
概要
临床特征。
15q24 微缺失综合征的特征包括全面发育迟缓; 轻度至重度(通常至少中度)智力障碍; 面部畸形;手脚,眼睛和生殖器的 先天性畸形;关节松弛;以及发育迟缓和生长不旺。 不太常见的症状包括:癫痫发作;传导性和感音神经性听力丧失; 尿道下裂和/或小阴茎。男性和女性同等 受累。
通过在15q24 染色体上证实 杂合缺失来进行确诊,最常见的是使用全基因组和靶向分子生物学方法确定参考基因组(NCBI Build 36 / hg18)中72.2-73.3Mb之间的1.1Mb区域的序列拷贝数的缺失。
管理。
症状的治疗:语言,职业和物理治疗;眼科,心脏,神经学症状的常规治疗;个体化学习方案。
监管:日常儿科护理; 常规发育评估; 监测具体的医疗问题。
诊断
临床诊断
15q24 微缺失综合征的临床表现种类多样。 发育迟缓和智力障碍是最恒有的特征; 然而,没有单一的临床特征可以确立临床诊断。
在发育迟缓或智力障碍的患者中应该考虑该诊断的特征包括:
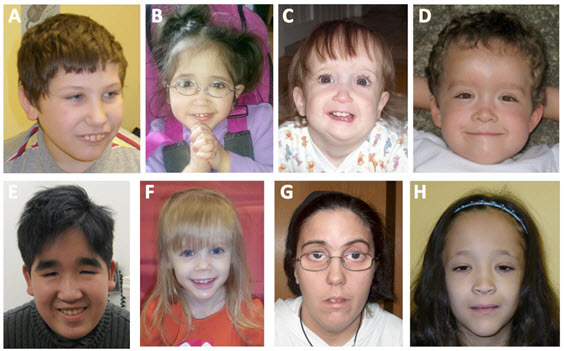
- 异常面部特征,特别是前发际高,眼深凹以及三角脸(参见 图1)
- 语言发育显著迟缓或丧失
- 肌张力低下
- 关节松弛
- 眼部异常,尤其是斜视
- 手脚异常:第五指短;显著缩短的第四掌骨和较短第五掌骨;拇指异常,比如拇指近端植入
- 发育迟缓和生长不旺
- 听力:传导性和感音神经性听力丧失
- 生殖器异常:男性尿道下裂或小阴茎,女性阴唇粘连
检测
细胞遗传学检测。通过染色体G显带或其他常规细胞遗传学条带技术的常规分析,不能鉴定出15q24微缺失。
分子遗传学检测
关键区域。15q24微缺失的诊断是通过在 染色体15q24上证明 杂合缺失来确定的。 目前鉴定的大多数15q24缺失涉及参考基因组(NCBI Build 36 / hg18 breakpoints B and C in Figure 2)72.2-73.3Mb之间1.1Mb区域。 缺失的实际大小和断点在患者之间不同,大多数缺失是由于区段 复制区域间的 非等位基因同源重组(NAHR)而发生的。
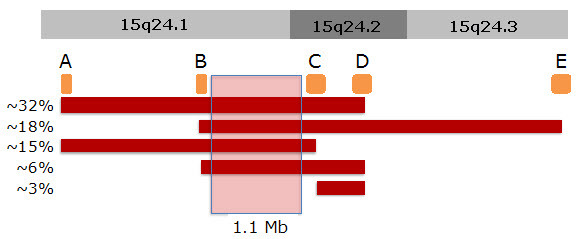
值得注意的是,通过确定“非典型”缺失的患者, 关键区域定位被进一步精确,其中一些患者仅涉及部分甚至不涉及所提出的1.1Mb关键区域,但仍然似乎是致病的。
- 携带有所提出的1.1Mb 关键区域之外的两名 新发突变缺失患者,具有轻微的发育迟缓,且都有比较正常的语言能力; 他们都具有 畸形特征,其中之一具有手部异常[ Mefford et al 2012]。
还报道了两例在 图2断点B和断点C之间仅涉及800 kb[ Andrieux et al 2009]和500 kb [ Ng et al 2011]的区域的大片段缺失病例。这两名患者的缺失表明 关键区域可能小于1.1Mb,但其较大的缺失任然很难知道哪些基因引起哪些特征。
未来鉴定较小的非典型缺失将继续精细 关键区域和 基因型- 表型 的相关性。
基因。15q24 微缺失综合征的认知特征,包括发育迟缓和严重的语言问题,主要是由于1.1Mb 关键区域的基因缺失引起的。在该区域内没有单一 基因的致病变异被鉴定为导致相似 表型。
临床检验
缺失/重复分析。15q24微缺失可以通过任何分子生物学方法确定缺失区域内的序列拷贝数来检测。全基因组和靶向途径方法都可以用。
表格 1.
检测结果的解释。取决于鉴定 缺失的初始检测方法,可能需要通过独立的方法验证该缺失。 如果已经使用高密度 基因组芯片技术来鉴定缺失,那么该缺失的验证可能不是必需的,因为超过50-100个相邻靶点不可能偶然地显示异常拷贝数。
检验策略
以先证者确立诊断需要检测15q24 微缺失综合征中常见的1.1-Mb最小关键区域缺失。
注意: 缺失不能通过常规 染色体分析来鉴定。
高危怀孕的 胎儿诊断和植入前遗传诊断(PGD)需要事先确定 先证者的 缺失和/或父母中平衡异位 携带状态。目前所有已知的案例都来源于 新发突变;尚未有父母携带平衡重排的报道,也没有轻微 受累的15q24微缺失患者有后代的报道。 然而,由于仅研究了有限数量的患者,父母中的平衡重排仍然是可能的。
临床特征
临床描述
15q24 微缺失综合征在临床上可识别的 表型包括:发育迟缓/智力障碍,面部畸形( 图 1), 先天性畸形和生长迟缓( 表 2)。男性和女性同等 受累。
智力障碍和发育迟缓。迄今为止报道的涉及1.1-Mb 关键区域的 缺失的所有患者都有轻度至重度的智力障碍(ID)。发育迟缓通常是完全性的,包括总体运动能力,语言能力和认知能力。 运动迟缓最可能与肌张力低下的程度有关,大多数情况下为轻度至中度。 语言和认知缺陷通常更为严重。 大多数 受累的患者在语言学习方面有严重的发育迟缓。认知能力从轻度到重度的智力障碍间分布; 大多数具有至少中度的智力障碍。
15q24 微缺失综合症患者的异常面部特征很普遍,包括前发际高,眼深凹和三角脸。其他常见的报道特征包括长或突出人中,下唇丰厚,内眦赘皮和尖下巴。59%的患者据报道出现眼部异常,斜视的发生频率最高。 其他罕见的异常包括虹膜缺损,脉络膜视网膜病变和远视。
59%的患者据报道出现手指异常。这些症状包括第五指短,显著缩短的第四掌骨和较短的第五掌骨;拇指异常,比如拇指近端植入,先天性手指屈曲,第五脚趾发育不良和脚趾并指畸形。
大约30%的患者据报道出现生殖器异常,最常见的症状是尿道下裂和小阴茎。
患病率
15q24 微缺失综合征的确切患病率未知;但它显然很少见。 迄今为止,全世界已有大约30名 受累的患者被报道。
15q24 微缺失综合征的大规模调查估计,其在孤独症谱系障碍患者的频率为0.1-0.2%[ McInnes et al 2010]。
在一个大数据量研究中,通过临床芯片比较基因组杂交研究,15q24微缺失在3:10,000-4:10,000的个人中被鉴定发现 [ Mefford et al 2012]。
遗传相关疾病
综合征性小眼症/Matthew Wood综合征(隐性)( OMIM)。 STRA6中的隐性致病变异导致了伴有眼部异常的严重智力障碍综合征,症状包括小眼畸形和/或无眼畸形,膈疝,肺发育不良,心脏缺陷和身材矮小[ Golzio et al 2007, Pasutto et al 2007]。
先天性肾上腺功能不全(隐性)( OMIM)。在几名患有 先天性肾上腺功能不全同时伴有部分或完全46,XY性反转的患者中报道了 CYP11A1的复合 杂合或 纯合致病突变 [ Katsumata et al 2002, Hiort et al 2005, Al Kandari et al 2006, Kim et al 2008, Rubtsov et al 2009]。还报道了一例仅在该基因中存在一个致病突变的病例[ Tajima et al 2001]。
MPI-CDG( CDG-1b;隐性)( OMIM)。 这种 常染色体隐性遗传病是由定位于15q24微缺失关键区域内的 MPI基因 的 复合杂合或 纯合致病突变所导致的[ Jaeken et al 1998, Schollen et al 2000]。 MPI-CDG的特征包括慢性腹泻,生长不旺,蛋白丢失性肠病和凝血功能障碍。 与其他类型的CDG不同,MPI-CDG通常不涉及中枢神经系统。(参见 糖基化先天疾病概述)
对应 重复。报道了几例携带有包含15q24 微缺失综合征的1.1-Mb 关键区域的15q24区域重复病例。目前尚不清楚15q24重复是否表现出特异的临床 表型[ El-Hattab et al 2010]。两例不相关患者的病例,报道了具有类似于15q24微缺失综合征的一些症状,包括发育迟缓,手指异常和畸形特征[ Kiholm Lund et al 2008, El-Hattab et al 2009]。相比之下,另外两名更严重 受累,且具有另一拷贝数改变的患者只有一个症状——发育迟缓[ El-Hattab et al 2010]。三例遗传背景已知的病例,其重复遗传自轻度受累或健康的父母。鉴于一些受累患者的父母表型和发现的其他拷贝数变化,这些重复可能是导致异常表型,或显示出外显率降低和/或 表现度差异的因素之一。
15q24重复的区域在1.1-Mb关键 缺失区域远侧的病例也有报道,且可能导致可变的异常表型。两个报道家系具有携带这种重复的多个 受累的患者; 这种重复的 杂合个体显示了不同程度的发育迟缓,肌张力低下和 畸形特征; 其中一个家庭也有个体患有自闭症谱系障碍 [ El-Hattab et al 2009, Roetzer et al 2010]。
鉴定诊断
发育迟缓和儿童期肌张力低下是15q24 微缺失综合征最常见的症状,其分子 细胞遗传学分析中常见且是相对非特异性的指征。然而,特征性面部 畸形特征和手部及生殖器异常的并发症状可能提示特别考虑15q24微缺失综合征。
Other diagnoses that may be considered in 受累的individuals include the following:
受累患者可能考虑的其他诊断包括:
- Angelman 综合征.Angelman综合征中也可见的15q24 微缺失综合征中所见的临床特征包括小头畸形,失语,生长发育迟缓,癫痫,以及异常快乐性格。
- Prader-Willi综合征(PWS)。 PWS也可能出现的15q24 微缺失综合征的临床特征包括严重的新生儿肌张力低下,癫痫,全面发育迟缓,斜视,上斜睑裂和隐睾。 然而,与PWS相反,15q24微缺失综合征尚未报道儿童期食欲旺盛和中枢性肥胖,并且行为障碍和睡眠紊乱也较少见。
管理
初步诊断后评估
要确定被诊断为15q24 微缺失综合征的个体的疾病程度和医疗管理需要,应考虑进行以下评估:
- 眼科检查
- 正规听力学评估以评估感音神经性听力损失
- 心脏评估
- 具有小头畸形和/或神经学症状的患者的脑成像
- 泌尿生殖器异常检查(比如:男性尿道下裂或隐睾)
- 综合发育评估
- 如有异常运动或怀疑癫痫应考虑神经科转诊
- 与临床遗传学家或遗传咨询师讨论结果
对症治疗
有如下对症治疗:
- 眼科,心脏,神经学症状的常规治疗
- 语言,职业和物理治疗
- 量身定制的个体化学习方案
没有特殊的抗癫痫药或抗精神病药物。
监测
适当的监管包括:
- 常规儿科护理
- 常规发育评估
- 监测具体的医疗问题
孕期管理
如果已知胎儿有15q24 微缺失综合征,推荐使用胎儿超声心动图和超声检查尝试观察腭裂并寻找膈肌疝。 密切监测子宫内发育迟缓是必要的。关于15q24微缺失综合征的发育结果和医疗并发症的咨询是合适。在良好的新生儿重症监护病房分娩是最优的选择,因为分娩后可能会发生呼吸道并发症和喂养困难。
遗传咨询
遗 传咨询是向个人和家庭提供关于遗 传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分涉及遗传风险评估和使用家族史及遗传检测来明确家庭成员的遗传状况的内容。 本节并不意味着能解决患者面临的所有个人,文化或伦理问题,或替代遗传学专业人士的咨询。-ED。
家庭成员风险
先证者的父母
- 未来怀孕的复发风险很低(大概<1%),但其风险仍大于普通人群,因为其父母可能有以下一种的情况:
- 生殖细胞嵌合
- 涉及15q24区域的染色体平衡重排
先证者的同胞
先证者的后代。无确诊15q24微缺失综合征患者的生育报道; 然而,15q24微缺失综合征的成人资料有限。 具有15q24缺失综合征的患者在每次生育中预计有50%的概率遗传自身携带的缺失。
遗传相关问题咨询
计划生育
- 遗传风险确定的最佳时间和产前检测可行性的讨论应在怀孕之前。 同样地,关于通过检测以确定无症状家族成员的遗传状况的决定最好是在怀孕前进行。
DNA样本保存是以备日后使用而做的DNA的储备(通常从白细胞中提取)。因为检测方法和我们对基因,基因变异以及疾病的理解可能会在未来有所改善,所以考虑对受累人群推荐存储DNA。
产前检测
Prenatal testing may be offered to unaffected parents who have had a child with the 15q24 微缺失综合征because of the 再发风险(probably <1%) associated with the possibility of 胚系嵌合. 由于存在生殖细胞嵌合可能性相关的复发风险(可能<1%),可以建议生育有15q24微缺失综合征但未受累的父母做产前检测。
产前检测在技术上是可行的。通常在约15至18周妊娠期间通过羊膜穿刺术或在约10至12周的妊娠期间通过绒毛膜穿刺获得胎儿细胞,可以使用分子遗传学检测中描述的方式使用芯片比较基因组杂交或靶向缺失分析技术进行分析。
注意:妊娠年龄指从末次正常月经的第一天或通过超声测量来计算月经周。 植入前遗传诊断(PGD)可以是已经确诊缺失的一些家庭的选择。
资源
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- My46Trait Profile
- Chromosome Disorder Outreach (CDO)PO Box 724Boca Raton FL 33429-0724Phone: 561-395-4252 (Family Helpline)Email: info@chromodisorder.org
- Unique: The Rare Chromosome Disorder Support GroupG1 The StablesStation Road WestOxted Surrey RH8 9EEUnited KingdomPhone: +44 (0) 1883 723356Email: info@rarechromo.org; ra rechromo@aol.com
分子遗传学
分子遗传学和OMIM表中的信息可能与GeneReview中的其他信息不同:表可能包含更多最新信息。-ED。
表 A.
| Gene | Chromosome Locus | Protein |
|---|---|---|
| 未知 | 15q24 | 未知 |
分子遗传病理学
15q24区域的特征为多个区段 重复(SD)区或低拷贝重复 [ Bailey et al 2002, El-Hattab et al 2009] (参见 图 2)。 片段重复区会促进非等位基因同源重组(NAHR),从而导致其涉及区域的微缺失或重复。 已经确定了五个SD区(A,B,C,D,E)在产生复发性15q24微缺失中发挥作用,最常见的缺失发生在A区和D区(3.1Mb),B区和E 区(3.8Mb) ,以及A区和C区(2.6Mb)之间。 位于B区和C区之间的关键区域被确定为常见缺失中最小的覆盖区域; 迄今为止,还没有仅仅只涉及关键区域的缺失病例报告。
在B区和C区之间的1.1-Mb关键区域包含26个在RefSeq数据库中被详细描述的基因。 该区域一个或多个基因的单倍剂量不足被认为会导致15q24微缺失综合征的表型; 然而,还没有能鉴定出该区域的某个基因致病突变可引起相似表型。 基于已知功能的候选基因如下
参考文献
Literature Cited
- al Kandari H, Katsumata N, Alexander S, Rasoul MA. Homozygous mutation of P450 side-chain cleavage enzyme gene (CYP11A1) in 46,XY patient with adrenal insufficiency, complete sex reversal, and agenesis of corpus callosum. J Clin Endocr Metab.2006; 91:2821–6.[ PubMed : 16705068]
- Andrieux J, Dubourg C, Rio M, Attie-Bitach T, Delaby E, Mathieu M, Journel H, Copin H, Blondeel E, Doco-Fenzy M, Landais E, Delobel B, Odent S, Manouvrier-Hanu S, Holder-Espinasse M. Genotype-phenotype correlation in four 15q24 deleted patients identified by array-CGH. Am J Med Genet A.2009; 149A:2813–9.[ PMC free article : PMC2874573] [ PubMed : 19921647]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science.2002; 297:1003–7.[ PubMed : 12169732]
- El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, Patel A, Scaglia F, Lupski JR, Cheung SW, Stankiewicz P. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum Genet.2009; 126:589–602.[ PMC free article : PMC3669685] [ PubMed : 19557438]
- El-Hattab AW, Zhang F, Maxim R, Christensen KM, Ward JC, Hines-Dowell S, Scaglia F, Lupski JR, Cheung SW. Deletion and duplication of 15q24: molecular mechanisms and potential modification by additional copy number variants. Genet Med.2010; 12:573–86.[ PubMed : 20860070]
- Golzio C, Martinovic-Bouriel J, Thomas S, Mougou-Zrelli S, Grattagliano-Bessieres B, Bonniere M, Delahaye S, Munnich A, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Batich T, Etchevers HC. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am J Hum Genet.2007; 80:1179–87.[ PMC free article : PMC1867105] [ PubMed : 17503335]
- Hiort O, Holterhus PM, Werner R, Marschke C, Hoppe U, Partsch CJ, Riepe FG, Achermann JC, Struve D. Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY sex reversal, and severe adrenal failure. J Clin Endocrinol Metab.2005; 90:538–41.[ PubMed : 15507506]
- Jaeken J, Matthijs G, Saudubray J-M, Dionisi-Vici C, Bertini E, de Lonlay P, Henri H, Carchon H, Schollen E, Van Schaftingen E. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet.1998; 62:1535–9.[ PMC free article : PMC1377152] [ PubMed : 9585601]
- Katsumata N, Ohtake M, Hojo T, Ogawa E, Hara T, Sato N, Tanaka T. Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J Clin Endocr Metab.2002; 87:3808–13.[ PubMed : 12161514]
- Kiholm Lund AB, Hove HD, Kirchhoff M. A. 15q24 microduplication, reciprocal to the recently described 15q24 microdeletion, in a boy sharing clinical features with 15q24 microdeletion syndrome patients. Eur J Med Genet.2008; 51:520–6.[ PubMed : 18755302]
- Kim CJ, Lin L, Huang N, Quigley CA. AvRuskin TW, Achermann JC, Miller WL. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J Clin Endocr Metab.2008; 93:696–702.[ PMC free article : PMC2266942] [ PubMed : 18182448]
- Klopocki E, Graul-Neumann LM, Grieben U, Tönnies H, Ropers HH, Horn D, Mundlos S, Ullmann R. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH. Eur J Pediatr.2008; 167:903–8.[ PMC free article : PMC2757600] [ PubMed : 17932688]
- Ng ISL, Chin WH, Lim ECP, Tan EC. An additional case of the recurrent 15q24.1 microdeletion syndrome and review of the literature. Twin Res Hum Genet.2011; 14:333–9.[ PubMed : 21787116]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet.2008; 82:477–88.[ PMC free article : PMC2426913] [ PubMed : 18252227]
- Masurel-Paulet A, Callier P, Thauvin-Robinet C, Chouchane M, Mejean N, Marle N, Mosca AL, Ben Salem D, Giroud M, Guibaud L, Huet F, Mugneret F, Faivre L. Multiple cysts of the corpus callosum and psychomotor delay in a patient with a 3.1 Mb 15q24.1q24.2 interstitial deletion identified by array-CGH. Am J Med Genet A.2009; 149A:1504–10.[ PubMed : 19533778]
- McInnes LA, Nakamine A, Pilorge M, Brandt T, Jiménez González P, Fallas M, Manghi ER, Edelmann L, Glessner J, Hakonarson H, Betancur C, Buxbaum JD. A large-scale survey of the novel 15q24 microdeletion syndrome in autism spectrum disorders identifies an atypical deletion that narrows the critical region. Mol Autism.2010; 1:5.[ PMC free article : PMC2907565] [ PubMed : 20678247]
- Mefford HC, Rosenfeld JA, Shur N, Slavotinek AM, Cox VA, Hennekam RC, Firth HV, Willatt L, Wheeler P, Morrow EM, Cook J, Sullivan R, Oh A, McDonald MT, Zonana J, Keller K, Hannibal MC, Ball S, Kussmann J, Gorski J, Zelewski S, Banks V, Smith W, Smith R, Paull L, Rosenbaum KN, Amor DJ, Silva J, Lamb A, Eichler EE. Further clinical and molecular delineation of the 15q24 microdeletion syndrome. J Med Genet.2012; 49:110–8.[ PMC free article : PMC3261729] [ PubMed : 22180641]
- Ng ISL, Chin WH, Lim ECP, Tan EC. An additional case of the recurrent 15q24.1 microdeletion syndrome and review of the literature. Twin Res Hum Genet.2011; 14:333–9.[ PubMed : 21787116]
- Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, Brasch F, Schirmer-Zimmermann H, Tolmie JL, Chitayat D, Houge G, Fernández-Martínez L, Keating S, Mortier G, Hennekam RC, von der Wense A, Slavotinek A, Meinecke P, Bitoun P, Becker C, Nürnberg P, Reis A, Rauch A. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet.2007; 80:550–60.[ PMC free article : PMC1821097] [ PubMed : 17273977]
- Roetzer KM, Schwarzbraun T, Obenauf AC, Hauser E, Speicher MR. Further evidence for the pathogenicity of 15q24 microduplications distal to the minimal critical regions. Am J Med Genet A.2010; 152A:3173–8.[ PubMed : 21108404]
- Rubtsov P, Karmanov M, Sverdlova P, Spirin P, Tiulpakov A. A novel homozygous mutation in CYP11A1 gene is associated with late-onset adrenal insufficiency and hypospadias in a 46,XY patient. J Clin Endocr Metab.2009; 94:936–9.[ PubMed : 19116240]
- Schollen E, Dorland L, de Koning TJ, Van Diggelen OP, Huijmans JGM, Marquardt T, Babovic-Vuksanovic D, Patterson M, Imtiaz F, Winchester B, Adamowicz M, Pronicka E, Freeze H, Matthijs G. Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Hum Mutat.2000; 16:247–52.[ PubMed : 10980531]
- Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, Striano P, Coppola A, Regan R, Price SM, Knoers NV, Eis PS, Brunner HG, Hennekam RC, Knight SJ, de Vries BB, Zuffardi O, Eichler EE. Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet.2007; 16:567–72.[ PubMed : 17360722]
- Tajima T, Fujieda K, Kouda N, Nakae J, Miller WL. Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocr Metab.2001; 86:3820–5.[ PubMed : 11502818]
- Van Esch H, Backx L, Pijkels E, Fryns JP. Congenital diaphragmatic hernia is part of the new 15q24 microdeletion syndrome. Eur J Med Genet.2009; 52:153–6.[ PubMed : 19233321]
Suggested Reading
- Cushman LJ, Torres-Martinez W, Cherry AM, Manning MA, Abdul-Rahman O, Anderson CE, Punnett HH, Thurston VC, Sweeney D, Vance GH. A report of three patients with an interstitial deletion of chromosome 15q24. Am J Med Genet A.2005; 137:65–71.[ PubMed : 16007617]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev.2009; 19:196–204.[ PMC free article : PMC2746670] [ PubMed : 19477115]
- Muntoni F. Journey into muscular dystrophies caused by abnormal glycosylation. Acta Myol.2004; 23:79–84.[ PubMed : 15605948]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet.2006; 38:1038–42.[ PubMed : 16906162]
Chapter Notes
Author Notes
Dr Mefford’s Laboratory website: depts.washington.edu/meflab
Acknowledgments
Revision History
- 23 February 2012 (me) Review posted live
- 1 September 2011 (hm) Original submission