
概述
临床特点. TUBB4A相关脑白质营养不良是一组表型谱系疾病,其MRI最严重可表现为伴基底节和小脑萎缩的髓鞘形成低下(hypomyelination with atrophy of the basal ganglia and cerebellum,H-ABC),最轻可为孤立的髓鞘形成低下。 逐渐出现的神经系统表现累及锥体束(痉挛状态、深腱反射活跃和Babinski征阳性)、锥体外系(僵硬、肌张力障碍、舞蹈手足徐动症、动眼危象和口 周运动障碍) 、小脑(共济失调、意向性震颤、辨距不良)和 延髓功能(构音障碍、发音困难和吞咽功能)均受累。 认知受到不同程度的损害,通常比运动功能 受累程度轻。 通常情况下,MRI表现为H-ABC的患者在儿童早期(一岁至三岁)发病,MRI表现为孤立性髓鞘形成低下的患者在儿童晚期或成年后发病。 病情进展速度随疾病严重程度而变化。
管理. 对症治疗:对导致功能受限的痉挛进行医疗管理和物理治疗;对肌张力障碍进行医疗管理,并且-当医疗管理效果不佳时-可能需要手术干预。吞咽功能障碍的患者可能需要使用胃造瘘管进行喂养。按常规方式治疗癫痫、便秘和胃食管反流病。
预防继发性并发症:补充钙和维生素D预防骨质疏松;注意行动减少患者的皮肤护理,并经常更换体位,防止出现褥疮;每年进行流感疫苗接种;使用常规跌倒预防策略、自适应设备(如轮椅和助步器)和物理治疗来帮助预防继发性损伤。
监测:常规评估(1)吞咽和喂养,以减少误吸风险;(2)营养,预防营养不良;(3)矫形和关节完整性,以检测关节脱位和脊柱侧凸。 至少每年进行一次:(1)医学评估,以评估体重和药物;(2)职业治疗、物理治疗、言语治疗、康复医学等方面的专家评估。 每年进行神经学评估以检测出现的并发症。
遗传咨询. TUBB4A相关脑白质营养不良为常染色体显性遗传疾病。 大部分受累的个体患病是由de novo致病性变异引起的。若先证者的父母无临床症状,那么先证者同胞受累的风险低,但仍大于一般人群,因为先证者的父母一方可能存在胚系嵌合或体细胞和胚系嵌合。 尚无TUBB4A相关脑白质营养不良患者生育的报道。
视野
诊断
提示性表现
当患者具有以下临床和脑MRI表现时,应考虑TUBB4A相关脑白质营养不良,这些表现描述了两种髓鞘形成低下表型。
临床表现
- 婴儿或儿童期发病
- 运动发育迟缓
- 存在锥体征和锥体外系体征
- 步态共济失调和小脑功能障碍
- 构音障碍、失音或“耳语”发音困难
脑MRI表现
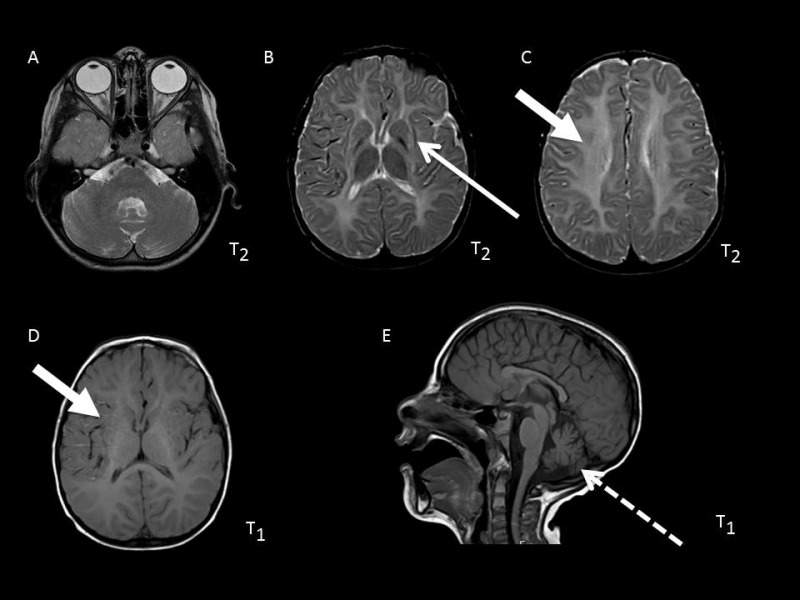
- 伴基底节、小脑萎缩的髓鞘形成低下(H-ABC) [van der Knaap et al 2002、Sasaki et al 2009、Blumkin et al 2014、Ferreira et al 2014、Hamilton et al 2014、Sagnelli et al 2016、Tonduti et al 2016]的特点为(图 1):
- 基底节进行性萎缩,以新纹状体(即壳核和尾状核)为主,常伴随着壳核体积的明显减小(随病情进展可消失),尾状核头部受累程度较轻。丘脑和苍白球通常不会受累。 需注意的是,尽管许多H-ABC表型患者到2岁时壳核的变化已很明显,但在一些儿童中这种变化可能直到儿童晚期才出现。
- 大脑白质区域,包括幕上白质、胼胝体和内囊在内,出现弥漫性髓鞘形成低下,表现为T2加权像轻度高信号、T1加权像典型的等信号或轻度高信号
- 与灰质结构相比,小脑白质T1加权像呈等信号或轻度高信号或低信号。蚓部受累为主的小脑萎缩是H-ABC常见的、但并非必不可少的临床特征。
建立诊断
当先证者具有特征性临床和MRI表现以及分子遗传学检测(见表 1)发现杂合TUBB4A 致病性变异,则可明确TUBB4A相关脑白质营养不良的诊断。
根据患者表型,可选择的分子遗传学检测方法包括基因靶向检测(多基因包或单基因检测)和基因组检测(全基因组测序)。
基因靶向检测需要临床医生确定可能涉及哪些基因,而基因组检测则不需要。 由于TUBB4A相关脑白质营养不良的表型较为宽泛,因此具有提示性表现中所描述的特征性表现的儿童可能通过靶向基因检测(见选项 1)而获得诊断,而那些表型轻微、难以与许多其他遗传性髓鞘形成低下性脑白质营养不良相鉴别的儿童更可能通过基因组检测而明确诊断(见选项 2)。
临床特征
临床描述
TUBB4A相关脑白质营养不良通常在1岁至3岁之间起病;表型严重的患者可在几个月龄时起病[van der Knaap et al 2002],而一些孤立的髓鞘形成低下患者可在儿童晚期或成年后起病[Hamilton et al 2014、Pizzino et al 2014、Shimojima et al 2015]。
本病为进展性病程;进展速度随疾病严重程度而变化。男性和女性同等受累。
临床表现包括以下内容。
运动发育迟缓. 有些儿童在一段正常的运动发育期后出现倒退[van der Knaap et al 2002]。
认知功能障碍. 认知受到不同程度的影响,但通常不如运动功能受累严重。学习困难较常见;社会意识通常不受累。
锥体束受累. 通常在儿童早期出现双侧或单侧上运动神经元功能障碍(痉挛状态、深腱反射活跃和 Babinski征阳性)[Mercimek-Mahmutoglu et al 2005, Wakusawa et al 2006]。
值得注意的是,一些存在杂合TUBB4A致病性变异和痉挛性截瘫的患者的脑MRI上显示轻度白质改变[Kancheva et al 2015],这扩大了TUBB4A相关脑白质营养不良的表型谱。但是迄今为止,对未分类的遗传性痉挛性截瘫患者进行分子遗传学检测尚未普遍检测出杂合TUBB4A致病性变异[Kumar et al 2015]。
锥体外系受累是由新纹状体受累引起的,包括僵硬、肌张力障碍、舞蹈手足徐动症、动眼危象和口周运动障碍。锥体外系特征 - 特别是偏身肌张力障碍 - 可能是本病的首发表现。锥体外系特征可能因体位变化或视觉和听觉刺激而加重。
步态功能障碍. 虽然部分患者可获得独立行走的能力,但许多患者从不来不能独走。步态不稳和跌倒较常见。
小脑征包括共济失调、意向性震颤、辨距不良和眼球震颤。
构音障碍、发音困难和吞咽功能障碍. 随着病情进展,患者逐渐出现交流和喂养困难,许多患者需要胃造瘘管进行喂养。
其他不太常见的表现(在病情严重者中出现)包括以下方面[van der Knaap et al 2002、Sasaki et al 2009、Simons et al 2013、Ferreira et al 2014、Hamilton et al 2014]:
小头
矮小
癫痫
视力差
听力丧失
由于运动功能障碍和体位不当导致的脊柱侧凸和关节脱位
低促性腺激素性性腺功能减退症(在1个患者中出现)[Tonduti et al 2016]
I
TUBB4A致病性变异嵌合体者. 值得注意的是,少数TUBB4A相关髓鞘形成低下患者的无症状父母是TUBB4A致病变异的嵌合体[Simons et al 2013]。
神经生理学研究
脑电图(EEG)通常正常或表现出慢背景波。
肌电图(EMG)和神经传导研究(NCS)正常。
脑干诱发电位(BSEP)通常延迟。
视觉诱发电位(VEP)通常正常。
体感诱发电位(SEP)在某些患者中存在传导延迟[van der Knaap et al 2002]。
实验室结果. 脑脊液(CSF)分析通常正常[van der Knaap et al 2002]。值得注意的是,在一些患者中可观察到CSF中5-甲基四氢叶酸降低,同时伴血浆中叶酸正常、CSF中5-MTHF还原酶正常或CSF中高香草酸降低[Tomás-Vila et al 2014, Tonduti et al 2016];这些异常并未被广泛描述[Mercimek-Mahmutoglu & Stockler-Ipsiroglu 2007],因此认为它们不是原发性代谢缺陷。
基因型-表型关联
c.745G>A. 对具有经典H-ABC MRI表现的患者进行初始研究发现,所有患者都有这个TUBB4A致病性变异[Simons et al 2013]。此后发现具有经典H-ABC表型(表 2)的患者不只存在这一种变异,也有其他致病性变异[Hamilton et al 2015]。
c.785G>A和c.1228G>A. 在一些不伴基底节萎缩的髓鞘形成低下患者中发现了这些变异[Ferreira et al 2014, Hamilton et al 2014, Miyatake et al 2014]。
患病率
本病确切的患病率尚未知;迄今为止已有71个受累个体被报道。
遗传相关(等位基因)疾病
除了本GeneReview中描述的杂合TUBB4A致病性变异引起的脑白质营养不良,TUBB4A致病性变异c.4C>G还可引起成年发病的喉发音困难或耳语发音困难(也称为DYT4肌张力障碍),该病脑MRI正常。受累个体在病程后期会出现全身性肌张力障碍和共济失调步态[Hersheson et al 2013]。有时会出现吞咽困难。
尽管Erro等人认为两种TUBB4A相关脑白质营养不良表型(H-ABC和孤立的髓鞘形成低下)和DYT4代表了TUBB4A相关疾病谱,但与脑白质营养不良不同的是,DYT4肌张力障碍有典型的常染色体显性遗传阳性家族史[Erro et al 2015a、Erro et al 2015b]。
鉴别诊断
在鉴别诊断时应考虑到儿童早期发病、和/或锥体外系征的髓鞘形成低下性脑白质营养不良。见脑白质营养不良概述。
佩梅病(PMD)是由PLP1基因内致病性变异或大片段的PLP1缺失/重复引起的X连锁疾病。它通常在婴儿期或儿童早期起病,表现为眼球震颤、上运动神经元功能障碍、步态共济失调和锥体束征。脑MRI显示弥漫性髓鞘形成低下,但没有TUBB4A相关脑白质营养不良-伴基底节、小脑萎缩的髓鞘形成低下(H-ABC)经典的小脑和基底节萎缩。
佩梅样病(PMLD1)(OMIM 608804)是由GJC2双等位基因致病性变异引起的常染色体隐性遗传疾病。PMLD1通常在儿童早期出现类似于H-ABC表型的症状,包括:发育迟缓、言语延迟、锥体和锥体外系受累、小脑征和智力完好。与PMD患者很相似,受累的儿童在疾病早期出现眼震[Uhlenberg et al 2004],尽管TUBB4A相关脑白质营养不良也有眼震的描述,但并不典型。
Pol III相关脑白质营养不良是由三个基因(POLR3A、POLR3B或POLR1C)中的某一个基因的双等位基因致病性变异引起的常染色体隐性遗传疾病。临床表现与TUBB4A相关脑白质营养不良类似,包括痉挛、步态共济失调、锥体外系运动障碍和小脑征。Pol III相关脑白质营养不良的其他表现包括异常牙列和低促性腺激素性性腺功能减退,TUBB4A相关脑白质营养不良通常没有这些症状。
SOX10相关脑白质营养不良/周围和中枢脱髓鞘、Waardenburg综合征和先天性巨结肠症(PCWH)(OMIM 609136)是由SOX10杂合致病性变异引起的常染色体显性遗传疾病。临床表现与TUBB4A相关脑白质营养不良有重叠,包括发育迟缓、痉挛、步态共济失调和锥体外系运动障碍。 PCWH的其他表现包括:
周围神经系统受累(感觉丧失)
游离唾液酸贮积障碍是由SLC17A5双等位基因致病性变异引起的常染色体隐性遗传性神经退行性疾病,SLC17A5变异导致唾液酸储存和运输缺陷。 最严重的类型(婴儿型游离唾液酸贮积病)临床表现包括粗糙的面部特征和非神经系统症状,如肝脾肿大和心脏肥大,较轻的类型(Salla疾病)临床表现与H-ABC类似,表现为伴痉挛、锥体外系运动障碍和癫痫发作的进行性神经系统恶化。
管理
初诊后评估
为明确TUBB4A相关脑白质营养不良患者的疾病严重程度和治疗需求,建议进行以下评估[Van Haren et al 2015]:
由儿科神经学家评估是否有发育迟缓、痉挛和锥体外系运动障碍表现
评估发育里程碑和认知功能
物理治疗师评估功能障碍和设备需求
评估言语(交流)和喂养(吞咽)
听力评估
骨科评估是否有脊柱侧弯和/或关节畸形的表现,特别是有严重肌张力障碍的患者
向临床遗传学家和/或遗传咨询师进行咨询
对症治疗
虽然TUBB4A相关脑白质营养没有治愈性治疗方法,但可以通过以下方式改善患者生活质量。
影响功能的痉挛会导致关节挛缩和脊柱侧弯;两者都需要物理治疗(拉伸和定位)和医疗管理。 可以口服GABA激动剂如巴氯芬和地西泮。在某些情况下,可考虑使用鞘内巴氯芬泵。 肌内注射肉毒杆菌毒素可能对局灶性痉挛有帮助。
肌张力障碍可以用以下方式进行管理:
当肌张力障碍与痉挛有关时,可使用巴氯芬或肌内注射肉毒杆菌毒素;
苯海索或丁苯那嗪;
当通过医学管理难以治疗肌张力障碍时,可考虑使用巴氯芬泵。 值得注意的是,迄今为止尚未在TUBB4A相关脑白质营养不良中进行深部脑刺激研究。
吞咽功能障碍患者可能需要胃造瘘管进行喂养,以减少误吸的风险。
构音障碍患者可能需要增强的交流工具。
有癫痫发作时应使用抗痫药物。
便秘通常是神经功能障碍和肠道蠕动差引起的,可以通过饮食、泻药和大便软化剂治疗。
胃食管反流病较常见,在疼痛评估时应考虑到这一点。
通过使用助步器或轮式移动设备和其他必要的设备提高患者的实践能力。
经常需要学校的安排,如个性化的教育计划。有了这样的安排,很多经典型H-ABC表型的孩子许多年都表现得和同年级水平一样或接近同年级水平,尽管后期可能会出现认知能力下降。
家庭支持和倡导团体可以为受累的个体提供必要的社会心理支持。
继发性并发症的预防
以下建议-基于共识-是针对所有脑白质营养不良患者制定的[Van Haren et al 2015]。
补充钙和维生素D预防骨质疏松
皮肤护理并经常更换体位,以帮助预防行动减少患者出现褥疮
每年进行流感疫苗接种
防摔策略、自适应设备(如轮椅和助步器)和物理治疗(增加力量),以帮助预防继发性损伤
监测
适当的监测内容如下:
常规评估患者的吞咽、喂养和营养,以减少误吸风险、预防营养不良
至少每年进行1次:
医疗评估,包括体格检查,以评估体重和药物
职业治疗、物理治疗、言语治疗和康复医学等方面专家的评估
骨科医生评估脊柱侧凸和关节脱位
每年进行一次神经系统估评估,评估患者的症状和任何新出现的并发症
仍在研究阶段的治疗方法
搜索ClinicalTrials.gov获取多种疾病和病症的临床研究信息。
Genetic Counseling遗传咨询
遗传咨询是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识、符合个人情况的决定。 以下几部分内容涉及遗传风险评估和根据家族史和遗传学检测来确定家庭成员的遗传状态。本部分内容的目的并不是为了解决患者可能面临的所有个人、文化或伦理问题,或者替代遗传学专业人员的咨询工作。—ED
家庭成员风险
先证者的父母
先证者的同胞
先证者的子代. TUBB4A相关脑白质营养不良患者的每个孩子都有50%的概率遗传TUBB4A致病性变异;但是,尚无TUBB4A相关脑白质营养不良患者生育的报道。
其他家庭成员. 鉴于迄今为止报道的大多数TUBB4A相关脑白质营养不良先证者患病均是由de novo TUBB4A致病性变异引起的,推测其他家庭成员的患病风险低。
遗传咨询相关问题
家庭计划
最好在怀孕前明确遗传风险并讨论产前检测的适用性。
DNA存储即储存DNA(通常从白细胞中提取)以备将来使用。 检测方法以及我们对基因、等位基因变异和疾病的理解在未来都可能会有所改善,因此受累的个体应考虑储存DNA。
资源
GeneReviews工作人员选择了以下对患者及其家庭有帮助的疾病特异性和/或综合支持组织和/或注册机构。GeneReviews不对其他组织提供的信息负责。关于选择标准的信息,点击这里。
- 澳大利亚脑白质营养不良支持组织Nerve Centre54 Railway RoadBlackburn Victoria 3130AustraliaPhone电话: 1800-141-400 (toll free); +61 3 98452831Fax传真: +61 3 95834379Email邮箱: mail@alds.org.au
- 欧洲脑白质营养不良协会(ELA)2, rue Mi-les-VignesB.P. 61024Laxou Cedex 54521FrancePhone电话: 03833093 34Fax传真: 03833000 68Email邮箱: ela@ela-asso.com
- 美国脑白质营养不良基金会(ULF)224 North Second StreetSuite 2DeKalb IL 60115Phone电话: 800-728-5483 (toll-free); 815-748-3211Fax传真: 815-748-0844Email邮箱: office@ulf.org
- 髓鞘疾病生物治疗项目Email邮箱: myelindisorders@cnmc.org
分子遗传学
分子遗传学部分和OMIM表内的信息可能与GeneReview中的其他部分内容不同:表格内可能包含更多最近的信息。—ED
表 A.
TUBB4A相关脑白质营养不良:基因和数据库
表 B.
OMIM中TUBB4A相关脑白质营养不良条目(查看OMIM中的全部内容)
基因结构. 可变剪接导致存在多个转录本,编码不同的异型体(见表 A,基因的详细信息)。转录本NM_006087.3 长2583 bp,包含4个外显子 [Lee et al 1984]。
致病性变异. 见表 2 和表 3 (pdf)致病性变异列表。
c.745G>A是最常见的TUBB4A致病性变异;它始终与经典表型伴基底节、小脑萎缩的髓鞘形成低下(H-ABC)相关联。
c.4C>G变异与脑白质营养不良无关,但可引起DYT4(“遗传性耳语发音障碍”)[Hersheson et al 2013]。
表 2.
在此GeneReview中讨论的TUBB4A致病性变异
DNA核苷酸改变 | 预测的蛋白质改变 | 参考序列 |
|---|---|---|
| c.4C>G 1 | p.Arg2Gly | NM_006087-.3 NP_006078-.2 |
| c.730G>A 2, 3 | p.Gly244Ser | |
| c.745G>A 2, 3, 4, 5, 6 | p.Asp249Asn | |
| c.785G>A 2, 3, 4 | p.Arg262His | |
| c.1228G>A 2, 4 | p.Glu410Lys |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员未独立验证变异的分类。
关于变异命名的注释:GeneReviews使用的是人类基因突变协会(varnomen-.hgvs.org)标准命名方法。关于命名的解释见快速参考。
1.
2.
3.
4.
5.
6.
要查看其他变异的信息,见 表 3(pdf)。
正常基因产物. 转录本NM_006087.3编码含有444个氨基酸的β-微管蛋白异构体。β-微管蛋白可以与α-微管蛋白结合形成异二聚体,形成组装微管的共聚物。微管是细胞骨架的重要组成部分,在许多细胞过程中起重要作用,如有丝分裂、运动和运输。
TUBB4A主要在CNS,特别是在小脑、壳核和白质中表达[Hersheson et al 2013, Nogales 2001]。
异常基因产物.TUBB4A致病性变异导致β-微管蛋白结构变化,研究者认为此变化影响微管的聚合或稳定性[Savage et al 1994]。
参考资料
引用文献
- Blumkin L, Halevy A, Ben-Ami-Raichman D, Dahari D, Haviv A, Sarit C, Lev D, van der Knaap MS, Lerman-Sagie T, Leshinsky-Silver E. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics. 2014;15:107-13. [PubMed: 24526230]
- Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81:1169-85. [PMC free article: PMC2276340] [PubMed: 17999358]
- Carvalho D, Santos S, Martins B, Marques FP. TUBB4A novel mutation reinforces the genotype-phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum. Brain. 2015;138:e327. [PubMed: 25168210]
- Erro R, Hersheson J, Ganos C, Mencacci NE, Stamelou M, Batla A, Thust SC, Bras JM, Guerreiro RJ, Hardy J, Quinn NP, Houlden H, Bhatia KP. H-ABC syndrome and DYT4: Variable expressivity or pleiotropy of TUBB4 mutations? Mov Disord. 2015a;30:828-33. [PubMed: 25545912]
- Erro R, Hersheson J, Houlden H, Bhatia KP. A novel TUBB4A mutation suggests that genotype-phenotype correlation of H-ABC syndrome needs to be revisited. Brain. 2015b;138:e370. [PubMed: 25614026]
- Ferreira C, Poretti A, Cohen J, Hamosh A, Naidu S. Novel TUBB4A mutations and expansion of the neuroimaging phenotype of hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Am J Med Genet A. 2014;164A:1802-7. [PubMed: 24706558]
- Hamilton EM, Polder E, Vanderver A, Naidu S, Schiffmann R, Fisher K, Raguž AB, Blumkin L. H-ABC Research Group, van Berkel CG, Waisfisz Q, Simons C, Taft RJ, Abbink TE, Wolf NI, van der Knaap MS. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain. 2014;137:1921-30. [PMC free article: PMC4345790] [PubMed: 24785942]
- Hamilton EM, Wolf NI, van der Knaap MS. Reply: A novel TUBB4A mutation suggests that genotype-phenotype correlation of H-ABC syndrome needs to be revisited. Brain. 2015;138:e371. [PubMed: 25619510]
- Hersheson J, Mencacci NE, Davis M, MacDonald N, Trabzuni D, Ryten M, Pittman A, Paudel R, Kara E, Fawcett K, Plagnol V, Bhatia KP, Medlar AJ, Stanescu HC, Hardy J, Kleta R, Wood NW, Houlden H. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol. 2013;73:546-53. [PMC free article: PMC3698699] [PubMed: 23424103]
- Inoue K, Shilo K, Boerkoel CF, Crowe C, Sawady J, Lupski JR, Agamanolis DP. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation. Ann Neurol. 2002;52:836-42. [PubMed: 12447940]
- Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L, Tournev I, Jordanova A. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov Disord. 2015;30:854-8. [PubMed: 25772097]
- Kumar KR, Vulinovic F, Lohmann K, Park JS, Schaake S, Sue CM, Klein C. Mutations in TUBB4A and spastic paraplegia. Mov Disord. 2015;30:1857-8. [PubMed: 26477786]
- Lee MG, Loomis C, Cowan NJ. Sequence of an expressed human beta-tubulin gene containing ten Alu family members. Nucleic Acids Res. 1984;12:5823-36. [PMC free article: PMC320034] [PubMed: 6462917]
- Mercimek-Mahmutoglu S, Stockler-Ipsiroglu S. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome. Tohoku J Exp Med. 2007;211:95-6. [PubMed: 17202777]
- Mercimek-Mahmutoglu S, van der Knaap MS, Baric I, Prayer D, Stoeckler-Ipsiroglu S. Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Report of a new case. Neuropediatrics. 2005;36:223-6. [PubMed: 15944912]
- Miyatake S, Osaka H, Shiina M, Sasaki M, Takanashi J, Haginoya K, Wada T, Morimoto M, Ando N, Ikuta Y, Nakashima M, Tsurusaki Y, Miyake N, Ogata K, Matsumoto N, Saitsu H. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology. 2014;82:2230-7. [PubMed: 24850488]
- Nogales E. Structural insight into microtubule function. Annu Rev Biophys Biomol Struct. 2001;30:397-420. [PubMed: 11441808]
- Pizzino A, Pierson TM, Guo Y, Helman G, Fortini S, Guerrero K, Saitta S, Murphy JL, Padiath Q, Xie Y, Hakonarson H, Xu X, Funari T, Fox M, Taft RJ, van der Knaap MS, Bernard G, Schiffmann R, Simons C, Vanderver A. TUBB4A de novo mutations cause isolated hypomyelination. Neurology. 2014;83:898-902. [PMC free article: PMC4153852] [PubMed: 25085639]
- Purnell SM, Bleyl SB, Bonkowsky JL. Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy. Pediatr Neurol. 2014;50:608-11. [PMC free article: PMC4029864] [PubMed: 24742798]
- Sagnelli A, Magri S, Farina L, Chiapparini L, Marotta G, Tonduti D, Consonni M, Scigliuolo GM, Benti R, Pareyson D, Taroni F, Salsano E, Di Bella D. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and FDG-PET findings. J Neurol. 2016;263:591-3. [PubMed: 26810722]
- Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev. 2009;31:582-7. [PubMed: 18851904]
- Savage C, Xue Y, Mitani S, Hall D, Zakhary R, Chalfie M. Mutations in the Caenorhabditis elegans beta-tubulin gene mec-7: effects on microtubule assembly and stability and on tubulin autoregulation. J Cell Sci. 1994;107:2165-75. [PubMed: 7983175]
- Shimojima K, Okumura A, Ikeno M, Nishimura A, Saito A, Saitsu H, Matsumoto N, Yamamoto T. A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease. Brain Dev. 2015;37:281-5. [PubMed: 24974158]
- Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, Tonduti D, Schmidt JL, Chudnow RS, van Coster R, Lagae L, Kisler J, Sperner J, van der Knaap MS, Schiffmann R, Taft RJ, Vanderver A. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013;92:767-73. [PMC free article: PMC3644625] [PubMed: 23582646]
- Tomás-Vila M, Menor F, Ley-Martos M, Jumillas-Luján MJ, Marco-Hernández AV, Barbero P. Hypomyelination with atrophy of the basal ganglia and cerebellum. Contribution of two new cases to a recently reported entity. Rev Neurol. 2014;58:161-5. [PubMed: 24504878]
- Tonduti D, Aiello C, Renaldo F, Dorboz I, Saaman S, Rodriguez D, Fettah H, Elmaleh M, Biancheri R, Barresi S, Boccone L, Orcesi S, Pichiecchio A, Zangaglia R, Maurey H, Rossi A, Boespflug-Tanguy O, Bertini E. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur J Paediatr Neurol. 2016;20:323-30. [PubMed: 26643067]
- Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloğlu H, Nürnberg P, Hübner C, Weschke B, Gärtner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251-60. [PMC free article: PMC1216059] [PubMed: 15192806]
- van der Knaap MS, Naidu S, Pouwels PJ, Bonavita S, van Coster R, Lagae L, Sperner J, Surtees R, Schiffmann R, Valk J. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol. 2002;23:1466-74. [PubMed: 12372733]
- Van Haren K, Bonkowsky JL, Bernard G, Murphy JL, Pizzino A, Helman G, Suhr D, Waggoner J, Hobson D, Vanderver A, Patterson MC. GLIA Consortium. Consensus statement on preventive and symptomatic care of leukodystrophy patients. Mol Genet Metab. 2015;114:516-26. [PubMed: 25577286]
- Wakusawa K, Haginoya K, Kitamura T, Togashi N, Ishitobi M, Yokoyama H, Higano S, Onuma A, Nara T, Iinuma K. Effective treatment with levodopa and carbidopa for hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J Exp Med. 2006;209:163-7. [PubMed: 16707859]
章节注释
修订记录
2016年11月3日(bp)审阅稿在线发布
2016年1月2日(av)提交原文