
简介
临床特征.
表异构酶缺乏半乳糖血症(GALE 型) 表现为三种形式:
- 全身性者在所有测试组织中酶活性显着降低。
- 外周型者红细胞(RBC)和循环白细胞酶活性缺乏,但在所有其他组织中正常或接近正常。
- 中间型者红细胞和循环白细胞酶活性缺乏,并且在其他测试细胞中低于正常水平的50%。
患有全身性表异构酶缺乏半乳糖血症的婴儿会发现常规乳饮食(含有乳糖,半乳糖和葡萄糖的二糖)有临床表现;包括张力减退,喂养不良,呕吐,体重减轻,黄疸,肝肿大,肝功能障碍,氨基酸尿症和白内障,及时从他们的饮食中去除半乳糖/乳糖可以解决或预防这些急性症状。相反,具有外周型或中间型的新生儿即使在常规牛奶饮食中通常也保持无症状,并且仅通过生化测试来鉴定,通常在新生儿筛查计划中。
诊断/检测。表异构酶缺乏半乳糖血症的诊断建立在RBC中GALE活性受损的 先证者和/或分子遗传学检测中GALE中双等位基因的致病变异的鉴定中。 RBC中GALE酶活性损伤的程度不能区分临床上严重全身型、外周型或中间型的表异构酶缺乏。需要进一步测试其他细胞类型,如受激活的白细胞或EBV转染的淋巴母细胞,以进行区分。
管理。表型的治疗:通过半乳糖/乳糖限制饮食预防或纠正全身性表异构酶缺乏半乳糖血症的急性和潜在致死症状。注意:受影响的个体可能需要微量环境来源的半乳糖:婴儿应该喂食含有微量半乳糖或乳糖的配方奶粉(例如大豆配方奶粉)。建议继续限制大龄儿童的乳制品。相反,患有外周型表异构酶缺乏的半乳糖血症的婴儿被认为无论饮食如何都保持无症状;具有中间型表异构酶缺乏症的婴儿半乳糖血症可能从早期膳食半乳糖/乳糖限制长期受益,但这机理仍然不清楚。
预防主要表现:在全身型表异构酶缺乏症中,限制饮食中的半乳糖/乳糖可以纠正或预防疾病的急性体征和症状(肝功能不全,肾功能不全和轻度白内障),但不能纠正或预防发育迟缓或学习障碍。在一些孩子身上观察到由于难以区分表异构酶缺乏性半乳糖血症的外周型或中间型,建议对所有GALE缺乏的婴儿进行饮食限制半乳糖/乳糖,一旦确认更准确的诊断,放宽限制。
监测:监测血红蛋白gal-1P(半乳糖-1-磷酸)或尿液半乳糖醇,特别是如果要使饮食正常化。可接受水平的红细胞gal-1P尚不清楚,但经典半乳糖血症的数据估计<3.5 mg / 100 mL(正常≤1.0mg/ 100 mL)。其他需要监控的是生长和发育的阶段参数。
需要避免的药剂/环境:全身表异构酶缺乏症患者限制膳食中的半乳糖/乳糖,从婴儿至终身。
对有风险的亲属进行评估:在等待表异构酶缺乏半乳糖血症的诊断检测结果时,应从出生开始对每个有风险的新生儿同胞进行治疗; 分子遗传学检测检测(如果已知家族中的致病变异)或RBC中GALE酶活性的测量(如果家族中的致病变异未知)可以进行。
遗传咨询。表异构酶缺乏半乳糖血症以 常染色体隐性遗传方式遗传。在受孕时,受累的个体每个完全同胞有25%的机会受到影响,50%的几率成为无症状携带者,25%的机会正常。如果已知该家族的致病变异,则可以对有风险的家庭成员进行携带者测试,并对可能增加风险的怀孕进行产前检测。
诊断
表异构酶缺乏半乳糖血症(GALE缺乏性半乳糖血症)一种包含三种形式:
- 全身型, 在所有测试组织中酶活性显著降低。
- 外周型,红细胞(RBC)和循环白细胞酶活性缺乏,但在所有其他组织中正常或接近正常。
- 中间型, RBC和循环白细胞酶活性缺乏,并且在其他测试细胞中低于正常水平的50%。
提示性结果
个体(正常牛奶饮食)具有以下新生儿筛查结果,临床特征和支持性实验室检查结果应怀疑表异构酶缺乏半乳糖血症:
新生儿筛查
- 在 新生儿筛查计划包括测量总半乳糖(gal + gal-1P)和GALT酶活性(参见 Galactosemia):
- 总半乳糖(半乳糖和半乳糖-1-磷酸的总和)升高;
和 - GALT酶活性正常.
- 在仅测量GALT酶活性低的状态下,受累的婴儿对于半乳糖血症将具有正常的新生儿筛查结果。
临床特征
- 肌张力低下
- 喂养困难
- 呕吐
- 体重减轻
- 黄疸
- 肝肿大
- 肝功能不全
- 白内障
- 没有临床发现(外周型或中间型)
支持性实验室发现
- 红细胞半乳糖 -1-磷酸浓度升高的(正常0-1.0 mg / 100 mL RBC):
- 在具有全身型表异构酶缺乏半乳糖血症中,高达170mg / 100mL
- 而在外周型和中间型表异构酶缺乏半乳糖血症中,则>30 mg/100 mL
- 尿半乳糖浓度高达116 mmol / L(2.09 g / 100 mL,对照<30 mg / 100 mL)
- 尿液中有非葡萄糖还原物质(代表尿液半乳糖)
- 尿中半乳糖醇浓度升高(年龄<1岁,肌酐<94.7 mmol / mol, 1-6岁则<45.3 mmol / mol,年龄> 6岁则<18.4 mmol / mol,)
- 氨基酸尿症
- 正常的GALT酶活性
建立诊断
通过下列之一在先证者中建立表异构酶缺乏半乳糖血症的诊断:
- 通过传统的分光光度法测定,红细胞(RBC)中血红蛋白(Hb)GALE酶活性0.0-8.0μmol/ hr / g(正常17.1-40.1μmol/ hr / g Hb)
和 / 或
- 使用液相色谱/串联质谱法测定红细胞中<0.5μmol/ hr / g Hb GALE酶活性(正常值为2.3-12.7μmol/ hr / g Hb) [Chen et al 2014]
和 / 或
- 单基因测试。如果仅发现一种或没有致病性变异,则首先进行GALE的序列分析,然后进行基因靶向 deletion/duplication analysis。
- 还可以考虑包括GALE和其他感兴趣基因的表型靶向检测(参见 Differential Diagnosis)。注意:(1)小组中包含的基因和用于每个基因的测试的诊断敏感性因实验室和随时间而变化。 (2)小组中使用的方法可以包括 序列分析, deletion/duplication analysis和/或其他基于非测序的测试。Table 1.
用于表异构酶缺乏半乳糖血症的分子遗传学检测
| 基因 1 | 检测方法 | 检测方法致病变异占比 2 |
|---|---|---|
| GALE | 测序 3 | 14/16 alleles and 13/14 alleles (~90%) 4 |
| 基因靶向deletion/duplication analysis 5 | 无报道 6 |
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics
- 3.
- 4.
全基因测序能揭示了大多数具有生物化学证实的GALE缺陷的人GALE致病变异(例如, Park et al [2005], Openo et al [2006],在Fridovich-Keil & Walter [2008]中综述); 然而,由于所研究的等位基因数量少且诊断的生化复杂性,这种估计可能随时间而变化。
- 5.
基因靶向deletion/duplication analysis检测基因内缺失或重复。 使用的方法可以包括: quantitative PCR,长程PCR和多重连接依赖性探针扩增(MLPA)和设计用于检测单 外显子缺失或重复的基因靶向微阵列。
- 6.
据报道,没有任何涉及GALE的缺失或重复导致表异构酶缺乏半乳糖血症。
附加测试
可以在成纤维细胞或成淋巴细胞中测量GALE酶活性,以帮助区分表异构酶缺乏半乳糖血症的全身型,外周型和中间型; 然而,据作者所知,此测试目前尚未在临床上提供。
临床特征
临床表现
由GALE酶活性降低引起的表异构酶缺乏型半乳糖血症的临床严重程度[Fridovich-Keil & Walter 2008]范围在可能是致命的[Holton et al 1981, Henderson et al 1983, Walter et al 1999, Sarkar et al 2010]与良性之间[Gitzelmann 1972]。
表异构酶缺乏型半乳糖血症可以通过酶活性水平分为以下三种形式:全身型,外周型和中间型(见Diagnosis) [Openo et al 2006]。注意:在所有三种形式中,外周循环的红细胞和白细胞中GALE酶活性缺乏。
全身型表异构酶缺乏型半乳糖血症与中间型或轻型表异构酶缺乏型半乳糖血症之间的关键区别在于具有全身表异构酶缺乏型的半乳糖血症患者在正常乳汁饮食中有临床发现,而具有中间型或轻型表异构酶缺乏型半乳糖血症患者在临床上保持良好,至少在新生儿期。
全身型表异构酶缺乏型半乳糖血症 患有全身型表异构酶缺乏型半乳糖血症的婴儿通常会出现 classic galactosemia症状:肌张力减退,喂养不良,呕吐,体重减轻,黄疸,肝肿大,肝功能不全(如血清转氨酶显着升高),氨基酸尿症和白内障。及时从饮食中去除半乳糖/乳糖可以解决或预防这些急性症状 [Walter et al 1999, Sarkar et al 2010] (see Management).。
全身型表异构酶缺乏型半乳糖血症患者的长期结果信息是有限的:报告此型人数不到10人[Walter et al 1999, Sarkar et al 2010]。有些人表现出长期并发症,这些并发症在儿童早期就已经明显(包括感觉神经性听力损伤以及身体和认知发育迟缓和/或学习困难),而另一些则没有。值得注意的是,大多数报告的个体都是近亲婚配的父母所生,这引起了人们的担忧,即除了GALE的其他 常染色体隐性遗传等位基因的纯合性可能是一些(如果不是大多数)报道的长期并发症的基础。那些长期随访的少数患有全身型表异构酶缺乏型半乳糖血症的个体表现出明显正常的青春期,没有明显的卵巢早衰证据[Walter et al 1999]。
外周型表异构酶缺乏型半乳糖血症 具有轻型的新生儿即使在普通的牛奶饮食中也通常是无症状的;这些婴儿仅在生化检测 新生儿筛查后升高的总半乳糖后才被发现。
患有外周型表异构酶缺乏型半乳糖血症的儿童即使保持正常的乳饮食,也似乎无症状。
中间型表异构酶缺乏型半乳糖血症 具有中间型的新生儿即使在常规乳饮食中也通常无症状,并且仅通过新生儿筛查鉴定。长期预后仍不清楚。一个未受到半乳糖/乳糖饮食限制的受累的患儿经历了运动和认知发展的延迟,在幼儿时期变得明显[Alano et al 1998, Openo et al 2006]。已知具有中间型表异构酶缺乏型半乳糖血症的所有其他个体已经通过膳食半乳糖/乳糖限制进行治疗,至少在婴儿期,并且迄今为止已经被跟踪的那些人似乎在临床上保持良好。
病理生理学 半乳糖通过包含酶半乳糖激酶(GALK,EC 2.7.1.6),半乳糖-1-磷酸尿苷转移酶(GALT,EC 2.7.7.12)和尿苷二磷 酸-半乳糖-4-表异构酶(GALE,EC 5.1.3.2)Leloir途径在人和其他物种中代谢。 。Figure 1所示,GALE催化了将UDP-半乳糖转化为UDP-葡萄糖的该途径中的必要步骤。 GALE是一种可逆酶,当UDP-半乳糖的其他来源受到限制时,它还催化UDP-葡萄糖合成UDP-半乳糖。在Leloir途径之外发挥作用,GALE还互相转换UDP-N-乙酰半乳糖胺和UDP-N-乙酰氨基葡萄糖。所有这四种UDP-糖都是人体糖蛋白和糖脂生物合成的必需底物。
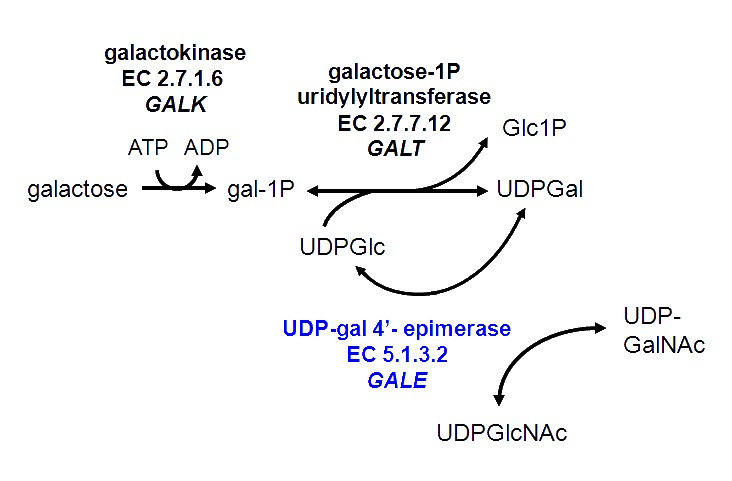
Figure 1
Leloir pathway
在classic galactosemia ,与表异构酶缺乏型半乳糖血症相关的白内障被认为是由眼晶状体中的半乳糖醇积聚引起的;其他急性表现有可能是由gal-1P(半乳糖-1-磷酸)或其他代谢物的组织积累引起的,但未证实。
暴露于半乳糖的表异构酶缺乏型半乳糖血症患者表现出UDP-半乳糖(UDP-gal)的异常积累。然而,由于人类需要GALE来进行UDP-gal和UDP-N-乙酰半乳糖胺(UDP-galNAc)的内源性生物合成,因此这些化合物的生产不足可导致至少部分表异构酶缺乏的病理生理学,特别是子宫,导致糖蛋白和糖脂(包括脑苷脂)的缺乏或异常产生。
基因型 - 表型相关性
由于临床上分子证实的表异构酶缺乏性半乳糖血症的个体数量不足以确定基因型/表型相关性,因此对转化的淋巴母细胞或其他“非外周”细胞类型的研究是区分生物化学不同形式的表异构酶缺乏的唯一方法[Mitchell et al 1975, Openo et al 2006]。
命名法
一些作者将不同形式的半乳糖血症称为I型,II型和III型半乳糖血症,其中:
I型半乳糖血症是指GALT deficiency
II型半乳糖血症是指GALK缺乏症
III型半乳糖血症是指GALE缺乏症(表异构酶缺乏,半乳糖血症)
流行
目前尚无真正的患病率数据。全身型表异构酶缺乏型半乳糖血症非常罕见;然而,新生儿筛查检测到的表异构酶缺乏型半乳糖血症可能在非洲裔美国婴儿中大约为1:6,700,在美国欧洲血统婴儿中大约为1:70,000[Alano et al 1997, Fridovich-Keil & Walter 2008]。
遗传相关(等位)疾病
除了在本GeneReview中讨论的表型之外,没有已知的表型与GALE中的致病变异相关。
鉴别诊断
GALT缺乏症。 半乳 糖-1-磷酸尿苷转移酶缺乏(GALT)引起的半乳糖血症可分为三种临床/生化表型:(1)典型的半乳糖血症; (2)临床变异性半乳糖血症; (3)Duarte(生化变异)半乳糖血症。 该分类基于:残留的红细胞GALT酶活性; 在乳糖限制饮食中观察到的半乳糖代谢物(例如,红细胞半乳糖-1-磷酸和尿液半乳糖醇)的水平; 最重要的是,受累的个体可能会发生急性和慢性长期并发症。 GALT中的双等位基因致病变异是致病原因; 遗传是 常染色体隐性遗传。
- Classic galactosemia半乳糖血症可导致危及生命的并发症,包括喂养问题,发育障碍,肝细胞损伤,出血和未经治疗的婴儿的大肠杆菌败血症。 如果在出生后的前十天提供乳糖限制性饮食,新生儿症状通常会很快消退,并且可以预防肝功能衰竭,败血症和新生儿死亡的并发症; 然而,尽管从幼年时期接受了充分的治疗,患有典型半乳糖血症的儿童仍然存在发育迟缓,言语问题(称为儿童时期的言语失调和构音障碍)以及运动功能异常的风险增加。 绝大多数患有典型半乳糖血症的女性表现出卵巢功能不全(POI)。
- Clinical variant galactosemia可导致未经治疗的婴儿出现危及生命的并发症,包括喂养问题,发育迟缓,包括肝硬化在内的肝细胞损伤和出血。 它可以在具有低残留GALT酶活性的任何血统的个体中发生,但可能在非洲裔美国人和南非的非洲本地人中高频率发生 p.Ser135Leu GALT等位基因相关的疾病。 新生儿筛查 (NBS)可能会遗漏患有临床变异性半乳糖血症的人,因为高半乳糖血症并不像经典的半乳糖血症那样明显。 与典型的半乳糖血症一样,如果在出生后的头几天提供乳糖限制性饮食,通常会预防严重的急性新生儿并发症。 患有临床变异性半乳糖血症的受治疗个体的长期预后也可能较轻微。
- Duarte variant galactosemia (biochemical variant galactosemia). 患有母乳或含乳糖配方的Duarte变异半乳糖血症的人通常(尽管不总是)无症状,至少在婴儿期。
半乳糖激酶(GALK)缺乏症(OMIM) 对于患有白内障的健康个体,半乳糖的血浆浓度增加和半乳糖醇的尿排泄增加,应考虑半乳糖激酶(GALK)缺乏症。受影响的个体具有正常的GALT酶活性,并且无积累gal-1P。白内障是由晶状体中半乳糖代谢物(半乳糖醇)的积累引起的。半乳糖醇不溶于酒精,其导致细胞内渗透压和肿胀增加,质膜氧化还原电位丧失,随后细胞死亡。检测溶血产物中降低的GALK酶活性能确诊。 GALK1中的双等位基因致病变异是病因[Kolosha et al 2000, Hunter et al 2001]; 为常染色体隐性遗传。大多数人群中GALK缺乏的患病率尚不清楚;然而,最近德国的一项研究报告了大约1:40,000的患病率,这与同一人群中典型的半乳糖血症的患病率类似 [Hennermann et al 2011]。在其他人群中,患病率可能低得多。
其他。 许多其他罕见疾病,包括以下疾病,也会导致婴儿食用牛奶的血液或尿液中的半乳糖或半乳糖代谢物升高:
- 门体静脉分流术
- 肝动静脉畸形
- Fanconi-Bickel综合征(OMIM) 由于SLC2A2中的 双等位基因的的致病变异。 患有这种病症的个体具有肝肾糖原积累,葡萄糖和半乳糖的利用受损以及近端肾小管肾病。
- 由PGE1中的 双等位基因的致病变异引起的先天性糖基化1T型 (OMIM) [Tegtmeyer et al 2014].。 具有这种病症的个体可以在RBC中表现出轻度增加的半乳糖-1-磷酸水平。 他们出生时可能有腭裂/双歧悬雍垂,并可能出现间歇性低血糖,扩张性心肌病,运动不耐受,血清肌酸激酶增加和肝脏疾病。
处理
初步诊断后的评估
为了确定被诊断患有表异构酶缺乏半乳糖血症的个体的疾病程度和需求,建议进行以下评估:
- 测量身高,体重和头围
- 营养和喂养评估
- 神经系统检查
- 发育评估
- 肝功能检查(血清AST,ALT,白蛋白,总蛋白,总胆红素和结合胆红素,凝血酶原时间和部分促凝血酶原激酶时间)
- 眼科咨询评估白内障
- 咨询临床遗传学家
治疗表型
全身表异构酶缺乏半乳糖血症 限制半乳糖/乳糖饮食预防或纠正全身性表异构酶缺乏半乳糖血症的急性和潜在致命症状。这意味着将婴儿从母乳或乳基配方转换为仅含有微量半乳糖或乳糖的配方,如大豆配方奶粉。值得注意的是,一些经典半乳糖血症的婴儿是处方元素配方,其半乳糖含量甚至低于大豆配方奶粉。对于具有全身性表异构酶缺乏半乳糖血症的婴儿,不应规定元素配方,因为GALE酶是UDP-半乳糖的内源性生物合成所必需的;也就是说,具有表异构酶缺乏半乳糖血症患者可能需要微量环境来源的半乳糖。然而,最佳所需的半乳糖摄入量仍然未知。
对于具有全身性表异构酶缺乏半乳糖血症的年龄较大的儿童,乳糖/乳糖的饮食持续限制。
注意:一些(但不是全部)医生建议患有典型半乳糖血症的个体也要戒除含有微量半乳糖/乳糖的非乳制品(例如一些水果和蔬菜,器官肉);这种更严格的饮食限制可能不适用于全身性差向异构酶缺乏症半乳糖血症患者。
在全身性表异构酶缺乏半乳糖血症中,半乳糖血症限制饮食中的半乳糖/乳糖似乎可以纠正或预防该疾病的急性体征和症状:肝功能障碍,肾功能不全和轻度白内障。据推测,如在经典的半乳糖血症中,饮食治疗不能纠正由于长期半乳糖暴露导致的严重组织损伤(例如,肝硬化或成熟白内障)。不能通过饮食限制半乳糖/乳糖来解决的成熟白内障可能需要手术切除。
外周型表异构酶缺乏半乳糖血症 患有外周型表异构酶缺乏半乳糖血症不需要任何饮食限制。
中间型表异构酶缺乏症半乳糖血症 具有中度表异构酶缺乏症半乳糖血症的个体通常用膳食半乳糖/乳糖限制治疗,至少在婴儿期。他们可能有(尚未知)长期并发症风险,包括学习障碍和/或白内障。因此,对于这些婴儿,继续母乳喂养或接触含有高水平半乳糖/乳糖的乳基配方可能是不可取的;但是,没有足够的数据来提出建议。
预防主要表型
在全身性表异构酶缺乏半乳糖血症中,半乳糖/乳糖的饮食限制可防止早期喂养问题,呕吐,体重增加不良,肝功能障碍和白内障。
在全身性表异构酶缺乏半乳糖血症,治疗无症状新生儿的挑战是,可能需要数月才能获得用于区分外周型差向异构酶缺乏症半乳糖血症和中间型表异构酶缺乏半乳糖血症的测试结果(参见建立诊断, Additional Testing);此外,可能无法进行此类测试。因此,最保守的方法是建议所有患有表异构酶缺乏半乳糖血症的婴儿对半乳糖/乳糖的饮食限制,一旦确认更准确的诊断就放宽限制。
监测
以下是适当的:
- 监测血1-磷酸半乳糖或尿液半乳糖醇,特别是如果要使饮食正常化。 GALE缺乏症中可接受的1-磷酸半乳糖水平尚不清楚,但根据经典半乳糖血症的经验估计红细胞中<3.5 mg / 100 mL。
- 跟踪生长。
- 监控发育的阶段性;根据需要提出支持性干预。
要避免的药物/情况
具有全身性表异构酶缺乏半乳糖血症的人群应该使用半乳糖/乳糖限制性饮食,从婴儿至终身。
具有中间型表异构酶缺乏半乳糖血症的人可以短期或长期置于半乳糖/乳糖限制性饮食中。在半乳糖激发后(例如,正常饮食2周)评估血1-磷酸半乳糖和/或尿半乳糖醇可以帮助确定个体是否应该长时间保持在半乳糖/乳糖限制饮食中。
风险亲属的评估
如果尚未进行产前检查(参见Genetic Counseling),应从出生开始对每个有风险的新生儿同胞进行治疗,直至获得诊断检测结果。诊断评估可包括:
- 如果已知家族中的致病变异,则进行分子遗传学检测;
- 如果家族中的致病变异未知,则测量红细胞中的GALE酶活性。
注意:如果担心产前检测的可靠性,可能会在进行诊断检测时给予大豆配方奶粉。
有关为遗传咨询目的测试有风险亲属的相关问题,请参阅Genetic Counseling。
正在观察的疗法
在ClinicalTrials.gov中搜索有关各种疾病和病症的临床研究信息。注意:这种疾病可能没有临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。 本节不是为了解决个人可能面临的所有个人,文化和伦理问题,也不是用遗传专业人员代替咨询。-编者。
遗传模式
表异构酶缺乏半乳糖血症遗传模式为常染色体隐性遗传.
对家庭成员的风险
先证者的父母
- 受累的孩子的父母通常是杂合子(即GALE致病性变异的携带者)。
- 杂合子(携带者)是无症状的,并且没有发生全身性表异构酶缺乏半乳糖血症症状的风险。
先证者的同胞
- 在受孕时,受累的个体的每个完全同胞有25%的机会受到影响,有50%的几率成为无症状的携带者,25%的机会正常。
- 杂合子(携带者)是无症状的,并且没有发生全身性表异构酶缺乏半乳糖血症症状的风险。
- [Walter et al 1999]的数据表明, 家族中鉴定的表异构酶缺乏半乳糖血症的亚型应该“持续运行”,这意味着如果一个同胞全身性表异构酶缺乏半乳糖血症,那么该家族中其他受累的同胞可能具有全身性表异构酶缺乏半乳糖血症。如果一个同胞具有外周型表异构酶缺乏半乳糖血症,该家族中的其他同胞可能具有相同的形式。
先证者的后代。由受累的个体与未受影响的非携带者伴侣的所有后代都是GALE中 致病性变异的肯定杂合子(携带者)。
其他家庭成员。假设没有其他的半乳糖血症家族史, 先证者父母的每个完全同胞在GALE中成为致病性变异的携带者的风险为50%。
携带者(杂合子)检测
使用分子遗传学检测 对有风险的亲属进行携带者检测需要事先鉴定家族中的GALE致病变异。
虽然检测携带者的生化检测也是可能的,但对照和携带者GALE酶活性的范围重叠,因此使分子遗传学检测成为携带者检测的首选方法。
注意:携带者是这种 常染色体隐性遗传的杂合子,并且没有发生全身性表异构酶缺乏半乳糖血症症状的风险。
相关的遗传咨询问题
有关为早期诊断和治疗目的Evaluation of Relatives at Risk,请参阅管理,评估风险亲属。
家庭计划
DNA库是DNA的存储(通常从白细胞中提取),以备将来使用。 因为测试方法和我们对基因,等位基因变异和疾病的理解将来可能会有所改善,所以应该考虑 受累的个体的DNA银行。
产前检查和植入前遗传学诊断
分子遗传学测试。 一旦在 受累的家庭成员中鉴定出GALE致病变异体,就可以选择产前检测和 植入前遗传诊断以增加表异构酶缺乏半乳糖血症的风险。
资源
GeneReviews的工作人员选择了以下疾病特异性和/或orumbrella支持组织和/或登记处,以使这种疾病患者及其家人受益。 GeneReviews不对其他组织提供的信息负责。 有关selectioncriteria的信息,请单击here.
- Save Babies Through Screening Foundation, Inc.P. O. Box 42197Cincinnati OH 45242Phone: 888-454-3383Email: email@savebabies.org
- The Galactosemia FoundationP.O. Box 2401Mandeville LA 70471Phone: 866-900-7421Email: president@galactosemia.org
- Association for Neuro-Metabolic Disorders (ANMD)5223 Brookfield LaneSylvania OH 43560-1809Phone: 419-885-1809; 419-885-1497Email: volk4olks@aol.com
- Children Living with Inherited Metabolic Diseases (CLIMB)United KingdomPhone: 0800-652-3181Email: info.svcs@climb.org.uk
分子遗传
Molecular Genetics和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能包含更多最新信息。 - 编者。
Table B.
OMIM条目用于表异构酶缺乏半乳糖血症 (View All in OMIM)
基因结构.GALE长度超过4 kb,有11个编码外显子,共编码348个氨基酸的蛋白质。已经鉴定了编码相同蛋白质的多个可变剪接转录物。转录物NM_000403.3代表具有1647个核苷酸和12个外显子的最长转录物。有关基因和蛋白质信息的详细摘要,请参见Table A,基因。
致病等位基因变异。基于本综述的原则,如果已经显示变异在任何细胞类型或测定系统中降低GALE表达,稳定性或催化功能,则认为变异是致病性的,无论它们是否已知会引起临床特征。大多数鉴定出具有表异构酶缺乏半乳糖血症患者在临床上无症状,但确实有GALE序列变异解释或可能解释其生化结果。
致病性变异,c.280G> A,已在 纯合性状态中被鉴定为具有严重的,全身型表异构酶缺乏半乳糖血症[Wohlers et al 1999]。就UDP-galNA代谢而言,这种致病变异残余约5%~25%的酶活性 [Wohlers et al 1999, Wohlers & Fridovich-Keil 2000]。
已经显示其他致病变异在体外或模型系统中引起GALE酶活性的中度至严重降低(例如,c.269G> A或c.548T> C [Quimby et al 1997, Wohlers et al 1999, Timson 2005] )但迄今为止,这些等位基因仅在杂合子或复合杂合子中被鉴定出来。
除了c.280G> A之外,对于表观上严重的纯合性致病变异的体内结果是未知的。值得注意的是,尚无在外周细胞中GALE酶完全无效无活性的个体报道:生化推理[Kalckar 1965]以及果蝇[Sanders et al 2010, Daenzer et al 2012] 和C. elegans [Brokate-Llanos et al 2014] 的GALE损伤模型表明 ,GALE酶活性的完全丧失可能导致人类和其他后生动物的无法生存 。
还必须考虑两个GALE等位基因之间相互作用的可能性。例如,在c.548T> C致病性变异的酵母模型系统中描述了部分显性负效,其表现出低GALE酶活性,并且c.101A> G致病性变异,其显示单独表达时略微降低GALE酶活性 [Quimby et al 1997]。
特定人群中常见的GALE变异等位基因包括:
- c.55C> T,c.715C> T和c.905G> A致病变异约占无症状韩国人中报道的具有外周表异构酶缺乏半乳糖血症的等位基因67%[Park et al 2005]。
- c.770A> G和c.956G> A致病变异与非洲裔美国人的外周型表异构酶缺乏半乳糖血症有关 [Alano et al 1997, Fridovich-Keil & Walter 2008]。
Table 2.
选定的GALE等位变异
| 变异分类 | DNA 核酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|---|
| 外周型 | c.505C>T | p.Arg169Trp | NM_000403.3 NP_000394.2 |
| c.715C>T | p.Arg239Trp | ||
| c.770A>G | p.Lys257Arg | ||
| c.905G>A | p.Gly302Asp | ||
| c.956G>A | p.Gly319Glu | ||
| 疑似中间型 | c.101A>G | p.Asn34Ser 1 | |
| 已报道和疑似全身型 | c.269G>A | p.Gly90Glu | |
| c.280G>A | p.Val94Met | ||
| c.548T>C | p.Leu183Pro 1 |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference。- 1.
正常基因产物。 由GALE编码的尿苷二磷 酸-半乳糖-4-表异构酶缺乏(在UniProt中称为尿苷二磷 酸-葡萄糖-4-表异构酶缺乏)蛋白质具有348个氨基酸。
异常基因产物。 缺乏GALE活性,无论是全身性还是外周性,都会导致表异构酶缺乏性半乳糖血症。
参考文献
Literature Cited
- Alano A, Almashanu S, Chinsky JM, Costeas P, Blitzer MG, Wulfsberg EA, Cowan TM. Molecular characterization of a unique patient with epimerase-deficiency galactosaemia. J Inherit Metab Dis. 1998;21:341 - 50. [PubMed: 9700591]
- Alano A, Almashanu S, Maceratesi P, Reichardt J, Panny S, Cowan TM. UDP-galactose-4-epimerase deficiency among African-Americans: evidence for multiple alleles. J Invest Med. 1997;45:191A.
- Brokate-Llanos AM, Monje JM, Murdoch Pdel S, Muñoz MJ. Developmental defects in a Caenorhabditis elegans model for type III galactosemia. Genetics. 2014;198:1559 - 69. [PMC free article: PMC4256771] [PubMed: 25298520]
- Chen J, Meyers GA, Bennett MJ. An interference-free two-step enzyme assay with UPLC-tandem mass spectrometric product measurement for the clinical diagnosis of uridine diphosphate galactose-4-epimerase deficiency. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;2014;959:5 - 9. [PubMed: 24732214]
- Daenzer JM, Sanders RD, Hang D, Fridovich-Keil JL. UDP-galactose 4'-epimerase activities toward UDP-Gal and UDP-GalNAc play different roles in the development of Drosophila melanogaster. PLoS Genet. 2012;8:e1002721. [PMC free article: PMC3359975] [PubMed: 22654673]
- Fridovich-Keil JL, Walter JH. Galactosemia. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 72. McGraw-Hill. 2008.
- Gitzelmann R. Deficiency of uridine diphosphate galactose 4-epimerase in blood cells of an apparently healthy infant. Preliminary communication. Helv Paediatr Acta. 1972;27:125 - 30. [PubMed: 4644860]
- Henderson MJ, Holton JB, MacFaul R. Further observations in a case of uridine diphosphate galactose-4-epimerase deficiency with a severe clinical presentation. J Inherit Metab Dis. 1983;6:17 - 20. [PubMed: 6408303]
- Hennermann JB, Schadewaldt P, Vetter B, Shin YS, Mönch E, Klein J. Features and outcome of galactokinase deficiency in children diagnosed by newborn screening. J Inherit Metab Dis. 2011;34:399 - 407. [PubMed: 21290184]
- Holton JB, Gillett MG, MacFaul R, Young R. Galactosaemia: a new severe variant due to uridine diphosphate galactose-4-epimerase deficiency. Arch Dis Child. 1981;56:885 - 7. [PMC free article: PMC1627389] [PubMed: 7305435]
- Hunter M, Angelicheva D, Levy HL, Pueschel SM, Kalaydjieva L. Novel mutations in the GALK1 gene in patients with galactokinase deficiency. Hum Mutat. 2001;17:77 - 8. [PubMed: 11139256]
- Kalckar HM. Galactose metabolism and cell "sociology". Science. 1965;150:305 - 13. [PubMed: 5319435]
- Kolosha V, Anoia E, de Cespedes C, Gitzelmann R, Shih L, Casco T, Saborio M, Trejos R, Buist N, Tedesco T, Skach W, Mitelmann O, Ledee D, Huang K, Stambolian D. Novel mutations in 13 probands with galactokinase deficiency. Hum Mutat. 2000;15:447 - 53. [PubMed: 10790206]
- Mitchell B, Haigis E, Steinmann B, Gitzelmann R. Reversal of UDP-galactose 4-epimerase deficiency of human leukocytes in culture. Proc Natl Acad Sci U S A. 1975;72:5026 - 30. [PMC free article: PMC388868] [PubMed: 1748]
- Openo KK, Schulz JM, Vargas CA, Orton CS, Epstein MP, Schnur RE, Scaglia F, Berry GT, Gottesman GS, Ficicioglu C, Slonim AE, Schroer RJ, Yu C, Rangel VE, Keenan J, Lamance K, Fridovich-Keil JL. Epimerase-deficiency galactosemia is not a binary condition. Am J Hum Genet. 2006;78:89 - 102. [PMC free article: PMC1380226] [PubMed: 16385452]
- Park HD, Park KU, Kim JQ, Shin CH, Yang SW, Lee DH, Song YH, Song J. The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patients. Genet Med. 2005;7:646 - 9. [PubMed: 16301867]
- Quimby BB, Alano A, Almashanu S, DeSandro AM, Cowan TM, Fridovich-Keil JL. Characterization of two mutations associated with epimerase-deficiency galactosemia, by use of a yeast expression system for human UDP-galactose-4-epimerase. Am J Hum Genet. 1997;61:590 - 8. [PMC free article: PMC1715948] [PubMed: 9326324]
- Sanders RD, Sefton JM, Moberg KH, Fridovich-Keil JL. UDP-galactose 4' epimerase (GALE) is essential for development of Drosophila melanogaster. Dis Model Mech. 2010;3:628 - 38. [PMC free article: PMC2931539] [PubMed: 20519568]
- Sarkar M, Bose SS, Mondal G, Chatterjee S. Generalized epimerase deficiency galactosemia. Indian J Pediatr. 2010;77:909 - 10. [PubMed: 20725869]
- Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, Raymond K, He P, Ichikawa M, Veltman J, Huijben K, Shin YS, Sharma V, Adamowicz M, Lammens M, Reunert J, Witten A, Schrapers E, Matthijs G, Jaeken J, Rymen D, Stojkovic T, Laforêt P, Petit F, Aumaître O, Czarnowska E, Piraud M, Podskarbi T, Stanley CA, Matalon R, Burda P, Seyyedi S, Debus V, Socha P, Sykut-Cegielska J, van Spronsen F, de Meirleir L, Vajro P, DeClue T, Ficicioglu C, Wada Y, Wevers RA, Vanderschaeghe D, Callewaert N, Fingerhut R, van Schaftingen E, Freeze HH, Morava E, Lefeber DJ, Marquardt T. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med. 2014;370:533 - 42. [PMC free article: PMC4373661] [PubMed: 24499211]
- Timson DJ. Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase. FEBS J. 2005;272:6170 - 7. [PubMed: 16302980]
- Walter JH, Roberts RE, Besley GT, Wraith JE, Cleary MA, Holton JB, MacFaul R. Generalised uridine diphosphate galactose-4-epimerase deficiency. Arch Dis Child. 1999;80:374 - 6. [PMC free article: PMC1717903] [PubMed: 10086948]
- Wohlers TM, Christacos NC, Harreman MT, Fridovich-Keil JL. Identification and characterization of a mutation, in the human UDP-galactose-4-epimerase gene, associated with generalized epimerase-deficiency galactosemia. Am J Hum Genet. 1999;64:462 - 70. [PMC free article: PMC1377755] [PubMed: 9973283]
- Wohlers TM, Fridovich-Keil JL. Studies of the V94M-substituted human UDPgalactose-4-epimerase enzyme associated with generalized epimerase-deficiency galactosaemia. J Inherit Metab Dis. 2000;23:713 - 29. [PubMed: 11117433]
本章节的注解
致谢
作者非常感谢将我们对表异构酶缺乏症半乳糖血症的认识带到目前状态的许多患者,家庭,医疗保健专业人员和科学家的时间和精力。 JLFK还感激美国国立卫生研究院和半乳糖血症基金会的资助。
作者历史
- 16 June 2016 (ma) 系统性更新发布到公开网页上
- 24 October 2013 (me) 系统性更新发布到公开网页上
- 25 January 2011 (me) 内容发布到公开网页上
- 31 August 2010 (jfk) 最早稿件