
摘要
临床特征.
威廉斯综合征(WS)的特征为心血管疾病(弹性蛋白动脉病,外周肺动脉狭窄,瓣上主动脉瓣狭窄,高血压),特征面相,结缔组织异常,智力障碍(通常较为轻微),特别的认知状况,独特性格特征,生长异常和内分泌异常(高钙血症、高钙尿、甲状腺功能减退和青春期提前)。婴儿期喂养困难常常导致体重增加不良。张力减退和关节可过度伸展会导致运动发育延迟。
诊断/测试.
临床诊断标准可用于威廉斯综合征;但是,诊断需要复发性7q11.23连续基因缺失的检测,而该缺失区域为包含了弹性蛋白基因(ELN)的威廉斯综合征的关键区域(WBSCR)。可运用荧光原位杂交 (FISH) 或缺失/重复测试来检测该连续基因缺失。
管理. 对症治疗:针对发育障碍的早期干预方案,特别教育方案和专业训练;方案中包含语言的、体能的、职业的、喂养的以及感觉整合各方面疗法。心理学和精神疾病的评估和治疗提供个性化的行为咨询和药物治疗,尤其是对注意力缺失障碍和焦虑。主动脉瓣狭窄或肺动脉狭窄、二尖瓣闭锁不全或肾动脉狭窄,这些情况需要进行手术。高血钙症的治疗包括调整饮食、口服糖皮质激 素、 以及磷酸盐静脉注射。可咨询肾病专家以治疗肾钙质沉着症、持续性的高血钙症以及高钙尿症。高血压、远视和复发性中耳炎的治疗与一般人群的治疗没有区别。如 有错牙畸形应考虑整牙术。伴有喂养困难的婴儿可能从喂养疗法中获益。便秘应该在各个年龄段都需积极的管理。青春期过早启动可用促性腺激素释放激素激动剂进 行 治疗。
次级并发症的预防:规范运动练习幅度来预防或改善关节挛缩;麻醉镇静和手术前进行麻醉咨询和心电图检查。
监测:每年应进行医疗评估、视力检查、听力评估、两臂的血压测量、点尿样的钙/肌氨酸比值检测、尿检等。每4-6个月,应对2岁以下的儿童应进行血清钙检测。甲状腺功能应每年检查一次,直到三岁,之后每两年检查一次。对所有人的额外定期评估包括:每两年对钙的血清浓度进行测量;在前五年,至少每年都对弹性蛋白动脉病的心脏病学评估,此后每2-3年进行一次评估;每十年进行一次肾和膀胱超声检查。在成年期的额外的周期性评估包括:口服葡萄糖耐量;二尖脱垂的心脏评估,主动脉闭锁不全,高血压, 长QT间隔和动脉狭窄;以及白内障的眼科评估。需规避的物品及环境:需避免服用儿童复合维生素片,因为所有的儿科复合维生素制剂都含有维生素D。
诊断
临床诊断的标准对威廉斯综合征(WS)有效 [Preus 1984, Committee on Genetics 2001, Committee on Genetics 2002].
威廉斯综合征表型是多变的,不依靠单一临床特征来确定诊断。
提示性表型
若伴有以下特征,应被怀疑为威廉斯综合征(WS)。
心血管疾病(弹性动脉病).任何动脉都可能变狭窄。主动脉瓣上狭窄(SVAS)是最具临床意义和最共同的心血管特征,75%的受累者具有该特征。外周肺动脉狭窄(PPS)常见于婴儿期。
- 结缔组织异常,嗓音嘶哑,腹股沟/脐部疝气,肠/膀胱分支,直肠脱垂,关节限制或松弛,以及柔软、松弛的皮肤。
- 智力障碍. 大多数人都有一定程度的智力缺陷,从严重到轻微。有些人智力一般。
- 特殊的认知情况. 语言上的短期记忆优势和极弱的空间认知能力较为典型。威廉斯综合症的认知状态与智商无关。
- 独特人格. 过度友好、共情、普遍性焦虑症、特定恐惧症和注意力缺乏都是常见的。
- 发育异常.
生长模式的特点是:在头4年出现产前发育不足、婴儿时期生长不旺(70%)、贫乏的体重增长和线性生长;线性生长的比率是正常儿童的75%;以及短暂的青春期快速生长期。成人的平均身高低于百分之三。
- 内分泌紊乱. 该发现包括特异性高钙血症、高钙尿症、甲状腺机能减退、亚临床甲状腺功能减退和青春期提前。特别是在成人中,观察到异常的口服葡萄糖耐量测试、肥胖和糖尿病的发生频率增加。
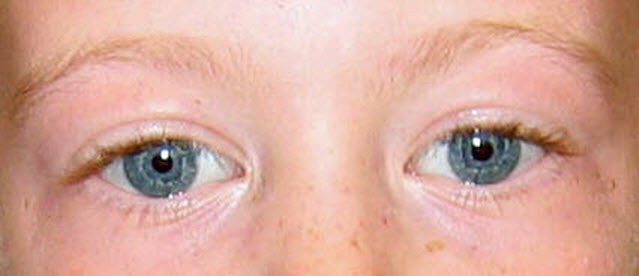
图1.
请注意,在威廉斯综合症患者中的星状虹膜模式。

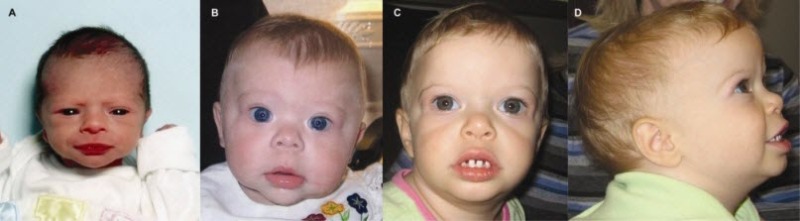
图3.
患有威廉斯综合症的儿童通常都有内眦赘皮,饱满的脸颊,和小的、间隔大的牙齿,这些症状在以下这些年龄的孩子中被观察到:A,新生儿;B,10个月;C和D,21个月。
建立诊断
威廉斯综合症(WS)的确立是在7q11.23号染色体上检测到1.5-1.8-Mb的杂合的微缺失。在此,WS被定义为在参考基因组(NCBI构建grch37/hg19)中的chr7:72,744,454-74,142,513 的位置上出现的复发性1.5-1.8-Mb缺失。
注:本区域内显著较大或更小的缺失的表型可能在临床上与WS不同(参见遗传相关疾病)。
尽管有几个值得关注的基因(例如:ELN)包含在1.5-1.8-Mb常见微缺失中,但是没有单一的基因的致病性变异导致WS(参见分子遗传学,缺失区域中的兴趣基因)。
基因组检测方法包括染色体微阵列(CMA)或者通过荧光原位杂交(FISH)得到的目标缺失分析,该方法决定序列拷贝数。注:WS不能通过G-显带染色体或其他常规的细胞遗传显带技术分析来鉴定。
Table 1.表1.
用于威廉斯综合征的基因组测试
| 缺失 1 | ISCA ID 2 | 区域位置 3, 4 | 测试方法 | 测试敏感度 | |
|---|---|---|---|---|---|
| 先证者 | At-risk family members有风险的家庭成员 | ||||
| 在7q11.23上1.5-1.8-Mb的杂合缺失 | ISCA-37392 | GRCh37/hg19 chr7: 72,744,454-74,142,513 | CMA 5 | 100% | 100% |
| FISH | 100% | 100% 6 | |||
基因组变异的标准化的临床注释和翻译来自临床基因组资源(ClinGen)项目(从前的细胞基因组阵列(ISCA)联盟的国际标准)
基因组坐标表示由ClinGen指定的7q11.23重复性微缺失相关的最小缺失大小。根据测试实验室所使用的芯片设计,缺失的坐标可能会有轻微的变化。注意,从这些基因组位置计算的微缺失的大小可能与预期的微缺失大小不同,因为在断点附近出现了片段性重复。在这个区域内显著的更大或更小的微缺失的表型可能与复发性的7q11.23微缺失有临床上的区别(见基因相关疾病)。
本区域内值得关注的基因请参阅分子遗传学
使用寡核苷酸微阵列或SNP基因型微阵列的染色体微阵列分析(CMA). 目前临床上使用的芯片覆盖7q11.23区域。
对于先症者还没有用FISH或CMA检测出缺失的有携带风险的家属进行FISH检测是不合适的
对有携带风险亲属的评估. FISH能够用于鉴定先证者有携带风险亲属的威廉斯综合征。只有当亲属有威廉斯综合征的迹象或症状时,才会对父母样本进行检测(见遗传咨询)。
测试特征. 关于测试特征的信息,包括敏感性和特异性,请参阅临床效用基因卡[Koehler et al 2014]
临床特征
临床描述
婴儿期.
威廉斯综合症(WS)的婴儿通常是预产期后出生的,与家庭其他成员相比显得较小。喂养困难导致体重不增加的现象很常见,包括胃食管(G-E)反流、吮吸和吞咽障碍、对食物厌恶和呕吐。长时间的绞痛(>4个月)可能与G-E返流、慢性便秘和/或先天性的高钙血症有关。其他的医学问题通常在第一年出现,包括斜视、慢性中耳炎、直肠脱垂、脐疝和/或腹股沟疝,心血管疾病[Morris et al 1988]。患WS的婴儿张力减退,通常有能过度扩展的关节,从而导致了运动发育指标的延迟。行走通常在24个月时开始。言语也发育迟缓,但随后会相对加强。所有年龄段的人都有运动困难。
认知能力.
患WS的人,75%会有智力障碍,通常是轻度的。WS病人的认知情况很独特,这包括口头短期记忆和语言的优势,但在视觉空间的建立方面极其薄弱[Mervis et al 2000]。因此,孩子们在口头测试上的得分要比在视觉空间构建上的测试要高[Greer et al 1997, Mervis et al 1998]。在智商上,没有性别差异,并且在儿童时期智商是稳定的[Mervis et al 2012b]。
在学业上,WS患者在阅读方面的表现相对较好,成年人也可以阅读高中水平的书籍,尽管可达到的水平层次变化范围很广。阅读技能与认知能力相关,而并不与语言相关技能的相关[Levy et al 2003]。尽管许多患WS的成年人能够完成简单的加法,但是在写作、绘画和数学方面的困难是很明显的。儿童和成年人与智商对应的适应性行为都低于预期 [Davies et al 1997, Howlin & Udwin 2006, Mervis & Pitts 2015],并且对成年人的能力有负面影响。
独特的个性和行为.
WS的性格特征包括过度友好、社交不抑制、过度共情、注意力问题和非社交性焦虑[Einfeld et al 2001, Doyle et al 2004, Morris 2010, Muñoz et al 2010]。其他常见的行为问题包括:感官调节/感觉处理困难、情绪调节困难、偏执、特定恐惧症(80%) [Dykens 2003, Laws & Bishop 2004, John & Mervis 2010, Pitts et al 2016]。有些人有与自闭症谱系障碍有重叠的症状,比如有限制的兴趣爱好,以及重复性的行为[Klein-Tasman et al 2009]。与患其他疾病的儿童相比,以下行为,患WS儿童发生率高:共情心、合群性、与人亲近、紧张、敏感和“可见性”(容易被注意到) [Klein-Tasman & Mervis 2003]。在儿童中,注意力缺陷障碍发生在65%的WS中,而焦虑症发生在57%的WS中(通常是特定的恐惧症) [Leyfer et al 2006]。焦虑在一生中很常见;对焦虑症的纵向研究表明80%的患者都有这种症状[Woodruff-Borden et al 2010]。
睡眠.
报告称65%的WS患者有睡眠问题,包括入眠时间的增加和睡眠效率的降低[Goldman et al 2009, Mason et al 2011]。夜间褪黑素峰反常或缺失已经在最近的研究中被记载 [Sniecinska-Cooper et al 2015, Santoro et al 2016]。
心血管疾病.
弹性动脉病存在于75%-80%的受影响个体中,并且可能会影响任何动脉 [Morris et al 1988, Pober et al 2008, Del Pasqua et al 2009, Collins et al 2010b]。
外周肺动脉狭窄(PPS)在婴儿时期很常见,但通常随着时间的推移而改善。
最常见的动脉病是主动脉瓣上狭窄(SVAS),它可能会随着时间而恶化,尤其是在生命的前5年[Collins et al 2010b]。最大的病态是由主动脉瓣上狭窄引起的,这可能是一个离散的沙漏型狭窄或是弥漫性主动脉瓣狭窄。如果不进行治疗,动脉阻力的增加会导致左心压力升高、心脏肥大和心脏衰竭。中间主动脉综合征很少发生,这包括胸和腹主动脉的弥漫性狭窄,但很难治疗,可能需要重新干预[Radford & Pohlner 2000]。
合并患有SVAS和PPS(双心室流出道梗阻)的个体可能会伴有双侧心室肥大和高血压,这将增加心肌缺血、节奏障碍和猝死的危险[Pham et al 2009]。冠状动脉狭窄可能与WS患者的猝死有联系 [Bird et al 1996]。在其中的293名患者中,猝死的发生率为1/1000,比年龄匹配的人群高出25到100倍[Wessel et al 2004]。修正的QT延长已经在13.6%的WS患者中有报道;推荐对复极化异常进行筛查 [Collins et al 2010a]。
对于WS患者,麻醉和镇静与包括心脏停搏的不良事件风险增加有关[Burch et al 2008, Olsen et al 2014]。因此,已有镇静和麻醉风险评估和管理指南 [Burch et al 2008, Matisoff et al 2015, Latham et al 2016]。
在有WS的人中,高血压的流行率是40%-50%。高血压可能出现在任何年龄段 [Broder et al 1999, Giordano et al 2001, Eronen et al 2002, Bouchireb et al 2010],可能在某些病人的高血压可能是肾脏动脉狭窄的继发症状[Deal et al 1992]。血管硬度的增加已在WS中有报道,对抗高血压药物有疗效反应[Kozel et al 2014]。
在成年人中,有二尖瓣脱垂和主动脉瓣不全的报道[Morris et al 1990, Kececioglu et al 1993, Collins et al 2010a]。
肠系膜狭窄可能导致腹痛。
神经血管异常很少被报道,但可能会导致中风[Ardinger et al 1994, Soper et al 1995, Cherniske et al 2004]。
眼睛,耳朵,鼻子,咽喉. 泪管阻塞、远视(67%)和斜视(50%)在WS患者中是常见的[Kapp et al 1995, Weber et al 2014]。成人中已有发生白内障的报道[Cherniske et al 2004]。慢性中耳炎被认为占受累个体的50%。对声音的过度敏感是很常见的(90%),报道称,,患WS的人在比正常人能承受的音量低20分贝(cb)会就感到不适 [Gothelf et al 2006]。有很多关于对某种声音有特殊恐惧的报道[Levitin et al 2005]。
已经观察到渐进的感觉神经性听力丧失;有63%的儿童和92%的成年人都有轻到中度的听力损失 [Gothelf et al 2006, Marler et al 2010]。在成人中,轻度到中-高频的感觉神经听力丧失是常见的,而耳屎则会过度积聚 [Cherniske et al 2004]。
大多数人的嗓音都是沙哑或低沉的;这是由于弹性蛋白缺乏导致的声带的异常[Vaux et al 2003]。
牙齿问题包括小牙,牙釉质发育不全,咬合不正 [Hertzberg et al 1994]。40%的WS患者缺少一个或更多的永久性牙齿[Axelsson et al 2003]。
肠胃困难.有WS的人都有感观防御,包括在听觉上 [Van Borsel et al 1997] 和触觉上。对食物质地的挑剔会使婴儿时期从母乳或配方奶粉到固体食物的转化上出现问题。
慢性腹痛是儿童和成人的常见症状;可能原因包括G-E返流、食管裂孔疝、消化性溃疡、胆结石、憩室炎、缺血性肠病、慢性便秘、以及焦虑的躯体化。憩室炎的发病率在患WS的青少年[Stagi et al 2010] 和成年人中有增加 [Partsch et al 2005]。便秘的并发症可能包括直肠脱垂、痔疮或肠穿孔。
高钙血症可能导致易怒、呕吐、便秘和肌肉痉挛;在婴儿时期更常见但也可能再次出现在成人时期中[Morris et al 1990, Pober et al 1993]。
尿道畸形.尿频和遗尿(50%)在WS儿童患者中很常见。50%的患者有肾动脉狭窄,尿路结构异常占患者的10%,膀胱憩室占50%,而肾钙质沉着症低于5% [Pober et al 1993, Pankau et al 1996, Sforzini et al 2002, Sammour et al 2006, Sammour et al, 2014]。膀胱容量减少,60%的患者逼尿肌过度活跃[Sammour et al 2006]。白天的尿禁平均发生在4岁,夜间尿禁在10岁时为50%。据估计,成人中有3%的人发生了夜间遗尿 [von Gontard et al 2016]。
骨骼肌/神经系统问题.张力减退和关节松弛致使幼儿用异常的补偿姿势来获得稳定。患WS年龄较大的儿童和成人典型的具有张力亢进和深部腱反射亢进的特点。脚跟线的逐渐收紧和腿筋的出现,导致青春期僵硬和笨拙的步态、驼背和脊柱前凸[Morris et al 1988, Kaplan et al 1989]。18%的人出现脊柱侧凸 [Morris et al 2010]。10%的人有桡尺骨骨性联接[Morris & Carey 1990]。良好的运动功能受损,导致在各种年龄段的工具使用和书写的困难。
成年人的小脑信号包括共济失调、测距不良和震颤 [Pober & Morris 2007]。
神经影像. 脑部核磁共振成像显示,脑容量缩小,脑灰质体积减少,特别是在顶骨和枕骨区域,以及脑回的复杂度增加 [Jackowski et al 2009, Eisenberg et al 2010]。在一些受累个体中发现有后窝尺寸减小,加上小脑大小没受影响从而可能会导致Chiari 1型畸形, [Pober & Filiano 1995, Mercuri et al 1997]。 生长发育 就以家庭背景而言,患WS的个人都较矮。已有针对WS的特定生长曲线[Morris et al 1988, Saul et al 1988, Martin et al 2007]。在70%的婴儿中观察到体重增加不良。生长的特征是:在头4年中,产前发育不足、体重增长不佳和线性增长不佳,其线性生长率是正常儿童的75%,以及短暂的青春期发育突增。成人的平均身高低于第三百分位数。在年龄较大的儿童和成人均会肥胖[Cherniske et al 2004]。青春期可能会提早发生,18%的病患表现有中枢性性早熟[Partsch et al 2002]。用促性腺激素释放激素来激素抑制在青春期早期或早熟的女孩中有很好地耐受,被治疗的女孩比没被治疗的WS患者要高[Spielmann et al 2015]。
高血钙症.15%-50%的WS患者有先天高钙血症,并且在头两年最常出现症状(易怒,呕吐,便秘) [Martin et al 1984, Morris et al 1988, Kim et al 2016]。高钙血症与脱水、高钙尿和肾钙质沉着症相关;与正常人相比,在所有年龄段WS病患有较高的血清钙中值[Sindhar et al 2016]。高钙血症的病因尚不清楚[Stagi et al 2016]。 内分泌问题. 内分泌异常包括甲状腺功能减退(10%)和过早的(尽管不是早熟)青春期(50%) [Kim et al 2016]。亚临床甲状腺机能减退(伴有正常的T3/T4水平的TSH升高)发生在31%的人中,在儿童中发生的频率比成人高[Palacios-Verdú et al 2015]。在青少年中,葡萄糖耐受不良的发生率为26%[Stagi et al 2014],其中63%发生在年轻成人中 [Masserini et al 2013]。在WS成人中观察到的有异常的口服葡萄糖耐量测试和糖尿病的发生频率增高[Cherniske et al 2004]。其他
- 头发过早灰白 [Morris et al 1988].
- 柔软松弛的皮肤较为典型.
- 具独特的面部特征但随着年龄的增长而变化(见提示性发现).
基因型-表型 相关性
WBSCR的7q11.23的重复缺失包含了1.55兆碱基(Mb)(占WS患者的90%-95%)或1.84 兆碱基Mb(占WS患者的5%-10%)[Bayés et al 2003, Palacios-Verdú et al 2015]。
- 高血压在那些NCF1为半合子的WS患者中比较少见,NCF1位于WBSCR的旁边低拷贝重复序列的一个区域 [Del Campo et al 2006]。
- 相比与有典型1.5-1.8-Mb WBSCR缺失的病患,有较大缺失(>2-4 Mb) 的病人具有较严重的更低的认知能力的表型[Stock et al 2003, Marshall et al 2008]。
在WBSCR中较短的缺失导致不同程度的表型,具体取决于缺失的程度。
- 具有部分WBSCR缺失的人,包括通常的端粒断点(包括GTF2I)具有典型的WS特性,包括智力障碍[Botta et al 1999, Heller et al 2003]。
- 那些携带部分WBSCR缺失但不包括GTF2I缺失的病人——包括一些有新发的短的缺失和携带“SVAS +”的家族——不表现有智力障碍,但常表现出WS认知模式[Morris et al 2003]。在两个家庭中,ELN的缺失和另外一种基因,LIMK1,与WS认知模式有关,但与智力障碍或WS的其他特征无关[Frangiskakis et al 1996]。另一个有类似缺失的家庭没有WS认知模式[Tassabehji et al 1998]。
在WBSCR中缺失的可能是母本或父本起源[Ewart et al 1993a, Dutly & Schinzel 1996, Urbán et al 1996]。在某些家系中,表型与父母本来源无关[Wu et al 1998],而小头畸形在其他研究中表现与WBSCR缺失的母本起源有关[Del Campo et al 2006]。
外显率
外显率是100%;表型特征的表达是可变的。
命名法
对WS的第一个描述是不完整的,因为它们受制于个人的主诉或观察者的医学专科局限。因此,肾病学家和内分泌学家对“先天性婴儿高钙血症”(IHC)进行了描述,心脏病学家报道了“主动脉瓣上狭窄综合症SASS)。
早期的报道还提到了与传说中的精灵相似的异常的面部特征:一段时间内,“威廉姆斯精灵综合征”一词被使用。
在Williams [1961] 和Beuren [1962]的报道之后,这一状况在美国被称为威廉姆斯综合症,在欧洲称为威廉斯-伯恩综合症。
患病率
在挪威的一项关于WS的研究报告显示该病的患病率为1:7500[Strømme et al 2002]。
遗传相关(等位基因)失调
常染色体显性皮肤松弛症是由ELN致病性移码突变引起的,该突变对弹性纤维结构有一定的显性负效应 [Tassabehji et al 1998, Zhang et al 1999, Sugitani et al 2012]。常染色体隐性皮肤松弛症在一个ELN外显子 12中带有纯合子隐性致病性变异的叙利亚家庭中有报道[Mégarbané et al 2009]。
常染色体显性的主动脉瓣上狭窄(SVAS)是由ELN突变或其内含子缺失引起的[Ewart et al 1993b, Olson et al 1993, Morris & Mervis 2000, Micale et al 2010]。伴有常染色体显性SVAS的个体通常只有结缔组织异常,没有WS。
常染色体显性“SVAS+”是由包含了ELN和其他相邻基因的WBSCR中的缺失引起的。有这些短缺失的家庭成员有SVAS而不是典型的WS;然而,他们与WS患者共享一些表型特征,包括视觉空间构造方面的困难 [Morris & Mervis 2000, Morris et al 2003]。
7q11.23重复综合症(威廉斯综合征区域重复综合症)是由1.5 mb区域重复引起的,该区域在WS中是最普遍的缺失。典型的颅面特征包括:大头、前额突出或宽阔、直眉、深眼、长睫毛、低挂/或低插入鼻柱、短人中、高腭穹、轻微的面部不对称、上唇薄、耳部螺旋重叠过度和小下颌。最重要的临床发现表达性语言有严重缺陷,包括一种语音障碍,这与WS个体所表现出来的语言展示的相对强度形成对比。许多人存在ADHD和/或焦虑、典型分离或社交焦虑。许多儿童在单足站立及前后脚连接走直线时有困难[Somerville et al 2005, Berg et al 2007, Van der Aa et al 2009, Dixit et al 2013, Morris et al 2015]。据报道,主动脉膨胀率为46.2%[Morris et al 2015]。
鉴别诊断
威廉斯综合征(WS)应与其他以发育迟缓、注意力缺陷多动障碍、身材矮小、独特面相及/或先天性心脏病为特点的综合征相区别。这些包括:努南综合征、22q11缺失(迪乔治综合征)、史密斯氏综合征、歌舞伎综合症和胎儿酒精综合征。
患者的主动脉瓣上狭窄(SVAS)应得到评估,从而决定是否 WS或常染色体显性SVAS(OMIM 185500)为合适的诊断。
Table 2表2.
在威廉斯综合征鉴别诊断时需要考虑的疾病
| 疾病 | 基因 | 遗传模式 | 疾病的临床表征 | ||
|---|---|---|---|---|---|
| 最常见的先天心脏病 | 特殊的面部特征 | 额外特征 | |||
| 威廉斯综合征 | 见注脚1 | AD | 主动脉瓣上狭窄 |
|
|
| Noonan syndrome努南综合征 | PTPN11SOS1RAF1RIT1KRASNRASBRAFMAP2K1 | AD | 肺动脉瓣狭窄 |
|
|
| 22q11.2缺失综合征 | 见脚注2 | AD | 圆锥动脉干畸形 |
|
|
| 史密斯氏综合征 | RAI1 3 | 见脚注3 | 心隔膜缺损 |
|
|
| 歌舞伎综合征 | KMT2D | AD 4 | 主动脉缩窄 |
|
|
| KDM6A | XL 5 | ||||
| 胎儿酒精综合征 | NA 6 | 心隔膜缺损 |
|
| |
管理
初步诊断后评估
为了确定一个被诊断为威廉斯综合症(WS)的个体的疾病程度和需要,并指导医疗管理,建议进行以下评估[Committee on Genetics 2001, Committee on Genetics 2002]:
- 完全的体格和神经病学的检查
- 在威廉斯综合征发育图表上测绘生长发育参数
- 心脏病学评估
- 一位在WS治疗上有治疗经验的心脏病专家的全面评估
- 四肢血压测量
- 超声波心动图,包括多普勒研究
- 心电图
- Additional cardiovascular imaging studies (computed tomography, magnetic resonance angiography, or cardiac catheterization) may be required in individuals with diminished pulses, bruits, or signs of diffuse thoracic aortic stenosis. 在有脉冲减少,杂音,或胸主动脉瓣狭窄的迹象的个体中有必要进行额外的心血管造影研究(计算机断层扫描,磁共振血管造影术,或心导管术)。
- 泌尿系统评估
- 膀胱和肾的超声检查
- BUN和肌酸酐的血清浓度
- 尿分析
- 钙测定
- 钙或电离钙的血清浓度
- 对尿样本的钙/肌酸酐测定(正常值见Sargent et al [1993] 。)
- 甲状腺功能测试
- 眼科评估
- 基线的听力学评估
- 多学科的发展评估,包括对运动、言语、语言、个人社会、一般认知和职业技能的评估
- 包括注意力,焦虑,和适应性能力的行为评估
- 向临床遗传学家和/或遗传咨询师咨询
临床治疗
认知/行为.
发育障碍应通过早期干预计划、特殊教育计划和职业培训来帮助。推荐的治疗方法包括言语/语言、体能和职业治疗。考虑马术治疗(在言语、体能和/或职业治疗中使用类似马的运动)。
- 口头表达上的优势可以帮助学习空间能力。
- 在阅读教学中推荐声学方式[John & Mervis 2010].
- 日常生活技能的掌握有利于成人的安康,应该被鼓励。
以心理评估,多导睡眠图,和精神病学评估个人引导治疗。
- 幼儿的行为可使用基于应用行为分析的技术来处理[Mervis & John 2010]。
行为咨询和精神药物治疗通常用于管理行为问题,特别是注意力缺陷障碍和焦虑,50%这样的问题需用药物治疗
- 自我镇静技术能够管理焦虑.
心血管.
对20%-30%的病患有SVAS的手术矫正[Kececioglu et al 1993, Bruno et al 2003, Collins et al 2010b]。有需要对冠状瓣膜不全或肾动脉狭窄进行手术治疗。
高血压按通常医疗方法治疗。在一个研究中成功使用了钙通道阻滞剂[Bouchireb et al 2010]。抗高血压治疗也能改善血管硬化[Kozel et al 2014]。推荐一位熟悉治疗WS的心脏病专家对心血管系统进行终生监测。
高血钙症. 高血钙的管理包括以下方面:
- 应对脱水状态进行评估;指示增加水的摄取量。
- 饮食应该在营养学家的帮助下进行调整,这样钙的摄入量就不会超过每日推荐摄入量(RDI)的100% [Ross et al 2011]。如果血清钙的浓度维持升高,膳食钙应该减少;但是必须对钙的血清浓度进行监测。应该建议父母不要在没有医疗监督的情况下限制钙的饮食摄入量。
应该避免含有维生素D的补充。如果怀疑维生素D缺乏,那么在开始治疗前检查维生素D水平是很重要的,并且在治疗期间必须对钙水平进行监测。在威廉姆斯综合症中,从肠道中吸收的钙会增加(原因不明),而维生素D会促进钙的吸收。
- 难治性高血钙症可用口服类固醇激素片.
- 静脉注射氨羟二磷酸二钠已经被成功的用于治疗有严重症状的高血钙症的婴儿[Cagle et al 2004, Oliveri et al 2004]。
对治疗持续性高钙血症、高钙尿和/或肾钙质沉着症应建议转诊给内分泌学家和/或肾脏科医师。
眼睛,耳朵,鼻子,和咽喉.
使用矫正镜片治疗远视;斜视的治疗方法是对一只眼进行修补或手术修补;多管狭窄的治疗方式与普通人群相同。
复发性的中耳炎可以用鼓膜切开术治疗。
当能预期有高噪音水平时,对耳朵进行保护可以对付对声音的超敏。
牙科护理可能需要对日常的刷牙和使用牙线进行辅助。在青少年和成人中,牙齿的清洗频率应增加到每四月一次。咬合不正的治疗应考虑到畸齿矫正。
胃肠道.
婴幼儿喂养问题的治疗和儿童和成人的腹痛取决于病因(如:G-E返流,高钙血症,食管裂孔疝和/或憩室炎)。婴儿经常受益于喂食疗法。
所有年龄段的便秘必须积极管理,因为会给早发性憩室病/憩室炎增加风险。治疗通常包括在渗透式的泻药治疗后对水和纤维的饮食增加。严重的腹部疼痛可能是指憩室炎和/或肠穿孔,它可能在WS患者的年轻阶段发生。
尿路异常. 患有发热性尿路感染的患者很可能需要对尿道下尿路进行调查来直接治疗,如使用排空的膀胱尿道造影照片。
内分泌. 过早的青春期可用促性腺激素释放激素刺激剂治疗[Partsch et al 2002, Pober 2010]。使用口服甲状腺素治疗甲状腺功能减退;亚临床甲状腺机能减退症通常只需跟踪观察,但不需要治疗。二级并发症的预防
以下是指症:
- 通过运动和平衡的饮食来避免胰岛素抵抗/糖尿病
- 通过调整运动幅度来预防或改善关节挛缩。
- 因为患者有双心室流出道梗阻,因此心肌功能不全的风险增加,尤其受麻醉诱发 [Horowitz et al 2002] ,由于WS患者在药物镇静或麻醉时发生不良事件的风险增加,因此外科手术时需要麻醉咨询。关于给WS患者的镇静和麻醉风险评估和麻醉管理已有指南出版 [Burch et al 2008, Matisoff et al 2015, Latham et al 2016]。在手术前需进行心电图检查。
- 对心肌功能不全和心脏骤停的风险的意识;对于手术程序,选择一个对心肺复苏有准备的医疗中心。
监控
表3.
对威廉斯综合征的监控
| 区间/ 年龄 | 测试/测量 |
|---|---|
| 在婴儿期-学步期 |
|
| 每年/所有年龄段1 |
|
| 每2年 |
|
| 每10年 |
|
| 成人期 |
|
除非另有注明
如果正常,OGTT应该每5年进行重复一次。
需避免的制剂/处境
患WS儿童不应补充多种维生素,因为所有小儿科多种维生素均包含维生素D。
怀孕管理
患WS妇女怀孕属高风险。他们应该对怀孕引起的高血压、心律不齐和心力衰竭的发生进行监控。由于尿路感染的风险增加,定期的尿分析应该在妊娠后期进行。建议对对胎儿进行超声监控 [Lin et al 2008]。
在研疗法
搜索ClinicalTrials.gov网站来获得众多有关疾病和条件临床研究的信息。注:可能没有对这疾病的临床试验。
遗传咨询
遗 传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于知情,以及符合个人情况的决定。 接着几个段落是涉及遗传风险评估, 根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的咨 询工作。-ED.
遗传模式
威廉斯综合征是常染色体显性遗传,大多数缺失为新发;极少有父母到孩子的传递报道 [Morris et al 1993, Sadler et al 1993, Mulik et al 2004]。
家庭成员的风险
先证者的父母
- 大多数情况下,WS患者的父母不受累。
- 在父母中没有发现WS的临床表型的情况下,对父母测试在先证者中出现的7q11.23微缺失是不必要的。
先证者的同胞.先证者同胞的患病风险基于父母的遗传状态:
先证者的后代
- WS患者的每一个孩子都有50%的机会遗传7q11.23微缺失。
遗传咨询相关问题
计划生育
- 遗传风险测定及产前检测讨论的最佳时间是怀孕前
- 对患有WS的年轻成年人讨论生育WS孩子的风险的遗传咨询(包括下一代风险及生育选择)是合适的
产前筛查
已知会有威廉斯综合征风险的怀孕. 产前利用基因组检测,检测在先证者中发现的 7q11.23微缺失,可以用于以下一些情况:
- 父母之一有7q11.23微缺失。
未知会有威廉斯综合征风险的怀孕.
CMA在未知有增加风险的怀孕中应用可能观察到复发性的微缺失。低风险妊娠没有产前指示。
注:产前检查结果不能可靠地预测表型。
并不清楚是否将会增加WS风险的妊娠的产前检查可能但很少被使用,因为大多数病例是在一个家庭中发生的,而且在低风险妊娠中没有产前指标。
着床前遗传学诊断可能对一些7q11.23微缺失已被CMA鉴定的家庭来说是一个选择。
资源
为了该病患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对该病的,或者包括其他GeneReviews疾病的患者支持组织和患者注册组织。GeneReviews不为其他组织提供的信息承担责任。选择这些组织的标准,请点击获取详细信息。
- Canadian Association for Williams SyndromePO Box 26206Richmond British Columbia V6Y 3V3CanadaPhone: 604-214-0132Email: cawbc@yahoo.com
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- Williams Syndrome Association (WSA)570 Kirts BoulevardSuite 223Troy MI 48084-4156Phone: 800-806-1871 (toll-free); 248-244-2229Fax: 248-244-2230Email: info@williams-syndrome.org
- Williams Syndrome Registry175 Cambridge StreetRoom 502Boston MA 02114Phone: 617-726-5318Fax: 617-724-1911Email: WSRegistry@partners.org
分子遗传学
在下面的分子生物学表和OMIM表内的信息可能和GeneReview里其他部分的信息不一致:表格里的信息可能包含更多较新的内容-EDTable A.表A
威廉斯综合征:基因和数据库
表B.
威廉斯缺失综合征OMIM词条(在OMIM中可查看所有)
分子遗传学发病机制
导致WS的7q11.23的缺失和相反的7q11.23的重复都是由该区域的基因组结构所介导的。7q11.23区域的两侧是低拷贝重复,这些重复易受非等位基因同源重组作用。在95%的WS患者中,缺失包含1.55 Mb;5%患者中,它包含1.84 Mb[Bayés et al 2003];缺失是在低拷贝重复(LCRs)块之间由非等位同源重组来介导的,而缺失的大小则反映涉及哪些LCRs块。
研究表明,在大约25%的病例中,未受累的父母的原染色体有包含7q11.23的7号染色体的倒位[Osborne et al 2001, Bayés et al 2003, Hobart et al 2010]。大约6%的普通人也有这种倒位多态性 [Hobart et al 2010]。有这种倒位的个体有1/1750的机会怀一个患WS的孩子[Hobart et al 2010]。这种倒位并不引起临床症状[Tam et al 2008]。
有两个关于WS患者的同胞的报告;在一个家庭中,缺失发生在具有包含7q11.23倒位的父亲的染色体上;而在另一种情况下,由于没有发现包含7q11.23的倒位,所以可能是母系胚系镶嵌性的结果[Scherer et al 2005]。
3个基因,GTF2I,GTF2IRD1和GTF2IRD2,在7q11.23区域和临近的LCR的一端。这些TFII-I基因家族的成员很可能在WS表型中扮演重要的角色,因为他们均能与各种启动子的基部和上游调节位点结合。这些转录因子蛋白参与了复杂的蛋白质相互作用,并在信号传导中发挥了作用。家族中的每一种蛋白质都有异构体,在不同的组织中有不同的表达模式,这就增加了这些基因的半合子状态可能促成WS表型的许多不同方面的可能性 [Hinsley et al 2004, Jackson et al 2005]。
ELN
致病变异. ELN致病变异通常导致常染色体显性SVAS [Li et al 1997]。ELN致病变异也在先天性皮肤松弛症中有报道[Tassabehji et al 1998, Zhang et al 1999]。
Normal 基因产物 正常基因产物. ELN基因的产物是属于结构蛋白质的弹性蛋白,它是一种存在于许多组织中的弹性纤维的主要成分。
异常基因产物. ELN的缺失是结缔组织异常的原因,包括WS患者的心血管疾病 [Ewart et al 1993a]。
在7q11.23微缺失区域中的其他值得关注的基因
- LIMK1 (Lim激酶1),在大脑中表达。LIMK1基因缺失与WS患者的视觉结构空间异常有联系 [Frangiskakis et al 1996, Morris et al 2003, Hoogenraad et al 2004]。Lim激酶1有两个LIM基序和一个可能涉及到细胞内信号的蛋白质激酶区域。
- GTF2I (普通转录因子 II, I) (OMIM 601679).GTF2I编码转录因子TFII-I[Pérez Jurado et al 1998, Danoff et al 2004],作为在细胞核中的一种可诱导的多功能转录因子[Roy 2012]和作为细胞质中的激动剂诱导钙进入的调节因子[Caraveo et al 2006]。“SVAS+”家族的缺失定位表明,该基因的缺失对智商有负面影响[Morris et al 2003]。这基因的重复与分离焦虑症有关[Mervis et al 2012a]。GTF2I单体型与孤独症谱系障碍的严重程度相关[Malenfant et al 2012],在一般人群中社会焦虑程度较低,社会沟通能力降低[Crespi & Hurd 2014],并影响特质焦虑和大脑对厌恶社会暗示的反应之间的相关性[Jabbi et al 2015]。
- STX1A(突触融合蛋白 1A) (OMIM 186590),涉及神经递质的释放和胰岛素的分泌。可能在WS患者中的糖尿病中扮演角色 [Osborne et al 1997, Lam et al 2005]。突触融合蛋白 1A通过蛋白与蛋白间的相互作用调整神经递质的释放。
- BAZ1B ( bromo结构域相邻的亮氨酸拉链结构 1B)。BAZ1B蛋白是核染色质重塑复合体的一部分。由于它结合维生素D受体,因此被推断它在WS中的高血钙症中起作用 [Meng et al 1998a]。
- CLIP2 (又名 CYLN2;细胞质连接体 2). CLIP2蛋白在大脑中大量表达,与膜微管相互作用,被推测涉及到WS的小脑异常[Hoogenraad et al 1998, Hoogenraad et al 2004] (OMIM 603432)。
- GTF2IRD1. 属于TFII-1转录家族,该蛋白与WS患者的颅面特征有关 [Osborne et al 1999, Tassabehji et al 2005]。
- NCF1 (嗜中性粒细胞胞质因子1)。编码NADPH氧化酶系统的成分。半合子状态与WS中高血压的风险减小相关。它在将近40%的WS患者中缺失 [Del Campo et al 2006]。
对于7q11.23微缺失中的剩余基因,与WS表型的相关性未知:
- RFC2 (复制因子 C, 亚基 2) (OMIM 600404),涉及DNA延伸[Peoples et al 1996]。RFC 2是涉及DNA延伸的引物识别蛋白。
- FZD9 (frizzled 9) (OMIM 601766),与果蝇frizzle基因同源[Wang et al 1997];它编码跨膜细胞表面受体,该受体结合Wnt蛋白可改变beta连环蛋白信号通路。Frizzle是区域受体家族的成员。
- FKBP6, 同源于免疫亲和素的FK-506结合蛋白类 [Meng et al 1998b]
- TBL2 [Meng et al 1998a]
- MLXIPL ( WS-基本螺旋-环-螺旋 亮氨酸拉链) [Meng et al 1998a, Cairo et al 2001]
- VPS37D [Schubert 2009]
- DNAJC30 编码热休克蛋白 [Merla et al 2002].
- BCL7B, [Meng et al 1998a]
- ABHD11 [Merla et al 2002]
- CLDN4 [Paperna et al 1998] (OMIM 602909)
- CLDN3 [Paperna et al 1998] (OMIM 602910)
- LAT2 [Brdicka et al 2002]
- WBSCR11 [Osborne et al 1999]
- WBSCR22 [Merla et al 2002]
- WBSCR23, WBSCR27 and和 WBSCR28 [Schubert 2009]
- GTF2IRD2, 有时在WS中缺失 [Makeyev et al 2004, Tipney et al 2004]
参考文献
已发表的指南/共识声明
- 美国儿科学会遗传学委员会.儿童威廉斯综合征健康监护.2001.在线可用.访问3-15-17.
文献引用
- Ardinger RH Jr, Goertz KK, Mattioli LF. Cerebrovascular stenoses with cerebral infarction in a child with Williams syndrome. Am J Med Genet. 1994;51:200–2. [PubMed: 8074144]
- Axelsson S, Bjornland T, Kjaer I, Heiberg A, Storhaug K. Dental characteristics in Williams syndrome: a clinical and radiographic evaluation. Acta Odontol Scand. 2003;61:129–36. [PubMed: 12868685]
- Bayés M, Magano LF, Rivera N, Flores R, Pérez Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 2003;73:131–51. [PMC free article: PMC1180575] [PubMed: 12796854]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet AL, Cheung SW. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–41. [PubMed: 17666889]
- Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26:1235–40. [PubMed: 13967885]
- Bird LM, Billman GF, Lacro RV, Spicer RL, Jariwala LK, Hoyme HE, Zamora-Salinas R, Morris C, Viskochil D, Frikke MJ, Jones MC. Sudden death in Williams syndrome: report of ten cases. J Pediatr. 1996;129:926–31. [PubMed: 8969740]
- Botta A, Sangiuolo F, Calza L, Giardino L, Potenza S, Novelli G, Dallapiccola B. Expression analysis and protein localization of the human HPC-1/syntaxin 1A, a gene deleted in Williams syndrome. Genomics. 1999;62:525–8. [PubMed: 10644452]
- Bouchireb K, Boyer O, Bonnet D, Brunelle F, Decramer S, Landthaler G, Liutkus A, Niaudet P, Salomon R. Clinical features and management of arterial hypertension in children with Williams-Beuren syndrome. Nephrol Dial Transplant. 2010;25:434–8. [PubMed: 19815602]
- Brdicka T, Imrich M, Angelisova P, Brdickova N, Horvath O, Spicka J, Hilgert I, Luskova P, Draber P, Novak P, Engels N, Wienands J, Simeoni L, Osterreicher J, Aguado E, Malissen M, Schraven B, Horejsi V. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. J Exp Med. 2002;196:1617–26. [PMC free article: PMC2196071] [PubMed: 12486104]
- Broder K, Reinhardt E, Ahern J, Lifton R, Tamborlane W, Pober B. Elevated ambulatory blood pressure in 20 subjects with Williams syndrome. Am J Med Genet. 1999;83:356–60. [PubMed: 10232742]
- Bruno E, Rossi N, Thuer O, Cordoba R, Alday LE. Cardiovascular findings, and clinical course, in patients with Williams syndrome. Cardiol Young. 2003;13:532–6. [PubMed: 14982294]
- Burch TM, McGowan FX Jr, Kussman BD, Powell AJ, DiNardo JA. Congenital supravalvular aortic stenosis and sudden death associated with anesthesia: what's the mystery? Anesth Analg. 2008;107:1848–54. [PubMed: 19020129]
- Cagle AP, Waguespack SG, Buckingham BA, Shankar RR, Dimeglio LA. Severe infantile hypercalcemia associated with Williams syndrome successfully treated with intravenously administered pamidronate. Pediatrics. 2004;114:1091–5. [PubMed: 15466114]
- Cairo S, Merla G, Urbinati F, Ballabio A, Reymond A. WBSCR14, a gene mapping to the Williams--Beuren syndrome deleted region, is a new member of the Mlx transcription factor network. Hum Mol Genet. 2001 Mar 15;10:617–27. [PubMed: 11230181]
- Caraveo G, van Rossum DB, Patterson RL, Snyder SH, Desiderio S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science. 2006;314:122–5. [PubMed: 17023658]
- Cherniske EM, Carpenter TO, Klaiman C, Young E, Bregman J, Insogna K, Schultz RT, Pober BR. Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A. 2004;131:255–64. [PubMed: 15534874]
- Collins RT 2nd, Aziz PF, Gleason MM, Kaplan PB, Shah MJ. Abnormalities of cardiac repolarization in Williams syndrome. Am J Cardiol. 2010a;106:1029–33. [PubMed: 20854969]
- Collins RT 2nd, Kaplan P, Somes GW, Rome JJ. Long-term outcomes of patients with cardiovascular abnormalities and Williams syndrome. Am J Cardiol. 2010b;105:874–8. [PubMed: 20211336]
- Committee on Genetics. American Academy of Pediatrics: Health care supervision for children with Williams syndrome. Pediatrics. 2001;107:1192–204. [PubMed: 11331709]
- Committee on Genetics. American Academy of Pediatrics: Health care supervision for children with Williams syndrome. Pediatrics. 2002;109:329. [Erratum]
- Crespi BJ, Hurd PL. Cognitive-behavioral phenotypes of Williams syndrome are associated with genetic variation in the GTF2I gene, in a healthy population. BMC Neurosci. 2014;15:127. [PMC free article: PMC4247780] [PubMed: 25429715]
- Danoff SK, Taylor HE, Blackshaw S, Desiderio S. TFII-I, a candidate gene for Williams syndrome cognitive profile: parallels between regional expression in mouse brain and human phenotype. Neuroscience. 2004;123:931–8. [PubMed: 14751286]
- Davies M, Howlin P, Udwin O. Independence and adaptive behavior in adults with Williams syndrome. Am J Med Genet. 1997;70:188–95. [PubMed: 9128941]
- Deal JE, Snell MF, Barratt TM, Dillon MJ. Renovascular disease in childhood. J Pediatr. 1992;121:378–84. [PubMed: 1517911]
- Del Campo M, Antonell A, Magano LF, Muñoz FJ, Flores R, Bayés M, Perez Jurado LA. Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension. Am J Hum Genet. 2006;78:533–42. [PMC free article: PMC1424678] [PubMed: 16532385]
- Del Pasqua A, Rinelli G, Toscano A, Iacobelli R, Digilio C, Marino B, Saffirio C, Mondillo S, Pasquini L, Sanders SP, de Zorzi A. New findings concerning cardiovascular manifestations emerging from long-term follow-up of 150 patients with the Williams-Beuren-Beuren syndrome. Cardiol Young. 2009;19:563–7. [PubMed: 19941695]
- Dixit A, McKee S, Mansour S, Mehta S, Tanteles G, Anastasiadou V, Patsalis P, Martin K, McCullough S, Suri M, Sarkar A. 7q11.23 microduplication: a recognizable phenotype. Clin Genet. 2013;83:155–61. [PubMed: 22369319]
- Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124A:263–73. [PubMed: 14708099]
- Dutly F, Schinzel A. Unequal interchromosomal rearrangements may result in elastin gene deletions causing the Williams-Beuren syndrome. Hum Mol Genet. 1996;5:1893–8. [PubMed: 8968740]
- Dykens EM. Anxiety, fears, and phobias in persons with Williams syndrome. Dev Neuropsychol. 2003;23:291–316. [PubMed: 12730029]
- Einfeld SL, Tonge BJ, Rees VW. Longitudinal course of behavioral and emotional problems in Williams syndrome. Am J Ment Retard. 2001;106:73–81. [PubMed: 11246715]
- Eisenberg DP, Jabbi M, Berman KF. Bridging the gene-behavior divide through neuroimaging deletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. Neuroimage. 2010;53:857–69. [PMC free article: PMC2916965] [PubMed: 20206275]
- Eronen M, Peippo M, Hiippala A, Raatikka M, Arvio M, Johansson R, Kahkonen M. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet. 2002;39:554–8. [PMC free article: PMC1735199] [PubMed: 12161592]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993a;5:11–6. [PubMed: 7693128]
- Ewart AK, Morris CA, Ensing GJ, Loker J, Moore C, Leppert M, Keating M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci U S A. 1993b;90:3226–30. [PMC free article: PMC46272] [PubMed: 8475063]
- Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Proschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 1996;86:59–69. [PubMed: 8689688]
- Giordano U, Turchetta A, Giannotti A, Digilio MC, Virgilii F, Calzolari A. Exercise testing and 24-hour ambulatory blood pressure monitoring in children with Williams syndrome. Pediatr Cardiol. 2001;22:509–11. [PubMed: 11894156]
- Goldman SE, Malow BA, Newman KD, Roof E, Dykens EM. Sleep patterns and daytime sleepiness in adolescents and young adults with Williams syndrome. J Intellect Disabil Res. 2009;53:182–8. [PMC free article: PMC5490443] [PubMed: 19067782]
- Gothelf D, Farber N, Raveh E, Apter A, Attias J. Hyperacusis in Williams syndrome: characteristics and associated neuroaudiologic abnormalities. Neurology. 2006;66:390–5. [PubMed: 16476938]
- Greer MK, Brown FR 3rd, Pai GS, Choudry SH, Klein AJ. Cognitive, adaptive, and behavioral characteristics of Williams syndrome. Am J Med Genet. 1997;74:521–5. [PubMed: 9342204]
- Heller R, Rauch A, Luttgen S, Schroder B, Winterpacht A. Partial deletion of the critical 1.5 Mb interval in Williams-Beuren syndrome. J Med Genet. 2003;40:e99. [PMC free article: PMC1735549] [PubMed: 12920091]
- Hertzberg J, Nakisbendi L, Needleman HL, Pober B. Williams syndrome--oral presentation of 45 cases. Pediatr Dent. 1994;16:262–7. [PubMed: 7937257]
- Hinsley TA, Cunliffe P, Tipney HJ, Brass A, Tassabehji M. Comparison of TFII-I gene family members deleted in Williams-Beuren syndrome. Protein Sci. 2004;13:2588–99. [PMC free article: PMC2286546] [PubMed: 15388857]
- Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, Rios CM, Kimberley KW, Gregg RG, Bray-Ward P. Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:220–8. [PMC free article: PMC2946898] [PubMed: 20425783]
- Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays. 2004;26:141–50. [PubMed: 14745832]
- Hoogenraad CC, Eussen BH, Langeveld A, van Haperen R, Winterberg S, Wouters CH, Grosveld F, De Zeeuw CI, Galjart N. The murine CYLN2 gene: genomic organization, chromosome localization, and comparison to the human gene that is located within the 7q11.23 Williams syndrome critical region. Genomics. 1998;53:348–58. [PubMed: 9799601]
- Horowitz PE, Akhtar S, Wulff JA, Fadley FA, Halees ZA. Coronary artery disease and anesthesia-related death in children with Williams syndrome. J Cardiothorac Vasc Anesth. 2002;16:739–41. [PubMed: 12486657]
- Howlin P, Udwin O. Outcome in adult life for people with Williams syndrome-- results from a survey of 239 families. J Intellect Disabil Res. 2006;50:151–60. [PubMed: 16403203]
- Jabbi M, Chen Q, Turner N, Kohn P, White M, Kippenhan JS, Dickinson D, Kolachana B, Mattay V, Weinberger DR, Berman KF. Variation in the Williams syndrome GTF2I gene and anxiety proneness interactively affect prefrontal cortical response to aversive stimuli. Transl Psychiatry. 2015;5:e622. [PMC free article: PMC4564573] [PubMed: 26285132]
- Jackowski AP, Rando K, Maria de Araújo C, Del Cole CG, Silva I, Tavares de Lacerda AL. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur J Paediatr Neurol. 2009;13:305–16. [PubMed: 18722146]
- Jackson TA, Taylor HE, Sharma D, Desiderio S, Danoff SK. Vascular endothelial growth factor receptor-2: counter-regulation by the transcription factors, TFII-I and TFII-IRD1. J Biol Chem. 2005;280:29856–63. [PubMed: 15941713]
- John AE, Mervis CB. Sensory modulation impairments in children with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:266–76. [PMC free article: PMC2997471] [PubMed: 20425786]
- Kaplan P, Kirschner M, Watters G, Costa MT. Contractures in patients with Williams syndrome. Pediatrics. 1989;84:895–9. [PubMed: 2797983]
- Kapp ME, von Noorden GK, Jenkins R. Strabismus in Williams syndrome. Am J Ophthalmol. 1995;119:355–60. [PubMed: 7503839]
- Kececioglu D, Kotthoff S, Vogt J. Williams-Beuren syndrome: a 30-year follow-up of natural and postoperative course. Eur Heart J. 1993;14:1458–64. [PubMed: 8299625]
- Klein-Tasman BP, Mervis CB. Distinctive personality characteristics of 8-, 9-, and 10-year-olds with Williams syndrome. Dev Neuropsychol. 2003;23:269–90. [PubMed: 12730028]
- Klein-Tasman BP, Phillips KD, Lord CE, Mervis CB, Gallo F. Overlap with the autism spectrum in young children with Williams syndrome. J Dev Behav Pediatr. 2009;30:289–99. [PMC free article: PMC2763277] [PubMed: 19668090]
- Kim Y-M, Cho JH, Kang E, Kim G-H, Seo E-J, Lee BH, Choi J-H, Yoo H-W. Endocrine dysfunctions in children with Williams-Beuren syndrome. Ann Pediatr Endocrinol Metab. 2016;21:15–20. [PMC free article: PMC4835556] [PubMed: 27104174]
- Koehler U, Pabst B, Pober B, Kozel B. Clinical utility gene card for: Williams–Beuren Syndrome. Eur J Hum Genet. 2014;22(9) [7q11.23] [PMC free article: PMC4135419] [PubMed: 24569604]
- Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. 2014;63:74–9. [PMC free article: PMC3932371] [PubMed: 24126171]
- Lam PP, Leung YM, Sheu L, Ellis J, Tsushima RG, Osborne LR, Gaisano HY. Transgenic mouse overexpressing syntaxin-1A as a diabetes model. Diabetes. 2005;54:2744–54. [PubMed: 16123365]
- Latham GJ, Ross FJ, Eisses MJ, Richards MJ, Geiduschek JM, Joffe DC. Perioperative morbidity in children with elastin arteriopathy. Paediatr Anaesth. 2016;26:926–35. [PubMed: 27397140]
- Laws G, Bishop D. Pragmatic language impairment and social deficits in Williams syndrome: a comparison with Down's syndrome and specific language impairment. Int J Lang Commun Disord. 2004;39:45–64. [PubMed: 14660186]
- Levitin DJ, Cole K, Lincoln A, Bellugi U. Aversion, awareness, and attraction: investigating claims of hyperacusis in the Williams syndrome phenotype. J Child Psychol Psychiatry. 2005;46:514–23. [PubMed: 15845131]
- Levy Y, Smith J, Tager-Flusberg H. Word reading and reading-related skills in adolescents with Williams syndrome. J Child Psychol Psychiatry. 2003;44:576–87. [PubMed: 12751849]
- Leyfer OT, Woodruff-Borden J, Klein-Tasman BP, Fricke JS, Mervis CB. Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:615–22. [PMC free article: PMC2561212] [PubMed: 16823805]
- Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1021–8. [PubMed: 9215670]
- Lin AE, Basson CT, Goldmuntz E, Magoulas PL, McDermott DA, McDonald-McGinn DM, McPherson E, Morris CA, Noonan J, Nowak C, Pierpont ME, Pyeritz RE, Rope AF, Zackai E, Pober BR. Adults with genetic syndromes and cardiovascular abnormalities: clinical history and management. Genet Med. 2008;10:469–94. [PMC free article: PMC2671242] [PubMed: 18580689]
- Makeyev AV, Erdenechimeg L, Mungunsukh O, Roth JJ, Enkhmandakh B, Ruddle FH, Bayarsaihan D. GTF2IRD2 is located in the Williams-Beuren syndrome critical region 7q11.23 and encodes a protein with two TFII-I-like helix-loop-helix repeats. Proc Natl Acad Sci U S A. 2004;101:11052–7. [PMC free article: PMC503739] [PubMed: 15243160]
- Malenfant P, Liu X, Hudson ML, Qiao Y, Hrynchak M, Riendeau N, Hildebrand MJ, Cohen IL, Chudley AE, Forster-Gibson C, Mickelson EC, Rajcan-Separovic E, Lewis ME, Holden JJ. Association of GTF2i in the Williams-Beuren syndrome critical region with autism spectrum disorders. J Autism Dev Disord. 2012;42:1459–69. [PubMed: 22048961]
- Marler JA, Sitcovsky JL, Mervis CB, Kistler DJ, Wightman FL. Auditory function and hearing loss in children and adults with Williams syndrome: cochlear impairment in individuals with otherwise normal hearing. Am J Med Genet C Semin Med Genet. 2010;154C:249–65. [PMC free article: PMC2913545] [PubMed: 20425785]
- Marshall CR, Young EJ, Pani AM, Freckmann ML, Lacassie Y, Howald C, Fitzgerald KK, Peippo M, Morris CA, Shane K, Priolo M, Morimoto M, Kondo I, Manguoglu E, Berker-Karauzum S, Edery P, Hobart HH, Mervis CB, Zuffardi O, Reymond A, Kaplan P, Tassabehji M, Gregg RG, Scherer SW, Osborne LR. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet. 2008;83:106–11. [PMC free article: PMC2443840] [PubMed: 18565486]
- Martin ND, Smith WR, Cole TJ, Preece MA. New height, weight and head circumference charts for British children with Williams syndrome. Arch Dis Child. 2007;92:598–601. [PMC free article: PMC2083767] [PubMed: 17301110]
- Martin ND, Snodgrass GJ, Cohen RD. Idiopathic infantile hypercalcaemia--a continuing enigma. Arch Dis Child. 1984;59:605–13. [PMC free article: PMC1628928] [PubMed: 6465928]
- Mason TB, Arens R, Sharman J, Bintliff-Janisak B, Schultz B, Walters AS, Cater JR, Kaplan P, Pack AI. Sleep in children with Williams Syndrome. Sleep Med. 2011;12:892–7. [PMC free article: PMC3210863] [PubMed: 21940205]
- Masserini B, Bedeschi MF, Bianchi V, Scuvera G, Beck-Peccoz P, Lalatta F, Selicorni A, Orsi E. Prevalence of diabetes and pre-diabetes in a cohort of Italian young adults with Williams syndrome. Am J Med Genet A. 2013;161A:817–821. [PubMed: 23495209]
- Matisoff AJ, Olivieri L, Schwartz JM, Deutsch N. Risk assessment and anesthetic management of patients with Williams syndrome: a comprehensive review. Paediatr Anaesth. 2015;25:1207–15. [PubMed: 26456018]
- Mégarbané H, Florence J, Sass JO, Schwonbeck S, Foglio M, de Cid R, Cure S, Saker S, Mégarbané A, Fischer J. An autosomal-recessive form of cutis laxa isdue to homozygous elastin mutations, and the phenotype may be modified by a heterozygous fibulin 5 polymorphism. J Invest Dermatol. 2009;129:1650–5. [PubMed: 19194475]
- Meng X, Lu X, Li Z, Green ED, Massa H, Trask BJ, Morris CA, Keating MT. Complete physical map of the common deletion region in Williams syndrome and identification and characterization of three novel genes. Hum Genet. 1998a;103:590–9. [PubMed: 9860302]
- Meng X, Lu X, Morris CA, Keating MT. A novel human gene FKBP6 is deleted in Williams syndrome. Genomics. 1998b;52:130–7. [PubMed: 9782077]
- Mercuri E, Atkinson J, Braddick O, Rutherford MA, Cowan FM, Counsell SJ, Dubowitz LM, Bydder G. Chiari I malformation in asymptomatic young children with Williams syndrome: clinical and MRI study. Eur J Paediatr Neurol. 1997;1:177–81. [PubMed: 10728215]
- Merla G, Ucla C, Guipponi M, Reymond A. Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum Genet. 2002;110:429–38. [PubMed: 12073013]
- Mervis CB, Dida J, Lam E, Crawford-Zelli NA, Young EJ, Henderson DR, Onay T, Morris CA, Woodruff-Borden J, Yeomans J, Osborne LR. Duplication of GTF2I results in separation anxiety in mice and humans. Am J Hum Genet. 2012a;90:1064–70. [PMC free article: PMC3370270] [PubMed: 22578324]
- Mervis CB, John AE. Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am J Med Genet C Semin Med Genet. 2010;154C:229–48. [PMC free article: PMC2922953] [PubMed: 20425784]
- Mervis CB, Kistler DJ, John AE, Morris CA. Longitudinal assessment of intellectual abilities of children with Williams syndrome: multilevel modeling of performance on the Kaufman Brief Intelligence Test-Second Edition. Am J Intellect Dev Disabil. 2012b;117:134–55. [PMC free article: PMC3334347] [PubMed: 22515828]
- Mervis CB, Morris CA, Bertrand J, Robinson BF. Williams syndrome: findings from an integrated program of research. In: Tager-Flusberg H, ed. Neurodevelopmental Disorders. Cambridge, MA: MIT Press; 1998:65-110.
- Mervis CB, Pitts CH. Children with Williams syndrome: Developmental trajectories for intellectual abilities, vocabulary abilities, and adaptive behavior. Am J Med Genet C Semin Med Genet. 2015;169:158–71. [PMC free article: PMC5007950] [PubMed: 25989316]
- Mervis CB, Robinson BF, Bertrand J, Morris CA, Klein-Tasman BP, Armstrong SC. The Williams syndrome cognitive profile. Brain Cogn. 2000;44:604–28. [PubMed: 11104544]
- Micale L, Turturo MG, Fusco C, Augello B, Pérez Jurado LA, Izzi C, Digilio MC, Milani D, Lapi E, Zelante L, Merla G. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet. 2010;18:317–23. [PMC free article: PMC2987220] [PubMed: 19844261]
- Morris CA. The behavioral phenotype of Williams syndrome: A recognizable pattern of neurodevelopment. Am J Med Genet C Semin Med Genet. 2010;154C:427–31. [PubMed: 20981771]
- Morris CA, Carey JC. Three diagnostic signs in Williams syndrome. Am J Med Genet Suppl. 1990;6:100–1. [PubMed: 2118769]
- Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113:318–26. [PubMed: 2456379]
- Morris CA, Leonard CO, Dilts C, Demsey SA. Adults with Williams syndrome. Am J Med Genet Suppl. 1990;6:102–7. [PubMed: 2118770]
- Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet. 2000;1:461–84. [PubMed: 11701637]
- Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L, Kimberley KW, Stock AD. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet. 2003;123A:45–59. [PubMed: 14556246]
- Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, Osborne LR. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am J Med Genet A. 2015;167A:2916–35. [PMC free article: PMC5005957] [PubMed: 26333794]
- Morris CA, Pani AM, Mervis CB, Rios CM, Kistler DJ, Gregg RG. Alpha 1 antitrypsin deficiency alleles are associated with joint dislocation and scoliosis in Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:299–306. [PMC free article: PMC2911626] [PubMed: 20425789]
- Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am J Med Genet. 1993;47:478–81. [PubMed: 8256809]
- Mulik VV, Temple KI, Howe DT. Two pregnancies in a woman with Williams syndrome. BJOG. 2004;111:511–2. [PubMed: 15104621]
- Muñoz KE, Meyer-Lindenberg A, Hariri AR, Mervis CB, Mattay VS, Morris CA, Berman KF. Abnormalities in neural processing of emotional stimuli in Williams syndrome vary according to social vs.non-social content. Neuroimage. 2010;50:340–6. [PMC free article: PMC3013360] [PubMed: 20004252]
- Oliveri B, Mastaglia SR, Mautalen C, Gravano JC, Pardo Argerich L. Long-term control of hypercalcaemia in an infant with williams-Beuren syndrome after a single infusion of biphosphonate (Pamidronate). Acta Paediatr. 2004;93:1002–3. [PubMed: 15303821]
- Olsen M, Fahy CJ, Costi DA, Kelly AJ, Burgoyne LL. Anaesthesia-related haemodynamic complications in Williams syndrome patients: a review of one institution's experience. Anaesth Intensive Care. 2014;42:619–24. [PubMed: 25233176]
- Olson TM, Michels VV, Lindor NM, Pastores GM, Weber JL, Schaid DJ, Driscoll DJ, Feldt RH, Thibodeau SN. Autosomal dominant supravalvular aortic stenosis: localization to chromosome 7. Hum Mol Genet. 1993;2:869–73. [PubMed: 8364568]
- Osborne LR, Campbell T, Daradich A, Scherer SW, Tsui LC. Identification of a putative transcription factor gene (WBSCR11) that is commonly deleted in Williams-Beuren syndrome. Genomics. 1999;57:279–84. [PubMed: 10198167]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–5. [PMC free article: PMC2889916] [PubMed: 11685205]
- Osborne LR, Martindale D, Scherer SW, Shi XM, Huizenga J, Heng HHQ, Costa T, Pober B, Lew L, Brinkman J, Rommens J, Koop B, Tsui LC. Identification of genes from a 500-kb region at 7q11.23 that is commonly deleted in Williams syndrome patients. Genomics. 1996;36:328–36. [PubMed: 8812460]
- Osborne LR, Soder S, Shi XM, Pober B, Costa T, Scherer SW, Tsui LC. Hemizygous deletion of the syntaxin 1A gene in individuals with Williams syndrome. Am J Hum Genet. 1997;61:449–52. [letter] [PMC free article: PMC1715888] [PubMed: 9311751]
- Palacios-Verdú MG, Segura-Puimedon M, Borralleras C, Flores R, Del Campo M, Campuzano V, Pérez-Jurado LA. Metabolic abnormalities in Williams-Beuren syndrome. J Med Genet. 2015;52:248–55. [PubMed: 25663682]
- Pankau R, Partsch CJ, Winter M, Gosch A, Wessel A. Incidence and spectrum of renal abnormalities in Williams-Beuren syndrome. Am J Med Genet. 1996;63:301–4. [PubMed: 8723124]
- Paperna T, Peoples R, Wang YK, Kaplan P, Francke U. Genes for the CPE receptor (CPETR1) and the human homolog of RVP1 (CPETR2) are localized within the Williams-Beuren syndrome deletion. Genomics. 1998;54:453–9. [PubMed: 9878248]
- Partsch CJ, Japing I, Siebert R, Gosch A, Wessel A, Sippell WG, Pankau R. Central precocious puberty in girls with Williams syndrome. J Pediatr. 2002;141:441–4. [PubMed: 12219071]
- Partsch CJ, Siebert R, Caliebe A, Gosch A, Wessel A, Pankau R. Sigmoid diverticulitis in patients with Williams-Beuren syndrome: relatively high prevalence and high complication rate in young adults with the syndrome. Am J Med Genet A. 2005;137:52–4. [PubMed: 16007633]
- Peoples R, Perez-Jurado L, Wang YK, Kaplan P, Francke U. The gene for replication factor C subunit 2 (RFC2) is within the 7q11.23 Williams syndrome deletion. Am J Hum Genet. 1996;58:1370–3. [letter] [PMC free article: PMC1915059] [PubMed: 8651315]
- Pérez Jurado LA, Wang YK, Peoples R, Coloma A, Cruces J, Francke U. A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet. 1998;7:325–34. [PubMed: 9466987]
- Pham PP, Moller JH, Hills C, Larson V, Pyles L. Cardiac catheterization and operative outcomes from a multicenter consortium for children with Williams syndrome. Pediatr Cardiol. 2009;30:9–14. [PubMed: 19052807]
- Pitts CH, Klein-Tasman BP, Osborne JW, Mervis CB. Predictors of specific phobia in children with Williams syndrome. J Intellect Disabil Res. 2016;60:1031–42. [PMC free article: PMC5026631] [PubMed: 27545817]
- Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–52. [PubMed: 20089974]
- Pober BR, Filiano JJ. Association of Chiari I malformation and Williams syndrome. Pediatr Neurol. 1995;12:84–8. [PubMed: 7748369]
- Pober BR, Johnson M, Urbán Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest. 2008;118:1606–15. [PMC free article: PMC2358987] [PubMed: 18452001]
- Pober BR, Lacro RV, Rice C, Mandell V, Teele RL. Renal findings in 40 individuals with Williams syndrome. Am J Med Genet. 1993;46:271–4. [PubMed: 8488870]
- Pober BR, Morris CA. Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:280–90. [PubMed: 17639596]
- Preus M. The Williams syndrome: objective definition and diagnosis. Clin Genet. 1984;25:422–8. [PubMed: 6723102]
- Radford DJ, Pohlner PG. The middle aortic syndrome: an important feature of Williams' syndrome. Cardiol Young. 2000;10:597–602. [PubMed: 11117392]
- Richter-Cook NJ, Dever TE, Hensold JO, Merrick WC. Purification and characterization of a new eukaryotic protein translation factor. Eukaryotic initiation factor 4H. J Biol Chem. 1998;273:7579–87. [PubMed: 9516461]
- Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. Washington DC: National Academies Press. 2011.
- Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene. 2012;492:32–41. [PMC free article: PMC3246126] [PubMed: 22037610]
- Sadler LS, Robinson LK, Verdaasdonk KR, Gingell R. The Williams syndrome: evidence for possible autosomal dominant inheritance. Am J Med Genet. 1993;47:468–70. [PubMed: 8256806]
- Sammour ZM, Gomes CM, de Bessa J Jr, Pinheiro MS, Kim CA, Hisano M, Bruschini H, Srougi M. Congenital genitourinary abnormalities in children with Williams-Beuren syndrome. J Pediatr Urol. 2014;10:804–9. [PubMed: 24582571]
- Sammour ZM, Gomes CM, Duarte RJ, Trigo-Rocha FE, Srougi M. Voiding dysfunction and the Williams-Beuren syndrome: a clinical and urodynamic investigation. J Urol. 2006;175:1472–6. [PubMed: 16516025]
- Santoro SD, Giacheti CM, Rossi NF, Campos LM, Pinato L. Correlations between behavior, memory, sleep-wake and melatonin in Williams-Beuren syndrome. Physiol Behav. 2016;159:14–9. [PubMed: 26976740]
- Sargent JD, Stukel TA, Kresel J, Klein RZ. Normal values for random urinary calcium to creatinine ratios in infancy. J Pediatr. 1993;123:393–7. [PubMed: 8355114]
- Saul RA, Stevenson RE, Rogers RC, et al. Growth references from conception to adulthood. Proc Greenwood Genet Ctr. 1988;1S:204–9.
- Scherer SW, Gripp KW, Lucena J, Nicholson L, Bonnefont JP, Perez-Jurado LA, Osborne LR. Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children. Hum Genet. 2005;117:383–8. [PMC free article: PMC2896963] [PubMed: 15933846]
- Schubert C. The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 2009;66:1178–97. [PubMed: 19039520]
- Sforzini C, Milani D, Fossali E, Barbato A, Grumieri G, Bianchetti MG, Selicorni A. Renal tract ultrasonography and calcium homeostasis in Williams-Beuren syndrome. Pediatr Nephrol. 2002;17:899–902. [PubMed: 12432430]
- Sindhar S, Lugo M, Levin MD, Danback JR, Brink BD, Yu E, Dietzen DJ, Clark AL, Purgert CA, Waxler JL, Elder RW, Pober BR, Kozel BA. Hypercalcemia in patients with Williams-Beuren syndrome. J Pediatr. 2016;178:254–60.e4. [PMC free article: PMC5085847] [PubMed: 27574996]
- Sniecinska-Cooper AM, Iles RK, Butler SA, Jones H, Bayford R, Dimitriou D. Abnormal secretion of melatonin and cortisol in relation to sleep disturbances in children with Williams syndrome. Sleep Med. 2015;16:94–100. [PubMed: 25441742]
- Somerville MJ, Mervis CB, Young EJ, Seo EJ, del Campo M, Bamforth S, Peregrine E, Loo W, Lilley M, Perez-Jurado LA, Morris CA, Scherer SW, Osborne LR. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N Engl J Med. 2005;353:1694–701. [PMC free article: PMC2893213] [PubMed: 16236740]
- Spielmann S, Partsch CJ, Gosch A, Pankau R. Treatment of central precocious puberty and early puberty with GnRH analog in girls with Williams-Beuren syndrome. J Pediatr Endocrinol Metab. 2015;28:1363–7. [PubMed: 26197460]
- Soper R, Chaloupka JC, Fayad PB, Greally JM, Shaywitz BA, Awad IA, Pober BR. Ischemic stroke and intracranial multifocal cerebral arteriopathy in Williams syndrome. J Pediatr. 1995;126:945–8. [PubMed: 7776102]
- Stagi S, Lapi E, Cecchi C, Chiarelli F, D'Avanzo MG, Seminara S, de Martino M. Williams-Beuren syndrome is a genetic disorder associated with impaired glucose tolerance and diabetes in childhood and adolescence: new insights from a longitudinal study. Horm Res Paediatr. 2014;82:38–43. [PubMed: 24925026]
- Stagi S, Lapi E, Chiarelli F, de Martino M. Incidence of diverticular disease and complicated diverticular disease in young patients with Williams syndrome. Pediatr Surg Int. 2010;26:943–4. [PubMed: 20652262]
- Stagi S, Manoni C, Scalini P, Chiarelli F, Verrotti A, Cecchi C, Lapi E, Giglio S, Romano S, de Martino M. Bone mineral status and metabolism in patients with Williams-Beuren syndrome. Hormones (Athens) 2016;15:404–12. [PubMed: 27394705]
- Stock AD, Spallone PA, Dennis TR, Netski D, Morris CA, Mervis CB, Hobart HH. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet A. 2003;120A:320–5. [PubMed: 12838549]
- Strømme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–71. [PubMed: 12088082]
- Sugitani H, Hirano E, Knutsen RH, Shifren A, Wagenseil JE, Ciliberto C, Kozel BA, Urbán Z, Davis EC, Broekelmann TJ, Mecham RP. Alternative splicing and tissue-specific elastin misassembly act as biological modifiers of human elastin gene frameshift mutations associated with dominant cutis laxa. J Biol Chem. 2012;287:22055–67. [PMC free article: PMC3381164] [PubMed: 22573328]
- Tam E, Young EJ, Morris CA, Marshall CR, Loo W, Scherer SW, Mervis CB, Osborne LR. The common inversion of the Williams-Beuren syndrome region at 7q11.23 does not cause clinical symptoms. Am J Med Genet A. 2008;146A:1797–806. [PMC free article: PMC2886033] [PubMed: 18553513]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M, Donnai D. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–7. [PubMed: 16293761]
- Tassabehji M, Metcalfe K, Hurst J, Ashcroft GS, Kielty C, Wilmot C, Donnai D, Read AP, Jones CJP. An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum Mol Genet. 1998;7:1021–8. [PubMed: 9580666]
- Tipney HJ, Hinsley TA, Brass A, Metcalfe K, Donnai D, Tassabehji M. Isolation and characterisation of GTF2IRD2, a novel fusion gene and member of the TFII-I family of transcription factors, deleted in Williams-Beuren syndrome. Eur J Hum Genet. 2004;12:551–60. [PubMed: 15100712]
- Urbán Z, Helms C, Fekete G, Csiszár K, Bonnet D, Munnich A, Donis-Keller H, Boyd CD. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet. 1996;59:958–62. [PMC free article: PMC1914803] [PubMed: 8808614]
- Van Borsel J, Curfs LM, Fryns JP. Hyperacusis in Williams syndrome: a sample survey study. Genet Couns. 1997;8:121–6. [PubMed: 9219010]
- Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009;52:94–100. [PubMed: 19249392]
- Vaux KK, Wojtczak H, Benirschke K, Jones KL. Vocal cord abnormalities in Williams syndrome: a further manifestation of elastin deficiency. Am J Med Genet A. 2003;119A:302–4. [PubMed: 12784297]
- von Gontard A, Niemczyk J, Borggrefe-Moussavian S, Wagner C, Curfs L, Equit M. Incontinence in children, adolescents and adults with Williams syndrome. Neurourol Urodyn. 2016;35:1000–5. [PubMed: 26370069]
- Wang YK, Samos CH, Peoples R, Perez-Jurado LA, Nusse R, Francke U. A novel human homologue of the Drosophila frizzled wnt receptor gene binds wingless protein and is in the Williams syndrome deletion at 7q11.23. Hum Mol Genet. 1997;6:465–72. [PubMed: 9147651]
- Weber SL, Souza RB, Ribeiro LG, Tavares MF, Goldchmit M. Williams syndrome: ophthalmological examination and review of systemic manifestations. J Pediatr Ophthalmol Strabismus. 2014;51:209–13. [PubMed: 24779423]
- Wessel A, Gravenhorst V, Buchhorn R, Gosch A, Partsch CJ, Pankau R. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A. 2004;127A:234–7. [PubMed: 15150772]
- Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24:1311–8. [PubMed: 14007182]
- Woodruff-Borden J, Kistler DJ, Henderson DR, Crawford NA, Mervis CB. Longitudinal course of anxiety in children and adolescents with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:277–90. [PMC free article: PMC2914498] [PubMed: 20425787]
- Wu YQ, Sutton VR, Nickerson E, Lupski JR, Potocki L, Korenberg JR, Greenberg F, Tassabehji M, Shaffer LG. Delineation of the common critical region in Williams syndrome and clinical correlation of growth, heart defects, ethnicity, and parental origin. Am J Med Genet. 1998;78:82–9. [PubMed: 9637430]
- Zhang MC, He L, Giro M, Yong SL, Tiller GE, Davidson JM. Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN). J Biol Chem. 1999;274:981–6. [PubMed: 9873040]
章节注释
Acknowledgments致谢
The author's research was supported by grant NS35102 from the National Institute of Neurological Disorders and Stroke, the Williams Syndrome Association, and the Las Vegas Pediatric Research Fund of the University of Nevada. The research has advanced thanks to the work of multiple talented scientist-collaborators over the past 30 years; current co-investigators include Dr. Carolyn B. Mervis of the University of Louisville, Department of Psychology and Dr. Lucy Osborne of the University of Toronto Department of Molecular Genetics. Also, thanks must be given to the individuals who have participated in Williams syndrome research for their commitment to the research as well as giving generously of their time.
作者的研究得到了来自美国国家神经障碍与中风研究所、威廉斯综合症协会以及内华达大学拉斯维加斯儿科研究基金的资助。这项研究的进展得益于过去30年多名有才华的科学家合作者的工作;目前的合作研究人员包括路易斯维尔大学的Carolyn B.Mervis博士、心理学系和多伦多大学分子遗传学系的Lucy Osborne博士。同时,也要感谢参与威廉斯综合症研究的个人,感谢他们对这项研究的承诺,以及慷慨地付出他们的时间。
Revision History修订记录
- 23 March 2017 (ha) Comprehensive update posted live2017年3月23日 (ha)全面更新发布
- 13 June 2013 (me) Comprehensive update posted live2017年6月13日 (me)全面更新发布
- 21 April 2006 (me) Comprehensive update posted to live Web site2006年4月21日 (me)网站发布全面更新
- 22 August 2003 (me) Comprehensive update posted to live Web site2003年8月22日 (me)网站发布全面更新
- 9 April 1999 (me) Review posted to live Web site1999年4月9日 (me)综述在线网络上传
- 7 January 1999 (cm) Original submission19991月7日(cm)原始稿件上传