
诊断
疑似诊断
当患者具备以下临床 [Biancheri et al 2007]或头颅MRI特征性异常表现 [Rossi et al 2008]时应怀疑髓鞘形成低下与先天的白内障(HCC)。
临床表现
- 双侧先天的白内障(一例病人有青少年型白内障 [Ugur & Tolun 2008]; 一例病人三岁时发现的轻度晶状体浑浊 [Biancheri et al 2011].)
- 典型表现:生后第一年内精神运动发育正常,逐渐缓慢出现进行性神经系统损害如:
- 共济失调
- 痉挛
- 丧失行走能力
- 轻到重度认知损害
- 少见表现 [Biancheri et al 2011]:
- 早发严重型:新生儿期肌张力低下及喂养困难,生后第一月内发育迟缓,儿童早期需要依赖轮椅
- 晚发轻型:生后两年内发育正常,之后出现急性运动功能倒退
MRI 表现
- 所有患者均出现弥漫的幕上白质异常信号
- 髓鞘形成低下相一致的异常脑白质信号表现:
- 一些患者T2加权像较高信号相对应的T1加权像低信号密度区域与不同程度的脑白质水含量增加区域相一致(图 3)
- 年长患者白质容积丢失及胶质细胞增生 (图 4)
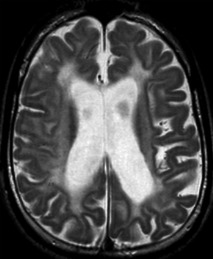
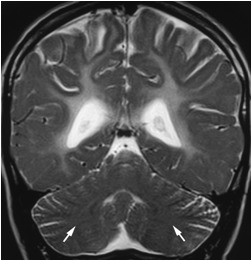
- 部分患者小脑半球的髓质中心表现有轻度增高T2加权像信号密度,与相邻的皮层灰质平行,并导致灰白质分界模糊 (图 5)
- 皮层及深部灰质结构正常
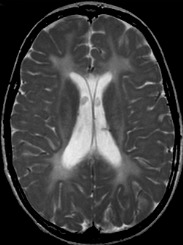
图 1.
轴位T2加权像显示与髓鞘形成低下一致的幕上白质弥漫高信号。
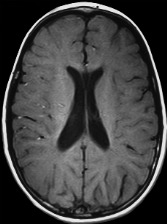
图 2.
轴位T1加权像显示与髓鞘形成低下一致的白质弥漫性等信号,与相邻灰质分界不清。
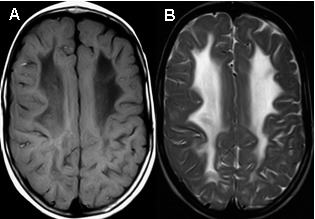
确立诊断
先证者 HCC的诊断的建立应用分子遗传学检测 明确FAM126A双等位基因的致病性变异(见 表 1)。
分子检测方法包括 单基因 检测、 表型靶向检测与 更为全面基因组的 检测。
- 包括FAM126A 与其他感兴趣的基因表型靶向检测 (见鉴别诊断)。注意:(1)用于每个基因检测的基因包包含的基因与检测的诊断 敏感性依据不同实验室与时间有差异。(2)在GeneReview讨论中的一些基因包可能包含疾病不相关基因。因此,临床医生需要在合理价格的基础上,优选最可能检出致病基因的多基因包。
- 点击这里了解更多有关多基因包的信息。
表 1.
髓鞘形成低下与先天性白内障分子遗传学检测方法总结
2.
序列分析检测变异包括良性、可能良性、意义不确定、可能致病性或致病性。致病变异包括小的基因内部的缺失/插入、错义、无义以及剪接位点变异; 通常 外显子 或者 全基因缺失/重复不被检测 。有关 序列分析结果解读的相关问题 点击这里.。
通过FAM126A全编码区及外显子-内含子交界区测序分析检出所有先证者中错义及剪接位点变异 [Zara et al 2006, Biancheri et al 2011, Traverso et al 2013a, Traverso et al 2013b].
4.
基因组的 DNA编码区与侧翼 内含子的区 序列分析不能明确外显子或全基因水平的缺失/重复变异的测试,包括定量PCR,长片段PCR,多重连接依赖的探针扩增技术(MLPA),以及包括该基因或染色体片段的染色体芯片 (CMA)技术。
5.
来自一个大的近亲婚配的土耳其家族中先证者FAM126A相关分析明确了 纯合性缺失 [Ugur & Tolun 2008].
临床特征
临床描述
髓鞘形成低下与 先天的 白内障 (HCC) 表型在迄今发现的5个无关家系中10个受累的 患者中高度一致。
出生前/围产期.所有受累的 患者生前与围产期表现正常。
眼部. 首发症状表现为出生时或生后一月内出现的双侧先天的白内障。除一例患者在青少年期出现白内障之外,所有患者均在生后一月内接受眼部手术 [Ugur & Tolun 2008]。
精神运动发育 生后一岁内正常,发育落后多在一岁后出现 [Biancheri et al 2007]。12至24月龄之间获得辅助行走能力。所有患者均无法获得独立行走能力。缓慢进展性的神经系统损害逐渐转变为行走能力的丧失。多数患者在8岁至9岁之间必须依赖轮椅 [Biancheri et al 2007].
认知能力.多数患者认知能力随时间表现为不伴倒退的轻度到中度智力障碍。
神经系统表现. 临床检查显示如下发病症状:
可有癫痫发作,但并不是主要的临床特征。神经生理学检查可见如下发病症状:
运动神经传导速度:多数患者为轻微到明显减慢,在年长患者处于较低值
- 复合肌肉动作电位:幅度减低
- 肌电图:无自发性电活动的去神经征象
- 清醒期脑电图:不规则背景活动;可记录到多灶性痫样放电
- 脑干听诱发电位:可在大于两岁的患者中记录到I-V波传导时间延长
躯体感觉诱发电位:在所有受累的患者中均记录到中枢传导时间延长
- 闪光视觉诱发电位:正常
- 视网膜电位:正常
神经病理表现 - 对伴有外周神经系统病变的患者进行腓肠神经活检显示髓鞘化纤维数目轻到重度减少,伴部分轴突包裹髓鞘变薄或无髓鞘
- 偶见多余的与不规则折叠纤维髓鞘解聚
- 电镜证实部分轴突无髓鞘包裹及髓鞘化纤维变薄,部分被很少的施旺细胞突起包绕形成小的洋葱球样外观
整形外科, 患者有轻度进展的脊柱侧凸伴行走能力丧失 [Biancheri et al 2007]。
生存期不详,最年长存活受累的患者29岁。
导致FAM126A蛋白表达完全缺失的致病性变异与双侧白内障、中枢与周围神经神经系统髓鞘形成低下的完全表型相关。引起部分蛋白功能缺陷的致病性变异与不伴周围神经系统受累的较轻表型相关。一例外显子8与9缺失的一例患者无先天的白内障;但9岁时出现白内障。第二例患者表现为先天性单侧白内障,然而这个家族中超过2岁的4个存活孩子6岁后均不能借助辅具的行走[Ugur & Tolun 2008]。到目前为止由于报道的 HCC病例数较少,这些关系尚须进一步明确。
完全外显。
流行病学
HCC属罕见病,目前尚无流行病学研究。
遗传学相关(等位基因)病
除了GeneReview本章中讨论的疾病外,尚无其它表型 与FAM126A突变相关的疾病
鉴别诊断
先天的白内障联合大脑髓鞘形成低下是髓鞘形成低下与先天性白内障 (HCC)的典型表现。 然而,下列的其它髓鞘化低下性疾病仍需鉴别。
- 中枢神经系统髓鞘形成的PLP1-相关性疾病 包括一系列表型从佩梅病(PMD)到痉挛性截瘫2型(SPG2)的表型。经典髓鞘化低下疾病PMD在婴儿或者幼儿早期的典型症状包括眼震、肌张力低下及认知损害,并逐渐出现严重痉挛及共济失调。生存期缩短。SPG2主要表现为伴或不伴中枢神经系统受累的痉挛性截瘫与生存期正常。女性携带者可有轻到中度疾病表现。可见家族内表型变异,但家庭内征象通常相当一致。PLP1重复突变、缺失突变及单核苷酸变异是致病原因。遗传方式呈X-连锁。
- 髓鞘化低下性脑白质营养不良2型(HLD2)(OMIM608804)主要特征为早发眼球震颤、运动发育里程碑延迟、共济失调、进行性痉挛,部分发作性癫痫、轻度外周神经系统疾病以及MRI弥漫性髓鞘形成低下。其由编码连接蛋白46.6的GJA12 (GJC2)致病突变导致[Uhlenberg et al 2004]。遗传方式呈常染色体隐性遗传。
- 游离唾液酸代谢等位基因病(见游离唾液酸潴积病)Salla病、中间型严重Salla病以及婴儿游离唾液酸潴积病(ISSD)是由游离唾液酸在溶酶体中异常储积导致的一类神经变性病。表型最轻的Salla病主要特征为出生时外观与神经系统正常,接着缓慢出现进行性神经系统恶化,导致轻到中度神经运动发育倒退、痉挛、手足徐动症以及癫痫发作。Salla病中的基底节髓鞘化异常及胼胝体发育不良是持续存在和早期表现[Sonninen et al 1999]。可见小脑白质改变,能够解释共济失调[Linnankivi et al 2003, Biancheri et al 2004]。游离唾液酸潴积病中最严重的表型是ISSD,受累的患者可有严重发育落后、粗糙面容、肝脾肿大及心脏增大,儿童早期常常死亡。SLC17A5 致病性变异是原因. 遗传方式是 常染色体隐性遗传。
- Cockayne综合征(CS)是一个疾病谱,包括:CS I 型,即“经典型”;CS II型为严重型,患者出生时即起病(表型与脑-眼-面综合征 [COFS]或Pena-Shokeir综合征 II型重叠);CS III型表型较轻;以及着色性干皮病型CS(XP-CS)。
CS I 型特征为宫内生长正常,在两岁前出现生长及发育异常。在疾病进展过程中,患儿身长、体重及头围等明显低于第五百分位数。进行性视、听力损害,中枢及外周神经系统病变导致严重障碍,多在十到二十岁之间死亡。
CS II 型(严重型CS或早发型CS)患者出生时即表现生长迟缓,生后神经发育程度极低甚至无发育。可有先天性白内障或其他眼部结构性缺失。受累患儿生后早期表现脊柱(后凸或侧凸)及关节挛缩。多于7岁前死亡。
CS I型及II型均有特征性“虎斑样”皮层下白质脱髓鞘表现,多灶性钙沉积与神经元功能相对保留 [Itoh et al 1999]。致病基因为ERCC6或ERCC8基因突变。遗传方式为染色体隐性遗传。
- 毛发低硫性营养不良(Tay综合征)(OMIM 601675)特征性表现为生长倒退、智力障碍、小头,先天的鱼鳞病以及脆性毛发[van der Knaap & Valk 2005]。病因为ERCC3、GTF2H5或者ERCC2 突变导致DNA修复缺陷。遗传方式为常染色体隐性遗传。
见 脑白质营养不良、髓鞘形成低下:OMIM表型系列 OMIM查看基因相关表型。
管理
对症治疗
通常生后一月内进行白内障摘除术。支持治疗包括下列:
- 理疗以改善运动功能
- 特殊教育
如有癫痫发作,则进行抗癫痫药物治疗
预防并发症
尽管尚无根治方法,对症治疗可改善患者的生存质量。建议通过以下多系统治疗以预防:
- 痉挛:药物治疗,理疗
- 共济失调:康复治疗
- 癫痫:抗惊厥药物治疗
- 认知发育迟缓:在学校或工作中制定针对性方案
- 整形:积极预防/治疗矫形问题,如关节挛缩或脊柱侧凸
- 喂养困难:如有需要,进行吞咽功能检测或胃造瘘
监测
定期进行监测:
- 神经系统评估以明确神经系统并发症;
- 由理疗医师评估是否需要借助辅助设备;
- 如新生儿期未发现白内障,则需进行眼部检查。
研究性治疗
搜索ClinicalTrials.gov以获取大量疾病相关临床研究信息。 注意:目前可能尚无关于该病的临床研究。
遗传咨询
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
家族成员风险
先证者父母
先证者同胞
- 受孕时, 每个先证者的同胞有 25%机会受累的, 50% 机会为无症状 携带者, 25% 机会既非受累也非携带者。
- 一旦确定有风险的同胞非受累,他/她成为是一个FAM126A致病性变异携带者的机会是2/3。
先证者后代. HCC患者不生育。
先证者其他家族成员. 先证者父母的每个同胞均有50% 成为FAM126A致病性变异 携带者的风险。
相关遗传咨询问题
家庭计划
- 妊娠前是遗传风险评估、明确携带者与可行的产前检测方法讨论的最佳时机。
DNA库 是储存DNA为将来可能的用途。DNA储存 (经典从白细胞中提取) 有利于将来在测试方法以及对基因的了解,等位基因变异与疾病方面具有较大进展,应当建立个人银行 受累的个体的DNA库。
产前诊断与植入前遗传学诊断
一旦受累的家庭成员明确FAM126A致病性变异, 为高风险妊娠的产前诊断和植入前遗传诊断成为了可能。
Resources
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- National Library of Medicine Genetics Home Reference
- National Eye Institute31 Center DriveMSC 2510Bethesda MD 20892-2510
- Prevent Blindness America211 West Wacker DriveSuite 1700Chicago IL 60606Phone: 800-331-2020 (toll-free)Email: info@preventblindness.org
- Myelin Disorders Bioregistry ProjectEmail: myelindisorders@cnmc.org
分子遗传学
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. -ED.
表 A.
髓鞘形成低下与先天性白内障: 基因与数据库
表 B.
OMIM 髓鞘形成低下与先天性白内障OMIM 词条 (在中OMIM查看全部OMIM)
最常见的剪接体由不包含第11号外显子的521个氨基酸组成。而由于第11号外显子上的提前出现的终止密码子,包含第11号外显子的剪接体只包含399个氨基酸。请参见 表A获得有关基因及蛋白的详细描述见表 A,。基因
表 2.
选择FAM126A 变异
| 变异分类 | DNA核苷酸变异 (Alias 1) | 预测蛋白变异 (Alias 1) | 参考文献 | 参考序列 |
|---|---|---|---|---|
| 良性 | c.624A>G | p.(=) 2 (Ser208Ser) | -- | rs3735231 |
| 致病性 | c.51+1G>A (IVS2+1G>A) | -- | Zara et al [2006] | NM_032581-.3 NP_115970-.2 |
| c.414+1G>T (IVS5+1G>T) | -- | Zara et al [2006] | ||
| c.158T>C | p.Leu53Pro | Zara et al [2006] | ||
| (531-439_743+348del) | (Arg209fsTer213) | Ugur & Tolun [2008] |
变异分类注解: 表中所列举的变异由作者提供。Genereviews未对变异的致病性进行独立验证。
命名注解: GeneReviews 遵照人类基因组变异协会(varnomen-.hgvs.org)的标准命名法。见Quick Reference 中命名法的解释。
1.
变异命名未遵照当前命名规则。
2.
P.(=)指蛋白未被分析,但是预测无改变。
正常的基因产物. FAM126A编码未知功能的521个氨基酸组成的膜蛋白[Zara et al 2006]。该蛋白包含两个推断的跨膜结构域,但功能结构域尚不清楚。异常的基因产物. 剪接变异体(c.414+1G>T及c.51+1G>A)通过mRNA剪切改变,导致蛋白翻译的提前终止。错义变异c.158T>C不改变mRNA的表达,但是通过某些未知的细胞通路导致严重的蛋白缺陷。基因组的531-439_743+348del缺失预测导致308个氨基酸的缺失,后者变异的作用未经过免疫蛋白沉淀实验的验证。
References
Literature Cited
- Biancheri R, Rossi A, Mancini MG, Minetti C. Cerebellar white matter involvement in Salla disease. Neuroradiology. 2004;46:587鈥�8. [PubMed: 15179531]
- Biancheri R, Zara F, Bruno C, Rossi A, Bordo L, Gazzerro E, Sotgia F, Pedemonte M, Scapolan S, Bado M, Uziel G, Bugiani M, Lamba LD, Costa V, Schenone A, Rozemuller AJ, Tortori-Donati P, Lisanti MP, van der Knaap MS, Minetti C. Phenotypic characterization of hypomyelination and congenital cataract. Ann Neurol. 2007;62:121鈥�7. [PubMed: 17683097]
- Biancheri R, Zara F, Rossi A, Mathot M, Nassogne MC, Yalcinkaya C, Erturk O, Tuysuz B, Di Rocco M, Gazzerro E, Bugiani M, van Spaendonk R, Sistermans EA, Minetti C, van der Knaap MS, Wolf NI. Hypomyelination and congenital cataract: broadening the clinical phenotype. Arch Neurol. 2011;68:1191鈥�4. [PubMed: 21911699]
- Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326鈥�33. [PubMed: 10413020]
- Linnankivi T, Lönnqvist T, Autti T. A case of Salla disease with involvement of the cerebellar white matter. Neuroradiology. 2003;45:107鈥�9. [PubMed: 12592494]
- Rossi A, Biancheri R, Zara F, Bruno C, Uziel G, van der Knaap MS, Minetti C, Tortori-Donati P. Hypomyelination and congenital cataract: neuroimaging features of a novel inherited white matter disorder. AJNR Am J Neuroradiol. 2008;29:301鈥�5. [PubMed: 17974614]
- Sonninen P, Autti T, Varho T, Hämäläinen M, Raininko R. Brain involvement in Salla disease. AJNR Am J Neuroradiol. 1999;20:433鈥�43. [PubMed: 10219409]
- Timmons M, Tsokos M, Asab MA, Seminara SB, Zirzow GC, Kaneski CR, Heiss JD, van der Knaap MS, Vanier MT, Schiffmann R, Wong K. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67:2066鈥�9. [PMC free article: PMC1950601] [PubMed: 17159124]
- Traverso M, Assereto S, Gazzerro E, Savasta S, Abdalla EM, Rossi A, Baldassari S, Fruscione F, Ruffinazzi G, Fassad MR, El Beheiry A, Minetti C, Zara F, Biancheri R. Novel FAM126A mutations in hypomyelination and congenital cataract disease. Biochem Biophys Res Commun. 2013a;439:369鈥�72. [PubMed: 23998934]
- Traverso M, Yuregir OO, Mimouni-Bloch A, Rossi A, Aslan H, Gazzerro E, Baldassari S, Fruscione F, Minetti C, Zara F, Biancheri R. Hypomyelination and congenital cataract: identification of novel mutations in two unrelated families. Eur J Paediatr Neurol. 2013b;17:108鈥�11. [PubMed: 22749724]
- Ugur SA, Tolun A. A deletion in DRCTNNB1A associated with hypomyelination and juvenile onset cataract. Eur J Hum Genet. 2008;16:261鈥�4. [PubMed: 17928815]
- Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloğlu H, Nürnberg P, Hübner C, Weschke B, Gärtner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251鈥�60. [PMC free article: PMC1216059] [PubMed: 15192806]
- van der Knaap MS, Naidu S, Pouwels PJ, Bonavita S, van Coster R, Lagae L, Sperner J, Surtees R, Schiffmann R, Valk J. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol. 2002;23:1466鈥�74. [PubMed: 12372733]
- van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. 3 ed. Berlin, Germany: Springer; 2005.
- Zara F, Biancheri R, Bruno C, Bordo L, Assereto S, Gazzerro E, Sotgia F, Wang XB, Gianotti S, Stringara S, Pedemonte M, Uziel G, Rossi A, Schenone A, Tortori-Donati P, van der Knaap MS, Lisanti MP, Minetti C. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet. 2006;38:1111鈥�3. [PubMed: 16951682]
Chapter Notes
Acknowledgments
We thank the Cell Line and DNA Bank from Patients Affected by Genetic Diseases collection (dppm.gaslini.org/biobank), supported by the Italian Telethon, for allowing us to obtain samples.
Revision History
- 4 June 2015 (me) Comprehensive update posted live
- 27 October 2011 (cd) Revision: mutation scanning and deletion/duplication analysis no longer available clinically; 序列分析 now available clinically
- 27 January 2011 (cd) Revision: prenatal testing available clinically
- 16 November 2010 (me) Comprehensive update posted live
- 14 October 2008 (me) Review posted live
- 14 May 2008 (rb) Original submission