
概述
临床特征.
CASK-相关疾病包括女性和男性中发现的一系列表型谱。其中两个主要的临床表现是:
- 小头畸形伴桥脑和小脑发育不全 (MICPCH), 通常与CASK基因的致病性 功能丧失性 变异有关;
MICPCH通常在中度至重度智力障碍、进展性小头畸形伴或不伴眼科异常以及感觉神经性听力丧失的女性中发现。至今有53例MICPCH女性患者的报道,其中年龄最大为21岁。大多数患者可以独坐;20%-25%患者具备行走能力;大多数都几乎没有语言能力。神经学特征可能包括:轴向性肌张力减退、四肢肌张力亢进或痉挛、以及肌张力障碍或其它运动障碍。大约40%有癫痫表现。行力特征可能包括睡眠障碍、手部刻板动作和自咬。
至今只有7名男性患者被报道有严重 表型. 该队列的代表性不足可能是由于男性早期致死导致。该男性患者典型表现为智力障碍和MICPCH, 或早期婴儿癫痫性脑病 (Ohtahara综合征, West综合征, 或早期肌阵挛性癫痫).
在具有较轻微的 (i.e., 功能下降的) 致病性变异的个体或家庭中,临床 表型 通常是 X-linked 智力障碍 (XLID) 伴或不伴眼球震颤和其它临床特征。超过24位男性和9位女性患者被报道过。男性具有轻微到严重的智力障碍,伴或不伴眼球震颤和其它眼部特征。女性通常是正常的,有些表现出轻度智力障碍,伴或不伴眼部特征。
管理.
对症治疗: 治疗是对症治疗,也包括营养支持,对听力丧失的治疗和理疗的使用。对MICPCH患儿的癫痫管理是标准治疗,基于具体的癫痫类型和发作频率。
GeneReview Scope
| CASK-相关疾病: 所包含疾病 |
|---|
|
For synonyms and outdated names see Nomenclature.
诊断
CASK-相关疾病包含在女性和男性中有所不同的一系列的表型谱。
- 女性典型地具有中度-重度的智力障碍和进展性小头畸形伴桥脑和小脑发育不全 (MICPCH)。可能的症状还有眼科异常以及感觉神经性听力损失。
- 男性的表型较宽,从严重的(智力障碍和MICPCH,或早期婴儿癫痫性脑病[Ohtahara综合征, West 综合征或早期肌阵挛性癫痫])到轻微的(X-linked智力障碍[XLID] 伴或不伴眼球震颤和其它临床特征)[Najm et al 2008, Burglen et al 2012, Saitsu et al 2012, Takanashi et al 2012, Nakamura et al 2014].
CASK-相关疾病的正式的诊断标准还未确立。
有以下症状应怀疑CASK-相关疾病:
MICPCH女性患者
临床症状:
- OFC 在出生时为小头畸形 (OFC < -2 SD) 至正常
- 进展性小头畸形 (≤ -10 SD)
- 中度至重度的智力障碍
- 缺乏语言能力
- 肌张力减退,肌张力亢进,或以上均有 (四肢中枢性肌张力减退和亢进)
- 癫痫发作
- 行力异常
- 身材矮小
- 眼科症状: 视神经发育不全,视网膜病变和斜视
- 感觉神经性听力损失
- 面容:浓密的弓形眉毛,鼻梁和鼻尖宽,鼻子小或短,人中长或上颌突出,下巴短和大耳朵
MRI发现:
- 桥脑和小脑发育不全伴小脑弥漫性轻度至重度发育不全[Najm et al 2008] 成比例地累及半球和蚓部[Takanashi et al 2010, Moog et al 2011, Takanashi et al 2012] (Figure 1)
- 小脑半球可能不对称地 受累的 .
- 桥脑发育不全可能从轻微至重度伴相对稀疏的桥脑膨出.
- 正常或正常偏低的胼胝体伴低的大脑/胼胝体比率 [Takanashi et al 2010]
- 相关MRI发现: 轻度减少大脑皮质额叶区域中回旋的数量和复杂性。
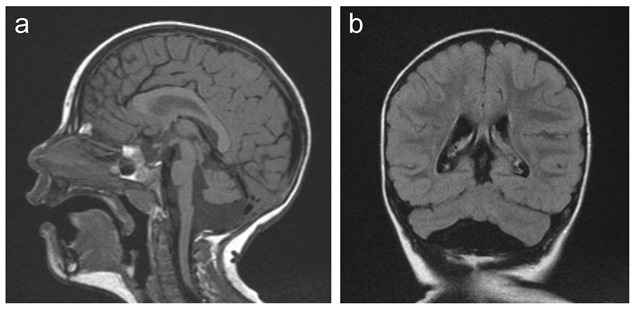
MICPCH男性患者
临床症状:
- 轻度至重度的产后小头畸形
- 严重的发育落后或无发育
- 早期难治性癫痫发作 (Ohtahara 综合征 [Saitsu et al 2012], West 综合征 [Takanashi et al 2012], 或肌阵挛癫痫 [Nakamura et al 2014])
MRI发现: 轻至重度桥小脑发育不全
X-linked 智力障碍(XLID) 伴或不伴眼球震颤的男性患者
临床症状:
- 轻至重度智力障碍
- 癫痫
- 先天性眼球震颤
- 震颤和步态不稳
MRI 症状: 小脑发育不全 (表现多变)
X-linked 智力障碍 (XLID) 伴或不伴眼球震颤的女性患者
大部分的临床症状:
- 智力正常至轻度智力障碍
- 正常至轻微的眼部症状包括 先天的 眼球震颤和斜视
- 无其它神经系统症状至轻度震颤或失神性癫痫
MRI发现: 未知
CASK-相关疾病的确诊是建立在女性检出CASK 杂合的 致病性变异 和男性检出CASK 半合子的 致病性变异 (Table 1). 对 先证者 分子遗传学检测 的方法可以是单个 基因 检测或使用 表型靶向检测.
单个 基因 检测. 为确诊MICPCH 先证者 (男性或女性):
- 若 序列分析未发现 致病性变异 ,应该进行 deletion/duplication analysis . 基因组重组,包括CASK 基因整体或部分的缺失,重复,和倒位,已知的大小从11 kb 至 4.5 Mb.
- 在女性中,从 deletion/duplication analysis 开始可能是合适的,因为它占了已发现致病性变异的一半 (Table 1).
- 在小部分的男性中,CASK 重组的 半合子的 或嵌合状态已报道过 [Saitsu et al 2012; K Kutsche, unpublished].
- 序列分析 和 deletion/duplication analysis 未发现 致病性变异 且仍高度怀疑CASK-相关疾病时,染色体 (核型 and FISH) 分析是合适的。已报道两名女性具有包含CASK 的Xp不平衡倒位 [Najm et al 2008; K Kutsche, unpublished].
多个-基因 panel. 考虑包含CASK 和其它感兴趣基因的 表型靶向检测 (see Differential Diagnosis).
Table 1.
CASK-相关疾病的分子遗传学检测
| 基因 1 | 检测方法 | 检出变异 2 | 不同表型/检测方法的变异检出率 3, 4 | |
|---|---|---|---|---|
| MICPCH | XLID +/- Nystagmus | |||
| CASK | 整个 基因 序列分析 5 | 变异 | 28/58 女性 6, 7, 8 6/49 男性 6, 7, 8, 9, 10 | 7 男性/505 (454 男性, 51 女性) 9, 11 |
| 缺失/重复 分析 12 | (多个)外显子 和整个-基因 缺失/重复 | 26/54 女性 6, 13, 14 1/20 男性 6 | 1 男性 15 | |
MICPCH = 小头畸形伴桥脑和小脑发育不全
XLID = X-linked 智力障碍
- 1.
- 2.
等位基因变异信息,见 Molecular Genetics .
- 3.
用于检测变异的检测方法的检出能力显示在 基因
- 4.
- 5.
- 6.
CASK 致病性变异/重组的嵌合可发生于男性和女性。致病性变异的嵌合难以检出因此可能导致检测结果存在假阴性。
- 7.
- 8.
在 受累的 女性和男性中, de novo 致病性的 功能丧失性 变异分布于整个 基因; 没有热点突变区域 [Najm et al 2008, Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Saitsu et al 2012, Takanashi et al 2012, Nakamura et al 2014].
- 9.
- 10.
CASK 体细胞 嵌合 致病性的 nonsense 变异在一名男性中被报道过 [Burglen et al 2012].
- 11.
在患XLID伴或不伴眼球震颤和/或其它额外临床表现的个体中,大部分的CASK 改变是 错义 和剪接变异 [Piluso et al 2009, Tarpey et al 2009, Hackett et al 2010].
- 12.
- 13.
女性的基因组重组包括整个或部分CASK的缺失和重复,大小从11 kb - 4.5 Mb [Froyen et al 2007, Hayashi et al 2008, Najm et al 2008, Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Saleem et al 2013]. 已知有两名女性具有破坏CASK的细胞遗传学上可见的Xp倒位 [Najm et al 2008; K Kutsche, unpublished].
- 14.
CASK 缺失 的嵌合已在一名MICPCH 和Ohtahara综合征男性的无症状母亲发现 [Saitsu et al 2012].
- 15.
临床特征
临床描述
智力障碍和小头畸形伴桥脑和小脑发育不全 (MICPCH)
Females. 至今共报道了53 名女性MICPCH患者,其中年龄最大的21岁。以下关于自然病史的信息来源于近期的综述 Moog et al [2011], Burglen et al [2012], 和Takanashi et al [2012] 除非另有说明。
出生时枕额周长 (OFC) 处于正常或正常稍低范围内的大约占 受累的 女性的三分之二;其余的表现为小头畸形 (OFC < -2 SD). 在第一年小头畸形总是很严重 (OFC -3.5 to -10 SD) ,经常在生命前四个月表现。
受累的女性在2-24个月时获得头部控制和进行眼神接触的能力。大多数 受累的 女性在7-36个月间可以独坐;然而,只有20%25%获得行走能力 (在18 至 72 个月间)。
大多数几乎没有语言能力;有些可以说一些词语。有一位个体可以说两个词语的句子。几乎所有的 受累的 女性智力发展都严重受损,仅有小部分达到中等智力。
相关的神经系统特征包括(轴向)肌张力低下,四肢肌张力过高(可能发展为痉挛)以及肌张力障碍或其他运动障碍。 将近40%观察到各种类型的癫痫发作;发病时间在出生至十岁之间。
MRI观察到的桥脑小脑发育不全的严重程度没有预后价值 [Moog et al 2011].
行为学 表型 可能包括睡眠障碍,手部刻板运动和自咬。
尚未报告 受累的 女性的死亡率。
其它发现:
- 出生身长正常。身材矮小在4岁时常见 [Moog et al 2011, Takanashi et al 2012].
- 经常观察到脊柱侧弯
- 能观察到各种眼科症状,特别是视神经发育不良,视网膜病和斜视。
- 先天性内脏异常 (e.g., 肾脏/泌尿科或心脏异常) 罕见; 没有特别的异常反复发生。
- 近期综述提示面部 表型 有浓密眉毛弓形,鼻梁和鼻尖宽,鼻孔小或短,上肢长或上颌突出,下巴小,耳朵大。
男性. 迄今为止,已有7位年龄在15岁以下的男性在CASK中有致病的功能丧失性 变异。 [Najm et al 2008 (patient 5), Burglen et al 2012 (patients 12, 13), Saitsu et al 2012 (patients 1, 2), Takanashi et al 2012 (patient 16), Nakamura et al 2014]. 受累的 男性的 表型 似乎代表了MICPCH表型谱中最严重的一端, CASK 致病性变异 嵌合的男性除外。
所有 受累的 男性都表现重度到极重度的发育落后或完全不发育,轻度至重度小头畸形 (OFC -2.7 to -6 SD), 和轻度至重度弥漫性桥脑小脑发育不全。
七个中有五个患有难治性癫痫发作。
- 两名诊断出大田原综合症(小儿癫痫性脑病并伴有爆发抑制)[Saitsu et al 2012].
- 一名从一出生开始就表现出频繁的肌阵挛(早期的肌阵挛性脑病),并在婴儿期出现爆发抑制性脑电图模式 [Jinnou et al 2012, Nakamura et al 2014] 另一例表现为每天痉挛和强直性癫痫发作,并伴有爆发抑制 [Burglen et al 2012].
- 一名诊断为West 综合征 [Takanashi et al 2012].
七个中有三个具有多种 (小的) 异常 [Burglen et al 2012, Saitsu et al 2012] ,一名有法洛四联症和肾积水 [Jinnou et al 2012, Nakamura et al 2014].
CASK 致病性 功能丧失性 变异在男性中可能也会导致围产期或早期致死。一名 受累的 男性在两周时去世 [Najm et al 2008 (patient 5)].
X-连锁智力障碍 (XLID) ± 眼球震颤
男性. 亚效的 CASK 致病性变异已在来自7个家庭的 24名 受累的 X-linked 智力障碍伴或不伴眼球震颤和/或其它异常的男性中被报道 [Piluso et al 2003, Piluso et al 2009, Tarpey et al 2009, Hackett et al 2010]. 他们具有轻度至重度 ID 伴或不伴有 先天的 眼球震颤和其它眼部症状,如斜视和视盘苍白。震颤和步态不稳可能是相关的。
在一个有三名 受累的 男性的家族中,其中两名表现为重度 ID, 先天的 肌张力减退,便秘,多动或攻击性行为和相对的大头畸形,两名表现感觉神经性耳聋;最初报道这个家族患有 FG syndrome [Piluso et al 2003, Piluso et al 2009].
OFC 通常是正常的。这种 表型 的男性脑干和小脑发育不全的发生率是未知的。MRI 或 CT 发现在三名男性和一名 杂合的 女性中报道;三名具有正常表现,一名男性患有小脑发育不全和重症肌无力,严重ID,小头畸形,眼球震颤,视盘苍白,身材矮小和面部畸形 [Hackett et al 2010 (family V, patient II-4)].
女性. 可获得九名 杂合的 女性的临床信息。其中4名是无症状的;其他5名表现为可变的临床特征:眼球震颤 (2), 震颤 (2), 轻微 (1) 或重度(1) ID, 和失神癫痫 (1).
基因型-表型关联
CASK 致病性 功能丧失性 变异通常导致小头畸形伴桥脑和小脑发育不全 (MICPCH) [Moog et al 2011, Hayashi et al 2012]. CASK 功能下降的 致病性变异通常导致男性 X-连锁智力障碍伴或不伴眼球震颤 [Hackett et al 2010].
在患有桥脑和小脑发育不全(MICPCH)的小头畸形患者中,临床特征与 功能丧失性 变异的特定类型(例如CASK功能丧失变异,包含CASK和/或连续基因的较大重排, 或基因内 失活 CASK变异)没有关联 [Moog et al 2011, Hayashi et al 2012].
X-linked 智力障碍伴或不伴眼球震颤的男性是由 功能下降的 CASK 致病性变异所致,该类型变异有害性比 失活 型致病性变异低。在四个具有眼球震颤的家族中,所有致病性变异均影响CASK蛋白的COOH-端, 提示眼球震颤表现可能存在 基因型-表型相关性 [Hackett et al 2010]. 近期,报告了新的 CASK-互作蛋白 FRMD7 (由 FRMD7编码) [Watkins et al 2013]. FRMD7 的致病性变异是 X-linked familial idiopathic infantile nystagmus的主要原因。这4个 CASK 致病性变异中有3个干扰了 CASK 与FRMD7的相互作用,支持了眼球震颤与影响CASK COOH-端的变异之前的关联。
外显率
在至今报道的53名女性和7名存活男性中,MICPCH 的外显率似乎是完全的。少数报道的男性具有特别严重的 表型 (参阅 Clinical Description), 除了那些CASK 致病性变异 体细胞嵌合 外。
XLID 伴或不伴眼球震颤的外显率未知。在至今报道的24名男性中外显率可能完全或很高,而在至今报道的9名 杂合的 女性中不完全外显且临床可变性高。
命名法
在一个检出CASK 致病性变异 且与FG 综合征 (FGS)-类似 表型 共分离的家庭被报道后,FG 综合征 4 (FGS4) 曾被提议作为一种 CASK-相关疾病 [Piluso et al 2003, Piluso et al 2009]. 然而,除了由复发性MED12致病性变异引起的FGS1外, (OMIM 300188, 305450, 309520) (参阅 MED12-Related Disorders), FGS的定义不明确。OMIM 将FGS4列在与MR, X-linked, 伴或不伴眼球震颤 (OMIM)同一个表型号下面。因此,将这个家庭纳入MR, X-连锁,伴或不伴眼球震颤,似乎更为合适。
发病率
MICPCH 和 CASK-相关的 X-linked 智力障碍伴或不伴眼球震颤的发病率均未知。
- MICPCH. 至今全世界范围内报道了53 名女性和7名男性; 另外作者已知还有另6名女性和3名男性。
- X-linked 智力障碍伴或不伴眼球震颤. 24名 受累的 男性和5名 杂合的 有症状的女性被报道过,包括FGS-类似 表型 的家庭[Piluso et al 2009, Tarpey et al 2009, Hackett et al 2010]. 通过对X 染色体 上718个基因的编码外显子进行测序而获得的第一批数据表明,CASK致病性变异导致的X连锁智力障碍伴或不伴眼球震颤可能占不到1%。 [Tarpey et al 2009].
遗传相关(等位基因)疾病
除本 GeneReview 所讨论的表型外,已知没有其它与CASK 突变相关的表型。
鉴别诊断
小头畸形伴桥脑和小脑发育不全(MICPCH)。 识别桥脑小脑畸形对于MICPCH的诊断至关重要。
桥脑和小脑发育不全 (PCH) 表现出与MICPCH重叠的特征但可以通过神经影像学和临床表现辨别。
在典型的PCH中 (see Pontocerebellar Hypoplasia Type 2 and Type 4) 小脑半球比蚓部受到的影响更大,导致在冠状图像中出现“蜻蜓”现象,而在CASK相关疾病中,可见“蝶形”图案,这是由于半球和蚓部的弥漫性发育不良所致。与MICPCH的女性相比,PCH2 / PCH4的个体的桥脑发育不全更为严重。胼胝体通常薄而发育不良。
PCH2的特征是全身性阵挛(“紧张不安”),伴缺乏自愿性运动发育以及后来出现的舞蹈症和痉挛,吞咽障碍,有时甚至癫痫发作。患有PCH2的个体通常生存至儿童时期。
PCH4的特征是羊水过多,挛缩,严重的全身性阵挛和中央呼吸衰竭,通常会导致新生儿死亡。
PCH2 和 PCH4 由TSEN54, TSEN34 或 TSEN2的 双等位基因的 致病性变异导致。
Ohtahara 综合征. 在两名 CASK-相关疾病的男性中被描述的Ohtahara 综合征 [Saitsu et al 2012] 也显示了弥漫性桥小脑发育不全的标志。Ohtahara综合征 - 早期婴幼儿抑制性爆发的癫痫性脑病 - 可能是由 STXBP1 或 ARX 的致病性变异导致 [Kato et al 2007, Saitsu et al 2008].
X-linked 智力障碍不伴眼球震颤 具有广泛的鉴别诊断,因为已知许多基因会导致非综合征和 综合征性 X-连锁智力障碍 [Lubs et al 2012].
一个患 X-linked 智力障碍的家族和一个 CASK 致病性变异 被报道具有FG 综合征 (FGS) 的 表型 [Piluso et al 2003, Piluso et al 2009]. 因为最初由 Opitz and Kaveggia [1974]描述的表型的广泛性,FGS 可能在临床诊断时存在困难。有FGS个体的家族被定位到X 染色体 的七个位点 (CASK 被列为 FGS4, OMIM). 然而,除了FGS1以外,早期定义表型很困难。FGS1是由MED12 (OMIM) 基因的复发性致病性变异所致。 (see MED12-Related Disorders).
智力障碍伴眼球震颤 可能在 X-linked 疾病 Allan Herndon-Dudley syndrome 中见到,它是由SLC16A2. 半合子的 致病性变异所致。患病个体表现为严重ID,小头畸形,神经系统症状(痉挛,肌张力障碍和共济失调),脊柱侧弯,大耳,和其它畸形。眼球震颤pathogenic variants in SLC16A2. 在某些病例中,眼球震颤也可能与X连锁智力障碍有关。
管理
初步诊断后的评估
为了确定诊断为CASK相关疾病的个体的疾病程度和需求,建议进行以下评估:
- 神经系统评估,包括脑电图
- 发展评估,包括大运动能力和精细运动能力以及语言
- 喂养评估
- 眼科检查
- 听力检查
- 骨科检查
- 心脏和肾脏的超声检查以评估罕见但可能的心脏或肾脏/泌尿科异常
- 临床遗传咨询
对症治疗
没有特定的治疗方法。
治疗是对症治疗,包括营养支持,听力损失治疗和物理治疗。
MICPCH小儿癫痫发作的治疗是标准治疗,并基于特定的癫痫发作类型和发作频率。
评估处于高风险的亲属
有关为 遗传咨询 目的而对高危亲属进行检测的相关问题,请参见Genetic Counseling 。
正在研究的疗法
搜索 ClinicalTrials.gov,以获取有关各种疾病和症状的临床研究信息。 注意:可能没有针对该疾病的临床试验。
遗传咨询
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
遗传模式
CASK-相关疾病以 X-linked 模式遗传。
家族成员风险 – X-连锁智力障碍 ± 眼球震颤
男性 先证者 的父母
男性 先证者 的同胞
- 先证者 同胞的风险取决于其母亲的遗传状态。
男性 先证者 的子代。从理论上讲,轻度 受累的 男性会将 致病性变异 遗传给所有女儿,而不会遗传给儿子。 然而,尚未有具有CASK致病变异的男性生育下一代的报道。
女性 先证者 的子代。具有CASK 致病性变异 的女性有50% 将变异遗传给每个孩子;遗传到该致病性变异的儿子将会是 受累的; 遗传到该致病性变异的女儿将不受累或有不同程度受累。
先证者的其他家族成员。若先证者父母一方也具有 致病性变异, 则他或她的女性家族成员可能有一定风险成为 杂合的 (无症状或有症状)而他或她的男性家族成员可能有 受累的 风险,取决于他们与先症者的遗传关系.
风险亲属的遗传携带者检测 需事先在家族中发现 致病性变异 。
相关遗传咨询问题
家庭计划
确定遗传风险和讨论产前检查可用性的最佳时间是在怀孕之前。
- 向携带者(无症状或有症状)或有携带者风险的年轻女性提供 遗传咨询 (包括对后代和生殖选择的潜在风险的讨论)是适当的。
DNA banking 是储存DNA(通常从白细胞中提取),以备将来使用。因为将来检测方法和我们对基因,等位基因变异和疾病的理解可能会改善,所以应考虑 受累的 患者的DNA储存。
资源
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- American Association on Intellectual and Developmental Disabilities (AAIDD)501 3rd Street NorthwestSuite 200Washington DC 20001Phone: 202-387-1968Fax: 202-387-2193Email: sis@aaidd.org
- Medline Plus
- National Center on Birth Defects and Developmental Disabilities1600 Clifton RoadMS E-87Atlanta GA 30333Phone: 800-232-4636 (toll-free); 888-232-6348 (TTY)Email: cdcinfo@cdc.gov
分子遗传学
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. - ED.
Table A.
CASK-相关疾病: 基因和数据库
| 基因 | 染色体位点 | 蛋白 | Locus Specific | HGMD |
|---|---|---|---|---|
| CASK | Xp11 | Peripheral plasma membrane protein CASK | CASK @ LOVD | CASK |
Table B.
CASK-相关疾病的OMIM 词条 (View All in OMIM)
分子遗传学致病机制
患有MICPCH的女性和男性中的CASK致病性变异是 失活 的,代表导致严重表型的 无效 等位基因 [Najm et al 2008, Moog et al 2011, Burglen et al 2012].。具有 半合子的 致病性 功能丧失性 变异的男性比女性有更严重的 受累的 。早期致死率可能是报告的男性人数少于预期的原因。
有人提出CASK-TBR1-RELN信号级联的干扰是MICPCH 表型 的基础 [Najm et al 2008]。CASK与 转录因子 TBR1形成复合物,该复合物诱导含有TBR1结合位点的基因的转录,包括RELN [Hsueh et al 2000, Hsueh 2006, Hsueh 2009]。RELN中的纯合致病变异与神经元迁移障碍,平脑畸形伴严重的小脑和海马发育不全 [Hong et al 2000].
Tbr1, Reln,和 Cask 突变体小鼠的脑畸形表现重叠提示Cask-Tbr1介导的靶基因调控对于皮层发育至关重要 [Bulfone et al 1998, Atasoy et al 2007].
近期,新的 CASK-互作蛋白质 FRMD7 (由 FRMD7 编码) 已被描述 [Watkins et al 2013]. CASK 与 FRMD7 的结合在动眼神经网络的发育中至关重要[Watkins et al 2013] 。FRMD7 致病性变异是导致 X-linked familial idiopathic infantile nystagmus 的主要原因。四个蛋白COOH-端的CASK 致病性变异中,有三个在患 X-linked 智力障碍和眼球震颤的男性中被描述 [Hackett et al 2010].
基因结构.CASK 包含 27 个外显子并经过可变 剪接, 得到编码不同异型体的三种不同的转录本。
- 转录本 1 代表最长的转录本并编码最长的蛋白质 (isoform 1)。
Ensembl database 列出了 22 种可能的 CASK transcript variants.
关于基因 和蛋白信息的详细概述,见 Table A, Gene.
Table 2.
CASK 转录本和异构体
| CASK Isoform | Transcript | Protein | Exons | # Amino Acids | Transcript Length |
|---|---|---|---|---|---|
| CASK isoform 1, variant 1 | NM_003688 | NP_003679 | 27 | 921 | 8298 bps |
| CASK isoform 2, variant 2 | NM_001126054 | NP_001119526 | 26 | 898 | 8229 bps |
| CASK isoform 3, variant 3 | NM_001126055 | NP_001119527 | 25 | 897 | 8226 bps |
根据人类参考基因组 GRCh37/hg19.
致病性变异
- MICPCH. 超过50 个不同的CASK 致病性变异已被描述 [Froyen et al 2007; Hayashi et al 2008; Najm et al 2008; Moog et al 2011; Burglen et al 2012; Hayashi et al 2012; Saitsu et al 2012; Takanashi et al 2012; Saleem et al 2013; Nakamura et al 2014; K Kutsche, unpublished].大多数是 私有的 变异;然而,一小部分复发性致病性变异已被报道,包含两个 nonsense 变异c.316C>T (p.Arg106Ter) 和 c.2074C>T (p.Gln692Ter) (见 Table 3)。致病性变异似乎在整个 基因 均匀分布。在任何人群中都没有发现主要的常见 致病性变异 。CASK 基因内的缺失和重复,从少数外显子的缺失或重复,到整个基因 的缺失,已在大约50% 受累的 个体中被描述;缺失可能也包含数量可变的侧翼基因。至今,发现了17个CASK 部分或完全缺失和5个CASK 部分重复 [Froyen et al 2007, Hayashi et al 2008, Najm et al 2008, Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Saitsu et al 2012, Takanashi et al 2012, Saleem et al 2013]. 这些部分缺失或重复似乎分布在整个基因。已知两种破坏CASK 的倒位 [Najm et al 2008; K Kutsche, unpublished].
- X-linked 智力障碍伴或不伴眼球震颤. 7个不同的 CASK 致病性变异和剪接变异c.2521-2A>T 已在该 表型 男性中被描述 [Piluso et al 2009, Tarpey et al 2009, Hackett et al 2010] (见 Table 3).
Table 3.
本篇GeneReview 中讨论的部分CASK 致病性变异
| 表型 | DNA核苷酸改变 | 蛋白质改变 | 参考序列 |
|---|---|---|---|
| MICPCH | c.316C>T | p.Arg106Ter | NM_003688 NP_003679 |
| c.2074C>T | p.Gln692Ter | ||
| X-linked 智力障碍 ± 眼球震颤 | c.83G>T | p.Arg28Leu | NM_001126055 NP_001119527 |
| c.802T>C | p.Tyr268His | ENST00000378163 ENSP00000367405 | |
| c.1186C>T | p.Pro396Ser | ||
| c.2129A>G | p.Asp710Gly | ||
| c.2183A>G | p.Tyr728Cys | ||
| c.2521-2A>T | |||
| c.2755T>C | p.Trp919Arg |
Note on variant classification: Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
Note on nomenclature: GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen
- .hgvs.org ). See Quick Reference for an explanation of nomenclature.
正常 基因产物.CASK 编码钙/钙调蛋白依赖性丝氨酸蛋白激酶,该蛋白激酶属于膜相关鸟苷酸激酶(MAGUK)蛋白家族 [Zheng et al 2011]。 该基因产物具有至少三个由可变 剪接 产生的 异型体 ,并在各种各样的组织中表达。但是,CASK主要在大脑中表达。CASK的最长亚型由921个氨基酸组成,并具有以下结构域:钙调蛋白依赖性激酶样 结构域 (CaMK);LIN-2和LIN-7相互作用(L27); PSD-95-Dlg-ZO1(PDZ); Src同源体3(SH3); 蛋白质4.1相互作用(4.1); 鸟苷酸激酶(GK)。
CASK 在大脑发育和突触功能中扮演关键角色 [Hsueh 2006, Hsueh 2009] :
- 突触前组织和调节神经递质的释放;
- 调节突触后部位的离子通道并维持脊柱形态;
- 进入神经元核并调节参与神经元发育的靶基因如Reln和Grin2b的表达。
异常 基因产物
- MICPCH 是由CASK基因的致病性的 功能丧失性 变异和重组导致。目前还不知道人类CASK基因的致病性变异是如果影响CASK的功能的。然而,致病性变异可能会导致CASK蛋白功能完成丧失或截短的CASK蛋白无法行使其功能。
- X-linked 智力障碍伴或不伴眼球震颤。错义突变和 非移码 缺失可能表达有残存功能的CASK蛋白。一部分蛋白-蛋白相关作用(e.g., CASK和脂蛋白-α之间的作用)可能会被干扰 [Wei et al 2011].最近,已报道描述了新的CASK-互作蛋白FRMD7 (由FRMD7编码) [Watkins et al 2013]. FRMD7 的致病性变异是 X-linked familial idiopathic infantile nystagmus 的主要原因。4个影响蛋白质COOH-末端区域的CASK 致病性变异已在X-linked 智力障碍伴或不伴眼球震颤的男性中被报告描述。4个中有3个变异干扰了CASK 和 FRMD7 的相互作用,支持了CASK 与 FRMD7结合在动眼神经网络发育中起重要作用的模型。 [Watkins et al 2013].
参考文献
引用的文章
- Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, Kavalali ET, Sudhof TC. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A. 2007;104:2525 - 30. [PMC free article: PMC1892970] [PubMed: 17287346]
- Bulfone A, Wang F, Hevner R, Anderson S, Cutforth T, Chen S, Meneses J, Pedersen R, Axel R, Rubenstein JL. An olfactory sensory map develops in the absence of normal projection neurons or GABAergic interneurons. Neuron. 1998;21:1273 - 82. [PubMed: 9883721]
- Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, Petit F, Afenjar A, Goizet C, Barresi S, Coussement A, Ioos C, Lazaro L, Joriot S, Desguerre I, Lacombe D, des Portes V, Bertini E, Siffroi JP, de Villemeur TB, Rodriguez D. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis. 2012;7:18. [PMC free article: PMC3351739] [PubMed: 22452838]
- Froyen G, Van Esch H, Bauters M, Hollanders K, Frints SG, Vermeesch JR, Devriendt K, Fryns JP, Marynen P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum Mutat. 2007;28:1034 - 42. [PubMed: 17546640]
- Hackett A, Tarpey PS, Licata A, Cox J, Whibley A, Boyle J, Rogers C, Grigg J, Partington M, Stevenson RE, Tolmie J, Yates JR, Turner G, Wilson M, Futreal AP, Corbett M, Shaw M, Gecz J, Raymond FL, Stratton MR, Schwartz CE, Abidi FE. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet. 2010;18:544 - 52. [PMC free article: PMC2987321] [PubMed: 20029458]
- Hayashi S, Mizuno S, Migita O, Okuyama T, Makita Y, Hata A, Imoto I, Inazawa J. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am J Med Genet. 2008;146A:2145 - 51. [PubMed: 18629876]
- Hayashi S, Okamoto N, Chinen Y, Takanashi J, Makita Y, Hata A, Imoto I, Inazawa J. Novel intragenic duplications and mutations of CASK in patients with mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH). Hum Genet. 2012;131:99 - 110. [PubMed: 21735175]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93 - 6. [PubMed: 10973257]
- Hsueh YP. The role of the MAGUK protein CASK in neural development and synaptic function. Curr Med Chem. 2006;13:1915 - 27. [PubMed: 16842202]
- Hsueh YP. Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann Neurol. 2009;66:438 - 43. [PubMed: 19847910]
- Hsueh YP, Wang TF, Yang FC, Sheng M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature. 2000;404:298 - 302. [PubMed: 10749215]
- Jinnou H, Okanishi T, Enoki H, Ohki S. Pontocerebellar hypoplasia type 3 with tetralogy of Fallot. Brain Dev. 2012;34:392 - 5. [PubMed: 21880448]
- Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, Tohyama J, Akasaka N, Hayasaka K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet. 2007;81:361 - 6. [PMC free article: PMC1950814] [PubMed: 17668384]
- Lubs HA, Stevenson RE, Schwartz CE. Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet. 2012;90:579 - 90. [PMC free article: PMC3322227] [PubMed: 22482801]
- Moog U, Kutsche K, Kortüm F, Chilian B, Bierhals T, Apeshiotis N, Balg S, Chassaing N, Coubes C, Das S, Engels H, Van Esch H, Grasshoff U, Heise M, Isidor B, Jarvis J, Koehler U, Martin T, Oehl-Jaschkowitz B, Ortibus E, Pilz DT, Prabhakar P, Rappold G, Rau I, Rettenberger G, Schluter G, Scott RH, Shoukier M, Wohlleber E, Zirn B, Dobyns WB, Uyanik G. Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet. 2011;48:741 - 51. [PubMed: 21954287]
- Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, Christian SL, Ullmann R, Kuechler A, Haas CA, Flubacher A, Charnas LR, Uyanik G, Frank U, Klopocki E, Dobyns WB, Kutsche K. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet. 2008;40:1065 - 7. [PubMed: 19165920]
- Nakamura K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Matsumoto N, Saitsu H. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 2014;36:272 - 3. [PubMed: 23623288]
- Opitz JM, Kaveggia EG. Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd. 1974;117:1 - 18. [PubMed: 4365204]
- Piluso G, Carella M, D'Avanzo M, Santinelli R, Carrano EM, D'Avanzo A, D'Adamo AP, Gasparini P, Nigro V. Genetic heterogeneity of FG syndrome: a fourth locus (FGS4) maps to Xp11.4-p11.3 in an Italian family. Hum Genet. 2003;112:124 - 30. [PubMed: 12522552]
- Piluso G, D'Amico F, Saccone V, Bismuto E, Rotundo IL, Di Domenico M, Aurino S, Schwartz CE, Neri G, Nigro V. A missense mutation in CASK causes FG syndrome in an Italian family. Am J Hum Genet. 2009;84:162 - 77. [PMC free article: PMC2668001] [PubMed: 19200522]
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, Okada I, Yoshimura Y, Hirai S, Kumada T, Hayasaka K, Fukuda A, Ogata K, Matsumoto N. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782 - 8. [PubMed: 18469812]
- Saitsu H, Kato M, Osaka H, Moriyama N, Horita H, Nishiyama K, Yoneda Y, Kondo Y, Tsurusaki Y, Doi H, Miyake N, Hayasaka K, Matsumoto N. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia. 2012;53:1441 - 9. [PubMed: 22709267]
- Saleem R, Setty G, Hussain N. MICrocephaly, disproportionate pontine and cerebellar hypoplasia syndrome: A clinico-radiologic phenotype linked to calcium/calmodulin-dependent serine protein kinase gene mutation. Indian J Hum Genet. 2013;19:104 - 7. [PMC free article: PMC3722619] [PubMed: 23901204]
- Takanashi J, Arai H, Nabatame S, Hirai S, Hayashi S, Inazawa J, Okamoto N, Barkovich AJ. Neuroradiologic features of CASK mutations. AJNR Am J Neuroradiol. 2010;31:1619 - 22. [PMC free article: PMC3756090] [PubMed: 20595373]
- Takanashi J, Okamoto N, Yamamoto Y, Hayashi S, Arai H, Takahashi Y, Maruyama K, Mizuno S, Shimakawa S, Ono H, Oyanagi R, Kubo S, Barkovich AJ, Inazawa J. Clinical and radiological features of Japanese patients with a severe phenotype due to CASK mutations. Am J Med Genet. 2012;158A:3112 - 8. [PubMed: 23165780]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535 - 43. [PMC free article: PMC2872007] [PubMed: 19377476]
- Watkins RJ, Patil R, Goult BT, Thomas MG, Gottlob I, Shackleton S. A novel interaction between FRMD7 and CASK: evidence for a causal role in idiopathic infantile nystagmus. Hum Mol Genet. 2013;22:2105 - 18. [PMC free article: PMC3633374] [PubMed: 23406872]
- Wei Z, Zheng S, Spangler SA, Yu C, Hoogenraad CC, Zhang M. Liprin-mediated large signaling complex organization revealed by the liprin-α/CASK and liprin-α/liprin-β complex structures. Mol Cell. 2011;43:586 - 98. [PubMed: 21855798]
- Zheng CY, Seabold GK, Horak M, Petralia RS. MAGUKs, synaptic development, and synaptic plasticity. Neuroscientist. 2011;17:493 - 512. [PMC free article: PMC3191319] [PubMed: 21498811]
章节注释
作者注释
章节注释我们对确定CASK相关疾病的表型谱和分子发病机制感兴趣。
致谢
We thank the families with individuals 受累的 by MICPCH participating in our research programs.
修订记录
- 26 November 2013 (me) Review posted live
- 28 February 2013 (kk) Original submission