:1285 - 6. [PMC free article: PMC5510872] [PubMed: 6134086]
- Butler MG, Sturich J, Lee J, Myers SE, Whitman BY, Gold JA, Kimonis V, Scheimann A, Terrazas N, Driscoll DJ. Growth standards of infants with Prader-Willi syndrome. Pediatrics. 2011;127:687 - 95. [PMC free article: PMC3065075] [PubMed: 21402637]
- Butler MG, Sturich J, Myers SE, Gold JA, Kimonis V, Driscoll DJ. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J Assist Reprod Genet. 2009;26:461 - 6. [PMC free article: PMC2767487] [PubMed: 19760168]
- Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with prader-willi syndrome. J Clin Endocrinol Metab. 2010;95:1131 - 6. [PMC free article: PMC2841537] [PubMed: 20061431]
- Cassidy SB, Driscoll DJ. Prader-Willi Syndrome. Eur J Hum Genet. 2009;17:3 - 13. [PMC free article: PMC2985966] [PubMed: 18781185]
- Cassidy SB, McCandless SM. Prader-Willi syndrome. In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. 3 ed. New York, NY: John Wiley & Sons; 2010:625-50.
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi Syndrome. Genet Med. 2012;14:10 - 26. [PubMed: 22237428]
- Cavaillé J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J, Hüttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci U S A. 2000;97:14311 - 6. [PMC free article: PMC18915] [PubMed: 11106375]
- Chai JH, Locke DP, Greally JM, Knoll JH, Ohta T, Dunai J, Yavor A, Eichler EE, Nicholls RD. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73:898 - 925. [PMC free article: PMC1180611] [PubMed: 14508708]
- Chamberlain SJ, Brannan CI. The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73:316 - 22. [PubMed: 11350123]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet. 1999;8:1025 - 37. [PubMed: 10332034]
- Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, Nakao M, Surti U, Chakravarti A, Ledbetter DH. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet. 1995;57:40 - 8. [PMC free article: PMC1801233] [PubMed: 7611294]
- Clarke DJ, Boer H, Whittington J, Holland A, Butler J, Webb T. Prader-Willi syndrome, compulsive and ritualistic behaviours: the first population-based survey. Br J Psychiatry. 2002;180:358 - 62. [PubMed: 11925360]
- Cox H, Bullman H, Temple IK. Maternal UPD(14) in the patient with a normal karyotype: clinical report and a systematic search for cases in samples sent for testing for Prader-Willi syndrome. Am J Med Genet A. 2004;127A:21 - 5. [PubMed: 15103712]
- Craig ME, Cowell CT, Larsson P, Zipf WB, Reiter EO, Albertsson Wikland K, Ranke MB, Price DA. Growth hormone treatment and adverse events in Prader-Willi syndrome: data from KIGS (the Pfizer International Growth Database). Clin Endocrinol (Oxf) 2006;65:178 - 85. [PubMed: 16886957]
- Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, Bosio L, Corrias A, Gargantini L, Salvatoni A, Tonini G, Trifirò G, Livieri C. Genetic Obesity Study Group of Italian Society of Pediatric endocrinology and diabetology (SIEDP). Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162:327 - 33. [PubMed: 12692714]
- Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med. 2002;8:643 - 4. [PubMed: 12091883]
- Dauvilliers Y, Baumann CR, Carlander B, Bischof M, Blatter T, Lecendreux M, Maly F, Besset A, Touchon J, Billiard M, Tafti M, Bassetti CL. CSF hypocretin-1 levels in narcolepsy, Kleine-Levin syndrome, and other hypersomnias and neurological conditions. J Neurol Neurosurg Psychiatry. 2003;74:1667 - 73. [PMC free article: PMC1757412] [PubMed: 14638887]
- De Cock VC, Diene G, Molinas C, Masson VD, Kieffer I, Mimoun E, Tiberge M, Tauber M. Efficacy of modafinil on excessive daytime sleepiness in Prader-Willi syndrome. Am J Med Genet A. 2011;155A:1552 - 7. [PubMed: 21671379]
- de Lind van Wijngaarden RF, Otten BJ, Festen DA, Joosten KF, de Jong FH, Sweep FC, Hokken-Koelega AC. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93:1649 - 54. [PubMed: 18303077]
- DelParigi A, Tschöp M, Heiman ML, Salbe AD, Vozarova B, Sell SM, Bunt JC, Tataranni PA. High circulating ghrelin: a potential cause for hyperphagia and obesity in prader-willi syndrome. J Clin Endocrinol Metab. 2002;87:5461 - 4. [PubMed: 12466337]
- Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, Chaste P, Siffroi JP, Chantot-Bastaraud S, Benyahia B, Trouillard O, Nygren G, Kopp S, Johansson M, Rastam M, Burglen L, Leguern E, Verloes A, Leboyer M, Brice A, Gillberg C, Betancur C. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66:349 - 59. [PubMed: 19278672]
- Desch L, Marle N, Mosca-Boidron AL, Faivre L, Eliade M, Payet M, Ragon C, Thevenon J, Aral B, Ragot S, Ardalan A, Dhouibi N, Bensignor C, Thauvin-Robinet C, El Chehadeh S, Callier P (2015) 6q16.3q23.3 duplication associated with Prader-Willi-like syndrome. Mol Cytogenet 25;8:42.
- Descheemaeker MJ, Govers V, Vermeulen P, Fryns JP. Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A. 2006;140:1136 - 42. [PubMed: 16646032]
- de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, Brady AF, Fairbrother UL, Dattani M, Keogh JM, Henning E, Yeo GS, O'Rahilly S, Froguel P, Farooqi IS, Blakemore AI. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18:3257 - 65. [PMC free article: PMC2722987] [PubMed: 19498035]
- de Vries BB, Fryns JP, Butler MG, Canziani F, Wesby-van Swaay E, van Hemel JO, Oostra BA, Halley DJ, Niermeijer MF. Clinical and molecular studies in fragile X patients with a Prader-Willi-like phenotype. J Med Genet. 1993;30:761 - 6. [PMC free article: PMC1016534] [PubMed: 8411072]
- De Waele K, Ishkanian SL, Bogarin R, Miranda CA, Ghatei MA, Bloom SR, Pacaud D, Chanoine JP. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur J Endocrinol. 2008;159:381 - 8. [PubMed: 18603572]
- Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, Tauber M., French Reference Centre for PWS. Endocrine disorders in children with Prader-Willi syndrome--data from 142 children of the French database. Horm Res Paediatr. 2010;74:121 - 8. [PubMed: 20395666]
- Driscoll DJ, Migeon BR. Sex difference in methylation of single-copy genes in human meiotic germ cells: implications for X chromosome inactivation, parental imprinting, and origin of CpG mutations. Somat Cell Mol Genet. 1990;16:267 - 82. [PubMed: 1694309]
- Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, Thompson R, Traylor R, Bejjani BA, Shaffer LG, Rosenfeld JA, Lamb AN, Sahoo T. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet. 2010;18:1196 - 201. [PMC free article: PMC2987474] [PubMed: 20588305]
- Dykens E, Shah B. Psychiatric disorders in Prader-Willi syndrome: epidemiology and management. CNS Drugs. 2003;17:167 - 78. [PubMed: 12617696]
- Dykens EM. Are jigsaw puzzle skills 'spared' in persons with Prader-Willi syndrome? J Child Psychol Psychiatry. 2002;43:343 - 52. [PubMed: 11944876]
- Dykens EM. Maladaptive and compulsive behavior in Prader-Willi syndrome: new insights from older adults. Am J Ment Retard. 2004;109:142 - 53. [PubMed: 15000675]
- Dykens EM, Cassidy SB, King BH. Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard. 1999;104:67 - 77. [PubMed: 9972835]
- Eiholzer U. Deaths in children with Prader-Willi syndrome. A contribution to the debate about the safety of growth hormone treatment in children with PWS. Horm Res. 2005;63:33 - 9. [PubMed: 15604598]
- Einfeld SL, Kavanagh SJ, Smith A, Evans EJ, Tonge BJ, Taffe J. Mortality in Prader-Willi syndrome. Am J Ment Retard. 2006;111:193 - 8. [PMC free article: PMC2422866] [PubMed: 16597186]
- Eldar-Geva T, Hirsch HJ, Rabinowitz R, Benarroch F, Rubinstein O, Gross-Tsur V. Primary ovarian dysfunction contributes to the hypogonadism in women with Prader-Willi Syndrome. Horm Res. 2009;72:153 - 9. [PubMed: 19729946]
- Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur J Endocrinol. 2010;162:377 - 84. [PubMed: 19946044]
- Erdie-Lalena CR, Holm VA, Kelly PC, Frayo RS, Cummings DE. Ghrelin levels in young children with Prader-Willi syndrome. J Pediatr. 2006;149:199 - 204. [PubMed: 16887433]
- EUCROMIC. Trisomy 15 CPM: probable origins, pregnancy outcome and risk of fetal UPD: European Collaborative Research on Mosaicism in CVS (EUCROMIC). Prenat Diagn. 1999;19:29 - 35. [PubMed: 10073903]
- Farholt S, Sode-Carlsen R, Christiansen JS, Østergaard JR, Høybye C. Normal cortisol response to high-dose synacthen and insulin tolerance test in children and adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2011;96:E173 - 80. [PubMed: 20980432]
- Feigerlová E, Diene G, Conte-Auriol F, Molinas C, Gennero I, Salles JP, Arnaud C, Tauber M. Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93:2800 - 5. [PubMed: 18460565]
- Fernández-Novoa MC, Vargas MT, Vizmanos JL, Garnacho C, Martínez JJ, Sanz P, Lluch D. Prader-Willi syndrome large deletion on two brothers. Is this the exception that confirm the rule? Rev Neurol. 2001;32:935 - 8. [PubMed: 11424049]
- Festen DA, de Weerd AW, van den Bossche RA, Joosten K, Hoeve H, Hokken-Koelega AC. Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. J Clin Endocrinol Metab. 2006;91:4911 - 5. [PubMed: 17003096]
- Fountain MD Jr, Schaaf CP. MAGEL2 and Oxytocin-Implications in Prader-Willi Syndrome and Beyond. Biol Psychiatry. 2015;78:78 - 80. [PubMed: 26092431]
- Gilmour J, Skuse D, Pembrey M. Hyperphagic short stature and Prader-Willi syndrome: a comparison of behavioural phenotypes, genotypes and indices of stress. Br J Psychiatry. 2001;179:129 - 37. [PubMed: 11483474]
- Glenn CC, Saitoh S, Jong MT, Filbrandt MM, Surti U, Driscoll DJ, Nicholls RD. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996;58:335 - 46. [PMC free article: PMC1914536] [PubMed: 8571960]
- Glenn CC, Deng G, Michaelis RC, Tarleton J, Phelan MC, Surh L, Yang TP, Driscoll DJ. DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diagn. 2000;20:300 - 6. [PubMed: 10740202]
- Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod. 1997;3:321 - 32. [PubMed: 9237260]
- Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS (2008) Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 93:4183-97. [PubMed: 18697869]
- Gray TA, Saitoh S, Nicholls RD. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc Natl Acad Sci U S A. 1999;96:5616 - 21. [PMC free article: PMC21909] [PubMed: 10318933]
- Gross-Tsur V, Hirsch HJ, Benarroch F, Eldar-Geva T. The FSH-inhibin axis in Prader-Willi syndrome: Heterogeneity of gonadal dysfunction. Reprod Biol Endocrinol. 2012;10:39. [PMC free article: PMC3472203] [PubMed: 22559970]
- Grugni G, Beccaria L, Corrias A, Crinò A, Cappa M, De Medici C, Di Candia S, Gargantini L, Ragusa L, Salvatoni A, Sartorio A, Spera S, Andrulli S, Chiumello G, Mussa A., Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED). Central adrenal insufficiency in young adults with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2013;79:371 - 8. [PubMed: 23311724]
- Grugni G, Marzullo P, Ragusa L, Sartorio A, Trifiro G, Liuzzi A, Crino A. Impairment of GH responsiveness to combined GH-releasing hormone and arginine administration in adult patients with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2006;65:492 - 9. [PubMed: 16984242]
- Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:E92. [PubMed: 11694676]
- Haqq AM, Farooqi IS, O'Rahilly S, Stadler DD, Rosenfeld RG, Pratt KL, LaFranchi SH, Purnell JQ. Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab. 2003a;88:174 - 8. [PubMed: 12519848]
- Haqq AM, Stadler DD, Rosenfeld RG, Pratt KL, Weigle DS, Frayo RS, LaFranchi SH, Cummings DE, Purnell JQ. Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2003b;88:3573 - 6. [PubMed: 12915638]
- Harpey JP, Heron D, Prudent M, Lesourd S, Henry I, Royer-Legrain G, Munnich A, Bonnefont JP. Recurrent meiotic nondisjunction of maternal chromosome 15 in a sibship. Am J Med Genet. 1998;76:103 - 4. [PubMed: 9508076]
- Hartley SL, Maclean WE Jr, Butler MG, Zarcone J, Thompson T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A. 2005;136:140 - 5. [PMC free article: PMC1896317] [PubMed: 15940679]
- Hayashi M, Miyata R, Tanuma N. Decrease in acetylcholinergic neurons in the pedunculopontine tegmental nucleus in a patient with Prader-Willi syndrome. Neuropathology. 2011;31:280 - 5. [PubMed: 20880323]
- Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V. Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with Prader-Willi syndrome. J Clin Endocrinol Metab. 2009;94:2262 - 8. [PubMed: 19401370]
- Ho AY, Dimitropoulos A. Clinical management of behavioral characteristics of Prader-Willi syndrome. Neuropsychiatr Dis Treat. 2010;6:107 - 18. [PMC free article: PMC2874334] [PubMed: 20505842]
- Holland AJ, Whittington JE, Butler J, Webb T, Boer H, Clarke D. Behavioural phenotypes associated with specific genetic disorders: evidence from a population-based study of people with Prader-Willi syndrome. Psychol Med. 2003;33:141 - 53. [PubMed: 12537045]
- Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398 - 402. [PubMed: 8424017]
- Horsthemke B, Buiting K. Imprinting defects on human chromosome 15. Cytogenet Genome Res. 2006;113:292 - 9. [PubMed: 16575192]
- Hosoki K, Kagami M, Tanaka T, Kubota M, Kurosawa K, Kato M, Uetake K, Tohyama J, Ogata T, Saitoh S. Maternal uniparental disomy 14 syndrome demonstrates prader-willi syndrome-like phenotype. J Pediatr. 2009 Dec;155(6):900 - 903.e1. [PubMed: 19800077]
- Höybye C. Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr. 2007;96:410 - 3. [PubMed: 17407467]
- Höybye C, Thoren M, Bohm B. Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader-Willi syndrome. J Intellect Disabil Res. 2005;49:245 - 52. [PubMed: 15816811]
- Iughetti L, Bosio L, Corrias A, Gargantini L, Ragusa L, Livieri C, Predieri B, Bruzzi P, Caselli G, Grugni G. Pituitary height and neuroradiological alterations in patients with Prader-Labhart-Willi syndrome. Eur J Pediatr. 2008;167:701 - 2. [PubMed: 17805568]
- Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of Angelman syndrome. Am J Hum Genet. 1999;65:1 - 6. [PMC free article: PMC1378067] [PubMed: 10364509]
- Kim SJ, Miller JL, Kuipers PJ, German JR, Beaudet AL, Sahoo T, Driscoll DJ. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20:283 - 90. [PMC free article: PMC3283188] [PubMed: 22045295]
- Kokkonen H, Leisti J. An unexpected recurrence of Angelman syndrome suggestive of maternal germ-line mosaicism of del(15)(q11q13) in a Finnish family. Hum Genet. 2000;107:83 - 5. [PubMed: 10982040]
- Kotzot D. Review and meta-analysis of systematic searches for uniparental disomy (UPD) other than UPD 15. Am J Med Genet. 2002;111:366 - 75. [PubMed: 12210294]
- Kozlov SV, Bogenpohl JW, Howell MP, Wevrick R, Panda S, Hogenesch JB, Muglia LJ, Van Gelder RN, Herzog ED, Stewart CL. The imprinted gene Magel2 regulates normal circadian output. Nat Genet. 2007;39:1266 - 72. [PubMed: 17893678]
- Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, Horsthemke B, Beaudet AL, Ledbetter DH. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet. 1996;66:77 - 80. [PubMed: 8957518]
- Kweh FA, Miller JL, Sulsona CR, Wasserfall C, Atkinson M, Shuster JJ, Goldstone AP, Driscoll DJ. Hyperghrelinemia in Prader-Willi syndrome begins in early infancy long before the onset of hyperphagia. Am J Med Genet A. 2015;167A:69 - 79. [PMC free article: PMC4391201] [PubMed: 25355237]
- Layton JB, Meier CR, Sharpless JL, Stürmer T, Jick SS. Brookhart MA1. Comparative Safety of Testosterone Dosage Forms. JAMA Intern Med. 2015;175:1187 - 96. [PMC free article: PMC4494981] [PubMed: 25962056]
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenen SB, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325 - 9. [PubMed: 7442771]
- Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O'Neill MA, Wevrick R. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum Mol Genet. 2005;14:627 - 37. [PubMed: 15649943]
- Liehr T, Brude E, Gillessen-Kaesbach G, König R, Mrasek K, von Eggeling F, Starke H. Prader-Willi syndrome with a karyotype 47,XY,+min(15)(pter->q11.1:) and maternal UPD 15--case report plus review of similar cases. Eur J Med Genet. 2005;48:175 - 81. [PubMed: 16053909]
- Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834 - 45. [PMC free article: PMC1734773] [PubMed: 11748306]
- Lukusa T, Fryns JP. Pure distal monosomy 10q26 in a patient displaying clinical features of Prader-Willi syndrome during infancy and distinct behavioural phenotype in adolescence. Genet Couns. 2000;11:119 - 26. [PubMed: 10893663]
- MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997;6:1873 - 8. [PubMed: 9302265]
- Macedo DB, Abreu AP, Reis AC, Montenegro LR, Dauber A, Beneduzzi D, Cukier P, Silveira LF, Teles MG, Carroll RS, Junior GG, Filho GG, Gucev Z, Arnhold IJ, de Castro M, Moreira AC, Martinelli CE Jr, Hirschhorn JN, Mendonca BB, Brito VN, Antonini SR, Kaiser UB, Latronico AC. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metab. 2014;99:E1097 - 103. [PMC free article: PMC4037732] [PubMed: 24628548]
- Maggouta F, Roberts SE, Dennis NR, Veltman MW, Crolla JA. A supernumerary marker chromosome 15 tetrasomic for the Prader-Willi/Angelman syndrome critical region in a patient with a severe phenotype. J Med Genet. 2003;40:e84. [PMC free article: PMC1735529] [PubMed: 12843333]
- Maillard AM, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S, Dukart J, Ferrari C, Conus P, Männik K, Zazhytska M, Siffredi V, Maeder P, Kutalik Z, Kherif F, Hadjikhani N, Beckmann JS, Reymond A, Draganski B, Jacquemont S. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry. 2015;20:140 - 7. [PMC free article: PMC4320286] [PubMed: 25421402]
- McCandless SE. Clinical report—health supervision for children with Prader-Willi syndrome. Pediatrics. 2011;127:195 - 204. [PubMed: 21187304]
- Miller J, Kranzler J, Liu Y, Schmalfuss I, Theriaque DW, Shuster JJ, Hatfield A, Mueller OT, Goldstone AP, Sahoo T, Beaudet AL, Driscoll DJ. Neurocognitive findings in Prader-Willi syndrome and early-onset morbid obesity. J Pediatr. 2006a;149:192 - 8. [PubMed: 16887432]
- Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. J Clin Endocrinol Metab. 2006b;91:413 - 7. [PubMed: 16317059]
- Miller JL, Angulo M. An open-label pilot study of N-acetylcysteine for skin-picking in Prader-Willi syndrome. Am J Med Genet A. 2014;164A:421 - 4. [PubMed: 24311388]
- Miller JL, Couch JA, Schmalfuss I, He G, Liu Y, Driscoll DJ. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in Prader-Willi syndrome. Am J Med Genet A. 2007;143A:476 - 83. [PubMed: 17103438]
- Miller JL, Goldstone AP, Couch JA, Shuster J, He G, Driscoll DJ, Liu Y, Schmalfuss IM. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am J Med Genet A. 2008;146A:570 - 7. [PubMed: 17431897]
- Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, Dykens E, Butler MG, Shuster JJ, Driscoll DJ. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A. 2011;155A:1040 - 9. [PMC free article: PMC3285445] [PubMed: 21465655]
- Miller SP, Riley P, Shevell MI. The neonatal presentation of Prader-Willi syndrome revisited. J Pediatr. 1999;134:226 - 8. [PubMed: 9931534]
- Milner KM, Craig EE, Thompson RJ, Veltman MW, Thomas NS, Roberts S, Bellamy M, Curran SR, Sporikou CM, Bolton PF. Prader-Willi syndrome: intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry. 2005;46:1089 - 96. [PubMed: 16178933]
- Myers SE, Whitman BY, Carrel AL, Moerchen V, Bekx MT, Allen DB. Two years of growth hormone therapy in young children with Prader-Willi syndrome: physical and neurodevelopmental benefits. Am J Med Genet A. 2007;143A:443 - 8. [PubMed: 17103437]
- Nagai T, Obata K, Ogata T, Murakami N, Katada Y, Yoshino A, Sakazume S, Tomita Y, Sakuta R, Niikawa N. Growth hormone therapy and scoliosis in patients with Prader-Willi syndrome. Am J Med Genet A. 2006;140:1623 - 7. [PubMed: 16770808]
- Nagai T, Obata K, Tonoki H, Temma S, Murakami N, Katada Y, Yoshino A, Sakazume S, Takahashi E, Sakuta R, Niikawa N. Cause of sudden, unexpected death of Prader-Willi syndrome patients with or without growth hormone treatment. Am J Med Genet A. 2005;136:45 - 8. [PubMed: 15937939]
- Nevsimalova S, Vankova J, Stepanova I, Seemanova E, Mignot E, Nishino S. Hypocretin deficiency in Prader-Willi syndrome. Eur J Neurol. 2005;12:70 - 2. [PubMed: 15613151]
- Norton ME, Brar H, Weiss J, Karimi A, Laurent LC, Caughey AB, Rodriguez MH, Williams J 3rd, Mitchell ME, Adair CD, Lee H, Jacobsson B, Tomlinson MW, Oepkes D, Hollemon D, Sparks AB, Oliphant A, Song K. Non-Invasive Chromosomal Evaluation (NICE) Study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207:137.e1 - 8. [PubMed: 22742782]
- Nyunt O, Cotterill AM, Archbold SM, Wu JY, Leong GM, Verge CF, Crock PA, Ambler GR, Hofman P, Harris M. Normal cortisol response on low-dose synacthen (1 microg) test in children with Prader Willi syndrome. J Clin Endocrinol Metab. 2010;95:E464 - 7. [PubMed: 20810574]
- Ohta T, Gray TA, Rogan PK, Buiting K, Gabriel JM, Saitoh S, Muralidhar B, Bilienska B, Krajewska-Walasek M, Driscoll DJ, Horsthemke B, Butler MG, Nicholls RD. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet. 1999;64:397 - 413. [PMC free article: PMC1377750] [PubMed: 9973278]
- Osório J. Growth and development: growth hormone therapy improves cognition in children with Prader-Willi syndrome. Nat Rev Endocrinol. 2012;8:382. [PubMed: 22565027]
- Papenhausen P, Schwartz S, Risheg H, Keitges E, Gadi I, Burnside RD, Jaswaney V, Pappas J, Pasion R, Friedman K, Tepperberg J. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A. 2011;155A:757 - 68. [PubMed: 21594998]
- Pravdivyi I, Ballanyi K, Colmers WF, Wevrick R. Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2-null mouse model of Prader-Willi syndrome. Hum Mol Genet. 2015;24:4276 - 83. [PubMed: 25926624]
- Priano L, Grugni G, Miscio G, Guastamacchia G, Toffolet L, Sartorio A, Mauro A. Sleep cycling alternating pattern (CAP) expression is associated with hypersomnia and GH secretory pattern in Prader-Willi syndrome. Sleep Med. 2006;7:627 - 33. [PubMed: 17023209]
- Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet. 2010 May 11;11:70. [PMC free article: PMC2877670] [PubMed: 20459762]
- Ramachandrappa S, Raimondo A, Cali AM, Keogh JM, Henning E, Saeed S, Thompson A, Garg S, Bochukova EG, Brage S, Trowse V, Wheeler E, Sullivan AE, Dattani M, Clayton PE, Datta V, Bruning JB, Wareham NJ, O'Rahilly S, Peet DJ, Barroso I, Whitelaw ML, Farooqi IS. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. 2013;123:3042 - 50. [PMC free article: PMC3696558] [PubMed: 23778139]
- Richer LP, Shevell MI, Miller SP. Diagnostic profile of neonatal hypotonia: an 11-year study. Pediatr Neurol. 2001;25:32 - 7. [PubMed: 11483393]
- Robinson WP, Dutly F, Nicholls RD, Bernasconi F, Peñaherrera M, Michaelis RC, Abeliovich D, Schinzel AA. The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet. 1998;35:130 - 6. [PMC free article: PMC1051217] [PubMed: 9580159]
- Rocha CF, Paiva CL. Prader-Willi-like phenotypes: a systematic review of their chromosomal abnormalities. Genet Mol Res. 2014;13:2290 - 8. [PubMed: 24737477]
- Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. 2000;44:25 - 30. [PubMed: 10711647]
- Runte M, Hüttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687 - 700. [PubMed: 11726556]
- Runte M, Varon R, Horn D, Horsthemke B, Buiting K. Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader-Willi syndrome. Hum Genet. 2005;116:228 - 30. [PubMed: 15565282]
- Sacco M, Di Giorgio G. Sudden death in Prader-Willi syndrome during growth hormone therapy. Horm Res. 2005;63:29 - 32. [PubMed: 15583479]
- Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719 - 21. [PMC free article: PMC2705197] [PubMed: 18500341]
- Saitoh S, Buiting K, Cassidy SB, Conroy JM, Driscoll DJ, Gabriel JM, Gillessen-Kaesbach G, Glenn CC, Greenswag LR, Horsthemke B, Kondo I, Kuwajima K, Niikawa N, Rogan PK, Schwartz S, Seip J, Williams CA, Nicholls RD. Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am J Med Genet. 1997;68:195 - 206. [PubMed: 9028458]
- Scaroni C, Ceccato F, Rizzati S, Mantero F. Concomitant therapies (glucocorticoids and sex hormones) in adult patients with growth hormone deficiency. J Endocrinol Invest. 2008;31:61 - 5. [PubMed: 19020389]
- Scerif M, Goldstone AP, Korbonits M. Ghrelin in obesity and endocrine diseases. Mol Cell Endocrinol. 2011;340:15 - 25. [PubMed: 21345363]
- Schaaf CP, Gonzalez-Garay ML, Xia F, Potocki L, Gripp KW, Zhang B, Peters BA, McElwain MA, Drmanac R, Beaudet AL, Caskey CT, Yang Y. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat Genet. 2013;45:1405 - 8. [PMC free article: PMC3819162] [PubMed: 24076603]
- Scheimann AO, Butler MG, Miller JL, Lee PD, Stevenson DA, Heinemann J, Driscoll DJ (2012) Letter to the editor: Long-term experience with duodenal switch in adolescents. Obes Surg. 22:517-8. [PubMed: 21874367]
- Schlumpf M, Eiholzer U, Gygax M, Schmid S, van der Sluis I, l'Allemand D. A daily comprehensive muscle training programme increases lean mass and spontaneous activity in children with Prader-Willi syndrome after 6 months. J Pediatr Endocrinol Metab. 2006;19:65 - 74. [PubMed: 16509530]
- Schrander-Stumpel CT, Curfs LM, Sastrowijoto P, Cassidy SB, Schrander JJ, Fryns JP. Prader-Willi syndrome: causes of death in an international series of 27 cases. Am J Med Genet A. 2004;124A:333 - 8. [PubMed: 14735579]
- Schulze A, Mogensen H, Hamborg-Petersen B, Graem N, Ostergaard JR, Brondum-Nielsen K. Fertility in Prader-Willi syndrome: a case report with Angelman syndrome in the offspring. Acta Paediatr. 2001;90:455 - 9. [PubMed: 11332942]
- Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med. 2001;3:206 - 11. [PMC free article: PMC3111049] [PubMed: 11388763]
- Shapira NA, Lessig MC, Lewis MH, Goodman WK, Driscoll DJ. Effects of topiramate in adults with Prader-Willi syndrome. Am J Ment Retard. 2004;109:301 - 9. [PubMed: 15176917]
- Shim JS, Lee SH, Seo SW, Koo KH, Jin DK. The musculoskeletal manifestations of Prader-Willi syndrome. J Pediatr Orthop. 2010;30:390 - 5. [PubMed: 20502241]
- Siemensma EP. Tummers-de Lind van Wijngaarden RF, Festen DA, Troeman ZC, van Alfen-van der Velden AA, Otten BJ, Rotteveel J, Odink RJ, Bindels-de Heus GC, van Leeuwen M, Haring DA, Oostdijk W, Bocca G, Mieke Houdijk EC, van Trotsenburg AS, Hoorweg-Nijman JJ, van Wieringen H, Vreuls RC, Jira PE, Schroor EJ, van Pinxteren-Nagler E, Willem Pilon J, Lunshof LB, Hokken-Koelega AC. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: a randomized controlled trial and longitudinal study. J Clin Endocrinol Metab. 2012;97:2307 - 14. [PubMed: 22508707]
- Sode-Carlsen R, Farholt S, Rabben KF, Bollerslev J, Schreiner T, Jurik AG, Christiansen JS, Höybye C. One year of growth hormone treatment in adults with Prader-Willi syndrome improves body composition: results from a randomized, placebo-controlled study. J Clin Endocrinol Metab. 2010;95:4943 - 50. [PubMed: 20702523]
- Soni S, Whittington J, Holland AJ, Webb T, Maina E, Boer H, Clarke D. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: implications for management and treatment. J Intellect Disabil Res. 2007;51:32 - 42. [PubMed: 17181601]
- Stagi S, Lapi E, Pantaleo M, Chiarelli F, Seminara S, de Martino M. Type II diabetes and impaired glucose tolerance due to severe hyperinsulinism in patients with 1p36 deletion syndrome and a Prader-Willi-like phenotype. BMC Med Genet. 2014;15:16. [PMC free article: PMC3916307] [PubMed: 24479866]
- Steinhausen HC, Eiholzer U, Hauffa BP, Malin Z. Behavioural and emotional disturbances in people with Prader-Willi Syndrome. J Intellect Disabil Res. 2004;48:47 - 52. [PubMed: 14675231]
- Stelzer Y, Sagi I, Yanuka O, Eiges R, Benvenisty N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat Genet. 2014;46:551 - 7. [PubMed: 24816254]
- Stevenson DA, Anaya TM, Clayton-Smith J, Hall BD, Van Allen MI, Zori RT, Zackai EH, Frank G, Clericuzio CL. Unexpected death and critical illness in Prader-Willi syndrome: report of ten individuals. Am J Med Genet A. 2004;124A:158 - 64. [PubMed: 14699614]
- Stevenson DA, Heinemann J, Angulo M, Butler MG, Loker J, Rupe N, Kendell P, Cassidy SB, Scheimann A. Gastric rupture and necrosis in Prader-Willi syndrome. J Pediatr Gastroenterol Nutr. 2007a;45:272 - 4. [PMC free article: PMC3241991] [PubMed: 17667731]
- Stevenson DA, Heinemann J, Angulo M, Butler MG, Loker J, Rupe N, Kendell P, Clericuzio CL, Scheimann AO. Deaths due to choking in Prader-Willi syndrome. Am J Med Genet A. 2007b;143A:484 - 7. [PMC free article: PMC3243066] [PubMed: 17036318]
- Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP. Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2004;89:4162 - 5. [PubMed: 15292365]
- Tauber M, Diene G, Molinas C, Hébert M. Review of 64 cases of death in children with Prader-Willi syndrome (PWS). Am J Med Genet A. 2008;146A:881 - 7. [PubMed: 18324685]
- Torrado M, Araoz V, Baialardo E, Abraldes K, Mazza C, Krochik G, Ozuna B, Leske V, Caino S, Fano V, Chertkoff L. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A. 2007;143A:460 - 8. [PubMed: 17163531]
- Tsuyusaki Y, Yoshihashi H, Furuya N, Adachi M, Osaka H, Yamamoto K, Kurosawa K. 1p36 deletion syndrome associated with Prader-Willi-like phenotype. Pediatr Int. 2010;52:547 - 50. [PubMed: 20113418]
- Varela MC, Kok F, Setian N, Kim CA, Koiffmann CP. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clin Genet. 2005;67:47 - 52. [PubMed: 15617548]
- Veltman MW, Craig EE, Bolton PF. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005;15:243 - 54. [PubMed: 16314754]
- Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF. Prader-Willi syndrome--a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry. 2004;13:42 - 50. [PubMed: 14991431]
- Vogels A, De Hert M, Descheemaeker MJ, Govers V, Devriendt K, Legius E, Prinzie P, Fryns JP. Psychotic disorders in Prader-Willi syndrome. Am J Med Genet A. 2004;127A:238 - 43. [PubMed: 15150773]
- West LA, Ballock RT. High incidence of hip dysplasia but not slipped capital femoral epiphysis in patients with Prader-Willi syndrome. J Pediatr Orthop. 2004;24:565 - 7. [PubMed: 15308908]
- Wey E, Bartholdi D, Riegel M, Nazlican H, Horsthemke B, Schinzel A, Baumer A. Mosaic imprinting defect in a patient with an almost typical expression of the Prader-Willi syndrome. Eur J Hum Genet. 2005;13:273 - 7. [PubMed: 15578038]
- Whittington JE, Holland AJ, Webb T. Ageing in people with Prader-Willi syndrome: mortality in the UK population cohort and morbidity in an older sample of adults. Psychol Med. 2015;45:615 - 21. [PubMed: 25088280]
- Whittington JE, Holland AJ, Webb T, Butler J, Webb T, Clarke D. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet. 2001;38:792 - 8. [PMC free article: PMC1734766] [PubMed: 11732491]
- Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H. Academic underachievement by people with Prader-Willi syndrome. J Intellect Disabil Res. 2004a;48:188 - 200. [PubMed: 14723660]
- Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H. Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. J Intellect Disabil Res. 2004b;48:172 - 87. [PubMed: 14723659]
- Wigren M, Hansen S. ADHD symptoms and insistence on sameness in Prader-Willi syndrome. J Intellect Disabil Res. 2005;49:449 - 56. [PubMed: 15882394]
- Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12:385 - 95. [PubMed: 20445456]
- Wolfgram PM, Carrel AL, Allen DB. Long-term effects of recombinant human growth hormone therapy in children with Prader-Willi syndrome. Curr Opin Pediatr. 2013;25:509 - 14. [PMC free article: PMC4396180] [PubMed: 23782572]
- Yang L, Zhan GD, Ding JJ, Wang HJ, Ma D, Huang GY, Zhou WH. Psychiatric illness and intellectual disability in the Prader-Willi syndrome with different molecular defects—a meta analysis. PLoS One. 2013;8:e72640. [PMC free article: PMC3743792] [PubMed: 23967326]
Chapter Notes
Revision History
- 4 February 2016 (ha) Comprehensive update posted live
- 23 January 2014 (cd) Revision: additional information about MAGEL2
- 11 October 2012 (me) Comprehensive update posted to live Web site
- 3 September 2009 (cd) Revision: added information about (i) case report of adrenocortical insufficiency in an individual with PWS and (ii) snoRNA HBII-85, which has been implicated as a cause of PWS
- 24 March 2008 (me) Comprehensive update posted to live Web site
- 12 July 2006 (sc) Revision: clarification of availability/reliability of 甲基化分析 done on CVS
- 16 June 2005 (me) Comprehensive update posted to live Web site
- 8 April 2004 (cd) Revision: quantitative PCR clinically available
- 1 May 2003 (me) Comprehensive update posted to live Web site
- 13 November 2000 (me) Comprehensive update posted to live Web site
- 6 October 1998 (pb) Review posted to live Web site
- Spring 1997 (sc) Original submission