
摘要
临床特征.
经典的Joubert综合征(JS)主要有三个特点:
- 一种独特的小脑和脑干畸形称为臼齿征(MTS)
- 肌张力低下
- 发育迟缓
这些发现通常伴有偶发性呼吸急促或呼吸暂停和/或非典型眼球运动。一般而言,呼吸异常随着年龄的增长而改善,躯干共济失调随着时间的推移而发展,运动技能的获得被延迟。认知能力是可变的,从严重的智力残疾到正常。其他发现可包括视网膜营养不良,肾脏疾病,眼部缺损,枕部脑膨出,肝纤维化,多指畸形,口腔错构瘤和内分泌异常。家庭内变异和家庭间变异均可见。
诊断/检测.
JS的临床诊断基于临床特征和MRI表现。迄今为止,已知34种基因中的致病变异会引起JS; 其中33个是常染色体,1个是X连锁的。通过检测33个常染色体隐性遗传JS相关基因之一中的双等位致病变异或一个X连锁JS相关基因中的杂合致病变异,可以在约62%-94%的临床诊断为JS的个体中建立JS的分子诊断。
治疗.
对症治疗: 婴儿和呼吸异常的儿童可能需要刺激性药物(如咖啡因); 补充氧气; 机械支撑; 或极少数情况下的气管造口术。其他干预措施可能包括口部运动障碍的言语治疗; 职业和物理治疗; 教育支持,包括视障者特别节目; 和胃造口管喂食。多指和有症状的上睑下垂和/或斜视可能需要手术。肾衰竭,终末期肾病,肝衰竭和/或纤维化均采用标准治疗方法。
监测: 年度评估生长,视力和肝肾功能; 定期神经心理学和发育测试。
应避免的药物/状况: 肾毒性药物,如肾功能不全患者的非甾体类抗炎药; 肝功能损害患者的肝毒性药物。
遗传咨询.
JS主要以常染色体隐性方式遗传。由OFD1中的致病变异引起的JS 以X连锁方式遗传。双基因遗传已被报道。
对于常染色体隐性遗传:在怀孕时,患者的每个同胞有25%的机会是患者,50%的几率是无症状的携带者,25%的机会不是患者且不是携带者。一旦在患者的家庭成员中确定了致病变异,就可以对高危家庭成员进行携带者检测,对高危妊娠进行产前检测,以及胚胎植入前遗传学诊断。对于已知患JS风险增加的妊娠,通过超声检查进行产前诊断(有或没有胎儿MRI)是成功的。
诊断
Joubert综合征(JS)的诊断标准仍在不断改进,但大多数作者一致认为神经放射学发现臼齿征是必须的 [Valente et al 2008, Parisi 2009, Brancati et al 2010].
Joubert综合征的诊断基于以下三个主要标准:
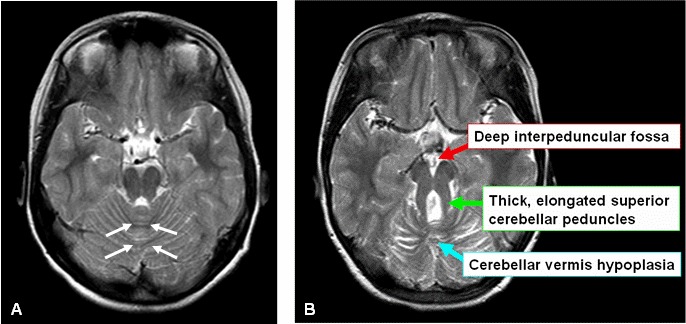
- 臼齿征。通过中脑和脑桥(峡部区域)的交界处的横断面中小脑蚓部发育不全和伴随脑干异常的MRI表现) [Maria et al 1997, Maria et al 1999b, Quisling et al 1999]。臼齿征包括异常深的大脑脚间窝; 突出的,直的,加厚的小脑上脚; 小脑蚓部和中线部分的发育不全(图1A,1B) [Maria et al 1999b]。建议使用高质量的MRI(厚度为3毫米)从中脑到脑桥穿过后颅窝,以及标准的横断,冠状和矢状切面。
- 婴儿期肌张力减退,随后发展为共济失调
- 发育迟缓/智力障碍
JS患者中经常发现的其他特征:
- 呼吸模式异常(交替呼吸急促和/或呼吸暂停)
- 异常的眼球运动,通常是动眼神经失用或视线追踪困难以及凝视和跟踪的急动 [Saraiva & Baraitser 1992, Steinlin et al 1997, Maria et al 1999b, Tusa & Hove 1999]
少于一半的JS患者中可能发生的其他症状包括视网膜营养不良,肾病,眼部缺损,枕部脑膨出,肝纤维化,多指畸形,口腔错构瘤和其他异常。术语“经典”或“纯粹的”JS已被用于指代JS而没有这些其他发现。然而,实际上在婴儿期或幼儿期被诊断患有经典JS的患者中有相当大比例的个体可能随着时间的推移表现出一种或多种这些症状。
建立诊断
JS的临床诊断是基于临床特征和MRI表现。迄今为止,已知34种基因中的致病变异会导致JS; 其中33个是常染色体,1个是X连锁的。通过鉴定33个常染色体隐性遗传JS相关基因之一中的双等位致病变异或一个X连锁JS相关基因中的杂合致病变异,可以在约62%-94%的临床诊断为JS的个体中建立JS的分子诊断。连接JS相关基因 [Bachmann-Gagescu et al 2015a] (参见 表 1a and 1b)。
分子遗传检测方法可以包括基因靶向检测(多个基因)和基因组检测(全基因组测序)的组合。基因靶向检测要求临床医生确定可能涉及哪些基因,而基因组检测则不需要。由于JS存在广泛的临床和遗传异质性, Vilboux et al [2017]建议从多个基因靶向检测开始,如果尚未建立分子诊断,则进行全外显子组测序。
- 多个靶向基因检测,包括34个JS基因和其他感兴趣的基因 (参见 遗传相关疾病). 注:(1)小组中包含的基因和每个基因所用测试的诊断灵敏度因实验室而异,并且可能随时间而变化。(2)一些多基因检测可能包括与本GeneReview中讨论的病症无关的基因; 因此,临床医生需要确定哪个多基因检测最有可能以最合理的成本鉴定病症的遗传病因,同时限制对不能解释潜在表型的基因中不明意义和致病变异的鉴定。(3)在一些实验室,小组选项可包括定制的实验室设计的小组和/或定制的表型聚焦外显子组分析,其中包括临床医生指定的基因。(4)小组检测中使用的方法可包括序列分析,缺失/重复分析和/或其他不基于测序的检测。对于这种疾病,建议检测包括缺失/重复分析 (参见 表 1)。
- 全基因组检测(临床可用时)包括全外显子组测序和基因组测序。有关全基因组检测的介绍,请单击此处。有关临床医生订购基因组测试的更多详细信息,请点击 此处.
注:虽然单基因检测或系列单基因检测很少有用,并且由于JS具有广泛的临床和遗传异质性,因此通常不推荐使用。如果适用可以首先在以下种族/血统的个体中对特定基因的致病变异进行针对性分析 :
- 德裔犹太人:p.Arg73Leu in TMEM216 [Edvardson et al 2010]
- 荷兰人:p.Arg2904Ter in CPLANE1 [Kroes et al 2016]
- 法裔加拿大人:CPLANE1,CC2D2A,NPHP1和TMEM231中的几种变异 [Srour et al 2015]
- 日本人: c.6012-12T>A in CEP290 [Suzuki et al 2016]
参见 表 1a 为JS的最常见的遗传病因(即表中任何一种基因的致病性变异可解释>1%的JS)。 表 1b 为JS的不太常见的遗传病因(本表中任何一种基因的致病性变异仅在少数家庭中报道)。
Table 1a.
Joubert综合征的分子遗传学:最常见的遗传病因
| 基因 1, 2 | 归因于该基因的致病变异在JS中的百分比% | 测试方法检测的致病变异比例3 | |
|---|---|---|---|
序列分析 4 | 靶向基因缺失/重复分析 5 | ||
| AHI1 | ~7%-10% 6, 7, 8 | >95% | 见脚注9 |
| C5orf42 | 8%-14% 7, 8, 10 | 100% | None reported |
| CC2D2A | ~8%-11% 7, 8, 11 | Close to 100% | 见脚注12 |
| CEP290 | 7%-10% 7, 8, 13, 14 | ~99% | 见脚注15 |
| CSPP1 | 2%-4% 7, 8, 16 | 100% | 未见报道 |
| INPP5E | 2%-4% 7, 8 | 100% | 未见报道 |
| KIAA0586 | ~2%-7% 8, 17 | 两个报道,一个复发性多外显子 缺失 18 | |
| MKS1 | ~2%-6% 7, 8, 19 | ~95% | 见脚注20 |
| NPHP1 | ~1%-2% 7, 8, 21, 22 | See footnote 22 | 20%-25% 22 |
| RPGRIP1L | 1%-4% 7, 8, 23 | 100% | 未见报道 |
| TCTN2 | ~1% 7 | 13/13 24 | 未见报道 |
| TMEM67 | ~6%-20% 7, 8, 9, 12, 25 | ~99% | 见脚注26 |
| TMEM216 | ~2%-3% 7, 8, 27 | 8/8 26 | 未见报道 |
- 6.
- 7.
Bachmann-Gagescu et al [2015a] 对来自375个家庭的440个个体检测了27个JS相关基因的致病变异。
- 8.
Vilboux et al [2017]使用27个基因检测小组和外显子组测序的组合,在86个测试家庭中的81个(94%)中鉴定了致病变异(总共100个个体)。
- 9.
- 10.
Kroes et al [2016] 评估了51个患有JS的北欧人群中的22个JS相关基因和599个额外的纤毛基因。与其他队列不同,该队列中的12%发现了CPLANE1致病变异。
- 11.
Gorden et al [2008], Doherty et al [2010]. 一个大型队列中CC2D2A致病变异的患病率为16/209(7.7%) [Bachmann-Gagescu et al 2012].
- 12.
- 13.
数据源自 Sayer et al [2006], Valente et al [2006b], Valente et al [2008], Travaglini et al [2009]和Bachmann-Gagescu et al [2015a] 持7%-10%。相比之下,北欧队列中51例只有一例呈阳性(2%) [Kroes et al 2016].
- 14.
Suzuki et al [2016] 报道在一个30个家庭(除了3个是日本人)的队列中变异分析的产率为83%,在TMEM67(26%的队列),CEP290(22%的队列)和OFD1,INPP5E,AHI1和CPLANE1(各占队列的7.4%)中鉴定出致病变异。
- 15.
一个报道 [Travaglini et al 2009]
- 16.
- 17.
在一个队列中,KIAA0586中的致病变异占了366个JS家庭的9个(2.5%) [Bachmann-Gagescu et al 2015b] ,但由于在一般人群中单碱基缺失(c.428delG)的频率很高 [Roosing et al 2015] 和广泛的临床表型 [Alby et al 2015, Malicdan et al 2015]],可能比以前更普遍。
- 18.
在KIAA0586中具有复合杂合致病变异的六个个体中的三个中,一个致病变异是包含外显子8-10的800bp缺失 [Malicdan et al 2015]。
- 19.
MKS1致病变异分为两个独立的系列:2/260个JS [Romani et al 2014] 和9/371个JS家庭 [Slaats et al 2016]。
- 20.
- 21.
M患有肾痨的个体可能更多
- 22.
纯合缺失与罕见的JS病例有关。单独的缺失/重复分析将检测杂合缺失但不检测NPHP1中的单核苷酸变异; 这种基因型预计很少见。常见的~290kb缺失是最常检测到的。.
- 23.
- 24.
- 25.
- 26.
一篇报道 [Khaddour et al 2007]
- 27.
患有JS的462个家庭中的14个(~3%)在TMEM216中具有致病变异 [Valente et al 2010].
- 28.
Table 1b.
分子遗传学Joubert综合征:不太常见的遗传病因
| 基因 1, 2, 3 | 简述 |
|---|---|
| ARL13B | 2个家庭;表型范围从经典JS到JS 合并枕叶脑膨出和色素性视网膜病变[Cantagrel et al 2008];没有缺失/重复报道。 |
| B9D1 | 2个家庭,都是JS的“纯粹”形式;该基因中的致病变异也会引起MKS。没有缺失/重复报道 [Romani et al 2014]。 |
| B9D2 | 2个家庭,都有多指和脑膨出,腭裂和舌头错构瘤;该基因中的致病变异也会引起MKS。没有缺失/重复报道[Bachmann-Gagescu et al 2015a]。 |
| C2CD3 | 1个系列中确定的2个家庭,具有提示OFD特征的腭裂和/或口腔系带。没有缺失/重复报道 [Bachmann-Gagescu et al 2015a]. |
| CEP41 | 基于对至少725个患有JS的个体进行筛选,其中许多人已被排除在已知JS相关基因中的致病变异之外,来自3个家系中具有JS的8个个体具有CEP41中的致病变异。仅有超过50%的患者表现出单侧或双侧的多指畸形。只有2人有视网膜疾病的证据,其中1人患有单侧眼缺损,单侧肾脏疾病和两性畸形,并在7天时死亡。在1个家庭中,所有5名男性患者都有小阴茎,2名有生长激素缺乏症。仅检测到剪切位点变异;没有缺失/重复报道 [Lee et al 2012a]。 |
| CEP104 | 3个家庭,都是JS的“纯粹”形式;没有缺失/重复报道 [Srour et al 2015]. |
| CEP120 | 491个JS患者中有4个在该基因中具有错义,移码,无义或剪切位点变异;表型范围从单纯JS到MKS,OFD和JS-JATD;没有报道大的缺失/重复 [Shaheen et al 2015b, Roosing et al 2016a]. |
| IFT172 | 440个JS患者中有1个在该基因中具有错义的致病变异 [Bachmann-Gagescu et al 2015a]。12个家系中有2个具有错义和/或截短致病变异的具有JS和JS-JATD(一个具有Mainzer-Saldino综合征的特征)的重叠特征,包括视网膜营养不良,肝纤维化,NPHP和小脑蚓部发育不全。没有缺失/重复报道 [Halbritter et al 2013]. |
| KIAA0556 | 该基因中的纯合截短致病变异在一个近亲家系的3个同胞中被鉴定;其中2个患有全垂体功能减退症(男性小阴茎,女性脑垂体发育不全 [Sanders et al 2015]。在另一个近亲家庭中,2个具有经典JS特征的同胞具有纯合截短的致病变异;没有缺失/重复报道 [Roosing et al 2016b]。 |
| KIF7 | 440个家系中有3个在该基因中具有致病变异 [Bachmann-Gagescu et al 2015a]。个体通常具有OFD特征,有或没有其他CNS发现,例如胼胝体发育不全,脑积水和巨头畸形 [Dafinger et al 2011, Putoux et al 2011]。多指和这些CNS发现的组合表明了肢端-胼胝体综合征和/或纤毛病综合征。无义和移码致病变异占主导地位;没有缺失/重复报道。 |
| OFD1 | X-连锁的;没有缺失/重复报道。该基因中的致病变异在4/440家庭中被鉴定 [Bachmann-Gagescu et al 2015a] 和2/250个家庭(2/84仅男性患病)[Coene et al 2009]。功能包括脑膨出,脑积水,巨头畸形,多小脑回,多指和视网膜疾病。1个家庭也有肾囊性病,脑积水,巨头畸形和多小脑回 [Field et al 2012]. |
| PDE6D | 在1个近亲家庭中,有3个同胞(具有纯合剪切位点变异),表型包括肾发育不全,视网膜营养不良,小眼病,眼部缺损和轴后多指 [Thomas et al 2014]. |
| POC1B | 该基因中的纯合致病性错义变异在具有LCA,扩大的多囊肾(类似于ADPKD而非NPHP)的伊拉克家庭中被鉴定,并且具有JS的经典特征和/或肝纤维化。值得注意的是,相同的纯合致病变异在具有JS的严重和缓慢进展的视锥杆细胞营养不良的家庭中被鉴定出来 [Beck et al 2014],没有缺失/重复报道。 |
| TCTN1 | 1/440家庭在该基因中具有致病变异 [Bachmann-Gagescu et al 2015a]。具有纯合剪切位点变异的两个同胞具有额颞叶巨脑回但没有视网膜或肾脏发现 [Garcia-Gonzalo et al 2011]。没有缺失/重复报道。 |
| TCTN3 | 1/440家庭在该基因中具有致病变异[Bachmann-Gagescu et al 2015a].1/58家庭(已排除已知的JS基因)具有双等位基因致病变异 [Thomas et al 2012].在具有严重的产前致死型的OFD IV型(Mohr-Majewski综合征)的5个家系中鉴定了纯合截短变异;然而,由于表型还包括轴后多指,囊性肾病,胆管增生和枕叶脑膨出,这是否代表一种OFD或MKS是有争议的。来自土耳其家庭的2名JS先证者,他们有一个纯合的错义变异,患有脊柱侧凸,多指畸形,口腔异常,马蹄肾和室间隔缺损 [Thomas et al 2012]。没有缺失/重复报道。 |
| TMEM107 | 在具有JS或“OFD VI”的238个个体中,1对具有该基因的纯合错义变异的近亲双胞胎表现出视网膜病和OFD的特征,包括轴后多指;另一名经典JS和视网膜病变的男性具有复合杂合致病变异[Lambacher et al 2016]。没有缺失/重复报道。 |
| TMEM138 | 1/440家庭在该基因中具有致病变异 [Bachmann-Gagescu et al 2015a]。来自8个近亲阿拉伯家庭的11个人患有眼缺损(6),视网膜营养不良(3),囊性肾或NPHP(3)。已观察到多指;1例MKS胎儿患有脑膨出[Lee et al 2012b]。没有缺失/重复报道。 |
| TMEM231 | 该基因中的致病变异可以解释一些具有法裔加拿大血统的JS的个体。2个家庭中的3个人具有严重的表型(不能行走,攻击性行为,缺乏独立的生活技能)。2个有肉眼可见的肾囊肿和视网膜疾病; 1个具有轴后多指和并指畸形 [Srour et al 2012a]。已经报道了该基因与其假基因之间的致病基因转换事件 [Maglic et al 2016]. |
| TMEM237 | 1/440家庭在该基因中具有致病变异 [Bachmann-Gagescu et al 2015a]。只有2/201个JS患者和90个MKS / JS患者有在这个基因中有致病变异[Huang et al 2011]。这种形式的JS最初被描述为哈特派人群中的MKS [Boycott et al 2007],其携带率估计为6% [Huang et al 2011]。脑膨出,脑积水和囊性肾病很常见。“牵牛花视神经异常”也已经在来自奥地利的具有双等位基因致病变异的大家庭中被报道[Janecke et al 2004, Huang et al 2011]。包含TMEM237外显子1和1a并延伸到相邻基因的24kb缺失已被鉴定 [Watson et al 2016]。 |
| TTC21B | 迄今为止,尚未报道具有该基因中双等位基因致病变异的JS个体。单个(杂合的)致病变异的意义是未知的。3个杂合子个体的临床信息未提供。参见 TTC21B, 致病变异 (pdf). 在753个具有纤毛病的个体的临床多样化队列中,5%在该基因中具有致病变异;然而,只有33%的人在不同的纤毛病基因中有第二个致病变异 [Davis et al 2011]. |
| ZNF423 | 1个近亲家庭表型为婴儿期发病的NPHP,小脑蚓部发育不全和内脏转位在该基因中具有纯合的致病错义变异;2/96其他JS个体在特定相互作用域中具有基因杂合突变,通过显性抑制机制导致(但未证实)功能丧失 [Chaki et al 2012]。没有缺失/重复报道。 |
该表中列出的任何一种基因的致病变异仅在少数家庭中报告(即可解释<1%的JS)。
ADPKD =常染色体显性多囊肾病
JS-JATD = Jeune窒息性胸部营养不良
LCA = Leber先天性黑朦
MKS =梅克尔综合征
NPHP =肾痨
OFD =口-面-指综合征- 1.
基因按字母顺序列出。
- 2.
参见 表 A. 基因和数据库 的 染色体 位点 和蛋白.
- 3.
临床特征
临床描述
经典Joubert综合征(JS)的特征在于三个主要发现:独特的小脑和脑干畸形,称为臼齿征(MTS),肌张力减退和发育迟缓。这些发现通常伴有偶发性呼吸急促或呼吸暂停和/或非典型眼球运动。一般而言,呼吸异常随着年龄的增长而改善,躯干共济失调随着时间的推移而发展,运动技能的获得被延迟。认知能力是可变的,从严重的智力残疾到正常。其他发现可包括视网膜营养不良,肾脏疾病,眼部缺损,枕部脑膨出,肝纤维化,多指畸形,口腔错构瘤和内分泌异常。表2将表型特征与基因联系起来; 表3将基因与表型特征联系起来。在JS中可以看到家庭内和家庭间的表型变异。
JS的许多临床特征在婴儿时期是显而易见的[Joubert et al 1969, Boltshauser & Isler 1977]。在所有临床亚型中都可以观察到眼球震颤,动眼神经失用和异常呼吸模式。大多数患有JS的儿童发生躯干性共济失调,并伴有肌张力减退,表现出运动技能获得延迟。
眼球震颤 许多患有Joubert综合征的儿童在出生时表现出水平眼球震颤,随着年龄的增长而改善。还观察到扭转和摆动旋转性眼球震颤。
动眼神经失用 通常在儿童而不是婴儿时期被发现,可能是由于对该发现的认识不足[Steinlin et al 1997]。许多患有动眼神经失用症的儿童表现出头部前顶作为其无法启动眼跳的补偿机制 [Hodgkins et al 2004, Khan et al 2008, Weiss et al 2009]。在2岁以下的婴幼儿中已经描述了水平头部震颤(即摇头) [Poretti et al 2014]。尽管出生时眼睛运动明显异常[M Parisi和A Weiss,个人观察],但由于视觉成熟,视力和功能性视力可随着年龄的增长而改善。
呼吸系统发现许多患有JS的儿童表现出呼吸暂停,呼吸急促或都有,有时交替出现,特别是在新生儿期 [Saraiva & Baraitser 1992, Steinlin et al 1997, Maria et al 1999a, Valente et al 2008]。虽然有些婴儿死于呼吸暂停,但偶发性呼吸暂停通常会随着年龄的增长而改善,并且可能完全消失[Maria et al 1999b]。患有JS的儿童患睡眠呼吸暂停的风险增加,包括中枢性(特别是婴儿期和儿童期)和阻塞性(特别是在晚期儿童/青春期与舌头肥大,肌张力减退和肥胖有关) [Parisi 2009]。使用经验证的睡眠问卷对JS患者自我报告的睡眠行为进行的一项调查显示,在接受调查的14名患者中, 6名有睡眠相关的呼吸障碍[Kamdar et al 2011]。一些患有由CEP290中的双等位基因致病变异引起的Leber先天性黑朦的个体也被发现在运动呼吸道纤毛中可能易于出现呼吸道症状,包括慢性鼻炎,复发性鼻窦炎和支气管炎[Papon et al 2010]。
中枢神经系统的发现
- 认知能力是可变的,从严重的智力残疾到正常的认知功能[Poretti et al 2009]; 一些人上过大学。当存在时,智力残疾通常处于中等范围[Steinlin et al 1997, Hodgkins et al 2004, Bulgheroni et al 2016, Summers et al 2017]。在110名JS 患者的研究中发现了小脑蚓部发育不全的严重程度与认知障碍之间的相关性 [Poretti et al 2017]。
- 言语失用,一个常见的发现,可能解释了语言理解和言语能力之间观察到的差异 [Hodgkins et al 2004, Braddock et al 2006]。
- 一些患病的个体存在异常的脑电图和/或癫痫发作 ; 确切的发病率未知 [Saraiva & Baraitser 1992]。一项研究发现,患有JS和异常脑电图的个体的认知障碍更严重[Summers et al 2017]。
- 据报道,一些患有JS的儿童患有自闭症 [Holroyd et al 1991, Ozonoff et al 1999]; 然而,最近的调查表明,许多这些行为障碍并不代表经典的自闭症谱系障碍 [Takahashi et al 2005]。
- 在一些儿童和青少年中存在行为问题,包括注意力不集中,多动症和非典型行为,例如发脾气 [Deonna & Ziegler 1993, Hodgkins et al 2004, Farmer et al 2006]。在一项针对54名JS患者的调查中,有近40%的人报告了情绪和行为问题[Bulgheroni et al 2016]。在对76个人的另一项调查中,行为问题更可能表现为内化(焦虑,抑郁)而非外化(攻击性,对立性蔑视) [Summers et al 2017]。
表2. Joubert综合征:临床亚型
临床亚型名称 | 除主要标准外的强制性特点1 | 强烈关联的特点2 | 其他名称 | 基因 |
经典或单纯Joubert综合症 | JS; JS类型A. | 许多基因 | ||
Joubert综合征与视网膜疾病(JS-Ret) | 视网膜营养不良(包括LCA) | JS类型B | AHI1 | |
Joubert综合征与肾脏疾病(JS-Ren) | NPHP(包括囊性肾病) | AHI1 | ||
Joubert综合征与眼肾疾病(JS-OR) | 视网膜营养不良(包括LCA);NPHP | 先天性肝纤维化(偶尔) | JS类型B;CORS;Senior-Løken综合征;Dekaban-Arima综合征 | AHI1 |
Joubert综合征与肝病(JS-H) | CHF | 眼缺损; NPHP | COACH综合征;Gentile综合征 | CC2D2A |
Joubert综合征与口-面-指特征(JS-OFD) | 舌头错构瘤; 口腔系带; 多指 3 | 唇裂/腭裂 | Varadi-Papp综合征; OFD VI; OFD IV;Mohr-Majewski综合征 | B9D2 |
具有肢端-胼胝体表型的Joubert综合征(JS-AC) | 胼胝体发育不全; 多指 3 | 脑积水 | Acrocallosal综合征 | KIF7 |
Joubert综合征与Jeune窒息性胸部营养不良特征(JS-JATD) | 骨骼发育不良(短肋骨,小胸,短肢,肾囊性疾病) | 多指3 ; 锥形骨骺; CHF | Jeune asphysiating胸部营养不良;Mainzer-Saldino综合征 | CEP120 |
改编自 Brancati et al [2010]。鉴于表型的极端临床异质性和许多特征起始年龄的变化,该分类方案不应被理解为确定的。
AC =肢端-胼胝体
CHF =先天性肝纤维化
COACH =小脑蚓部发育不全,智力发育不全,共济失调,眼缺损和肝纤维化
CORS =小脑-眼-肾综合征
H =肝脏
JATD = Jeune窒息性胸部营养不良
LCA = Leber先天性黑朦
NPHP =肾痨
OFD =口-面-指综合征
OR= 眼-肾
Ren =肾
Ret =视网膜
1、主要标准=臼齿征(MTS),肌张力减退,发育迟缓(DD)
2、其他特征包括脑膨出,轴后多指,其他结构性脑畸形(包括多小脑回),先天性心脏缺陷,先天性巨结肠疾病和局部缺陷可见于这些亚型,但不是主要特征。
3、多指通常是轴后,特别是手以及轴前,特别是脚。OFD VI综合征的独特之处:中间或中央多指与Y形掌骨。
具有视网膜疾病的Joubert综合征(JS-Ret)的特征在于色素性视网膜病变,其可能与经典的视网膜色素变性无法区分; 对于先天性失明的新生儿发病和类似于Leber先天性黑朦(LCA)的减弱或熄灭的视网膜电图,它偶尔可能是严重的[Tusa & Hove 1999]。然而,视网膜疾病可能不是进行性的,并不总是存在于婴儿期或幼儿期[Steinlin et al 1997]。对JSRD的235个家庭进行的一项调查发现30%的视网膜营养不良[Doherty 2009]。
- 眼部缺损最常被描述为脉络膜视网膜的 [Saraiva & Baraitser 1992, Parisi 2009] ,并且可能与肝纤维化有关,如COACH综合征变异[Doherty et al 2010]。一项调查描述了19%的JSRD家庭中具有眼部缺损 [Doherty 2009]。描述为“牵牛花视神经异常”的视网膜变化已经在具有双等位TMEM237基因致病变异的蒂罗尔地区的一个奥地利家庭中被描述[Janecke et al 2004, Huang et al 2011]。
- 其他可变的存在:
- 上睑下垂,斜视和/或弱视
- 第三神经麻痹 [Hodgkins et al 2004]
Joubert综合征伴肾病(JS-Ren)传统上有两种形式(肾痨和囊性发育不良); 然而,这些现在似乎是连续的,具体的肾脏表现随肾病的阶段而变化。青少年肾痨是慢性肾小管间质性肾病的一种形式,通常在生命的第一或第二个十年出现多饮,多尿,尿液浓缩缺陷,生长迟缓和/或贫血。终末期肾病的进展平均发生在13岁 [Hildebrandt et al 1998]。在超声检查中可见的肾脏变化发生在病程的后期,小的,有疤痕的肾脏,回声增加,皮质髓质连接处偶有囊肿,结果与囊性发育不良(即胎儿未成熟肾脏肾小叶中多个可变大小的囊肿)一致 [Saraiva & Baraitser 1992, Steinlin et al 1997, Satran et al 1999]。
除了肾痨和囊性发育不良之外,还报道了第二种类似于常染色体隐性遗传性多囊肾病(ARPKD)的肾病。
- 据报道,由双等位基因TMEM67致病变异引起的三个JS 患者具有更典型的ARPKD肾脏疾病,伴有扩大的弥漫性微囊性肾脏和早发性严重高血压以及先天性肝纤维化; 此外,他们表现出肾痨的慢性贫血 [Gunay-Aygun et al 2009]。
- 在哈特派人群中,大约70%由双等位基因TMEM237致病变异引起的JS先证者患有囊性肾病和肾功能异常,一些人患有高血压 [Boycott et al 2007, Huang et al 2011]。
据报道,有23% [Doherty 2009]和30%[Saraiva & Baraitser 1992]的JS患者同时患有肾病。随着群体年龄的增长,患病率可能会增加,因为肾脏疾病可能在儿童期和青春期发展 [Steinlin et al 1997]。
Joubert综合征伴有眼-肾疾病(JS-OR)。视网膜疾病和肾功能损害通常在同一个体中共同发生,许多JS相关基因与肾囊性疾病和视网膜营养不良有关,这种组合有时被称为Senior-Løken综合征 [Parisi 2009, Brancati et al 2010](表2)。在过去,JS-OR也被称为Dekaban Arima综合征(视网膜病变,囊性发育不良肾脏),其在产前或出生时可以是明显的。
Joubert综合征合并肝病(JS-H)。肝纤维化通常是进行性的,但在出生时很少有症状(参见先天性肝纤维化概述)。先天性肝纤维化是一种系统的发育障碍,其特征在于组织学上由导管板的重塑缺陷(导管板畸形),肝内门静脉的异常分支和门静脉的进行性纤维化。临床发现包括肝脏扩大,形态异常,肝细胞功能相对完好,门静脉高压导致脾肿大,脾功能亢进和胃食管静脉曲张。
在一个队列中,18%的JS患者观察到肝纤维化 [Doherty 2009]。
当存在于JS中时,肝纤维化通常与脉络膜视网膜病变相关,并且有时与肾病相关。眼缺损,认知障碍(“ Ò ligophrenia”),共济失调,小脑蚓部发育不全,和肝纤维化已被称为COACH综合征 [Satran et al 1999, Gleeson et al 2004, Doherty et al 2010].。
具有口-面-指特征的Joubert综合征(JS-OFD)。口腔检查结果可包括中线上唇裂,舌中线凹槽,牙槽嵴血肿(图2B),腭裂,口腔系带和舌状分叶或错构瘤。颅面特征通常包括眼距过宽或内眦距过宽,发育不全的鼻翼和小颌畸形。
多指见于8%-19%的先证者[Doherty 2009, Brancati et al 2010]。多指可以是单侧或双侧的,通常是轴后(图2C),虽然也经常报告脚趾的轴前多指(图2D)[Saraiva & Baraitser 1992]。
在一些具有JS的人中具有中央多指,其中额外的指出现在中心指之间并且通常伴有Y形掌骨,其中许多人具有口-面-指综合征VI / Varadi-Papp综合征[Gleeson et al 2004]。OFD VI现已被定义为JS的一种形式,需要MTS以及以下一个或多个特征:舌错构瘤/口腔系带/上唇缺口,近中多指畸形和下丘脑错构瘤[Poretti et al 2012]。
{kind=link}
{kind=link}
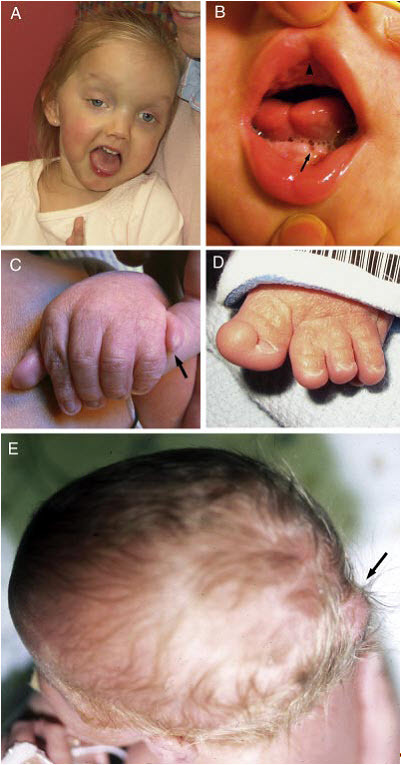
图2. JSRD的临床特征
A. 一名患有JS / COACH综合征的女孩在27个月时的面部特征显示前额宽阔,拱形眉毛,斜视,眼睑下垂(右眼)和张口,表明面部张力减弱
B. 口腔发现一个患有口-面-指综合征样特征的JS,表现为中线上唇裂(箭头),舌中线凹槽和下牙槽嵴凸起(箭头)
C. 一个 JS婴儿的左手有轴后多指(箭头)
D. 一个JS婴儿的左脚有和拇指的轴前多指
E. 从上看一个小枕骨头颅的婴儿,头骨的枕骨突出(箭头)
许可使用:Parisi [2009]。面部照片经家人许可使用。
具有肢端-胼胝体特征的Joubert综合征(JS-AC)。胼胝体发育不全在JS中很常见[Valente et al 2005]。在一项对20名JS患者的调查中,80%患有一定程度的胼胝体发育不全[Senocak et al 2010]。在具有双等位基因KIF7致病变异的患者中,胼胝体异常相对频繁[Bachmann-Gagescu et al 2015a],提示与肢端-胼胝体综合征(参见遗传相关疾病)重叠,其中也观察到多指和脑积水 [Putoux et al 2011]。
Joubert综合征与Jeune窒息性胸部营养不良(JS-JATD)。已报道几个患有JS的儿童具有JATD(参见遗传相关疾病)和相关的短肋胸部发育不良状况,Mainzer-Saldino综合征的特征,反映了这些疾病共同的纤毛起源 [Lehman et al 2010, Halbritter et al 2013, Shaheen et al 2015b]。
JS中的其他发现并非特定于给定子类
脊柱侧凸已被描述,很可能与早期张力减退有关。
已经描述了内分泌异常 ; 它们包括垂体激素功能障碍,从孤立的生长激素缺乏或甲状腺激素缺乏到男性更广泛的泛垂体功能减退或小阴茎[Delous et al 2007, Wolf et al 2007, Parisi 2009, Sanders et al 2015]。
JS可能会增加肥胖,这表明与纤毛病Bardet-Biedl综合征有关 ; INPP5E双等位基因致病变异在JS和MORM综合征(智力迟钝,肥胖,视网膜营养不良和小阴茎)的鉴定强化了这种关联[Bielas et al 2009, Jacoby et al 2009]。
典型的面部特征,包括具有双颞狭窄的长脸,高弓形眉毛,上睑下垂,具有前倾鼻孔的突出鼻梁,三角形嘴,凸颌和低耳朵[Maria et al 1999a](图2A); 然而,这些特征在婴儿期很难辨别,因此是非特异性的。尽管如此,许多观察家报告了“Joubert综合征相”[Braddock et al 2007]。具有双等位基因KIF7致病变异的颅面特征通常包括巨头畸形,额叶凸起,眼距过宽,高腭和小颌畸形 [Dafinger et al 2011, Putoux et al 2011]。
许多患有JS的人有心脏缺陷,在某些情况下与口-面-指综合征的特征相关,并且包括房间隔缺损,主动脉瓣异常和主动脉缩窄 [Bachmann-Gagescu et al 2015a]。
在某些个体中可以看到包括内脏转位在内的侧向缺陷 [Parisi 2009]。
先天性巨结肠症已在少数人中描述 [Brancati et al 2010]。
中耳感染可能导致听力损失 [Kroes et al 2010]。已经描述了感觉神经性听力损失。
舌头肥大。许多人有节奏性的舌头运动,可能导致舌头肥大。
其他CNS畸形
- 小脑半球肿大 [Poretti et al 2017],小脑异位 [Saraiva & Baraitser 1992] 和小脑叶片紊乱 [Senocak et al 2010, Poretti et al 2011]
- 第四脑室或后颅窝中的脑脊液异常积累,类似于Dandy-Walker畸形; 一项调查 [Maria et al 2001]约占10%,另一项调查约占42%[Poretti et al 2017]
- 枕部脑膨出(图2E)或脑膜膨出[Genel et al 2004]
- 异常的脑干和下丘脑错构瘤,尤其是口-面-指综合征VI型相关发现[Poretti et al 2011]
- 脑室扩大[Quisling et al 1999, Senocak et al 2010, Putoux et al 2011]或脑积水需要分流而没有Dandy-Walker畸形的典型征象[Genel et al 2004]
- 海马畸形/旋转不良,一项有限的调查 [Senocak et al 2010] 描述80%个体,两项较大调查中占15%-18% [Poretti et al 2011, Poretti et al 2017]
- 脑皮质异常包括异位,发育不良,巨脑回,多小脑回 [Gleeson et al 2004, Dixon-Salazar et al 2004]和神经上皮囊肿 [Marsh et al 2004, Senocak et al 2010]
- 神经病理学评估异常的脑桥,小脑和髓质的核和束[Doherty 2009]; 没有基于弥散张量成像的皮质脊髓和上小脑束的交叉[Poretti et al 2007]; 功能性MRI研究的运动任务期间的异常激活模式 [Parisi et al 2004b]
基因表型相关性
表3包括关于基因型-表型相关性的初步信息。
表3. 通过表型特征与JS相关的基因
基因 | 表型特征(除了臼齿征) | 等位基因/相关症状3 | |||||||
视网膜营养不良 | 眼缺损1 | 肾 | 眼-肾2 | 肝1 | 口 | 多指 | 其他 | ||
AHI1 | ++4 | (+) | +5 | + | (+) | 多小脑回6 | |||
CPLANE1 | (+) | +7 | +7 | 法裔加拿大8和荷兰人口的创始人效应9 | OFD VI | ||||
CC2D2A | + | + | + | + | +10 | 脑膨出,脑室扩大,癫痫发作11;法裔加拿大人口中较温和的表型12 | 梅克尔综合症13 | ||
CEP290 | ++ | + | ++ | ++14 | + | 脑膨出;心脏;内脏反转;其他15 | |||
CSPP1 | (+) | (+) | (+) | (+) | SNHL;胼胝体发育不全;脑膨出; 哈特派人群中的创始人变异16 | 梅克尔综合症,JATD17 | |||
INPP5E | + | + | + | (+) | + | MORM综合征18 | |||
KIAA0586 | (+) | + | (+) | (+) | 广泛的表型:严重的HLS(和腭裂)到JATD与短肋骨和狭窄的胸部到单纯JS19 | HLS,JATD | |||
MKS1 | (+) | 肾脏/肝脏的发现和PD在1个人中被描述20 | 梅克尔综合症 | ||||||
NPHP1 | + | ++ | + | 有时描述“轻度臼齿征”21 | 少年NPHP 1型,Cogan综合征 | ||||
RPGRIP1L | (+) | (+) | ++ | + | (+) | (+) | 脑膨出 | 梅克尔综合征,视网膜疾病22 | |
TCTN2 21 | 马蹄足23 | 梅克尔综合症24 | |||||||
TMEM67 | + 25 | + | ++26,27 | (+) | 脑膨出 | 梅克尔综合征28 | |||
TMEM21629 | (+) | (+) | ++ | + | (+) | + | + | 心脏发现;脑膨出 | 梅克尔综合症 |
(+)=功能不常见但已被描述
+=某些情况下存在特征
+ + =主要功能
HLS =脑积水综合征
NPHP =肾痨
LCA = Leber先天性黑朦
BBS = Bardet-Biedl综合征
MORM =精神发育迟滞,躯干肥胖,视网膜营养不良,小阴茎 [Jacoby et al 2009]
OFD =口-面-指综合征
PD =多指
JATD = jeune窒息性胸营养不良
SNHL =感觉神经性听力损失
1、可能包括COACH综合征:小脑蚓部发育不全,智力发育不全,共济失调,眼缺损和肝纤维化
2、这是指视网膜疾病加肾病; 过去使用的术语包括:Senior-Løken综合征(视网膜病变和青少年发病的肾痨); Dekaban-Arima综合征(视网膜病变,囊性发育不良肾脏)。
3、有关详细信息,请参阅遗传相关疾病。
4、具有双等位基因AHI1致病变异最常见的临床关联是视网膜营养不良,存在于约80% [Valente et al 2008]。已经描述了早发性先天性失明[Valente et al 2006a] 和肝脏受累[Vilboux et al 2017]。
5、还描述了与肾痨的一致的肾病 [Parisi et al 2006, Utsch et al 2006]。
6、Dixon-Salazar et al [2004], Gleeson et al [2004]
7、该表型最接近于纯粹或经典的Joubert综合征,其中几个个体表现出轴前,轴后和/或中间多指,一些具有视网膜受累[Srour et al 2015]或肝脏受累[Vilboux et al 2017]。患病的个体(年龄从1.5岁到52岁)都没有肾功能受损或肝病的证据[Srour et al 2012a, Srour et al 2012b, Srour et al 2015]。该基因中的致病变异也引起OFD VI,具有典型的轴前和/或中间多指和下丘脑错构瘤的特征 [Lopez et al 2014, Romani et al 2015]。
8、该基因中的致病变异是 Joubert et al [1969]描述的家庭中JS的病因。在魁北克省圣劳伦斯河下游地区发现的法裔加拿大人群中发现了几种致病变异 [Srour et al 2012b, Srour et al 2015]。
10、已经描述了肝脏受累 [Gorden et al 2008, Noor et al 2008]。
11、那些在CC2D2A中具有致病性变异的患者脑室扩大和癫痫发作的可能性增加[Bachmann-Gagescu et al 2012].。
13、等位基因缺失与Meckel综合征表型相关,和错义变异和/或低效等位基因与JS 相关[Tallila et al 2008, Mougou-Zerelli et al 2009]。
14、高达50%具有视网膜和肾脏受累的个体在CEP290中具有双等位基因致病变异[Valente et al 2008]。
15、表型谱非常广泛,包括先天性失明,眼部缺损,肾病,脑膨出,室间隔心脏病和内脏位置异常。
16、Shaheen et al [2014], Vilboux et al [2017]
17、该基因的致病变异表型已被描述,范围从轻度表型的经典JS,视网膜病变和感觉神经性听力丧失[Akizu et al 2014] 到具有Jeune骨骼发育不良特征的JS-JATD表型 [Tuz et al 2014]到致死性类MKS表型 [Shaheen et al 2014]。还描述了薄胼胝体,枕部脑膨出和异位 [Akizu et al 2014, Tuz et al 2014]。
19、该基因的致病变异引起广泛的纤毛病表型,从具有相对轻微表现和损伤的单纯JS [Bachmann-Gagescu et al 2015b, Roosing et al 2015]到Jeune窒息性胸部营养不良(小胸,短肋骨,身材矮小) [Alby et al 2015, Malicdan et al 2015] 到脑积水和胎儿死亡 [Alby et al 2015]。这种广泛的表型不能通过致病变异的性质来解释,因为许多患者由于移码,异常剪切或无义变异而具有纯合或复合杂合截短变异。
20、相比MKS中描述的更严重的变异, 由MKS1致病变异引起的具有JS的个体具有至少一种有部分功能的变异(例如,保留一些功能的错义变异)[Romani et al 2014, Slaats et al 2016] 。大多数患者具有相对温和的表型,特征为具有或不具有视网膜营养不良的经典JS。只有一个报告的个体(在该基因中9个致病变异中)具有肾回声,肝纤维化和轴后多指的额外特征 [Slaats et al 2016]。
21、在NPHP1和JS中具有双等位基因致病变异的一些个体具有独特的臼齿征:细长但薄的上小脑脚节和较弱的蚓部发育不全 [Parisi et al 2004a]。
22、RPGRIP1L致病变异也会导致Meckel综合征。值得注意的是,功能丧失致病变异预测为更严重(并且在许多情况下是致死的)Meckel表型[Delous et al 2007, Wolf et al 2007]。
23、已描述该基因中具有致病变异的个体的数量有限; 因此,表型谱是未知的 [Sang et al 2011]。
25、无论肝脏状态如何,TMEM67 中的双等位基因致病变异存在于53%的眼部缺损患者中 [Doherty et al 2010]。
26、TMEM67中的双等位基因致病变异占所有具有肝脏受累的JS的70% [Doherty et al 2010, Iannicelli et al 2010]。
27、Baala et al [2007], Brancati et al [2009], Doherty et al [2010], Vilboux et al [2017]
28、已经在具有致死形式的Meckel综合征的个体中发现TMEM67中更严重的功能丧失变异[Smith et al 2006],而具有部分功能的变异导致JS患有肝病或肾病和肝纤维化,不具有臼齿征及其他神经系统症状 [Otto et al 2009, Doherty et al 2010]。
29、肾痨和多指是常见的; 一些人具有OFD的特征 [Edvardson et al 2010, Valente et al 2010]。
命名法
术语“Joubert综合征和相关疾病”(JSRD)过去曾用于描述具有臼齿征的病症和经典Joubert综合征的临床特征,并且还具有其他器官参与。在旨在减少对混乱和不一致使用的名词的依赖的不断发展的命名法中,已经提出了至少八种共享三个主要发现的JS临床亚型(表2) [Brancati et al 2010]。最近,“Joubert综合症”已经成为描述所有形式的JS的公认术语。
在过去,以下一些疾病被描述为不同的综合征,但是最近的研究表明,许多患有这些疾病的个体表现出臼齿征[Satran et al 1999, Gleeson et al 2004]。这种常染色体隐性遗传疾病的例子包括:
- Dekaban-Arima综合征(视网膜病变,囊性发育不良肾脏) [Dekaban 1969]
- Senior-Løken综合征(SLS;视网膜病变和青少年发病的肾痨) [Løken et al 1961, Senior et al 1961]
- COACH综合征(小脑蚓部发育不全,智力发育不全,共济失调,眼缺损和肝纤维化)[Verloes & Lambotte 1989, Gentile et al 1996]
- Varadi-Papp综合征(口-面-指综合征VI [OFD VI])包括小脑蚓部发育不全,口腔系带,舌错构瘤和中线唇裂,以及具有Y形掌骨的中央多指的特征[Münke et al 1990]。已经描述了肾脏和心脏受累。
患病率
Joubert综合征(JS)的患病率尚未确定。许多作者描述的范围在1:80,000和1:100,000之间,但这可能低估 [Kroes et al 2007, Parisi et al 2007, Brancati et al 2010]。
在法裔加拿大人口中,JS的流行程度相对较高,其中有几个创始人变异。Joubert et al [1969] 首次描述的家庭可追溯到17世纪从法国移民到魁北克的创始人 [Badhwar et al 2000]。然而,在该家庭和其他家庭中,似乎在法裔加拿大人群中存在多种含有CPLANE1致病变异的单倍型。事实上,在35个法裔加拿大家庭中,在以下基因中33个(94%)中发现了致病变异(括号中给出的患病家庭数量):CPLANE1(14),CC2D2A(9),NPHP1(3),TMEM231(2); 和CEP290,TMEM67,TCTN1,OFD1,B9D1,C2CD3和CEP104(各1个)。许多法裔加拿大个体对于CPLANE1,CC2D2A,TMEM231或NPHP1中的不同致病变异是复合杂合的 [Srour et al 2012a, Srour et al 2012b, Srour et al 2015]。
CPLANE1中的另一种创始变异p.Arg2904Ter发生在荷兰人群中 [Kroes et al 2016]。
TMEM216创始人变异,p.Arg73Leu,在德裔犹太人群具有为1:92-1:100 的携带率[Edvardson et al 2010, Valente et al 2010]。
在加拿大的哈特派人群,十个相关个体有MKS样表型,包括脑膨出和囊性肾脏为TMEM237纯合无义致病变异(c.52C>T; p.Arg18Ter),携带率为6%[Huang et al 2011]。两个不同的哈特派家庭在CSPP1中具有相同的纯合致病性移码变异c.363_364delTA [Shaheen et al 2014],为单独的创始人变异。
在日本JS家庭的调查显示,在12个致病基因中6/27有CEP290致病突变c.6012-12 T>A; 7/27家庭在TMEM67中具有致病变异,但没有发现创始等位基因 [Suzuki et al 2016]。
相关遗传疾病
导致Joubert综合征(JS)的基因的致病变异也已经在具有与JS重叠的临床发现的疾病中被鉴定; 因此,在许多情况下,已经变得难以确定先前识别的病症是否真正不同于JS(即是等位基因病症)或者是JS谱的一部分(参见表3)。其中一些疾病的简要描述如下。
Acrocallosal综合征(ACLS)(OMIM 200990),一种常染色体隐性遗传疾病,其特征为巨头畸形,智力障碍,胼胝体发育不全和偶尔的后颅窝异常,眼球性高血压,双手多指畸形,以及足部的轴前多趾。据推测,ACLS是血小板综合征的等位基因。鉴定具有这两种疾病和KIF7致病变异的几个家庭证实了所提出的关联; 值得注意的是,有几位先证者在颅脑成像中有臼齿征(MTS)的证据,这表明ACLS和JS可能代表重叠的纤毛病[Putoux et al 2011]。
Bardet-Biedl综合征 (BBS)通常以常染色体隐性遗传方式遗传,其特征为视网膜视锥杆细胞营养不良,躯干肥胖,轴后多指,认知障碍,男性低促性腺激素性性腺功能减退,女性生殖器畸形以及可能包括的肾脏疾病结构畸形,肾发育不全,肾积水,囊性肾和肾小球肾炎。进行性视网膜损伤常导致失明; 肾功能衰竭可能导致严重的致死率。一些患病的个体有肝纤维化。尽管许多个体表现共济失调与协调性差,但小脑受累或结构畸形并不典型 [Baskin et al 2002]。已经描述了至少19种基因中的致病变异,所有这些基因都在初级纤毛中起作用。已显示CEP290,MKS1和NPHP1中的致病变异同时引起BBS和JS [Leitch et al 2008, Zaghloul & Katsanis 2009, Knopp et al 2015]。
Cogan综合征(OMIM 257550)是一种常染色体隐性遗传性家庭性先天性动眼神经失用症,其特征是水平自愿眼球运动缺陷伴有急动。动眼神经失用也是JS的常见表现。通过纤维追踪进行的详细神经影像学表明,与JS相比,Cogan综合征中的一些途径可能存在细微差异[Merlini et al 2010]。
一些患有Cogan综合征的个体也有小脑蚓部发育不全,伴有臼齿征的证据[Whitsel et al 1995, Sargent et al 1997],并偶尔有肾痨。已经在一些具有Cogan综合征的个体中鉴定了大约290-kb的NPHP1缺失和一个NPHP1序列变异的复合杂合变异[Saunier et al 1997, Betz et al 2000]。
Hydrolethalus综合征(HLS)(OMIM PS236680)是一种致死的常染色体隐性遗传疾病,与脑中线异常(通常为脑积水或无脑儿伴锁孔枕骨大孔),小颌畸形,双手轴后多指畸形和足轴前多趾畸形相关。在芬兰人群中,已经鉴定出HYLS1中的致病变异 [Mee et al 2005]。KIF7中的致病变异在近亲阿尔及利亚血统中被鉴定,其中四个患病胎儿具有与HLS一致的特征,但也具有与MTS类似的中脑-后脑畸形[Putoux et al 2011]。KIAA0586的致病变异已经在具有HLS的胎儿以及具有JS和多种纤毛病表型的个体中被描述[Alby et al 2015]。
Jeune窒息性胸部营养不良(JATD)是一种常染色体隐性遗传性骨骼发育不良,其特征为长而窄的胸部,身材矮小,四肢短,多指和肾囊性疾病,骨骼发现可能包括手足的锥形骺,不规则干骺端,缩短的髂骨和三叉戟形髋臼。它在婴儿期通常是致命的仅次于呼吸功能不全。已经鉴定了超过12种纤毛基因和/或基因座(包括几种鞭毛内转运蛋白)。已经在具有JATD的三个家庭中鉴定了TTC21B中的杂合致病变异,其中一个先证者证明了无效等位基因和低效等位基因的复合杂合性[Davis et al 2011]。CSPP1 [Tuz et al 2014] 和KIAA0586 [Malicdan et al 2015] 的致病变异已经在具有JS和JATD表现的个体中被鉴定。
Leber先天性黑朦 (LCA)是一种严重的视网膜营养不良,通常在出生后的第一年就会变得明显。视功能通常较差,常伴有眼球震颤,瞳孔反应缓慢或几乎没有,畏光,高度远视和圆锥角膜。视敏度很少超过20/400。一个典型的发现是Franceschetti视觉标志,包括眼睛戳,压,揉。眼底的外观变化很大。虽然视网膜最初可能看起来正常,但是在儿童期后期经常会观察到色素性视网膜病变,这种视网膜病变使人想起色素性视网膜炎。视网膜电图特征性地“不可检测”或严重不正常。至少17种基因中的致病变异引起LCA,CEP290致病变异约占LCA的20%,其中一个纯合内含子致病变异占欧洲队列中孤立先天性失明的至少20% [den Hollander et al 2006]。
Mainzer-Saldino综合征(MZSDS)是一种常染色体隐性遗传疾病,有视网膜营养不良,肾脏疾病(通常是肾痨)和指骨锥形骺的三个诊断标准。可变的发现包括小脑发育不全,狭窄的胸腔,肝纤维化和长头,与两种病症中描述的IFT140中的致病变异和JATD的特征显著重叠[Mainzer et al 1970, Perrault et al 2012]。鞭毛内运输装置的另一组成部分IFT172中的致病变异已经在具有JATD,MZSDS和JS的患者中被描述 [Halbritter et al 2013]。
已被用于包括Ellis-van Creveld综合征(EVC),短肋骨多发综合征(SRPS),JATD和MZSDS的术语是短肋胸部发育不良(SRTD)(OMIM PS208500),有或无多指畸形; 这些病症是常染色体隐性遗传性骨骼纤毛病,其特征是收缩的胸廓,短肋骨,缩短的管状骨,以及髋臼顶部的“三叉”外观。这些骨骼发育不良和某些形式的JS之间显然有很多重叠。
梅克尔综合征(OMIM PS249000)是一种常染色体隐性遗传病,其特征为囊性肾病三联征,后颅窝异常(通常是枕部脑膨出),以及导致肝纤维化和胆管增生的肝导管板畸形。多指相对常见。已经描述了一些人有小脑蚓部发育不全。梅克尔综合征在产前或围产期通常是致死的 [Kyttälä et al 2006, Smith et al 2006]。已经在Meckel综合征中鉴定了至少21种基因的致病变异[Knopp et al 2015]。至少18个基因CEP290,TMEM67,RPGRIP1L,CC2D2A,CEP41,MKS1,B9D1,B9D2,TMEM138,TMEM231,TCTN2,TCTN3,TMEM237,CPLANE1,CSPP1,CEP120,TMEM107和TMEM216的致病变异,在具有JS的个体中也被鉴定出 [Parisi 2009, Valente et al 2010, Thomas et al 2012, Romani et al 2014, Bachmann-Gagescu et al 2015a, Knopp et al 2015, Shaheen et al 2015a, Roosing et al 2016a, Slaats et al 2016]。在许多情况下,预测对蛋白质功能的更严重影响的致病变异如转录终止或缺失与致命的Meckel综合征表型相关,而较轻的致病变异如错义变异与JS相关[Romani et al 2014, Slaats et al 2016]。在一些家庭中,相同的致病变异可以在具有Meckel综合征的胎儿和具有JS的儿童中发现,显示这些疾病可以代表疾病谱[Valente et al 2010]。
MORM(精神发育迟缓,躯干肥胖,视网膜营养不良,小阴茎)综合征(OMIM 610156),一种常染色体隐性疾病,似乎与由INPP5E致病变异引起的Bardet-Biedl 综合征相关 [Bielas et al 2009, Jacoby et al 2009]。患有这种疾病的个体具有正常的生长参数和寿命,具有先天性非进行性视网膜营养不良和静态轻度至中度认知障碍; 与Bardet-Biedl综合征相反,没有多指,明显的性腺机能减退或明显的肾脏疾病[Hampshire et al 2006]。
肾痨是一种常染色体隐性肾病,其特征在于肾小管萎缩和进行性间质纤维化以及髓质囊肿的后期发展,其由至少19种基因的致病变异引起[Hildebrandt et al 2009, Hurd & Hildebrandt 2011, Wolf 2015]。终末期肾病的发病年龄可以变化,从而确定婴儿,少年和青年等亚型。大约25%的青少年肾病患者中发现纯合的,大约290-kb的 NPHP1缺失[Hoefele et al 2005, Saunier et al 2005, Hildebrandt et al 2009]并且是一小部分JS患者的致病原因。注意:最常见的形式青少年肾痨,也可以是JS的肾脏表现。相反,据估计10%的肾病患者有肾外发现,在某些情况下可能包括臼齿征 [Saunier et al 2005]。
口-面-指综合症描述了一组异质性疾病,其特征是面部,口腔异常(通常分叶状舌和口腔系带),以及多指畸形等异常。基于其他相关的临床特征,已经描述了至少13种临床亚型。这些特征也与Meckel综合征,短肋骨多发综合症和JS重叠。到目前为止为OFD鉴定的基因中,所有基因都具有纤毛作用,并且几个与JS重叠。
I型口-面-指综合征 (OFD1)与原发性纤毛功能障碍有关,其特征是以下异常:
- 口腔(舌裂,错构瘤或舌头脂肪瘤,硬腭或软腭裂,附属牙龈鞘,缺牙症和其他牙齿异常)
- 面部(眼距过宽或内眦距过宽,鼻翼发育不全,中唇裂或假性上唇裂,小颌畸形)
- 数字(不同程度的指过短,并指,和第五指的弯曲变形;重复拇趾[大脚趾]; 手轴前或轴后多指)
- 脑(脑内囊肿,胼胝体发育不全,伴有或不伴有Dandy-Walker畸形的小脑发育不全)
- 肾(多囊肾病)
高达50%的OFD1患者具有一定程度的智力残疾,通常是轻微的。几乎所有患者都是女性。然而,已经描述了患有OFD1的男性,主要是由患有OFD1的女性生育的畸形胎儿。
值得注意的是,表型谱得到了扩展,已认识到四个个体所描述的临床特征(胎儿水肿,黄疸,快速深腱反射,癫痫发作和左肺三叶) [Terespolsky et al 1995, Brzustowicz et al 1999]与OFD1中的致病变异相关[Budny et al 2006]。OFD1中的致病变异也已在具有JS的罕见男性和OFD的特征中描述[Coene et al 2009, Field et al 2012]。
口-面-指综合症IV型(OFD IV,Mohr-Majewski综合征)(OMIM 258860)的特征是拇指和轴后多指并指,胫骨发育不良,可变短肋骨,囊性肾和脑异常。TCTN3中的致病性截短变异在几个家系中被鉴定出来,这些家系具有严重的致死性OFD IV表型和长骨弯曲,囊性肾,枕骨性脑膨出和肝脏胆管增生,但没有短肋骨; 这些胎儿中的一些还显示出小脑蚓部发育不全,提示臼齿征 [Thomas et al 2012]。值得注意的是,这种表型与梅克尔综合征和JS重叠。
口-面-指综合症VI型(OFD VI,Varadi-Papp综合征)(OMIM 277170)。患有OFD VI的个体通常具有中心多指,其中额外的指出现在中心指之间,并且通常伴有Y形掌骨,以及小脑蚓部发育不全,口腔系带,舌裂或错构瘤(图2B),以及颅面特征包括眼距过宽和中线唇裂。已经描述了肾脏和心脏受累 [Münke et al 1990]。可能导致咀嚼,吞咽和呼吸问题。OFD VI被定义为JS的一种形式,需要MTS以及以下一个或多个特征:舌错构瘤/口腔系带/上唇缺口,中间多指和下丘脑错构瘤 [Poretti et al 2012]。一个小组鉴定了9/11 OFD VI 家系中CPLANE1的致病变异 [Lopez et al 2014]。轴前和/或中央多指和下丘脑错构瘤的特征更可能与CPLANE1致病变异相关,而舌错构瘤和舌系带与该基因的致病变异无关[Lopez et al 2014, Romani et al 2015]。另一组在OFD VI的17个个体中仅有2个发现了CPLANE1致病变异; 在OFD VI中也报道了TMEM216,TMEM107和OFD1中的致病变异[Romani et al 2015, Lambacher et al 2016]。
鉴别诊断
管理
初步诊断后的评估
为了确定被诊断患有Joubert综合征(JS)的个体的疾病程度,建议进行以下基本评估以确定患病的婴儿/儿童的疾病程度 [Parisi et al 2007] (全文)。建议由共识小组制定,并在Joubert综合症和相关疾病基金会网站上列出。
- 高质量的MRI扫描检查,以评估脑畸形,神经元迁移障碍或头颅畸形,如果在诊断时没有做到,可预示较差的预后或癫痫发作
- 基本神经系统评估,特别注意声调,呼吸模式(呼吸急促和呼吸暂停),眼球运动,发育和小脑功能
- 以多导睡眠图作为基本评估的睡眠史,特别是如果存在症状性呼吸暂停
- 通过语言治疗师和/或透视吞咽研究评估语言功能
- 使用适合年龄的工具进行发育评估
- 小儿眼科医生通过散瞳检查评估眼缺损和视网膜变化,以及斜视和上睑下垂,并考虑视觉诱发电位,视网膜电图和眼球运动测试等专业测试
- 腹部超声检查,以评估肝纤维化或肾囊肿和/或与肾痨相关的发现(例如皮质髓质分化丧失)
- 测试肾功能,包括血压,血尿素氮(BUN),血清肌酐浓度,全血细胞计数(CBC)和尿液分析测试浓缩能力(如果可行的话)
- 肝功能检查包括转氨酶血清浓度,白蛋白,胆红素和凝血酶原时间
- 对于患有小阴茎的男性或任何有生长激素缺乏迹象的儿童,对其他垂体异常进行内分泌评估
- 如果怀疑骨骼发育不良,如短肋多指或JATD,进行骨骼检查和/或肢体X线检查
- 咨询临床遗传学家,记录家庭史,评估生长和头部大小,并评估其他异常情况,包括多指畸形,畸形面部特征,舌肿瘤/分叶和小阴茎
治疗表现
呼吸
- 如果异常严重,应考虑对呼吸暂停异常的婴儿和儿童进行呼吸暂停监测。支持疗法可能包括刺激性药物,如咖啡因或补充氧气,特别是在新生儿期。
- 在某些情况下,通过使用以下方法,可以在有明显呼吸紊乱的婴儿的外科手术过程中进行麻醉管理:
- 不用阿片类药物的局部麻醉,以避免呼吸暂停发作的加剧[Vodopich & Gordon 2004];
- α-2激动剂如可乐定或右美托咪定可以避免呼吸抑制和阿片类药物的其他并发症,同时实现无运动 [Vodopich & Gordon 2004]。
- 在极少数情况下,可能会在患有严重呼吸功能障碍的儿童中考虑机械支持和/或气管切开术。
- 积极治疗中耳感染可以避免传导性听力损失。
肌张力减退的治疗干预
- 语言治疗师对语言障碍的适当管理和治疗
- 鼻胃管饲管或胃造口管放置用于喂养患有严重吞咽困难的儿童
- 通过早期干预计划进行职业,身体和言语治疗
- 个性化的教育评估和对学龄儿童的支持,以最大限度地提高学校的表现
- 适当年龄的定期神经心理学和发育测试
其他CNS畸形
- 神经外科咨询适用于有脑积水证据(头围快速增大和/或囟门膨出)的患者。注意:当JS发生脑积水时,很少需要分流。
- 后颅窝囊肿和液体收集很少需要干预。
- 脑膨出可能需要手术闭合。
- 癫痫发作应由神经科医生使用标准抗癫痫药物进行评估和治疗。
- 各种精神药物已被用于治疗Joubert综合征的行为并发症; 没有一种药物对所有儿童都有效。
眼科
- 根据需要手术治疗症状性上睑下垂,斜视或弱视
- 屈光不正的矫正镜片
- 虽然在这种疾病中缺乏特定的研究,但是对于动眼神经失用可用视觉治疗
- 当存在先天性失明或进行性视网膜营养不良时,对视力受损者进行干预
肾病
- 咨询肾脏病专家。
- 由肾痨引起的终末期肾病(ESRD)经常需要在青少年期间或之后进行透析和/或肾移植。
- ESRD的高血压,贫血和其他并发症需要特殊治疗。
肝纤维化
- 咨询胃肠病学家。
- 肝功能衰竭和/或纤维化应由胃肠病学家进行管理,并安排进行外科手术,如门静脉分流治疗食管静脉曲张和门静脉高压症。
- 有些人需要原位肝移植。
骨骼
- 多指的手术治疗
- 骨科专家对脊柱侧凸进行适当的医疗管理
其他
- 通过标准手术干预治疗口面裂。
- 损害正常吞咽或引起呼吸道阻塞的舌肿瘤可能需要手术切除。
- 老年人的阻塞性睡眠呼吸暂停和/或舌肥大的症状可能需要用多导睡眠图评估和/或耳鼻喉科医师评估腺样体切除术,扁桃体切除术或手术舌头缩小。有些孩子晚上使用BiPAP或C-PAP。
- 与内分泌科医生就月经不调和垂体激素缺乏(如激素替代)进行咨询。
- 应采取适当措施控制肥胖,包括饮食,运动和行为疗法
- 应采用常规疗法治疗先天性心脏缺陷和局部异常。
- 指出了先天性巨结肠疾病(如果存在)的手术矫正。
预防继发性并发症
对于具有结构性心脏异常的个体,对外科手术和牙科手术需预防性使用抗生素。
监控
由于没有统一可靠的特征可以预测Joubert综合征婴儿或幼儿可能出现的并发症,因此建议进行一些年度评估(参见Joubert Syndrome and Related Disorders Foundation网站):
- 小儿和神经系统评估和监测生长,性成熟,呼吸(包括呼吸暂停症状)和运动功能
- 酌情进行神经心理学和发育评估和测试
- 眼科评估视力,跟踪能力和视网膜营养不良的发展
- 腹部超声检查评估可能的肝肾异常
- 肝功能检查
- 肾功能的评估:血压的测量,血清BUN和肌酐的浓度,CBC,以及晨尿的尿液评估
要避免的药剂/情况
肾功能不全的个体应避免使用肾毒性药物,如非甾体类抗炎药。
肝功能受损的个体应避免使用肝毒性药物。
风险亲属的评估
具有与JS的个体相似的临床特征的同胞或亲属需要遗传咨询。如果已在先证者中鉴定出致病变异,在有症状亲属中检测这些致病变异是可行的。
有关为有风险亲属遗传咨询的问题,请参阅遗传咨询。
正在研究的疗法
搜索美国的ClinicalTrials.gov和欧洲的www.ClinicalTrialsRegister.eu,了解各种疾病和病症的临床研究信息。注意:这种疾病可能没有临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。以下部分涉及遗传风险评估以及使用家庭史和基因检测来阐明家庭成员的遗传状况。本节不是为了解决个人可能面临的所有个人,文化或道德问题,也不是为了替代与遗传专业人士的咨询。-ED。
遗传方式
Joubert综合征(JS)主要以常染色体隐性方式遗传。
OFD1 -相关JS为X连锁方式遗传(参见linkaway为X连锁遗传的讨论)。
对家庭成员的风险-常染色体隐性遗传
先证者的父母
- 患病儿童的父母是必然杂合子(即,JS相关基因中的一种致病变异的携带者)。
- 杂合子是无症状的,并且没有患上这种疾病的风险。
先证者的兄弟
- 在受孕时,患者的每个同胞有25%的机会受到影响,有50%的机会成为无症状的携带者,25%的机会不患病且不是携带者。
- 杂合子(携带者)是无症状的,并且没有患上这种疾病的风险。
先证者的后代
- 先证者的后代是JS相关基因中致病变异的必然杂合子(携带者)。
- 尽管据报道没有任何JS患者可以生育,但在这种情况下现在已知的广泛的认知障碍可能会增加有后代个体即将出现的可能性。
其他家庭成员。先证者父母的每个同胞都有50%的风险成为JS相关基因中致病变异的携带者。
有关家庭成员风险的信息 - X连锁遗传(OFD1相关)请点击此处(pdf)。
携带者(杂合子)检测
对有风险的亲属进行携带者检测需要事先确定家庭中与JS相关的致病变异。
相关的遗传咨询问题
有关为早期诊断和治疗目的评估有风险的亲属的信息,请参阅管理,评估风险亲属。
家庭计划
- 确定遗传风险的最佳时间,携带者状态的检测以及产前检测可用性的讨论是在怀孕前进行的。
- 向患者,携带者或有携带风险的年轻人提供遗传咨询(包括对后代和生殖选择的潜在风险的讨论)是适当的。
DNA库是存储DNA的(通常从白细胞中提取),以备将来使用。因为测试方法和我们对基因,等位基因变异和疾病的理解将来可能会有所改善,所以应该考虑为患者个体储存DNA。
产前检查和植入前遗传学诊断
分子遗传学检测 一旦在患病的家庭成员中发现了与JS相关的致病变异,就可以进行产前检测,并进行胚胎植入前遗传学诊断。
医疗专业人员和家庭内部关于使用产前检查的观点可能存在差异,特别是如果考虑将检测用于终止妊娠而不是早期诊断。虽然大多数中心会认为有关产前检测的决定是父母的选择,但对这些问题的讨论是恰当的。
产前影像 已报道使用超声检查确定结构性异常如脑膨出为妊娠25%风险的JS进行早期诊断[van Zalen-Sprock et al 1996, Wang et al 1999]。更典型的是,早在妊娠中期就通过产前超声检查后颅窝和/或肾脏(用于囊肿和扩大和/或高回声肾脏)和手指(用于多指)来完成胎儿的产前诊断 [Ní Scanaill et al 1999, Aslan et al 2002, Doherty et al 2005]。实际上,JS胎儿的产前超声检查结果相对非特异性,包括颈部透明带增厚,扩大的小脑池,小脑蚓部发育不全,枕部脑膨出和脑室扩大,在缺乏家庭史的情况下难以确诊JS。此外,小脑蚓部是一种相对较晚发育的结构,并且可能直到妊娠18周才覆盖第四脑室,使得在妊娠早期难以观察到臼齿征(MTS)[Bromley et al 1994]。使用2D超声和3D超声重建与表面渲染已经在几个早在22周内没有JS家庭史的胎儿中发现MTS [Quarello et al 2014]。
通过在妊娠11至12周开始的连续产前超声成像,在妊娠20周时对小脑和其他胎儿解剖结构进行详细评估,然后在妊娠20周-22周时进行胎儿MRI成像,可在有风险的胎儿中对JS进行准确的产前诊断 [Doherty et al 2005]。在一系列胎儿患有JS的风险为25%的12次妊娠中,一个中心能够在早在22周时根据胎儿脑部结构(包括MTS)的胎儿MRI检查正确诊断三个JS患病胎儿[Saleem & Zaki 2010]。在迄今为止最早报道的诊断中,通过妊娠17至18周的胎儿MRI在两个独立的高危妊娠中发现了MTS[Saleem et al 2011]。尽管包括胎儿MRI在内的产前成像对于后颅窝异常的诊断很有用,但其对JS诊断的敏感性和特异性尚不清楚,其使用尚未得到系统评估。
对于已经生育JS的孩子的一对夫妇来说,产前诊断Joubert综合征和相关疾病的发现(例如,脑膨出,肾囊性变,多指畸形或胎儿成像的后颅窝异常)是非常重要的; 然而,缺乏这些体征并不排除Joubert综合征和相关疾病的诊断,因为成像敏感性未知和家庭内变异。
资源
GeneReviews的工作人员选择了以下疾病特异性和/或支持组织和/或注册机构,以帮助患有此疾病的个人及其家人。GeneReviews不对其他组织提供的信息负责。有关选择标准的信息,请单击此处。
- Joubert综合症及相关疾病基金会
电话: 614-864-1362
- 电子邮件: info@jsrdf.org
- My46 Trait Profile
- 国立神经疾病和中风研究所(NINDS)
PO Box 5801
Bethesda MD 20824
电话: 800-352-9424(免费电话); 301-496-5751; 301-468-5981(TTY)
- 国家医学遗传图书馆
- 失用症孩子
1501 Reedsdale Street
Suite 202
Pittsburgh PA 15233
电话: 412-785-7072
- 纤毛病联盟
英国
电话: 44 20 7387 0543
- Joubert综合症与信息和家庭交流的联系(JS-LIFE Registry)
分子遗传
Molecular Genetics和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能包含更多最新信息。- ED。
表A. Joubert综合症:基因和数据库
基因 | 染色体位置 | 蛋白 | 特定位点的数据库 | HGMD | ClinVar |
AHI1 | 6q23.3 | Jouberin | AHI1 @ LOVD | AHI1 | AHI1 |
ARL13B | 3q11.1-q11.2 | ADP-ribosylation factor-like protein 13B | ARL13B database | ARL13B | ARL13B |
B9D1 | 17p11.2 | B9 domain-containing protein 1 | B9D1@ LOVD | B9D1 | B9D1 |
B9D2 | 19q13.2 | B9 domain-containing protein 2 | B9D2 | B9D2 | |
C2CD3 | 11q13.4 | C2 domain-containing protein 3 | C2CD3 | C2CD3 | |
CC2D2A | 4p15.32 | Coiled-coil and C2 domain-containing protein 2A | CC2D2A | CC2D2A | |
CEP41 | 7q32.2 | Centrosomal protein of 41 kDa | CEP41 | CEP41 | |
CEP104 | 1p36.32 | Centrosomal protein of 104 kDa | CEP104 | CEP104 | |
CEP120 | 5q23.2 | Centrosomal protein of 120 kDa | CEP120 | CEP120 | |
CEP290 | 12q21.32 | Centrosomal protein of 290 kDa | CEP290 | CEP290 | |
CPLANE1 | 5p13.2 | Ciliogenesis and planar polarity effector 1 | C5orf42@ LOVD | CPLANE1 | CPLANE1 |
CSPP1 | 8q13.1-q13.2 | Centrosome and spindle pole-associated protein 1 | CSPP1 | CSPP1 | |
IFT172 | 2p23.3 | Intraflagellar transport protein 172 homolog | IFT172 | IFT172 | |
INPP5E | 9q34.3 | 72 kDa inositol polyphosphate 5-phosphatase | INPP5E@ LOVD | INPP5E | INPP5E |
KIAA0556 | 16p12.1 | Protein KIAA0556 | KIAA0556 | KIAA0556 | |
KIAA0586 | 14q23.1 | Protein TALPID3 | KIAA0586 | KIAA0586 | |
KIF7 | 15q26.1 | Kinesin-like protein KIF7 | KIF7 @ LOVD | KIF7 | KIF7 |
MKS1 | 17q22 | Meckel syndrome type 1 protein | MKS1@ LOVD | MKS1 | MKS1 |
NPHP1 | 2q13 | Nephrocystin-1 | NPHP1@ LOVD | NPHP1 | NPHP1 |
OFD1 | Xp22.2 | Oral-facial-digital syndrome 1 protein | OFD1@ LOVD | OFD1 | OFD1 |
PDE6D | 2q37.1 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit delta | PDE6D | PDE6D | |
POC1B | 12q21.33 | POC1 centriolar protein homolog B | POC1B | POC1B | |
RPGRIP1L | 16q12.2 | Protein fantom | RPGRIP1L | RPGRIP1L | |
TCTN1 | 12q24.11 | Tectonic-1 | TCTN1@ LOVD | TCTN1 | TCTN1 |
TCTN2 | 12q24.31 | Tectonic-2 | TCTN2 | TCTN2 | |
TCTN3 | 10q24.1 | Tectonic-3 | TCTN3 | TCTN3 | |
TMEM67 | 8q22.1 | Meckelin | TMEM67@ LOVD | TMEM67 | TMEM67 |
TMEM107 | 17p13.1 | Transmembrane protein 107 | TMEM107 | TMEM107 | |
TMEM138 | 11q12.2 | Transmembrane protein 138 | TMEM138 | TMEM138 | |
TMEM216 | 11q12.2 | Transmembrane protein 216 | TMEM216 database | TMEM216 | TMEM216 |
TMEM231 | 16q23.1 | Transmembrane protein 231 | TMEM231 | TMEM231 | |
TMEM237 | 2q33.1 | Transmembrane protein 237 | TMEM237@ LOVD | TMEM237 | TMEM237 |
TTC21B | 2q24.3 | Tetratricopeptide repeat protein 21B | TTC21B | TTC21B | |
ZNF423 | 16q12.1 | Zinc finger protein 423 | ZNF423 | ZNF423 |
数据来自以下标准数据库:基因 来自HGNC; 染色体位点 来自OMIM; 蛋白质来自UniProt。有关提供链接的数据库(Locus Specific,HGMD,ClinVar)的说明,请单击 此处。
表B. OMIM中Joubert综合症条目(在OMIM查看全部)
JOUBERT SYNDROME 1; JBTS1 | |
ARIMA SYNDROME | |
OFD1 GENE; OFD1 | |
JOUBERT SYNDROME 10; JBTS10 | |
PHOSPHODIESTERASE 6D, cGMP-SPECIFIC, ROD, DELTA; PDE6D | |
ZINC FINGER PROTEIN 423; ZNF423 | |
NEPHROCYSTIN 1; NPHP1 | |
INTRAFLAGELLAR TRANSPORT 172, CHLAMYDOMONAS, HOMOLOG OF; IFT172 | |
JOUBERT SYNDROME 2; JBTS2 | |
JOUBERT SYNDROME 3; JBTS3 | |
ABELSON HELPER INTEGRATION SITE 1; AHI1 | |
ADP-RIBOSYLATION FACTOR-LIKE 13B; ARL13B | |
JOUBERT SYNDROME 4; JBTS4 | |
TECTONIC FAMILY, MEMBER 1; TCTN1 | |
MKS1 GENE; MKS1 | |
TRANSMEMBRANE PROTEIN 67; TMEM67 | |
CENTROSOMAL PROTEIN, 290-KD; CEP290 | |
KIAA0586 GENE; KIAA0586 | |
JOUBERT SYNDROME 5; JBTS5 | |
CENTROSOMAL PROTEIN, 41-KD; CEP41 | |
JOUBERT SYNDROME 6; JBTS6 | |
RPGRIP1-LIKE; RPGRIP1L | |
KINESIN FAMILY MEMBER 7; KIF7 | |
JOUBERT SYNDROME 7; JBTS7 | |
CENTROSOME SPINDLE POLE-ASSOCIATED PROTEIN 1; CSPP1 | |
B9 DOMAIN-CONTAINING PROTEIN 2; B9D2 | |
COILED-COIL AND C2 DOMAINS-CONTAINING PROTEIN 2A; CC2D2A | |
TETRATRICOPEPTIDE REPEAT DOMAIN-CONTAINING PROTEIN 21B; TTC21B | |
JOUBERT SYNDROME 9; JBTS9 | |
JOUBERT SYNDROME 8; JBTS8 | |
INOSITOL POLYPHOSPHATE-5-PHOSPHATASE, 72-KD; INPP5E | |
TRANSMEMBRANE PROTEIN 216; TMEM216 | |
CENTROSOMAL PROTEIN, 120-KD; CEP120 | |
NEPHRONOPHTHISIS 12; NPHP12 | |
TECTONIC FAMILY, MEMBER 2; TCTN2 | |
TECTONIC FAMILY, MEMBER 3; TCTN3 | |
B9 DOMAIN-CONTAINING PROTEIN 1; B9D1 | |
JOUBERT SYNDROME 13; JBTS13 | |
TRANSMEMBRANE PROTEIN 237; TMEM237 | |
JOUBERT SYNDROME 14; JBTS14 | |
TRANSMEMBRANE PROTEIN 138; TMEM138 | |
JOUBERT SYNDROME 15; JBTS15 | |
JOUBERT SYNDROME 16; JBTS16 | |
CILIOGENESIS AND PLANAR POLARITY EFFECTOR 1; CPLANE1 | |
JOUBERT SYNDROME 17; JBTS17 | |
POC1 CENTRIOLAR PROTEIN, CHLAMYDOMONAS, HOMOLOG OF, B; POC1B | |
JOUBERT SYNDROME 18; JBTS18 | |
NEPHRONOPHTHISIS 14; NPHP14 | |
TRANSMEMBRANE PROTEIN 231; TMEM231 | |
JOUBERT SYNDROME 20; JBTS20 | |
JOUBERT SYNDROME 21; JBTS21 | |
JOUBERT SYNDROME 22; JBTS22 | |
C2 CALCIUM-DEPENDENT DOMAIN-CONTAINING PROTEIN 3; C2CD3 | |
TRANSMEMBRANE PROTEIN 107; TMEM107 | |
JOUBERT SYNDROME 23; JBTS23 | |
KIAA0556 GENE; KIAA0556 | |
JOUBERT SYNDROME 24; JBTS24 | |
CENTROSOMAL PROTEIN, 104-KD; CEP104 | |
JOUBERT SYNDROME 25; JBTS25 | |
JOUBERT SYNDROME 26; JBTS26 | |
JOUBERT SYNDROME 27; JBTS27 | |
JOUBERT SYNDROME 28; JBTS28 | |
JOUBERT SYNDROME 31; JBTS31 |
分子遗传病因
已知致病变异引起Joubert综合征(JS)的所有基因定位于初级纤毛和/或基体和中心体,它们可能在这些细胞器的形成,形态和/或功能中起作用。纤毛是膜的毛发状突起,由基体固定。
运动纤毛具有9 + 2微管轴质结构,允许运动和液体流动; 它们存在于特定的细胞类型上,如呼吸道上皮细胞和精子。初级纤毛具有9 + 0微管结构并且通常是非运动的。初级纤毛在大多数细胞类型中发现,并且似乎在细胞化学,机械感觉和细胞信号传导中起作用,包括参与分化,细胞分裂和细胞极性的WNT,sonic hedgehog(SHH)和PDGF信号传导途径。
由纤毛功能中重要的一种或多种蛋白质的缺陷引起的纤毛病,具有许多特征,包括肾病,视网膜营养不良和多指 [Badano et al 2006综述]。纤毛缺陷与特定表型的关联尚未完全阐明,但在Joubert综合征中观察到的后脑畸形中,已知SHH信号传导对于神经管的背腹图案化和小脑颗粒细胞增殖是关键的 [Doherty 2009]。
注意:在这部分JS相关基因的致病变异可解释1%以上的JS(见表1a)。在一篇链接文件中JS相关基因的致病变异可解释不到1%的JS(参见表1b)。
AHI1
基因结构。 AHI1包含28个外显子和几种可变的剪切形式。最常见的转录本全长为5,528 bp。
致病变异。已经报道了纯合无义,错义和剪切变异,缺失和插入 [Dixon-Salazar et al 2004, Ferland et al 2004, Parisi et al 2006, Romano et al 2006, Utsch et al 2006]。(有关更多信息,请参阅表A,特定位置。)
正常基因产物。1196-氨基酸蛋白,AHI1(也称为jouberin)。该蛋白质包括一个卷曲螺旋结构域,一个SH3结构域和六个WD40重复序列,并介导多种功能,包括信号转导,RNA加工和囊泡运输。
异常基因产物。AHI1功能丧失导致Joubert综合征。在存活的Ahi1缺失小鼠品系中,表型范围从围产期致死表型到早期视网膜变性,光感受器感觉纤毛和外部区段的发育失败 [Westfall et al 2010]。
CPLANE1
基因结构。该参考序列(NM_023073.3)包含53个外显子。CPLANE1预测编码3,197个氨基酸的蛋白质(NP_075561.3) [Srour et al 2012b, Srour et al 2015]。
致病变异。在来自法裔加拿大血统的许多不相关家庭的14个患病的个体中发现了8种不同的致病变异,其中几种与不同的单倍型相关联,这代表了法裔加拿大人群中不同的创始效应 [Srour et al 2012b, Srour et al 2015]。许多患病的个体是两种不同致病变异的复合杂合子。已经在荷兰人群中描述了另一种创始变异(p.Arg2904Ter) [Kroes et al 2016],并且在具有OFD VI表型的那些中描述了致病变异[Lopez et al 2014, Romani et al 2015]。
表4. 在此GeneReview中讨论的CPLANE1致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.4006C>T | p.Arg1336Trp | |
c.4804C>T | p.Arg1602Ter | |
c.6354dupT | p.Ile2119TyrfsTer2 | |
c.6407delC | p.Pro2136HisfsTer31 | |
c.7400 + 1G>A | ||
c.7477C>T | p.Arg2493Ter | |
c.8710C>T | p.Arg2904Ter | |
c.4690G>A | p.Ala1564Thr | 见脚注1 |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
1、c.4690G> A(p.Ala1564Thr)变异发生在 Srour et al [2015]提出的交替外显子(外显子40a)中。
正常基因产物。编码的蛋白质具有跨膜蛋白和卷曲螺旋结构域的特征。蛋白质组学研究表明其与神经发育中重要的蛋白质相互作用。它似乎在多种组织中广泛表达,包括中枢神经系统,但对该基因知之甚少。
异常基因产物。预计疾病相关变异导致功能丧失,异常剪切,外显子跳跃(c.7400 + 1G> A)或蛋白质预测算法预测的错义变异具有破坏性 [Srour et al 2012b, Srour et al 2015]。
CC2D2A
基因结构。该基因包含38个外显子,编码1620个氨基酸的蛋白质,该蛋白质与RPGRIP1L编码的蛋白质的结构域一样。
致病变异。该基因的变异导致Meckel综合征和JSRD,包括COACH综合征变异; 缺失变异与更严重(通常是致死的)Meckel综合征表型相关[Mougou-Zerelli et al 2009]。在法裔加拿大人的CC2D2A中已经鉴定了几种致病变异和可能的创始效应[Srour et al 2012b, Srour et al 2015]。
正常基因产物。该蛋白质具有卷曲螺旋和C2钙结合结构域,并且似乎在纤毛形成中起关键作用。多个转录物来自可变剪切。CC2D2A定位于基体并与CEP290相互作用 [Gorden et al 2008]。
异常基因产物。CC2D2A蛋白的缺失导致人类疾病。
斑马鱼同源物的功能丧失导致前房囊肿(相当于肾囊肿)和其他与纤毛功能障碍一致的变化 [Gorden et al 2008]。
CEP290
基因结构。该基因包含54个外显子,跨越93.2kb的基因组DNA,转录本全长为7972bp。可变剪切导致几种不同的亚型。
致病变异。在CEP290中已经鉴定出超过100种不同的致病变异,其中绝大多数预测是截短的(112个中有40个无义和48个移码)。还鉴定了与JS相关的一个大的杂合部分缺失,但是大多数截短变异是由小的插入或缺失引起的。只有三种变异是错义;20种影响剪切[Coppieters et al 2010]。
与CEP290中的致病变异相关的表型谱是广泛的,包括LCA,肾痨,Senior-Løken综合征,JS,Meckel综合征和Bardet-Biedl综合征(参见表3)。虽然很难确定明确的基因型- 表型相关性,但已经描述了一些有限的关联,并在基因座特异性数据库CEP290base中进行了总结[Coppieters et al 2010]。
正常基因产物。 CEP290编码290kd的中心体蛋白(也称为nephrocystin-6),其包含2479个氨基酸残基。Nephrocystin-6是一种调节ATF4活性的中心体蛋白,ATF4是一种与肾囊肿形成有关的转录因子。该蛋白含有13个推定的卷曲螺旋结构域,与SMC同源的区域(染色体的结构维持)ATP酶,6个KID基序,3个原肌球蛋白同源结构域和ATP / GTP结合位点基序A。该蛋白定位于中心体和纤毛并具有N-糖基化,酪氨酸硫酸化,磷酸化,N-豆蔻酰化和酰胺化的位点。Nephrocystin-6也被证明可与其他JSRD相关蛋白相互作用,包括CC2D2A和meckelin [Gorden et al 2008, Leitch et al 2008, Tallila et al 2008]。
异常基因产物。CEP290功能丧失会导致疾病。斑马鱼的敲减实验导致小脑,肾和视网膜发育异常 [Sayer et al 2006]。有证据表明,这种蛋白在小鼠胚胎发生过程中在小脑中表达[Valente et al 2006b]。已经在rd16小鼠和阿比西尼亚猫中鉴定了两种在cep290中具有致病变异的自然产生的动物模型; 两者都表现出进行性视网膜变性但没有肾或小脑缺陷 [Coppieters et al 2010]。
CSPP1
基因结构。 CSPP1编码一个短的876个氨基酸亚型和一个长的1221个氨基酸亚型[Patzke et al 2006, Tuz et al 2014]。
致病变异。无义截短,移码截短和剪切位点变异构成已报道的致病变异的大部分并且分布在整个蛋白质中[Akizu et al 2014, Tuz et al 2014]。已经描述了导致异常剪切和引入下游移码的一种错义变异[Tuz et al 2014]。似乎没有任何明确的基因型-表型相关性来解释该基因中具有致病变异的个体的广泛表型。
表5. 在此GeneReview中讨论的CSPP1致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.363_364delTA | p.His121GlnfsTer22 |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
正常基因产物。该基因编码的中心体纺锤体相关蛋白1(CSPP1)为101.5 kd(长亚型为142 kd),含有5个卷曲螺旋结构域。它与中心体相互作用,在细胞周期进程和有丝分裂的纺锤体组织中发挥作用 [Patzke et al 2006]。
异常基因产物。来自患有CSPP1相关JS的患病个体的成纤维细胞显示出纤毛发生缺陷,纤毛较少和/或短纤维[Tuz et al 2014]。还注意到sonic hedgehog信号传导受损[Shaheen et al 2014]。
INPP5E
基因结构。 INPP5E包含9个外显子和3440bp的mRNA,编码644个氨基酸的蛋白质。
致病变异。该基因中催化活性磷酸酶结构域内的错义变异引起某些形式的JSRD [Bielas et al 2009]。在具有Bardet-Biedl综合征样MORM综合征的一个家庭中,鉴定的致病变异导致蛋白质的提前截短和末端18个氨基酸的缺失 [Jacoby et al 2009]。
正常基因产物。该基因编码的蛋白质是72-kd肌醇多磷酸5-磷酸酶(也称为肌醇1,4,5-三磷酸[InsP3] 5-磷酸酶),这种酶通过动员细胞内钙参与磷脂酰肌醇信号传导,并作为第二信使介导细胞对各种刺激的反应。这种酶定位于初级纤毛的中央核心,影响磷脂酰肌醇的代谢和稳定性 [Jacoby et al 2009]。
异常基因产物。JS相关致病变异损害酶的5-磷酸酶活性并改变纤毛磷脂酰肌醇比率,使纤毛不稳定。具有同源基因纯合缺失的小鼠在出生后不久就会死亡,并表现出眼球突出,多指,囊性肾,骨骼异常,腭裂和脑异常,如露脑畸形[Jacoby et al 2009]。末端18个氨基酸的缺失似乎影响了纤毛内蛋白质的定位[Jacoby et al 2009]。
KIAA0586
基因结构。 KIAA0586(TALPID3)包含34个外显子并编码的1644个氨基酸的蛋白质(其最长亚型),已报道至少6种亚型 [Roosing et al 2015]。
致病变异。导致广泛表型的致病变异通常是截短变异或偶尔是错义变异 [Alby et al 2015, Bachmann-Gagescu et al 2015b, Malicdan et al 2015, Roosing et al 2015]。
一种相对常见的致病变异(c.428delG)在一般人群中的频率预计为1/300 [Roosing et al 2015],并且在几个队列中,尚未鉴定出第二种可能的致病变异[Bachmann-Gagescu et al 2015b, Roosing et al 2015]。
值得注意的是 ,Malicdan et al [2015] 鉴定了KIAA0586存在一个复发多外显子缺失,导致蛋白质的提前终止 ; 目前尚不清楚是否在其他群体报告的队列中是否评估了如此大规模的基因内缺失。
表6. 在此GeneReview中讨论的KIAA0586致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.428delG | p.Arg143LysfsTer4 | NM_001244189.1 |
del exon 8-exon 10 |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
正常基因产物。 KIAA0586编码一种中心体蛋白,预测其具有四个卷曲螺旋结构域和一个富含C端脯氨酸的结构域。它是纤毛发生和hedgehog信号传导所必需的。鸡中的直系同源蛋白是TALPID3,它对于发育中的鸡的四肢,神经管和体节中的sonic hedgehog转导是必需的[Malicdan et al 2015]。
异常基因产物。KIAA0586协助在中心体远端环状结构的组装,以介导蛋白质运输到纤毛;KIAA0586的丢失导致纤毛囊泡的形成和中心体迁移的失败。在鸡胚,突变小鼠和斑马鱼胚胎中KIAA0586表达的破坏导致细胞缺乏初级纤毛并导致面部,肢体和神经管缺陷[Malicdan et al 2015]。一系列的KIAA0586致病变异全部发生在中心体定位所必需的高度保守结构域之前 [Malicdan et al 2015]。此外,来自KIAA0586致病变异患者的成纤维细胞表现出纤毛减少,纤毛较短,以及sonic hedgehog信号改变[Alby et al 2015, Malicdan et al 2015]。
MKS1
基因结构。 MKS1的长度为21170个碱基,包括18个外显子,并编码559个氨基酸的蛋白。多种转录物编码该基因的不同亚型。
致病变异。该基因已经描述了多种错义,无义和其他截短变异,包括11个JS的个体中的4个中检测到该基因的一个复发致病变异(p.Ser372del)[Romani et al 2014, Slaats et al 2016]。对于具有更严重的Meckel综合征表型的个体,预测MKS1变异比具有JS的那些更具破坏性,JS通常在该基因中携带至少一个非截短变异。
表7. 在此GeneReview中讨论的MKS1致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.1115_1117delCCT | p.Ser372del | NM_017777.3 |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
正常基因产物。由该基因编码的蛋白质定位于基体的过渡区,并且是在纤毛细胞中形成初级纤毛所必需的。该基因中更严重的变异导致1型Meckel综合征和13型Bardet-Biedl综合征。
异常基因产物。通过测试该基因中具有JS致病变异的个体的成纤维细胞,Slaats et al [2016] 观察到细胞纤毛数量正常或减少,其长度比来自对照个体的纤毛更多变。此外,关键纤毛蛋白ARL13B和INPP5E的分布也发生了改变; INPP5E通常以依赖ARL13B的方式沿着纤毛分布,需要功能性过渡区,似乎在这些个体中存在缺陷[Slaats et al 2016]。
NPHP1
基因结构。 NPHP1包含20个外显子; 其cDNA为3,713 bp。该基因位于两个大的反向重复元件侧翼区域,编码nephrocystin-1。
致病变异。除了纯合的290-kb缺失包含NPHP1和BENE基因的一部分之外 [Saunier et al 2000, Parisi et al 2004a],NPHP1中偶发的单核苷酸变异也被鉴定[Hoefele et al 2005]。(有关更多信息,请参阅表A.)一些比家庭性幼年型肾病1型或 Senior-Løken综合征表型更严重的个体(见表3)具有纯合的NPHP1缺失以及AHI1或CEP290的杂合变化,提示修饰基因的贡献 [Tory et al 2007]。
正常基因产物。Nephrocystin-1是一种733个氨基酸的蛋白质,具有src同源结构域3(SH3)结构域,可介导与其他蛋白质的相互作用。Nephrocystin似乎定位于细胞的原始纤毛,细胞-细胞粘附连接点和基体,可能在细胞分裂和细胞-细胞和细胞-基质粘附信号传导中起作用[Hildebrandt et al 2009]。Nephrocystin与AHI1蛋白以及蛋白质INVS,NPHP3和NPHP4相互作用,这些蛋白由在其他形式的肾痨中突变的基因编码。
异常基因产物。Nephrocystin -1与许多其他纤毛蛋白的结合及其定位于肾上皮中的纤毛/基体提示其在肾小管发育中的关键作用。
RPGRIP1L
基因结构。该基因包含26个外显子和3948bp,编码1315个氨基酸的蛋白质。
致病变异。已经确定了各种各样的错义,无义和剪切变异。一般而言,更严重的截短变异与致死的Meckel综合征表型相关,而不太严重的变异导致JSRD,包括COACH变异 [Delous et al 2007, Wolf et al 2007]。此外,p.Ala229Thr变异与由其他基因的致病变异引起的纤毛病患者的视网膜变性的发展相关 [Khanna et al 2009]。
表8. 在此GeneReview中讨论的RPGRIP1L致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.685G>A | p.Ala229Thr |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
正常基因产物。由该基因编码的蛋白质具有卷曲螺旋结构域,C2钙结合结构域,RPGR(视网膜色素变性GTP酶调节剂)相互作用结构域和中心体蛋白相关结构域。它可以定位于基体-中心体复合体或纤毛细胞中的初级纤毛和中心体。该蛋白质与nephrocystin-4相互作用,在一些形式的肾痨和Senior-Løken综合征中有缺陷[Arts et al 2007]。已经鉴定了两种转录物编码不同蛋白质亚型的RPGRIP1L。
异常基因产物。RPGRIP1L功能丧失与疾病有关。此外,p.Ala229Thr变化似乎改变了RPGRIP1L编码的蛋白质与RPGR蛋白质的相互作用,导致感光细胞的丧失 [Khanna et al 2009]。
TCTN2
基因结构。 TCTN2(构造家庭成员2)包含18个外显子并编码几种转录物,其中最长的是697个氨基酸。该基因编码N端信号肽和在果蝇直系同源物中保守的C末端跨膜结构域 [Reiter & Skarnes 2006]。
致病变异。该基因中的无义,移码和剪切位点变异与JSRD和MKS有关[Sang et al 2011, Shaheen et al 2011]。
正常基因产物。 Tectonic-2。在小鼠中,已知Tctn2蛋白调节hedgehog信号传导和纤毛发生。它与Mks1和Cc2d2a相互作用。
异常基因产物。TCNTN2功能丧失与疾病有关。已经提出涉及NPHP,JS和MKS蛋白的纤毛“相互作用组”的概念来解释纤毛结构的模块性质以及参与多种细胞过程的蛋白质相互作用的不同功能 [Sang et al 2011]。
TMEM67(MKS3)
基因结构。该基因包含28个外显子,跨越62.0kb的基因组DNA,转录本全长为3,467bp [Smith et al 2006]。至少存在一种29个外显子的可变剪切形式,长度为3,280bp,编码具有995个氨基酸残基的蛋白质[ Ensembl数据库 ]。
致病变异。在具有Joubert综合征和相关病症的个体中鉴定的致病变异包括导致异常转录物的剪切位点变异和错义变异,两者都可能是代表比导致Meckel综合征的严重致死变异更轻微表型的亚等位基因 [Smith et al 2006, Baala et al 2007]。该基因中的致病变异在具有JS和肝脏受累的个体(COACH变异)中特别普遍 [Iannicelli et al 2010]。
正常基因产物。Meckelin是一种995个氨基酸的蛋白质,计算分子量为108 kd,预计含有信号肽,至少两个富含半胱氨酸的重复序列,以及一个490个氨基酸的细胞外区域,其次是七个跨膜结构域和一个小的30个残基的细胞质尾 [Smith et al 2006]。该蛋白定位于肾和胆管上皮细胞和其他纤毛细胞的初级纤毛和质膜,并且已经显示与Meckel综合征中涉及的MKS1蛋白相互作用。Meckelin在早期纤毛发生过程中参与了中心体向顶端细胞表面的迁移,对于纤毛发育和功能至关重要[Dawe et al 2007]。
异常基因产物。TMEM67功能丧失与疾病有关。自发大鼠突变TMEM67单核苷酸变异wpk / wpk表现出多囊肾和脑积水伴有胼胝体发育不全[Smith et al 2006]。在鼠自发性缺失突变中观察到相当的表型,其通常因多囊肾在三周时死亡; 有些还患上脑积水 [Cook et al 2009]。
TMEM216
基因结构。 TMEM216包含六个外显子。最长的剪切亚型(NM_001173990)编码148个氨基酸的蛋白质。有多种剪切变异。TMEM216和TMEM138之间的23kb基因间区域似乎协调了这两种纤毛基因的表达,两个基因都可以引发JS [Lee et al 2012b]。
致病变异。致病变异包括错义,无义和剪切变异。一种常见变异(c.218G> T)导致蛋白质变化p.Arg73Leu,似乎是德裔犹太人群中的创始变异,携带频率为1:92至1:100 [Edvardson et al 2010, Valente et al 2010]。致病变异,其中许多被预测产生截短的蛋白质,也导致致死的梅克尔综合征表型 [Valente et al 2010]。
表9. 在此GeneReview中讨论的TMEM216致病变异
DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
c.218G>T | p.Arg73Leu |
关于变异分类的注释:表中列出的变异由作者提供。GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen .hgvs.org)的标准命名惯例。有关术语的解释,请参见快速参考。
正常基因产物。最长的亚型是跨膜蛋白216,一种含有四个疏水跨膜结构域的四跨膜蛋白。这些蛋白质似乎调节其他伴侣蛋白质的信号传导和运输特性,包括Wnt受体。TMEM216定位于初级纤毛的基部并与meckelin形成复合物,meckelin是由TMEM67编码的另一种JSRD缺陷的跨膜蛋白[Valente et al 2010]。此外,TMEM216和TMEM138是纤毛发生所必需的,因为每个都定位于不同的囊泡池,其携带从高尔基体到初级纤毛的纤毛组装所必需的蛋白质[Lee et al 2012b]。
异常基因产物。斑马鱼中tmem216的破坏导致原肠胚形成缺陷以及典型的纤毛功能改变的其他变化[Valente et al 2010]。
参考文献
出版的指南和共识
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Available online. 2007. Accessed 6-23-17. [PubMed: 17377524]
引用的文献
- Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, Sirelkhatim MO, Al-Hassnan Z, Alkuraya B, Mohamed JY, Al-Salem A, Alrashed M, Faqeih E, Softah A, Al-Hashem A, Wali S, Rahbeeni Z, Alsayed M, Khan AO, Al-Gazali L, Taschner PE, Al-Hazzaa S, Alkuraya FS. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012;20:420 - 7. [PMC free article: PMC3306854] [PubMed: 22353939]
- Akizu N, Silhavy JL, Rosti RO, Scott E, Fenstermaker AG, Schroth J, Zaki MS, Sanchez H, Gupta N, Kabra M, Kara M, Ben-Omran T, Rosti B, Guemez-Gamboa A, Spencer E, Pan R, Cai N, Abdellateef M, Gabriel S, Halbritter J, Hildebrandt F, van Bokhoven H, Gunel M, Gleeson JG. Mutations in CSPP1 lead to classical Joubert syndrome. Am J Hum Genet. 2014;94:80 - 6. [PMC free article: PMC3882909] [PubMed: 24360807]
- Alby C, Piquand K, Huber C, Megarbané A, Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G, Bessières B, El Chehadeh-Djebbar S, Laurent N, Faivre L, Sztriha L, Zombor M, Szabó H, Failler M, Garfa-Traore M, Bole C, Nitschké P, Nizon M, Elkhartoufi N, Clerget-Darpoux F, Munnich A, Lyonnet S, Vekemans M, Saunier S, Cormier-Daire V, Attié-Bitach T, Thomas S. Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am J Hum Genet. 2015;97:311 - 8. [PMC free article: PMC4573270] [PubMed: 26166481]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Märker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882 - 8. [PubMed: 17558407]
- Aslan H, Ulker V, Gulcan EM, Numanoglu C, Gul A, Agar M, Ark HC. Prenatal diagnosis of Joubert syndrome: a case report. Prenat Diagn. 2002;22:13 - 6. [PubMed: 11810643]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186 - 94. [PMC free article: PMC1785313] [PubMed: 17160906]
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, O'Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O'Day D, Alswaid A, Ramadevi A R, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla À, Ozyürek H, Haliloğlu G, Tuysuz B, Topçu M, Chance P, Parisi MA, Glass IA, Shendure J, Doherty D, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015a;52:514 - 22. [PMC free article: PMC5082428] [PubMed: 26092869]
- Bachmann-Gagescu R, Ishak GE, Dempsey JC, Adkins J, O'Day D, Phelps IG, Gunay-Aygun M, Kline AD, Szczaluba K, Martorell L, Alswaid A, Alrasheed S, Pai S, Izatt L, Ronan A, Parisi MA, Mefford H, Glass I, Doherty D. Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet. 2012;49:126 - 37. [PubMed: 22241855]
- Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, Boyle EA, Wilson M, Marques Lourenço C, Arslan M. University of Washington Center for Mendelian Genomics, Shendure J, Doherty D. KIAA0586 is mutated in Joubert syndrome. Hum Mutat. 2015b;36:831 - 5. [PMC free article: PMC4537327] [PubMed: 26096313]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125 - 48. [PubMed: 16722803]
- Badhwar A, Andermann F, Valerio RM, Andermann E. Founder effect in Joubert Syndrome. Ann Neurol. 2000;48:435 - 6.
- Baskin E, Kayiran SM, Oto S, Alehan F, Agildere AM, Saatci U. Cerebellar vermis hypoplasia in a patient with Bardet-Biedl syndrome. J Child Neurol. 2002;17:385 - 7. [PubMed: 12150587]
- Beck BB, Phillips JB, Bartram MP, Wegner J, Thoenes M, Pannes A, Sampson J, Heller R, Göbel H, Koerber F, Neugebauer A, Hedergott A, Nürnberg G, Nürnberg P, Thiele H, Altmüller J, Toliat MR, Staubach S, Boycott KM, Valente EM, Janecke AR, Eisenberger T, Bergmann C, Tebbe L, Wang Y, Wu Y, Fry AM, Westerfield M, Wolfrum U, Bolz HJ. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum Mutat. 2014;35:1153 - 62. [PMC free article: PMC4425427] [PubMed: 25044745]
- Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P, Hildebrandt F. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr. 2000;136:828 - 31. [PubMed: 10839884]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, Schurmans S, Dallapiccola B, Majerus PW, Valente EM, Gleeson JG. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032 - 6. [PMC free article: PMC2746682] [PubMed: 19668216]
- Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57 - 66. [PubMed: 576733]
- Boycott KM, Parboosingh JS, Scott JN, McLeod DR, Greenberg CR, Fujiwara TM, Mah JK, Midgley J, Wade A, Bernier FP, Chodirker BN, Bunge M, Innes AM. Meckel syndrome in the Hutterite population is actually a Joubert-related cerebello-oculo-renal syndrome. Am J Med Genet A. 2007;143A:1715 - 25. [PubMed: 17603801]
- Braddock BA, Farmer JE, Deidrick KM, Iverson JM, Maria BL. Oromotor and communication findings in joubert syndrome: further evidence of multisystem apraxia. J Child Neurol. 2006;21:160 - 3. [PubMed: 16566884]
- Braddock SR, Henley KM, Maria BL. The face of Joubert syndrome: a study of dysmorphology and anthropometry. Am J Med Genet Part A. 2007;143A:3235 - 42. [PubMed: 18000967]
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. [PMC free article: PMC2913941] [PubMed: 20615230]
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D'Arrigo S, Emma F, Fazzi E, Gallizzi R, Gentile M, Loncarevic D, Mejaski-Bosnjak V, Pantaleoni C, Rigoli L, Salpietro CD, Signorini S, Stringini GR, Verloes A, Zabloka D, Dallapiccola B, Gleeson JG, Valente EM, et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432 - 42. [PMC free article: PMC2635428] [PubMed: 19058225]
- Bromley B, Nadel AS, Pauker S, Estroff JA, Benacerraf BR. Closure of the cerebellar vermis: evaluation with second trimester US. Radiology. 1994;1994;193:761 - 3. [PubMed: 7972820]
- Brzustowicz LM, Farrell S, Khan MB, Weksberg R. Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson-Golabi-Behmel syndrome. Am J Hum Genet. 1999;65:779 - 83. [PMC free article: PMC1377986] [PubMed: 10441586]
- Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura M, Lenzner S, Latos-Bielenska A, Ropers HH. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171 - 8. [PubMed: 16783569]
- Bulgheroni S, D'Arrigo S, Signorini S, Briguglio M, Di Sabato ML, Casarano M, Mancini F, Romani M, Alfieri P, Battini R, Zoppello M, Tortorella G, Bertini E, Leuzzi V, Valente EM, Riva D. Cognitive, adaptive, and behavioral features in Joubert syndrome. Am J Med Genet A. 2016;170:3115 - 24. [PubMed: 27530364]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170 - 9. [PMC free article: PMC2495072] [PubMed: 18674751]
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, Gee HY, Ramaswami G, Hong CJ, Hamilton BA, Cervenka I, Ganji RS, Bryja V, Arts HH, van Reeuwijk J, Oud MM, Letteboer SJ, Roepman R, Husson H, Ibraghimov-Beskrovnaya O, Yasunaga T, Walz G, Eley L, Sayer JA, Schermer B, Liebau MC, Benzing T, Le Corre S, Drummond I, Janssen S, Allen SJ, Natarajan S, O'Toole JF, Attanasio M, Saunier S, Antignac C, Koenekoop RK, Ren H, Lopez I, Nayir A, Stoetzel C, Dollfus H, Massoudi R, Gleeson JG, Andreoli SP, Doherty DG, Lindstrad A, Golzio C, Katsanis N, Pape L, Abboud EB, Al-Rajhi AA, Lewis RA, Omran H, Lee EY, Wang S, Sekiguchi JM, Saunders R, Johnson CA, Garner E, Vanselow K, Andersen JS, Shlomai J, Nurnberg G, Nurnberg P, Levy S, Smogorzewska A, Otto EA, Hildebrandt F. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;2012;150:533 - 48. [PMC free article: PMC3433835] [PubMed: 22863007]
- Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B, van Wijk E, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FP, van Bokhoven H, de Brouwer AP. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465 - 81. [PMC free article: PMC2756557] [PubMed: 19800048]
- Cook SA, Collin GB, Bronson RT, Naggert JK, Liu DP, Akeson EC, Davisson MT. A mouse model for Meckel syndrome type 3. J Am Soc Nephrol. 2009;20:753 - 64. [PMC free article: PMC2663826] [PubMed: 19211713]
- Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010;31:1097 - 108. [PubMed: 20690115]
- Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC, Fabretti F, Janecke AR, Ebermann I, Nürnberg G, Nürnberg P, Zentgraf H, Koerber F, Addicks K, Elsobky E, Benzing T, Schermer B, Bolz HJ. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest. 2011;121:2662 - 7. [PMC free article: PMC3223820] [PubMed: 21633164]
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Gyapay G, Rieger S, Tönshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H, Gallagher TE, Lewis RA, Bergmann C, Otto EA, Saunier S, Scambler PJ, Beales PL, Gleeson JG, Maher ER, Attié-Bitach T, Dollfus H, Johnson CA, Green ED, Gibbs RA, Hildebrandt F, Pierce EA, Katsanis N, et al. TTC21B contributes both causal and modifying alleles across the cilopathy spectrum. Nat Genet. 2011;43:189 - 96. [PMC free article: PMC3071301] [PubMed: 21258341]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA. The Meckel-Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173 - 86. [PubMed: 17185389]
- Dekaban AS. Hereditary syndrome of congenital retinal blindness (Leber), polycystic kidneys and maldevelopment of the brain. Am J Ophthalmol. 1969;68:1029 - 37. [PubMed: 5362879]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Berthélémé JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Rüther U, Schneider-Maunoury S, Attié-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875 - 81. [PubMed: 17558409]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556 - 61. [PMC free article: PMC1559533] [PubMed: 16909394]
- Deonna T, Ziegler AL. Cognitive development and behavior in Joubert syndrome. Biol Psychiatry. 1993;33:854 - 5. [PubMed: 8373928]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979 - 87. [PMC free article: PMC1182159] [PubMed: 15467982]
- Doherty D. Joubert syndrome: Insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009;16:143 - 54. [PMC free article: PMC2804071] [PubMed: 19778711]
- Doherty D, Glass IA, Siebert JR, Strouse PJ, Parisi MA, Shaw DW, Chance PF, Barr M Jr, Nyberg D. Prenatal diagnosis in pregnancies at risk for Joubert syndrome by ultrasound and MRI. Prenat Diagn. 2005;25:442 - 7. [PubMed: 15966043]
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl W, Gentile M, Gorden NT, Hikida A, Knutzen D, Özyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA. Mutations in 3 genes (MKS3, RPGRIP1L, and CC2D2A) cause COACH syndrome/Joubert syndrome with congenital hepatic fibrosis. J Med Genet. 2010;47:8 - 21. [PMC free article: PMC3501959] [PubMed: 19574260]
- Edvardson S, Shaag A, Zenvirt S, Erlich Y, Hannon GJ, Shanske AL, Gomori JM, Ekstein J, Elpeleg O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86:93 - 7. [PMC free article: PMC2801745] [PubMed: 20036350]
- Farmer JE, Deidrick KM, Gitten JC, Fennell EB, Maria BL. Parenting stress and its relationship to the behavior of children with Joubert syndrome. J Child Neurol. 2006;21:163 - 7. [PubMed: 16566885]
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008 - 13. [PubMed: 15322546]
- Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, Gardner A, Glubb G, Hobson L, Corbett M, Friend K, Willis-Owen S, Gecz J. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20:806 - 9. [PMC free article: PMC3376274] [PubMed: 22353940]
- Frank V, Ortiz Brüchle N, Mager S, Frints SG, Bohring A, du Bois G, Debatin I, Seidel H, Senderek J, Besbas N, Todt U, Kubisch C, Grimm T, Teksen F, Balci S, Zerres K, Bergmann C. Aberrant splicing is a common mutational mechanism in MKS1, a key player in Meckel-Gruber syndrome. Hum Mutat. 2007;28:638 - 9. [PubMed: 17437276]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, García-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776 - 84. [PMC free article: PMC3145011] [PubMed: 21725307]
- Genel F, Atlihan F, Ozdemir D, Targan S. Development of hydrocephalus in a patient with Joubert syndrome. J Postgrad Med. 2004;50:153. [PubMed: 15235220]
- Gentile M, Di Carlo A, Susca F, Gambotto A, Caruso ML, Panella C, Vajro P, Guanti G. COACH syndrome: report of two brothers with congenital hepatic fibrosis, cerebellar vermis hypoplasia, oligophrenia, ataxia, and mental retardation. Am J Med Genet. 1996;64:514 - 20. [PubMed: 8862632]
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, Graham JM Jr, Maria BL, Barkovich AJ, Dobyns WB. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125 - 34. [PubMed: 14981712]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topçu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559 - 71. [PMC free article: PMC2668034] [PubMed: 18950740]
- Gunay-Aygun M, Parisi MA, Doherty D, Tuchman M, Tsilou E, Kleiner DE, Huizing M, Turkbey B, Choyke P, Guay-Woodford L, Heller T, Szymanska K, Johnson CA, Glass I, Gahl WA. MKS3-related ciliopathy with features of autosomal recessive polycystic kidney disease, nephronophthisis, and Joubert Syndrome. J Pediatr. 2009;155:386 - 92.e1. [PMC free article: PMC2925444] [PubMed: 19540516]
- Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, Krug P, Filhol E, Davis EE, Airik R, Czarnecki PG, Lehman AM, Trnka P, Nitschké P, Bole-Feysot C, Schueler M, Knebelmann B, Burtey S, Szabó AJ, Tory K, Leo PJ, Gardiner B, McKenzie FA, Zankl A, Brown MA, Hartley JL, Maher ER, Li C, Leroux MR, Scambler PJ, Zhan SH, Jones SJ, Kayserili H, Tuysuz B, Moorani KN, Constantinescu A, Krantz ID, Kaplan BS, Shah JV, Hurd TW, Doherty D, Katsanis N, Duncan EL, Otto EA, Beales PL, Mitchison HM, Saunier S, Hildebrandt F, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93:915 - 25. [PMC free article: PMC3824130] [PubMed: 24140113]
- Hampshire DJ, Ayub M, Springell K, Roberts E, Jafri H, Rashid Y, Bond J, Riley JH, Woods CG. MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur J Hum Genet. 2006;14:543 - 8. [PubMed: 16493448]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23 - 35. [PMC free article: PMC2807379] [PubMed: 19118152]
- Hildebrandt F, Nothwang HG, Vossmerbaumer U, Springer C, Strahm B, Hoppe B, Keuth B, Fuchshuber A, Querfeld U, Neuhaus TJ, Brandis M. Lack of large, homozygous deletions of the nephronophthisis 1 region in Joubert syndrome type B. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Pediatr Nephrol. 1998;12:16 - 9. [PubMed: 9502560]
- Hodgkins PR, Harris CM, Shawkat FS, Thompson DA, Chong K, Timms C, Russell-Eggitt I, Taylor DS, Kriss A. Joubert syndrome: long-term follow-up. Dev Med Child Neurol. 2004;46:694 - 9. [PubMed: 15473174]
- Hoefele J, Sudbrak R, Reinhardt R, Lehrack S, Hennig S, Imm A, Muerb U, Utsch B, Attanasio M, O'Toole JF, Otto E, Hildebrandt F. Mutational analysis of the NPHP4 gene in 250 patients with nephronophthisis. Hum Mutat. 2005;25:411. [PubMed: 15776426]
- Holroyd S, Reiss AL, Bryan RN. Autistic features in Joubert syndrome: a genetic disorder with agenesis of the cerebellar vermis. Biol Psychiatry. 1991;29:287 - 94. [PubMed: 2015333]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, Greenberg CR, McLeod DR, Bernier FP, Chudley AE, Müller T, Shboul M, Logan CV, Loucks CM, Beaulieu CL, Bowie RV, Bell SM, Adkins J, Zuniga FI, Ross KD, Wang J, Ban MR, Becker C, Nürnberg P, Douglas S, Craft CM, Akimenko MA, Hegele RA, Ober C, Utermann G, Bolz HJ, Bulman DE, Katsanis N, Blacque OE, Doherty D, Parboosingh JS, Leroux MR, Johnson CA, Boycott KM. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713 - 30. [PMC free article: PMC3234373] [PubMed: 22152675]
- Hurd TE, Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol. 2011;118:e9 - 14. [PMC free article: PMC2992643] [PubMed: 21071979]
- Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C, Ardissino GL, Bertini E, Boltshauser E, Castorina P, D'Arrigo S, Fischetto R, Leroy B, Loget P, Bonnière M, Starck L, Tantau J, Gentilin B, Majore S, Swistun D, Flori E, Lalatta F, Pantaleoni C, Penzien J, Grammatico P, Dallapiccola B, Gleeson JG, Attie-Bitach T, Valente EM, et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat. 2010;31:E1319 - 31. [PMC free article: PMC2918781] [PubMed: 20232449]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, Gergely F, Riley JH, Pérez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027 - 31. [PubMed: 19668215]
- Janecke AR, Müller T, Gassner I, Kreczy A, Schmid E, Kronenberg F, Utermann B, Utermann G. Joubert-like syndrome unlinked to known candidate loci. J Pediatr. 2004;144:264 - 9. [PubMed: 14760273]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813 - 25. [PubMed: 5816874]
- Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123:695 - 709. [PubMed: 22331178]
- Kamdar BB, Nandkumar P, Krishnan V, Gamaldo CE, Collop NA. Self-reported sleep and breathing disturbances in Joubert syndrome. Pediatr Neurol. 2011;45:395 - 9. [PubMed: 22115003]
- Khaddour R, Smith U, Baala L, Martinovic J, Clavering D, Shaffiq R, Ozilou C, Cullinane A, Kyttälä M, Shalev S, Audollent S, d'Humières C, Kadhom N, Esculpavit C, Viot G, Boone C, Oien C, Encha-Razavi F, Batman PA, Bennett CP, Woods CG, Roume J, Lyonnet S, Génin E, Le Merrer M, Munnich A, Gubler MC, Cox P, Macdonald F, Vekemans M, Johnson CA, Attié-Bitach T, et al. Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype-phenotype correlation. Mutation in brief #960. Online. Hum Mutat. 2007;28:523 - 4. [PubMed: 17397051]
- Khan AO, Oystreck DT, Seidahmed MZ, AlDrees A, Almalik SA, Alorainy IA, Salih MA. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008;115:2286 - 9. [PubMed: 19041481]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, MacDonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attié-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739 - 45. [PMC free article: PMC2783476] [PubMed: 19430481]
- Knopp C, Rudnik-Schöneborn S, Eggermann T, Bergmann C, Begemann M, Schoner K, Zerres K, Ortiz Brüchle N. Syndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol Cell Probes. 2015;29:299 - 307. [PubMed: 26003401]
- Kroes HY, Fransen van de Putte DE, Ravesloot CJ, Lindhout D. The birth prevalence of Joubert syndrome: a population based study in the Netherlands. Eur J Hum Genet 2007;15(Suppl 1).
- Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijman IJ, Kloosterman WP, Giles RH, Knoers NV, van Haaften G. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet. 2016;24:214 - 20. [PMC free article: PMC4717206] [PubMed: 25920555]
- Kroes HY, Van Zanten BG, De Ru SA, Boon M, Mancini GM, Van der Knaap MS, Poll-The BT, Lindhout D. Is hearing loss a feature of Joubert syndrome, a ciliopathy? Int J Pediatr Otorhinolaryngol. 2010;74:1034 - 8. [PubMed: 20591505]
- Kyttälä M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestilä M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38:155 - 7. [PubMed: 16415886]
- Lambacher NJ, Bruel AL, van Dam TJ, Szymańska K, Slaats GG, Kuhns S, McManus GJ, Kennedy JE, Gaff K, Wu KM, van der Lee R, Burglen L, Doummar D, Rivière JB, Faivre L, Attié-Bitach T, Saunier S, Curd A, Peckham M, Giles RH, Johnson CA, Huynen MA, Thauvin-Robinet C, Blacque OE. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol. 2016;18:122 - 31. [PubMed: 26595381]
- Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, Marsh SE, Olvera J, Brancati F, Iannicelli M, Ikegami K, Schlossman AM, Merriman B, Attié-Bitach T, Logan CV, Glass IA, Cluckey A, Louie CM, Lee JH, Raynes HR, Rapin I, Castroviejo IP, Setou M, Barbot C, Boltshauser E, Nelson SF, Hildebrandt F, Johnson CA, Doherty DA, Valente EM, Gleeson JG. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012a;44:193 - 9. [PMC free article: PMC3267856] [PubMed: 22246503]
- Lee JH, Silhavy JL, Lee JE, Al-Gazali L, Thomas S, Davis EE, Bielas SL, Hill KJ, Iannicelli M, Brancati F, Gabriel SB, Russ C, Logan CV, Sharif SM, Bennett CP, Abe M, Hildebrandt F, Diplas BH, Attié-Bitach T, Katsanis N, Rajab A, Koul R, Sztriha L, Waters ER, Ferro-Novick S, Woods CG, Johnson CA, Valente EM, Zaki MS, Gleeson JG. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science. 2012b;335:966 - 9. [PMC free article: PMC3671610] [PubMed: 22282472]
- Lehman AM, Eydoux P, Doherty D, Glass IA, Chitayat D, Chung BY, Langlois S, Yong SL, Lowry RB, Hildebrandt F, Trnka P. Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy. Am J Med Genet A. 2010;152A:1411 - 9. [PMC free article: PMC4048012] [PubMed: 20503315]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443 - 8. [PubMed: 18327255]
- Løken AC, Hanssen O, Halvorsen S, Jolster NJ. Hereditary renal dysplasia and blindness. Acta Paediatr. 1961;50:177 - 84. [PubMed: 13763238]
- Lopez E, Thauvin-Robinet C, Reversade B, Khartoufi NE, Devisme L, Holder M, Ansart-Franquet H, Avila M, Lacombe D, Kleinfinger P, Kaori I, Takanashi J, Le Merrer M, Martinovic J, Noël C, Shboul M, Ho L, Güven Y, Razavi F, Burglen L, Gigot N, Darmency-Stamboul V, Thevenon J, Aral B, Kayserili H, Huet F, Lyonnet S, Le Caignec C, Franco B, Rivière JB, Faivre L, Attié-Bitach T. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet. 2014;133:367 - 77. [PubMed: 24178751]
- Maglic D, Stephen J, Malicdan MC, Guo J, Fischer R, Konzman D, Mullikin JC, Gahl WA, Vilboux T, Gunay-Aygun M, et al. TMEM231 gene conversion associated with Joubert and Meckel-Gruber syndromes in the same family. Hum Mutat. 2016;37:1144 - 8. [PubMed: 27449316]
- Mainzer F, Saldino RM, Ozonoff MB, Minagi H. Familial nephropathy associated with retinitis pigmentosa, cerebellar ataxia and skeletal abnormalities. Am J Med. 1970;49:556 - 62. [PubMed: 4991086]
- Malicdan MC, Vilboux T, Stephen J, Maglic D, Mian L, Konzman D, Guo J, Yildirimli D, Bryant J, Fischer R, Zein WM, Snow J, Vemulapalli M, Mullikin JC, Toro C, Solomon BD, Niederhuber JE, Gahl WA, Gunay-Aygun M, et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J Med Genet. 2015;52:830 - 9. [PMC free article: PMC5517294] [PubMed: 26386044]
- Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999a;14:583 - 90. [PubMed: 10488903]
- Maria BL, Bozorgmanesh A, Kimmel KN, Theriaque D, Quisling RG. Quantitative assessment of brainstem development in Joubert syndrome and Dandy-Walker syndrome. J Child Neurol. 2001;16:751 - 8. [PubMed: 11669349]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. "Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423 - 30. [PubMed: 9373798]
- Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, Fennell E. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999b;14:368 - 76. [PubMed: 10385844]
- Marsh SE, Grattan-Smith P, Pereira J, Barkovich AJ, Gleeson JG. Neuroepithelial cysts in a patient with Joubert syndrome plus renal cysts. J Child Neurol. 2004;19:227 - 31. [PubMed: 15119486]
- Mee L, Honkala H, Kopra O, Vesa J, Finnilä S, Visapää I, Sang TK, Jackson GR, Salonen R, Kestilä M, Peltonen L. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum Mol Genet. 2005;14:1475 - 88. [PubMed: 15843405]
- Merlini L, Vargas MI, De Haller R, Rilliet B, Fluss J. MRI with fibre tracking in Cogan congenital oculomotor apraxia. Pediatr Radiol. 2010;40:1625 - 33. [PubMed: 20449733]
- Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, Babarit C, Romano S, Salomon R, Amiel J, Esculpavit C, Gonzales M, Escudier E, Leheup B, Loget P, Odent S, Roume J, Gérard M, Delezoide AL, Khung S, Patrier S, Cordier MP, Bouvier R, Martinovic J, Gubler MC, Boddaert N, Munnich A, Encha-Razavi F, Valente EM, Saad A, Saunier S, Vekemans M, Attié-Bitach T. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat. 2009;30:1574 - 82. [PMC free article: PMC2783384] [PubMed: 19777577]
- Münke M, McDonald DM, Cronister A, Stewart JM, Gorlin RJ, Zackai EH. Oral-facial-digital syndrome type VI (Varadi syndrome): further clinical delineation. Am J Med Genet. 1990;35:360 - 9. [PubMed: 2309783]
- Ní Scanaill S, Crowley P, Hogan M, Stuart B. Abnormal prenatal sonographic findings in the posterior cranial fossa: a case of Joubert's syndrome. Ultrasound Obstet Gynecol. 1999;13:71 - 4. [PubMed: 10201091]
- Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F, Paterson AD, Lutfullah M, Vincent JB, Ayub M. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet. 2008;82:1011 - 8. [PMC free article: PMC2427291] [PubMed: 18387594]
- Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Utsch B, Sayer JA, Wolf MT, Becker C, Nurnberg G, Nurnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F. Hypomorphic mutations in Meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet. 2009;46:663 - 70. [PubMed: 19508969]
- Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behavior in Joubert syndrome. J Child Neurol. 1999;14:636 - 41. [PubMed: 10511335]
- Papon JF, Perrault I, Coste A, Louis B, Gérard X, Hanein S, Fares-Taie L, Gerber S, Defoort-Dhellemmes S, Vojtek AM, Kaplan J, Rozet JM, Escudier E. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J Med Genet. 2010;47:829 - 34. [PubMed: 20805370]
- Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet Part C Semin Med Genet. 2009;151C:326 - 40. [PMC free article: PMC2797758] [PubMed: 19876931]
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004a;75:82 - 91. [PMC free article: PMC1182011] [PubMed: 15138899]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15:511 - 21. [PubMed: 17377524]
- Parisi MA, Doherty D, Eckert ML, Shaw DWW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334 - 9. [PMC free article: PMC2563230] [PubMed: 16155189]
- Parisi MA, Pinter JD, Glass IA, Field K, Maria BL, Chance PF, Mahurin RK, Cramer SC. Cerebral and cerebellar motor activation abnormalities in a subject with Joubert syndrome: A functional MRI study. J Child Neurol. 2004b;19:214 - 8. [PubMed: 15119482]
- Patzke S, Stokke T, Aasheim HC. CSPP and CSPP-L associate with centrosomes and microtubules and differently affect microtubule organization. J Cell Physiol. 2006;209:199 - 210. [PubMed: 16826565]
- Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90:864 - 70. [PMC free article: PMC3376548] [PubMed: 22503633]
- Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F, Il'yasov K, Huisman TA. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007;28:1929 - 33. [PubMed: 17898198]
- Poretti A, Christen HJ, Elton LE, Baumgartner M, Korenke GC, Sukhudyan B, Hethey S, Cross E, Steinlin M, Boltshauser E. Horizontal head titubation in infants with Joubert syndrome: a new finding. Dev Med Child Neurol. 2014;56:1016 - 20. [PubMed: 24814865]
- Poretti A, Dietrich Alber F, Brancati F, Dallapiccola B, Valente EM, Boltshauser E. Normal cognitive functions in joubert syndrome. Neuropediatrics. 2009;40:287 - 90. [PubMed: 20446224]
- Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 2011;32:1459 - 63. [PubMed: 21680654]
- Poretti A, Snow J, Summers AC, Tekes A, Huisman TA, Aygun N, Carson KA, Doherty D, Parisi MA, Toro C, Yildirimli D, Vemulapalli M, Mullikin JC, Cullinane AR, Vilboux T, Gahl WA, Gunay-Aygun M, et al. Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause. J Med Genet. 2017. Epub ahead of print. [PubMed: 28087721]
- Poretti A, Vitiello G, Hennekam RC, Arrigoni F, Bertini E, Borgatti R, Brancati F, D'Arrigo S, Faravelli F, Giordano L, Huisman TA, Iannicelli M, Kluger G, Kyllerman M, Landgren M, Lees MM, Pinelli L, Romaniello R, Scheer I, Schwarz CE, Spiegel R, Tibussek D, Valente EM, Boltshauser E. Delineation and diagnostic criteria of Oral-Facial-Digital Syndrome type VI. Orphanet J Rare Dis. 2012;7:4. [PMC free article: PMC3313869] [PubMed: 22236771]
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, Uz E, Buzas D, Gomes C, Patrier S, Bennett CL, Elkhartoufi N, Frison MH, Rigonnot L, Joyé N, Pruvost S, Utine GE, Boduroglu K, Nitschke P, Fertitta L, Thauvin-Robinet C, Munnich A, Cormier-Daire V, Hennekam R, Colin E, Akarsu NA, Bole-Feysot C, Cagnard N, Schmitt A, Goudin N, Lyonnet S, Encha-Razavi F, Siffroi JP, Winey M, Katsanis N, Gonzales M, Vekemans M, Beales PL, Attié-Bitach T. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43:601 - 6. [PMC free article: PMC3674836] [PubMed: 21552264]
- Quarello E, Molho M, Garel C, Couture A, Legac MP, Moutard ML, Bault JP, Fallet-Bianco C, Guibaud L. Prenatal abnormal features of the fourth ventricle in Joubert syndrome and related disorders. Ultrasound Obstet Gynecol. 2014;43:227 - 32. [PubMed: 23868831]
- Quisling RG, Barkovich AJ, Maria BL. Magnetic resonance imaging features and classification of central nervous system malformations in Joubert syndrome. J Child Neurol. 1999;14:628 - 35. [PubMed: 10511334]
- Reiter JF, Skarnes WC. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev. 2006;20:22 - 7. [PMC free article: PMC1356097] [PubMed: 16357211]
- Romani M, Mancini F, Micalizzi A, Poretti A, Miccinilli E, Accorsi P, Avola E, Bertini E, Borgatti R, Romaniello R, Ceylaner S, Coppola G, D'Arrigo S, Giordano L, Janecke AR, Lituania M, Ludwig K, Martorell L, Mazza T, Odent S, Pinelli L, Poo P, Santucci M, Signorini S, Simonati A, Spiegel R, Stanzial F, Steinlin M, Tabarki B, Wolf NI, Zibordi F, Boltshauser E, Valente EM. Oral-facial-digital syndrome type VI: is C5orf42 really the major gene? Hum Genet. 2015;134:123 - 6. [PMC free article: PMC4282684] [PubMed: 25407461]
- Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, Ruta R, Mancini F, Mazza T, Castellana S, Hanene B, Carluccio MA, Darra F, Máté A, Zimmermann A, Gouider-Khouja N, Valente EM. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis. 2014;9:72. [PMC free article: PMC4113192] [PubMed: 24886560]
- Romano S, Boddaert N, Desguerre I, Hubert L, Salomon R, Seidenwurm D, Bahi-Buisson N, Nabbout R, Sonigo P, Lyonnet S, Brunelle F, Munnich A, de Lonlay P. Molar tooth sign and superior vermian dysplasia: a radiological, clinical, and genetic study. Neuropediatrics. 2006;37:42 - 5. [PubMed: 16541367]
- Roosing S, Hofree M, Kim S, Scott E, Copeland B, Romani M, Silhavy JL, Rosti RO, Schroth J, Mazza T, Miccinilli E, Zaki MS, Swoboda KJ, Milisa-Drautz J, Dobyns WB, Mikati MA, İncecik F, Azam M, Borgatti R, Romaniello R, Boustany RM, Clericuzio CL, D'Arrigo S, Strømme P, Boltshauser E, Stanzial F, Mirabelli-Badenier M, Moroni I, Bertini E, Emma F, Steinlin M, Hildebrandt F, Johnson CA, Freilinger M, Vaux KK, Gabriel SB, Aza-Blanc P, Heynen-Genel S, Ideker T, Dynlacht BD, Lee JE, Valente EM, Kim J, Gleeson JG. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. Elife. 2015;4:e06602. [PMC free article: PMC4477441] [PubMed: 26026149]
- Roosing S, Romani M, Isrie M, Rosti RO, Micalizzi A, Musaev D, Mazza T, Al-Gazali L, Altunoglu U, Boltshauser E, D'Arrigo S, De Keersmaecker B, Kayserili H, Brandenberger S, Kraoua I, Mark PR, McKanna T, Van Keirsbilck J, Moerman P, Poretti A, Puri R, Van Esch H, Gleeson JG, Valente EM. Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J Med Genet. 2016a;53:608 - 15. [PMC free article: PMC5013089] [PubMed: 27208211]
- Roosing S, Rosti RO, Rosti B, de Vrieze E, Silhavy JL, van Wijk E, Wakeling E, Gleeson JG. Identification of a homozygous nonsense mutation in KIAA0556 in a consanguineous family displaying Joubert syndrome. Hum Genet. 2016b;135:919 - 21. [PMC free article: PMC4955754] [PubMed: 27245168]
- Saleem SN, Zaki MS. Role of MR imaging in prenatal diagnosis of pregnancies at risk for Joubert syndrome and related cerebellar disorders. AJNR Am J Neuroradiol. 2010;31:424 - 9. [PubMed: 19942698]
- Saleem SN, Zaki MS, Soliman NA, Momtaz M. Prenatal magnetic resonance imaging diagnosis of molar tooth sign at 17 to 18 weeks of gestation in two fetuses at risk for Joubert syndrome and related cerebellar disorders. Neuropediatrics. 2011;42:35 - 8. [PubMed: 21500139]
- Sanders AAWM, de Vrieze E, Alazami AM, Alzahrani F, Malarkey EB, Sorusch N, Tebbe L, Kuhns S, van Dam TJP, Alhashem A, Tabarki B, Lu Q, et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015;16:293. [PMC free article: PMC4699358] [PubMed: 26714646]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O'Toole JF, Held S, Reutter HM, Lane WS, Rafiq MA, Noor A, Ansar M, Devi AR, Sheffield VC, Slusarski DC, Vincent JB, Doherty DA, Hildebrandt F, Reiter JF, Jackson PK. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513 - 28. [PMC free article: PMC3383065] [PubMed: 21565611]
- Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726 - 31. [PubMed: 1341417]
- Sargent MA, Poskitt KJ, Jan JE. Congenital ocular motor apraxia: imaging findings. AJNR Am J Neuroradiol. 1997;18:1915 - 22. [PubMed: 9403454]
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459 - 69. [PubMed: 10508989]
- Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, Antignac C. Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet. 2000;66:778 - 89. [PMC free article: PMC1288163] [PubMed: 10712196]
- Saunier S, Morin G, Calado J, et al. Large deletions of the NPH1 region in Cogan syndrome (CS) associated with familial juvenile nephronophthisis (NPH). Am J Hum Genet. 1997;61:A346.
- Saunier S, Salomon R, Antignac C. Nephronophthisis. Curr Opin Genet Dev. 2005;15:324 - 31. [PubMed: 15917209]
- Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674 - 81. [PubMed: 16682973]
- Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol. 1961;52:625 - 33. [PubMed: 13910672]
- Senocak EU, Oğuz KK, Haliloğlu G, Topçu M, Cila A. Structural abnormalities of the brain other than molar tooth sign in Joubert syndrome-related disorders. Diagn Interv Radiol. 2010;16:3 - 6. [PubMed: 20108204]
- Shaheen R, Almoisheer A, Faqeih E, Babay Z, Monies D, Tassan N, Abouelhoda M, Kurdi W, Al Mardawi E, Khalil MM, Seidahmed MZ, Alnemer M, Alsahan N, Sogaty S, Alhashem A, Singh A, Goyal M, Kapoor S, Alomar R, Ibrahim N, Alkuraya FS. Identification of a novel MKS locus defined by TMEM107 mutation. Hum Mol Genet. 2015a;24:5211 - 8. [PubMed: 26123494]
- Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, AlQahtani K, Alkuraya FS. A. TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat. 2011;32:573 - 8. [PubMed: 21462283]
- Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, Superti-Furga A, Mitchison HM, Almoisheer A, Alamro R, Alshiddi T, Alzahrani F, Beales PL, Alkuraya FS, et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015b;24:1410 - 9. [PMC free article: PMC4321448] [PubMed: 25361962]
- Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, Ibrahim Khalil M, Al-Yacoub N, Davis EE, Mola NA, Szymanska K, Herridge W, Chudley AE, Chodirker BN, Schwartzentruber J, Majewski J, Katsanis N, Poizat C, Johnson CA, Parboosingh J, Boycott KM, Innes AM, Alkuraya FS. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet. 2014;94:73 - 9. [PMC free article: PMC3882732] [PubMed: 24360803]
- Slaats GG, Isabella CR, Kroes HY, Dempsey JC, Gremmels H, Monroe GR, Phelps IG, Duran K, Adkins J, Kumar SA, Knutzen DM, Knoers NV, Mendelsohn NJ, Neubauer D, Mastroyianni SD, Vogt J, Worgan L, Karp N, Bowdin S, Glass IA, Parisi MA, Otto EA, Johnson CA, Hildebrandt F, van Haaften G, Giles RH, Doherty D. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J Med Genet. 2016;53:62 - 72. [PMC free article: PMC5060087] [PubMed: 26490104]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH, Harris PC, Johnson CA. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38:191 - 6. [PubMed: 16415887]
- Sriganesh K, Vinay B, Jena S, Sudhir V, Saini J, Umamaheswara Rao GS. Anesthetic management of patients with Joubert syndrome: a retrospective analysis of a single-institutional case series. Paediatr Anaesth. 2014;24:1180 - 4. [PubMed: 25040301]
- Srour M, Hamdan FF, McKnight D, Davis E, Mandel H, Schwartzentruber J, Martin B, Patry L, Nassif C, Dionne-Laporte A, Ospina LH, Lemyre E, Massicotte C, Laframboise R, Maranda B, Labuda D, Décarie JC, Rypens F, Goldsher D, Fallet-Bianco C, Soucy JF, Laberge AM, Maftei C, Boycott K, Brais B, Boucher RM, Rouleau GA, Katsanis N, Majewski J, Elpeleg O, Kukolich MK, Shalev S, Michaud JL, et al. Joubert Syndrome in French Canadians and Identification of Mutations in CEP104. Am J Hum Genet. 2015;97:744 - 53. [PMC free article: PMC4667103] [PubMed: 26477546]
- Srour M, Hamdan FF, Schwartzentruber JA, Patry L, Ospina LH, Shevell MI, Désilets V, Dobrzeniecka S, Mathonnet G, Lemyre E, Massicotte C, Labuda D, Amrom D, Andermann E, Sébire G, Maranda B, Rouleau GA, Majewski J, Michaud JL, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet. 2012a;49:636 - 41. [PubMed: 23012439]
- Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D, Massicotte C, Dobrzeniecka S, Capo-Chichi JM, Papillon-Cavanagh S, Samuels ME, Boycott KM, Shevell MI, Laframboise R, Désilets V, Maranda B, Rouleau GA, Majewski J, Michaud JL, et al. Mutations in C5ORF42 cause Joubert syndrome in the French-Canadian population. Am J Hum Genet. 2012b;90:693 - 700. [PMC free article: PMC3322222] [PubMed: 22425360]
- Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-up in children with Joubert syndrome. Neuropediatrics. 1997;28:204 - 11. [PubMed: 9309710]
- Su Y, Xie J, Yu S, Luo H, Wu W, Xu Z. Clinical and genetic analysis for a Joubert syndrome family with CC2D2A gene mutations. Zhonghua Er Ke Za Zhi. 2015;53:431 - 5. [PubMed: 26310553]
- Summers AC, Snow J, Wiggs E, Liu AG, Toro C, Poretti A, Zein WM, Brooks BP, Parisi MA, Inati S, Doherty D, Vemulapalli M, Mullikin JC, Vilboux T, Gahl WA, Gunay-Aygun M, et al. Neuropsychological phenotypes of 76 individuals with Joubert syndrome evaluated at a single center. Am J Med Genet A. 2017. Epub ahead of print. [PubMed: 28497568]
- Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, Ieda D, Saitoh S, Hiyane M, Suzumura H, Yagyu K, Shiraishi H, Nakajima M, Fueki N, Habata Y, Ueda Y, Komatsu Y, Yan K, Shimoda K, Shitara Y, Mizuno S, Ichinomiya K, Sameshima K, Tsuyusaki Y, Kurosawa K, Sakai Y, Haginoya K, Kobayashi Y, Yoshizawa C, Hisano M, Nakashima M, Saitsu H, Takeda S, Matsumoto N. Molecular genetic analysis of 30 families with Joubert syndrome. Clin Genet. 2016;90:526 - 35. [PubMed: 27434533]
- Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, Raashid Y, Malik-Sharif S, Castle B, Ahmed M, Bennett C, Carlton R, Johnson CA. Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. 2012;1:18. [PMC free article: PMC3579735] [PubMed: 23351400]
- Takahashi TN, Farmer JE, Deidrick KK, Hsu BS, Miles JH, Maria BL. Joubert syndrome is not a cause of classical autism. Am J Med Genet A. 2005;132A:347 - 51. [PubMed: 15633174]
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82:1361 - 7. [PMC free article: PMC2427307] [PubMed: 18513680]
- Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet. 1995;59:329 - 33. [PubMed: 8599356]
- Thomas S, Legendre M, Saunier S, Bessières B, Alby C, Bonnière M, Toutain A, Loeuillet L, Szymanska K, Jossic F, Gaillard D, Yacoubi MT, Mougou-Zerelli S, David A, Barthez MA, Ville Y, Bole-Feysot C, Nitschke P, Lyonnet S, Munnich A, Johnson CA, Encha-Razavi F, Cormier-Daire V, Thauvin-Robinet C, Vekemans M, Attié-Bitach T. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet. 2012;91:372 - 8. [PMC free article: PMC3415538] [PubMed: 22883145]
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, Saada J, Perrault I, Amiel J, Litzler J, Filhol E, Elkhartoufi N, Kwong M, Casanova JL, Boddaert N, Baehr W, Lyonnet S, Munnich A, Burglen L, Chassaing N, Encha-Ravazi F, Vekemans M, Gleeson JG, Valente EM, Jackson PK, Drummond IA, Saunier S, Attié-Bitach T. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014;35:137 - 46. [PMC free article: PMC3946372] [PubMed: 24166846]
- Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, Antignac C, Salomon R, Saunier S. High NPHP1 and NPHP6 mutation rate in patiens with Joubert syndrome and nephronophthisis: Potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566 - 75. [PubMed: 17409309]
- Travaglini L, Brancati F, Attie-Bitach T, Audollent S, Bertini E, Kaplan J, Perrault I, Iannicelli M, Mancuso B, Rigoli L, Rozet JM, Swistun D, Tolentino J, Dallapiccola B, Gleeson JG, Valente EM, Zankl A, Leventer R, Grattan-Smith P, Janecke A, D'Hooghe M, Sznajer Y, Van Coster R, Demerleir L, Dias K, Moco C, Moreira A, Kim CA, Maegawa G, Petkovic D, Abdel-Salam GM, Abdel-Aleem A, Zaki MS, Marti I, Quijano-Roy S, Sigaudy S, de Lonlay P, Romano S, Touraine R, Koenig M, Lagier-Tourenne C, Messer J, Collignon P, Wolf N, Philippi H, Kitsiou Tzeli S, Halldorsson S, Johannsdottir J, Ludvigsson P, Phadke SR, Udani V, Stuart B, Magee A, Lev D, Michelson M, Ben-Zeev B, Fischetto R, Benedicenti F, Stanzial F, Borgatti R, Accorsi P, Battaglia S, Fazzi E, Giordano L, Pinelli L, Boccone L, Bigoni S, Ferlini A, Donati MA, Caridi G, Divizia MT, Faravelli F, Ghiggeri G, Pessagno A, Briguglio M, Briuglia S, Salpietro CD, Tortorella G, Adami A, Castorina P, Lalatta F, Marra G, Riva D, Scelsa B, Spaccini L, Uziel G, Del Giudice E, Laverda AM, Ludwig K, Permunian A, Suppiej A, Signorini S, Uggetti C, Battini R, Di Giacomo M, Cilio MR, Di Sabato ML, Leuzzi V, Parisi P, Pollazzon M, Silengo M, De Vescovi R, Greco D, Romano C, Cazzagon M, Simonati A, Al-Tawari AA, Bastaki L, Mégarbané A, Sabolic Avramovska V, de Jong MM, Stromme P, Koul R, Rajab A, Azam M, Barbot C, Martorell Sampol L, Rodriguez B, Pascual-Castroviejo I, Teber S, Anlar B, Comu S, Karaca E, Kayserili H, Yüksel A, Akcakus M, Al Gazali L, Sztriha L, Nicholl D, Woods CG, Bennett C, Hurst J, Sheridan E, Barnicoat A, Hennekam R, Lees M, Blair E, Bernes S, Sanchez H, Clark AE, DeMarco E, Donahue C, Sherr E, Hahn J, Sanger TD, Gallager TE, Dobyns WB, Daugherty C, Krishnamoorthy KS, Sarco D, Walsh CA, McKanna T, Milisa J, Chung WK, De Vivo DC, Raynes H, Schubert R, Seward A, Brooks DG, Goldstein A, Caldwell J, Finsecke E, Maria BL, Holden K, Cruse RP, Swoboda KJ, Viskochil D, et al. Expanding CEP290 mutational spectrum in ciliopathies. Am J Med Genet A. 2009;149A:2173 - 80. [PMC free article: PMC4340070] [PubMed: 19764032]
- Tusa RJ, Hove MT. Ocular and oculomotor signs in Joubert syndrome. J Child Neurol. 1999;14:621 - 7. [PubMed: 10511333]
- Tuz K, Bachmann-Gagescu R, O'Day DR, Hua K, Isabella CR, Phelps IG, Stolarski AE, O'Roak BJ, Dempsey JC, Lourenco C, Alswaid A, Bönnemann CG, Medne L, Nampoothiri S, Stark Z, Leventer RJ, Topçu M, Cansu A, Jagadeesh S, Done S, Ishak GE, Glass IA, Shendure J, Neuhauss SC, Haldeman-Englert CR, Doherty D, Ferland RJ. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014;94:62 - 72. [PMC free article: PMC3882733] [PubMed: 24360808]
- Utsch B, Sayer JA, Attanasio M, Rodrigues Pereira R, Eccles M, Hennies H-C, Otto EA, Hildebrandt F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21:32 - 5. [PubMed: 16240161]
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51:1 - 23. [PubMed: 18164675]
- Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G, Bertini E, Boltshauser E, Zaki MS, Abdel-Aleem A, Abdel-Salam GM, Bellacchio E, Battini R, Cruse RP, Dobyns WB, Krishnamoorthy KS, Lagier-Tourenne C, Magee A, Pascual-Castroviejo I, Salpietro CD, Sarco D, Dallapiccola B, Gleeson JG. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006a;59:527 - 34. [PubMed: 16453322]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, Adams M, Szymanska K, Mazzotta A, Lee JE, Tolentino JC, Swistun D, Salpietro CD, Fede C, Gabriel S, Russ C, Cibulskis K, Sougnez C, Hildebrandt F, Otto EA, Held S, Diplas BH, Davis EE, Mikula M, Strom CM, Ben-Zeev B, Lev D, Sagie TL, Michelson M, Yaron Y, Krause A, Boltshauser E, Elkhartoufi N, Roume J, Shalev S, Munnich A, Saunier S, Inglehearn C, Saad A, Alkindy A, Thomas S, Vekemans M, Dallapiccola B, Katsanis N, Johnson CA, Attié-Bitach T, Gleeson JG. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619 - 25. [PMC free article: PMC2894012] [PubMed: 20512146]
- Valente EM, Marsh SE, Castori M, Dixon-Salazar T, Bertini E, Al-Gazali L, Messer J, Barbot C, Woods CG, Boltshauser E, Al-Tawari AA, Salpietro CD, Kayserili H, Sztriha L, Gribaa M, Koenig M, Dallapiccola B, Gleeson JG. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann Neurol. 2005;57:513 - 9. [PubMed: 15786477]
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, Bertini E, Dallapiccola B, Gleeson JG, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006b;38:623 - 5. [PubMed: 16682970]
- van Zalen-Sprock RM, van Vugt JM, van Geijn HP. First-trimester sonographic detection of neurodevelopmental abnormalities in some single-gene disorders. Prenat Diagn. 1996;16:199 - 202. [PubMed: 8710771]
- Verloes A, Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am J Med Genet. 1989;32:227 - 32. [PubMed: 2929661]
- Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, Vemulapalli M, Mullikin JC, Malicdan MC, Gahl WA, Gunay-Aygun M, et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med. 2017. Epub ahead of print. [PubMed: 28125082]
- Vodopich DJ, Gordon GJ. Anesthetic management in Joubert syndrome. Paediatr Anaesth. 2004;14:871 - 3. [PubMed: 15385018]
- Wang P, Chang FM, Chang CH, Yu CH, Jung YC, Huang CC. Prenatal diagnosis of Joubert syndrome complicated with encephalocele using two-dimensional and three-dimensional ultrasound. Ultrasound Obstet Gynecol. 1999;14:360 - 2. [PubMed: 10623998]
- Watson CM, Crinnion LA, Berry IR, Harrison SM, Lascelles C, Antanaviciute A, Charlton RS, Dobbie A, Carr IM, Bonthron DT. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet. 2016;17:1. [PMC free article: PMC4700600] [PubMed: 26729329]
- Weiss AH, Doherty D, Parisi M, Shaw D, Glass I, Phillips JO. Eye movement abnormalities in Joubert syndrome. Invest Ophthalmol Vis Sci. 2009;50:4669 - 77. [PMC free article: PMC3919872] [PubMed: 19443711]
- Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci. 2010;30:8759 - 68. [PMC free article: PMC2923804] [PubMed: 20592197]
- Whitsel EA, Castillo M, D'Cruz O. Cerebellar vermis and midbrain dysgenesis in oculomotor apraxia: MR findings. AJNR Am J Neuroradiol. 1995;16:831 - 4. [PubMed: 7611051]
- Wolf MT. Nephronophthisis and related syndromes. Curr Opin Pediatr. 2015;27:201 - 11. [PMC free article: PMC4422489] [PubMed: 25635582]
- Wolf MT, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, Salomon R, Stallmach T, Sayer JA, Waldherr R, Griebel M, Oh J, Neuhaus TJ, Josefiak U, Antignac C, Otto EA, Hildebrandt F. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007;72:1520 - 6. [PubMed: 17960139]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428 - 37. [PMC free article: PMC2648685] [PubMed: 19252258]
建议阅读
- Fluss J, Blaser S, Chitayat D, Akoury H, Glanc P, Skidmore M, Raybaud C. Molar tooth sign in fetal brain magnetic resonance imaging leading to the prenatal diagnosis of Joubert syndrome and related disorders. J Child Neurol. 2006;21:320 - 4. [PubMed: 16900929]
- Helou J, Otto EA, Attanasio M, Allen SJ, Parisi M, Glass I, Utsch B, Hashmi S, Fazzi E. Mutation analysis of NPHP6/CEP290 in patients with Joubert-syndrome and Senior-Løken syndrome. J Med Genet. 2007;44:657 - 63. [PMC free article: PMC2597962] [PubMed: 17617513]
- Sturm V, Leiba H, Menke MN, Valente EM, Poretti A, Landau K, Boltshauser E. Ophthalmological findings in Joubert syndrome. Eye (Lond.) 2010;24:222 - 5. [PubMed: 19461662]
章节注释
作者
更新历史
- 29 June 2017 (bp) 系统性更新发布到公开网页上
- 11 April 2013 (cd/mp) 修订: 发现TMEM231 和TCTN3 突变导致JSRD; 阐明 TTC21B突变的功能;C5orf42 和CEP41可进行临床序列分析和缺失/重复分析; 修改图2
- 13 September 2012 (cd) 修订: TCTN1, TCTN2, TTC21B, 和TMEM13可进行临床序列分析; TMEM138可进行缺失/重复分析
- 14 June 2012 (cd/mp) 修订: TMEM216 创始突变c.218G>T 可进行临床靶向突变分析
- 24 May 2012 (cd/mp) 修订: Joubert 综合征 11, 15, 16, and 17 分别由 TTC21B, CEP41, TMEM138, 和 C5orf52 突变导致
- 29 March 2012 (me) 系统性更新发布到公开网页上
- 8 March 2007 (cd) 修订: TMEM67 (MKS3) 突变在3/22 JS个体中发现并不包含NPHP1 缺失; MKS3 是第六个JS位点
- 4 August 2006 (cd) 修订: 可进行 CEP290 突变的临床检测和产前检测
- 25 July 2006 (cd) 修订: AHI1可进行 临床序列分析; AHI1 和NPHP1可进行临床产前检测
- 30 June 2006 (ca) 修订: JTS个体中检测到 CEP290 (NPHP6) 突变
- 24 February 2006 (me) 系统性更新发布到公开网页上
- 9 July 2003 (me) 内容发布到公开网页上
- 27 January 2003 (mp) 最早稿件