
概述
临床特征.
原发性纤毛运动障碍 (primary ciliary dyskinesia,PCD)与内脏位置异常, 精子运动异常, 纤毛功能结构异常有关 导致粘液和细菌在呼吸道滞留,引起慢性 耳鼻窦肺部疾病.超过 75%的足月的患有PCD的新生儿有 ‘新生儿呼吸窘迫症’ 需要辅助供氧几天至几周. 慢性呼吸道感染, 发生在幼年早期,导致和成人表现相似的支气管扩张. 鼻塞和鼻窦炎, 发生在幼年早期, 持续到成人. 慢性/反复性耳道感染, 发生在大多数年幼儿童身上, 和短暂的或者后来不可逆的听力丧失有关. 全内脏反位(所有内脏器官镜像反转并没有明显的生理影响)表现在 40%-50% 的患有PCD的个体上; 内脏异位(正常情况下不对称的结构左右不协调 可以导致严重的畸形)大概有12%的表现.几乎所有患有PCD的男性 由于精子活动异常而不育.
诊断/检验.
PCD的诊断可以通过临床的 表型 和通过纤毛超微结构分析或通过 分子遗传学检测. 大概三分之二的先证者可以被诊断通过 表现出与PCD有联系的32个基因型中的一个 双等位基因的 致病变异型.
管理.
治疗临床表现: 积极的措施以使粘液更干净 (胸背叩击和体位引流, 震荡背心, 呼吸动作帮助远端呼吸道的清洁) 和迅速的抗生素疗法以治疗呼吸道细菌感染 (支气管炎, 鼻窦炎, 和中耳炎);并考虑肺叶切除以治疗局部支气管扩张; 肺移植以治疗终末期肺病; 鼻窦手术治疗广泛的鼻窦感染; 考虑用PE管放置法处理慢性中耳炎;按需要可用 语言障碍矫正和助听器. 按需要可用外科手术以治疗 先天的 心脏病. ICSI (胞质内的精子注射) 或通过捐精 (AIDS)人工授精以治疗男性不育.预防次级并发症: 日常免疫接种 (包括流感疫苗和肺炎双球菌疫苗) 防止呼吸感染; 宣传教育控制感染 包括注意洗手,避免接触病人 ,正确的 清洁/消毒 呼吸设备;呼吸道疾病早用抗生素(由之前的呼吸道状况决定).
监督: 肺脏学家跟踪监视肺功能和痰中的病原体并评估肺病程度/级数; 对于那些有慢性中耳炎的人,直到青少年前的日常听力评估.
需避免的药剂/环境: 止咳药; 接触呼吸病原, 烟草烟雾, 和其他空气污染以及呼吸道刺激.
遗传咨询.
PCD遗传方式为 常染色体隐性遗传 . 受累的 个体的父母必然是杂合子,因此携带一个 等位基因 和一个 致病性变异. 杂合子 (携带者) 无症状. 理论上,受累的个体的每个同胞有25%的概率 受累,有 50% 的概率是无症状的 携带者, 有 25% 的概率是既不受累也不是携带者. 如果家庭中的致病变异型已知,高危亲属的带菌体实验和有受累风险的孕期产前诊断是可行的.
诊断
推测性结论
原发性纤毛不动症 (PCD) 可通过以下(但不限于)的临床表现判断:
- 慢性的耳-鼻窦-肺疾病
- 慢性湿咳有痰
- 慢性喘气气困
- 肺功能测试有阻塞性肺病
- 患有PCD个体中常见病原持续扩大感染
- 胸片发现有慢性异常
- 胸部 CT 扫描发现支气管扩张
- 鼻窦x光照片发现有慢性全鼻窦炎
- 慢性中耳炎,大多数显著发生在学龄前儿童
- 新生儿呼吸窘迫, 尽管任期妊娠
- 从新生儿期开始慢性鼻塞
- 内脏位置异常 可能是一下任一种情况:
- 单侧性缺陷 (全内脏器官镜面反转但没有明显生理影响)
- 心房不定位
- 内脏异位 (正常情况下不对称器官左右不协调,通常临床分类为无脾 [主要表现为双侧右侧器官, 或右房异构] 或 多脾 [主要变现为双侧左侧器官或左房异构]) [Zhu et al 2006]
- 杵状指 典型与支气管扩张有关
- 男性不育
建立诊断
诊断一个 先证者 有原发性纤毛不动症可基于临床症状和 有已知的与PCD有关的32个基因型中双等位基因的 致病突变型 (见 Table 1A, Table 1B). 如果诊断不能通过 分子遗传学检测建立, 纤毛诊断 可以使用; 然而, 应该指出大概30%的患有PCD的个体纤毛没有微结构异常, 这是以前诊断的金标准.在很多病例中需要诊断测试面板 [Lucas & Leigh 2014].当需要用
分子遗传学检测 诊断PCD时, 临床医生需要考虑到大多数致病突变型出现在 Table 1A包含的基因中, 致病突变很少在 Table 1B包含的基因中, 超过三分之一的明确判断出有PCD的个体没有已知的可辨认的在32个基因中的致病突变型.
分子基因检测
分子检测方法可包括 系列单-基因 测试, 靶标分析 来监测 致病突变型, 使用 表型靶向检测, 和 基因组的 测试.
系列单-基因 测试 按致病突变最常发生的顺序检测 (Table 1A) (也就是说, DNAH5, DNAH11, CCDC39, DNAI1, CCDC40, CCDC103, SPAG1, ZMYND10, ARMC4, CCDC151, DNAI2, RSPH1, CCDC114, RSPH4A, DNAAF1, DNAAF2, 和 LRRC6). 通常从 序列分析开始测试基因, 接下来是基因靶向 缺失/复制分析 该基因 如果只辨认出一个 致病性变异 .
针对性分析在某个 基因 上的致病突变可在以下种族/祖先合适的个体先执行:
- p.Trp453Ter 于 DNAI2
- p.Tyr245Ter 于 C21orf59
- c.876_877delAT 于 CCDC65
- 贝多因人
- p.Asn150Ser 于 DNAL1
- p.Lys268del 于 RSPH9
- 阿米什人或门诺会
- p.Leu795Pro 于 DNAAF5 (原名 HEATR2)
- c.10815delT 于 DNAH5
- p.Gln1450Ter 于 DNAH5
- c.48+2dupT 于 DNAI1
- 法罗群岛. p.Lys308Ter 于 HYDIN
- 爱尔兰旅行者
- c.258_262dupGGCCC 于 CCNO
- c.166dupC 于 RSPH4A
- 3549-bp 大 缺失 于 DNAAF4 (原名 DYX1C1)
- 北荷兰省 (福伦丹). c.742G>A 于 CCDC114
- 英国-巴基斯坦人
- c.630delG 于 LRRC6
- p.Gln154Ter于RSPH4A
- c.383dupG 于 CCDC103
- p.His154Pro 于 CCDC103
- 西班牙波多黎各 c.921+3_6delAAGT于 RSPH4A
表型靶向检测 包括一些或全体32个已知与PCD有关联的基因 (见 Table 1A, Table 1B) 以及其他的感兴趣的基因也被考虑 (见 Differential Diagnosis) . 注意: 列入的基因和多基因面板的 敏感性 随实验室和时间的不同而不同.需要更多信息有关多
基因测试 (如果可用) 包括 外显子组测序, 基因组测序, 并且线粒体测序也要考虑如果系列单-基因 测试 (和/或 使用 表型靶向检测) 没能确切对有PCD特征的个体做出诊断. 了解更多信息有关综合 基因组的 测试 点 这.
Table 1A.
原发性纤毛不动症分子遗传学: 最常见的遗传原因
| 基因 1, 2 | 位置 | 所有此基因引起的PCD的比例 | |
|---|---|---|---|
| 观察(在无关系的受累个体中鉴定出双等位基因致病突变) 3, 4, 5 | 所有 PCD中的估计 6 | ||
| DNAH5 | CILD3 |
| 15%-29% |
| |||
| DNAH11 | CILD7 |
| 6%-9% |
| CCDC39 | CILD14 |
| 4%-9% 9 |
| |||
| DNAI1 | CILD1 |
| 2%-10% |
| CCDC40 | CILD15 |
| 3%-4% 9 |
| |||
| CCDC103 | CILD17 |
| <4% 10 |
| SPAG1 | CILD28 |
| <4% 10 |
| ZMYND10 | CILD22 |
| <2%-4% 10 |
| ARMC4 | CILD23 |
| <3% 10, 12 |
| CCDC151 | CILD30 |
| <3% 10 |
| DNAI2 | CILD9 |
| 2% 12 |
| RSPH1 | CILD24 |
| 2% 14 |
| CCDC114 | CILD20 |
| <2% 10, 12 |
| RSPH4A | CILD11 |
| 1%-2% 14 |
| |||
| DNAAF1 (LRRC50) | CILD13 |
| 1%-2% 15 |
| |||
| DNAAF2 (KTU) | CILD10 |
| <1%-2% 15 |
| |||
| LRRC6 | CILD19 |
| 1% 15 |
任何一种上表包括的基因的致病突变型占PCD ≥1% .
1.
2.
3.
引用所有PCD患者不管他们潜在的纤毛缺陷种类除非特殊说明.
4.
大多数致病突变检测通过 序列分析. 序列分析检测突变可以是良性的, 可能是良性的, 属于 意义不确定, 可能致病, 或致病的. 致病突变可能包括小基因内 缺失/插入 和 错义, 无义, 和 剪接位点 突变; 代表性地, 外显子 或 全-基因缺失/重复 没被检测到. 想知道更多序列测试结果的解释, 点 这.
5.
以下基因报道有基因内缺失或重复: DNAH5, CCDC40, SPAG1, ZMYND10, ARMC4, 和 DNAAF1. 可使用的基因靶向 缺失/重复 分析 方法包括 : 定量 PCR, 长片段 PCR, 多重连接探针扩增技术 (MLPA), 和基因-靶向 微阵列芯片设计来检测单-外显子缺失或重复.
6.
一些估算是基于纤毛结构缺陷推断(见 Ciliary Ultrastructural Analysis).
7.
65中24个 (37%) 人有外动力蛋白臂缺陷 [Hornef et al 2006]
8.
58中13个 (22%)人有正常的纤毛微结构[Knowles et al 2012]
9.
轴丝解体和内动力蛋白臂缺陷站所有PCD中的~14% .
10.
11.
63中10个 (~<15%)人有内+外动力蛋白臂缺损 [Knowles et al 2013c]
12.
外动力蛋白臂缺损占所有PCD中的 38.5% .
13.
47中2个 (4%) 人有外动力蛋白臂缺损 [Loges et al 2008]
14.
中央复合物缺损占有所PCD中的7% .
15.
内+外 动力蛋白臂缺损占所有PCD中的~10.5%.
Table 1B.
原发性纤毛不动症分子遗传学: 不太常见的遗传原因
| 基因 1, 2, 3 | 位置 | 意见 |
|---|---|---|
| C21orf59 | CILD26 | 295 4 个人中4个(<1%) 患有PCD 5 的无关人群 有 双等位基因的 致病突变型 [Austin-Tse et al 2013]. |
| CCDC65 (DRC2) | CILD27 | 295 4个人中2个 (<1%) 患有 PCD 5 的无关人群 有 双等位基因的 致病突变型 [Austin-Tse et al 2013]. |
| CCNO | CILD29 | 54 4个患有纤毛发育不全或发育较少的无关人群中10(~19%)个有 双等位基因的 致病突变型 [Wallmeier et al 2014]. 这与所有PCD 6中 <1% 一致. |
| DNAAF3 | CILD2 | 3/3 阿拉伯或巴基斯坦家庭之前映射该区域有 纯合性 致病突变型; 没有致病突变在其他111个家庭中检测到 [Mitchison et al 2012]. |
| DNAH1 | 2个来自沙特阿拉伯的同胞 (他们的父母是 近亲婚配的) 是 纯合性 错义 突变 p.Lys1154Gln [Imtiaz et al 2015]. | |
| DNAH8 | 1 个体有PCD是 纯合性 致病性变异 [Watson et al 2014]. | |
| DNAL1 | CILD16 | 患有PCD的个体来自 2个无关的 近亲婚配的 巴多因祖先的家庭是 纯合性 错义 致病突变型 (p.Asn150Ser) [Mazor et al 2011]. |
| DRC1 (CCDC164) | CILD21 | 3 有微管连接蛋白缺陷 7 的家庭有 双等位基因的致病突变型 [Wirschell et al 2013]. |
| DNAAF4 (DYX1C1) | CILD25 | 106 4 个有内外动力蛋白臂缺陷的无关系人群中10(<9%)个有 双等位基因的致病突变型 [Tarkar et al 2013]. 这与所有PCD 8中 <1% 一致. |
| DNAAF5 (HEATR2) | CILD18 |
|
| HYDIN | CILD5 |
|
| MCIDAS | 60 4 个 有纤毛发育不良或发育较少的无关人群中有4个(<7%) 有 双等位基因的致病突变型 [Boon et al 2014]. 这与所有PCD 6中 <1% 一致. | |
| NME8 (TXNDC3) | CILD6 | 1/41 具有PCD的无关人群有致病 无义 突变 在 反式 构型有一个 内含子的 突变表现出 剪接改变 [Duriez et al 2007]. |
| RSPH3 | 48个 有中央复合物 9 径向轴条蛋白缺损的无关人群中有5个(10%)有致病突变型 [Jeanson et al 2015]. | |
| RSPH9 | CILD12 | 48个 有中央复合物缺损 9的无关人群中有4个有致病突变型 [Kott et al 2013]; 184 个无关人群中没有人有 PCD 5 [Ziętkiewicz et al 2012]. 这与所有PCD 9中 <1% 一致. |
列在此表中的任一基因致病突变型只在一部分家庭中报导过 (也就是说, PCD中<1%).
1.
基因以字母的顺序排列.
2.
见 Table A. Genes and Databases 可知 染色体位点 和蛋白.
3.
4.
5.
引用所有的PCD患者不管他们潜在的纤毛缺陷
6.
在所有PCD中纤毛发育不全或发育较少是罕见的病例.
7.
微管连接蛋白(出现在外周微管二联体之间) 是动力蛋白调控复合物的一部分对轴丝弯曲有很重要的作用.
8.
内外动力蛋白臂缺损占所有PCD中的~10.5% .
9.
中央复合体缺损占所有PCD中的 7% .
纤毛微结构分析
透射电子显微镜鉴定纤毛微结构缺陷需要呼吸上皮活性组织检查 , 通常获得通过刷或刮鼻甲内表面或者通过支气管镜检刷支气管表面 [MacCormick et al 2002, Chilvers et al 2003]. 大约30%的临床表型 有PCD和鼻一氧化氮浓度低的群体有正常的纤毛微结构(其中很多诊断确认都是通过鉴定 双等位基因的 致病突变在 Table 1A 或 Table 1B所列的基因之一上) [Knowles et al 2013a].动力蛋白臂缺损经常由特定的突变
基因. 见 Table 2 (pdf). 最普遍的原发性纤毛不动症微结构缺损定义如下 (Figure 1) [Knowles et al 2013a, Davis et al 2015]:
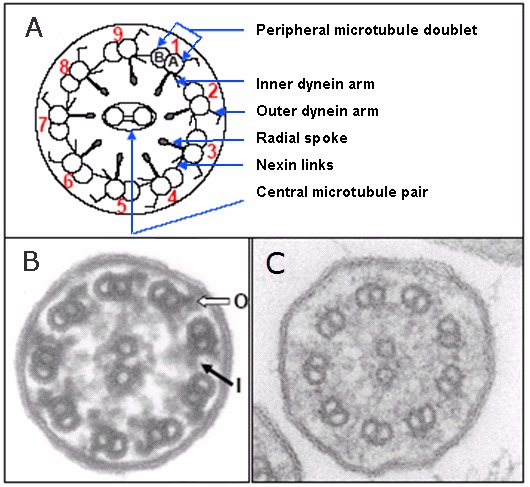
- 变短 和/或 缺失仅外周动力蛋白臂 (明确的微结构缺损中~55% , 或所有PCD中的 38.5% )
- 变短或缺失内外动力蛋白臂 (明确的显微结构缺损中~15% , 或所有PCD中的 10.5%)
- 微管 (轴丝) 解体与内动力蛋白臂和中央复合体缺失有关 (明确的微结构缺损中的5%-20%,或所有PCD中的 ~14% )
- 中央复合体缺失或破坏 (中央微管对和/或 径向轴条蛋白) (明确的微结构缺损中的~10% , 或所有PCD中的 7% )
- 变短和/或缺失仅内动力蛋白臂 (罕见)
- 纤毛稀少 (仅有很少的纤毛)有或没有正常的微结构 (罕见).
注意: (1) 评价纤毛微结构的专业知识是被需要的为了区别初级(基因)缺损和由于暴露在不同环境和传染性病原体产生的获得性缺损. (2) 经典的卡塔格内综合症可有内脏转位, 鼻窦炎, 和支气管扩张没有明显的微结构缺损被观察报道. 现在知道一些患有经典的卡塔格内综合症的家庭有 双等位基因的 致病突变于 DNAH11 (位点 CILD7).想了解更多信息有关于纤毛微结构病例与突变的
临床特征
临床描述
原发性纤毛不动症(PCD) 表现为: (1) 纤毛结构功能异常,基因缺陷,导致粘液和细菌滞留在呼吸道引起慢性的耳-鼻窦-肺疾病; 以及 (2)鞭毛的结构异常导致精子活动性能异常 .
肺病. 肺病的程度和严重度因人而异 [Marthin et al 2010]. 这种可变性一定程度上反应出 基因型 (见 基因型-表型相关性).超过75%足月的患有PCD的新生儿有呼吸窘迫需要辅助供氧几天至几周,然后,在这个年龄很少被诊断出 [Noone et al 2004, Ferkol & Leigh 2006, Mullowney et al 2014].慢性呼吸道感染在幼年早期出现
. 大多数儿童全年有慢性咳嗽; 慢性鼻窦炎和鼻塞 (经常伴有粘膜炎和明显的鼻引流)在生命的第一个月开始 经常在出生.痰中明显产出口咽的菌落 , 流感嗜血杆菌,肺炎双球菌, 和 金黄葡萄球菌 在幼年早期开始产生, 之后是绿脓杆菌(起初是光滑型后是粘液型)变得更加普遍. 尽管在儿童时期很罕见, 非结核分支杆菌感染发生在超过10%的成人上 [Noone et al 2004, Davis et al 2015].慢性呼吸道感染导致支气管扩张可能会出现在一些年幼儿童身上成人身上几乎都会发生
[Noone et al 2004, Brown et al 2008, Davis et al 2015].一部分有慢性呼吸道感染的成人在肺部有钙沉积
, 结果导致,咳出钙小石 (碎石) [Kennedy et al 2006].一些人在中年发展到肺病末期一部分人因此接受肺移植
.呼吸疾病攻击患有PCD的个体发生在幼年早期,得到合适的治疗肺病的恶化会减缓
.
鼻塞和鼻窦感染 在幼年早期发生并持续到成年 [Noone et al 2004, Leigh et al 2009].
慢性/反复性耳部感染, 在大部分患有PCD的幼童身上发生,到学龄期变得少发. 在很多婴儿和幼童身上, 慢性中耳炎会引起短暂的耳聋可能会影响说话能力. 如果不治疗, 中耳炎可能引起不可逆的耳聋 [Hadfield et al 1997, Majithia et al 2005].
不育. 几乎所有患有PCD的男性是不育的因其精子不具有正常的运动功能.一些患有的PCD的女性可以正常的生育
; 其他女性生育功能受损并且子宫外孕的风险增加因其输卵管处纤毛功能受损 [Afzelius 2004].
器官位置异常
- 全内脏反位 (所有的内脏器官镜面反转并没有明显的生理影响)在接近40%-50% 的患有PCD的个体中观察到.
- 内脏异位 (也叫做 "心房不定位") 在患有PCD个体中的大约12%表现出来 [Kennedy et al 2006, Shapiro et al 2014a]. 内脏异位,正常情况下不对称器官左右不协调, 不同于全内脏反位经常在临床上被诊断为无脾 (主要是双侧均系右侧结构,或右侧异构 ) 或多脾 (主要是双侧均系左侧结构,或左侧异构).对那些有内脏异位的患者, 先天的 心血管畸形是很常见很复杂的, 并且经常会致死. 特定的与内脏异位有关的心血管缺陷包括心房异构, 大血管转位, 右室双出口, 异常静脉回流, 下腔静脉缺如, 和两侧都有上腔静脉 [Zhu et al 2006].肺异构, 经常是无症状的, 可以是右侧异构(双侧三叶肺并有双侧动脉支气管) 或左侧异构 (两侧肺及其肺叶肺门都是正常的左肺的特征).胃可能居右; 肝可能居中, 或者左右叶可能颠倒.肠袢不正常旋转可能导致肠梗阻或肠扭结 (血管阻塞).中枢神经系统,骨骼 ,和生殖泌尿系统畸形也可能会发生 .
其他. 患有PCD的个体在罕见的情况下可能会有脑积水,反映出室管膜纤毛功能异常 [Wessels et al 2003, Kosaki et al 2004].
基因型-表型相关性
基因型-表型 相关性对大多数致病突变是不可获得的. 见 Table 2 (pdf) 可获得更多信息有关于突变 基因 和观察到的纤毛缺陷的相关性. 例如:
- 儿童具有 双等位基因的 致病突变在 CCDC39 或 CCDC40 上(与微管解体和内动力蛋白臂缺陷相关) 其肺功能更差 胸部CT可见更多支气管扩张, 重量更轻比起那些双等位基因致病突变在那些仅和外动力蛋白臂缺陷相关的基因或内外动力蛋白臂缺陷都相关的基因 [Davis et al 2015].
- 个体患有PCD有 双等位基因的 致病突变在 CCDC65, DRC1, HYDIN, RSPH1, RSPH4A, 和 RSPH9 上其微结构和正常的相比不够分立/清晰不能分辨.
- 个体有致病突变在编码中央复合体或径向轴条蛋白成分的基因上(比如, RSPH1, RSPH3, RSPH4A, RSPH9, 和 HYDIN)没有内脏位置异常.
命名法
现在称为原发性纤毛不动症(PCD)过去用来描述此病症的名字包括纤毛运动障碍综合症和阿西利亚综合症.
和全内脏反位相关的PCD称为卡塔格内综合症.
流行性
PCD在挪威和日本的群体中发生概率大概为1:16,000 , 此数值推测源于射线照相术的调查和具有支气管扩张临床证据的右位心 [Torgersen 1950, Katsuhara et al 1972]. 基于这些数据, 在美国患有PCD个体的总数约为12,000 到 17,000.在有很多
近亲婚配的地方病例在人群中发生的概率更高.比如:
- PCD 发生的概率是 1:400 在定居在北荷兰的渔村的福伦丹人中 [Onoufriadis et al 2013].
- 使用纤毛拍频测试, 盛行的PCD 发生概率在南亚人群中为1:2265 (主要祖先是巴基斯坦) 在英国 [O’Callaghan et al 2010].
- 在美国的阿米什人和门诺派教徒中PCD非常盛行 [Ferkol et al 2013].
相关基因(等位基因) 疾病
至今, PCD只有 与 32个在此GeneReview讨论的基因中的31个基因上的双等位基因的致病突变相关的 表型 . 双等位基因致病突变有一个例外, DNAH1, 也与非综合症性男性不育相关 [Ben Khelifa et al 2014].
鉴别诊断
慢性肺病和支气管扩张. 在评估原发性纤毛不动症(PCD)为了排除以下的失调症状合适的调查是需要的:
- 免疫缺陷,例如免疫球蛋白 G (IgG) 子类缺乏
- 过敏
- 胃食管返流疾病
- 韦格氏肉芽肿 (上下呼吸道疾病)
器官位置异常. 没能建立正常的左右不对称结构引起大范围的先天的 畸形包括全内脏反位和内脏异位综合症(多脾和无脾) 可能恰巧与心脏缺损相关. 超过80个基因, 包括PCD相关基因, 是发育内脏不对称结构所需的基因.大约 25% 的有全内脏反位的个体有 PCD [Zhu et al 2006]. 内脏异位患者中PCD患者的比例未知但是至少12%的PCD患者有内脏异位[Kennedy et al 2006, Shapiro et al 2014a].偶尔
, 致病突变在 RPGR (与 X-连锁 色素性视网膜炎相关)在男性 色素性视网膜炎 患者中被鉴定与PCD共分离 [Bukowy-Bieryłło et al 2013]; 也可见 Moore et al [2006] 并引用在其中.一个
致病性变异 于 OFD1 (与 1型 口-面-指 综合症 相关)也在患有X-联锁 智力障碍的家庭中被发现与PCD共分离 [Budny et al 2006].
处治
初步诊断后的评估
为了确定疾病程度评价和确立患有原发性纤毛不动症(PCD)的个体的需要 (PCD), 建议使用以下评估:
- 肺病
- 呼吸道状况(主要是痰的状况)来确定被感染的组织并指导抗菌治疗. 对年长的儿童和成人针对非结核分支杆菌的特定环境也包括在其中.
- 胸部x线照片 和/或胸部CT可用来确定呼吸道疾病和支气管扩张的分布位置和严重程度
- 肺部功能测试 (肺量测定法)来确定阻塞性损伤的严重程度
- 脉搏血氧仪,如果达到边缘线也需要隔夜饱和度研究
- 鼻塞和/或鼻窦炎. 鼻窦成像 (鼻窦x光或最好是鼻窦CT)
- 慢性/反复性耳部感染. 正式的听力评估 (见 Deafness and Hereditary Hearing Loss Overview 可知不同年龄所用的听力评估.)
- 其他. 临床的基因咨询
治疗症状
现在, 没有专门的治疗方案可以治疗纤毛功能不良. 此部分所述的治疗方法是根据经验得来的目的是治疗纤毛和精子鞭毛功能不良的后果. 几乎没有使用PCD专门的治疗方案的材料支持.
肺病. 处治PCD患者应包括积极的措施以提高粘液的清洁度, 预防呼吸道感染, 和治疗细菌感染.增加粘液清洁度的方法和治疗
囊胞性纤维症的方法相似, 包括胸背叩击和体位引流, 震荡背心, 和呼吸动作以帮助呼吸道远端的清洁. 因为咳嗽是有效的清洁方法, 病人应该被鼓励咳嗽并用其他帮助深呼吸和咳嗽的活动 (比如., 剧烈运动).常规免疫接种可保护免受呼吸道病原体的感染
:
- 百日咳
- b型流感嗜血杆菌
- 肺炎球菌疫苗
- 每年流感病毒疫苗
合适的抗生素疗法治疗呼吸道细菌感染 (支气管炎, 鼻窦炎, 和中耳炎) 是必要的以防止不可逆的损伤. 痰环境可以用来指导选择合适的抗菌疗法. 那些抗生素疗法一疗程完成后症状在几天或几周内反复的患者, 扩大抗生素的使用范围或者甚至要考虑预防性抗生素的范围. (考虑使用慢性抗生素疗法需要评估会产生多药耐药细菌的风险.)对患有局部支气管扩张的个体
, 可使用肺叶切除以减少其余留下来的肺叶的感染.这种方法,然而 ,是有争议的;决策过程也应该包括咨询PCD方面的专家.肺移植使用在肺病末期的患者上
.
鼻塞和鼻窦炎. 在一些有大范围鼻窦炎的患者身上, 鼻窦手术可以帮助改善减缓症状.
慢性/反复性耳部感染. 抗生素疗法无效果的中耳炎, PE管放置法可能会有帮助PE; 然而, 一些PCD患者使用PE管放置法后会有进攻性的耳液溢 [Hadfield et al 1997].语言障碍矫正和助听器对耳聋和语言迟缓的儿童来说可能是需要的
.
男性不育. 一对夫妇中男性有PCD引发的不育可以选择使用ICSI(卵母细胞胞浆内单精子注射)体外授精. 在这个过程中, 精子从精液中获取得到(在少精的男性中)或者从睾丸活检中提取(在阻梗性无精症男性患者中)精子通过体外授精注射到提取的卵子中 [Sha et al 2014].其他的方法是人工授精通过捐献精子
(AIDS).
器官位置异常. 典型地, 器官位置异常不需要介入除非有生理功能不良 (比如, 先天的 心脏病)需要手术介入 .
预防次级并发症
合适的预防方法:
- 日常免疫接种(包括流感疫苗和肺炎双球菌疫苗)防止呼吸道感染
- 有关感染控制的教育包括注意洗手,避免和病人接触,正确的清洁/消毒呼吸设备, 和早使用抗生素治疗呼吸疾病(由之前呼吸道状况决定)
监测
接下来需要由肺专家监测肺功能和痰中的病原体并评估肺病的程度/级数.对年幼的患有中耳炎的儿童
, 日常的听力评估是必要的, 并且应该持续到青少年期, 到青少年期听力通常是正常的 [Majithia et al 2005]. 通常, 耳部疾病在童年后期会改善听力筛查不是必须的.
需要避免的药剂/环境
不应该使用止咳药因为咳嗽对分泌物清除很重要.暴露在呼吸道病原体
, 烟草烟雾, 和其他污染物和刺激物可能会损坏呼吸道黏膜并且应该避免刺激粘液分泌.
对有风险亲属评估
应该对 先证者 年长和年幼的同胞评估为了尽早确认那些可以从开始实施治疗和预防措施受益的人.
- 如果家庭中的致病突变不是已知的,严格的临床病例史和身体检查并需要胸片和 鼻一氧化氮测量 来判断有风险同胞的疾病状态.
见 Genetic Counseling 可知有风险同胞的 遗传咨询 信息.
怀孕管理
对患有PCD的女性, 任何肺部感染和肺功能状况应该被PCD(或囊胞性纤维症)专家严格评估来决定孩子所承受的风险.
调查中的治疗方法
查 ClinicalTrials.gov 获取一系列疾病和病症的临床研究相关信息. 注意:不一定有此病的临床试验.
遗传咨询
遗传咨询是提供个体或家庭基因病性质,遗传,和影响信息的过程以帮助他们做出有医学指导的个人的决定. 接下来的部分探讨基因风险评估和使用的家庭史和基因测试确认家庭成员的基因状态. 这个部分无意解决任何人可能遇到的任何个人的,文化的,伦理的问题或者代替基因专家作出指导. —ED.
遗传方式
原发性纤毛不动症 (PCD) 以 常染色体隐性遗传 方式遗传.注意
:尽管其他的遗传方式在少数报道中也被提出 [Narayan et al 1994, Badano et al 2006, Moore et al 2006, Bukowy-Bieryłło et al 2013], 常染色体显性遗传 没有被任何随后的出版物报道或者在作者所实验的超过500个参与Genetic Disorders of Mucociliary Clearance Consortium (GDMCC)的病人中被观察到.
家庭成员的风险 — 常染色体隐性遗传
先证者的父母
- 杂合子是无症状的没有患有PCD的风险.
先证者的同胞
- 杂合子是无症状的没有患有PCD的风险.
相关的遗传咨询问题
见 处治, 有风险亲属评估 可获得有风险亲属的评估信息出于早期诊断治疗目的.
计划生育
DNA银行 是一个储存 DNA (主要是从白细胞中提取的) 以便将来使用. 因为测试方法和我们对基因的理解, 等位基因突变, 和疾病在将来很可能会增多, 应该考虑建设受累的 人群的DNA银行.
产前诊断和植入前遗传
一旦 受累的 家庭成员的PCD-关联致病突变被确认, 高患病风险的妊娠的产前诊断和PCD 植入前遗传诊断 是可行的.医学专家和家庭内使用产前诊断的观点可能不同, 尤其是如果测试是出于终止妊娠的目的而不是早期诊断的目的. 尽管大多数中心认为决定是否做产前诊断是父母选择, 这些事情的考虑还是合适的.
资源
GeneReviews 员工挑选出以下特定疾病和/或一般支持组织 和/或注册机构为了患有疾病的个体或他们的家庭的利益. GeneReviews 不对其他组织所提供的材料负责. 想了解有关挑选的标准的信息, 点 这.
- Genetic Disorders of Mucociliary Clearance Consortium (GDMCC)Cystic Fibrosis / Pulmonary Research & Treatment CenterMarsico Lung Institute7215 Marsico HallCB #7248Chapel Hill NC 27599-7248Fax: 919-966-7524; 919-843-5309Email: godwine@med.unc.edu; kelli_sullivan@med.unc.edu
- Kartagener's Syndrome and Primary Ciliary Dyskinesia FoundationL盲rchenweg 14Ettlingen 76275GermanyPhone: 07243 39338Email: info@kartagener-syndrome.de; rerafra@web.de
- PCD (Primary Ciliary Dyskinesia) Foundation10137 Portland Avenue SouthMinneapolis MN 55420Phone: 952-303-3155; 612-386-1261Fax: 952-303-3178Email: info@pcdfoundation.org
- Primary Ciliary Dyskinesia Family Support Group15 Shuttleworth GroveWavendon Gate Milton Keynes MK7 7RXUnited KingdomPhone: 01908 281635Email: chair@pcdsupport.org.uk
- American Lung Association1301 Pennsylvania Avenue NorthwestWashington DC 20004Phone: 800-548-8252 (Toll-free HelpLine); 800-586-4872 (toll-free); 202-785-3355Fax: 202-452-1805Email: info@lungusa.org
- Ciliopathy AllianceUnited KingdomPhone: 44 20 7387 0543
分子遗传学
分子遗传学和OMIM表格中的信息可能和 GeneReview别处的信息有区别: 表格可能含有更多最新的信息. -ED.
Table A.
原发性纤毛不动症: 基因和数据库
Table B.
OMIM 关于原发性纤毛不动症的条目 (View All in OMIM)
| 244400 | 纤毛不动症, 原发, 1; CILD1 |
| 603332 | 动力蛋白, 轴丝, 重链 1; DNAH1 |
| 603335 | 动力蛋白, 轴丝, 重链 5; DNAH5 |
| 603337 | 动力蛋白, 轴丝, 重链 8; DNAH8 |
| 603339 | 动力蛋白, 轴丝, 重链 11; DNAH11 |
| 603395 | 精子相关抗原 1; SPAG1 |
| 604366 | 动力蛋白, 轴丝, 中间链 1; DNAI1 |
| 605483 | 动力蛋白, 轴丝, 中间链 2; DNAI2 |
| 606763 | 纤毛不动症, 原发性, 2; CILD2 |
| 607070 | 含锌指MYND域蛋白 10; ZMYND10 |
| 607421 | 含硫氧还蛋白域蛋白 3; TXNDC3 |
| 607752 | 细胞周期蛋白 O; CCNO |
| 608644 | 纤毛不动症, 原发性, 3; CILD3 |
| 608646 | 纤毛不动症, 原发性, 4; CILD4 |
| 608647 | 纤毛不动症, 原发性, 5; CILD5 |
| 608706 | 动力蛋白, 轴丝, 组装因子 4; DNAAF4 |
| 609314 | 径向轴条头蛋白 1, 衣藻, 同系物; RSPH1 |
| 610062 | 动力蛋白, 轴丝, 轻链 1; DNAL1 |
| 610812 | 脑积水诱导, 小鼠, 同系物; HYDIN |
| 610852 | 纤毛不动症, 原发, 6; CILD6 |
| 611088 | 含卷曲螺旋域蛋白 65; CCDC65 |
| 611884 | 纤毛不动症, 原发, 7; CILD7 |
| 612274 | 纤毛不动症, 原发, 8; CILD8 |
| 612444 | 纤毛不动症, 原发, 9; CILD9 |
| 612517 | 动力蛋白, 轴丝, 组装因子 2; DNAAF2 |
| 612518 | 纤毛不动症, 原发, 10; CILD10 |
| 612647 | 径向轴条头蛋白 4, 衣藻, 同系物, A; RSPH4A |
| 612648 | 径向轴条头蛋白 9, 衣藻, 同系物; RSPH9 |
| 612649 | 纤毛不动症, 原发性, 11; CILD11 |
| 612650 | 纤毛不动症, 原发性, 12; CILD12 |
| 613190 | 动力蛋白, 轴丝, 组装因子 1; DNAAF1 |
| 613193 | 纤毛不动症, 原发性, 13; CILD13 |
| 613798 | 含卷曲螺旋域蛋白 39; CCDC39 |
| 613799 | 含卷曲螺旋域蛋白 40; CCDC40 |
| 613807 | 纤毛不动症, 原发性, 14; CILD14 |
| 613808 | 纤毛不动症, 原发性, 15; CILD15 |
| 614017 | 纤毛不动症, 原发性, 16; CILD16 |
| 614086 | 多纤毛分化和DNA合成相关细胞周期蛋白; MCIDAS |
| 614566 | 动力蛋白, 轴丝, 组装因子 3; DNAAF3 |
| 614677 | 含卷曲螺旋域蛋白 103; CCDC103 |
| 614679 | 纤毛不动症, 原发性, 17; CILD17 |
| 614864 | 动力蛋白, 轴丝, 组装因子 5; DNAAF5 |
| 614874 | 纤毛不动症, 原发性, 18; CILD18 |
| 614930 | 富含亮氨酸重复蛋白 6; LRRC6 |
| 614935 | 纤毛不动症, 原发性, 19; CILD19 |
| 615038 | 含卷曲螺旋域蛋白 114: CCDC114 |
| 615067 | 纤毛不动症, 原发性, 20; CILD20 |
| 615288 | 动力蛋白调控复合体, 亚基 1, 衣藻, 同系物; DRC1 |
| 615294 | 纤毛不动症, 原发性, 21; CILD21 |
| 615408 | ARMADILLO REPEAT-CONTAINING PROTEIN 4; ARMC4 |
| 615444 | 纤毛不动症, 原发性, 22; CILD22 |
| 615451 | 纤毛不动症, 原发性, 23; CILD23 |
| 615481 | 纤毛不动症, 原发性, 24; CILD24 |
| 615482 | 纤毛不动症, 原发性, 25; CILD25 |
| 615494 | 染色体 21 开放阅读框 59; C21ORF59 |
| 615500 | 纤毛不动症, 原发性, 26; CILD26 |
| 615504 | 纤毛不动症, 原发性, 27; CILD27 |
| 615505 | 纤毛不动症, 原发性, 28; CILD28 |
| 615872 | 纤毛不动症, 原发性, 29; CILD29 |
| 615876 | 径向轴条头蛋白 3, 衣藻, 同系物; RSPH3 |
| 615956 | 含卷曲螺旋域蛋白 151; CCDC151 |
| 616037 | 纤毛不动症, 原发性, 30; CILD30 |
分子遗传病理
纤毛, 细胞器几乎在所有细胞中存在, 从一个中心粒修饰形成的毛基体中放射出来. 不同种类的纤毛包括: 运动纤毛, 初级静纤毛, 和初级动纤毛 (比如说, 节点纤毛). 纤毛是复杂的, 高度保守的结构. 动纤毛大约由250个蛋白组成; 每个纤毛是 ‘9+2’ 结构有9个外周微管双联体包围中央微管对, 中央微管对也叫做轴丝 (Figure 1). 内外动力蛋白臂在外周微管上在透射电子显微镜纤毛横截面图像上能看到.内外动力蛋白臂组成轴丝中的微管双联体的连接桥梁并是纤毛震动的动力驱动蛋白
[Afzelius et al 2001, Afzelius 2004, Zariwala et al 2007]. 外动力蛋白臂由几条重链,中间链,和轻链组成 [Satir 1999]. 内动力蛋白臂非常复杂随着轴丝整个长度的不同而不同 [Perrone et al 2000, DiBella & King 2001].动纤毛上的缺陷和原发性纤毛不动症
(PCD)/卡塔格内综合症有关, 并且在 “初级” (感觉)纤毛上的缺陷和几个人类疾病相关包括 Bardet-Biedl 综合症 (纤毛的基体), 常染色体显性多囊肾病, 常染色体隐性多囊肾病 (肾单纤毛缺陷), 肾结核, Joubert 综合症, 色素性视网膜炎 (光感受器连接纤毛), 脑积水, 和 Alstrom 综合症 [Afzelius 2004, Badano et al 2006, Zariwala et al 2007, Leigh et al 2009, Tobin & Beales 2009]. 那些疾病大多数在基因上是杂合子, 还有很多基因需要确定 [Badano et al 2006, Tobin & Beales 2009].
PCD通过呼吸道纤毛以及精子的鞭毛结构,功能,和生物基因异常来判断. 动力蛋白臂缺少 (或较短) 发生在大约 55% 的有明确的微结构缺损的人中; 大约 10% 的人群有中央复合体或径向轴条或微管连接蛋白缺陷 (在外周微管二联体之间并是动力蛋白调控复合体的一部分对轴丝弯曲很重要). 一些PCD患者 (~30%) 没有明显的微结构缺陷 [Knowles et al 2013a, Davis et al 2015].50%的PCD患者有全内脏反位说明纤毛对胚胎形成左右不对称结构的诱导有重要作用
.点
这可获得生物模型的更多信息.到现在为止
, 32个引起 常染色体隐性遗传 PCD的基因上的致病突变已经被确认 (见 Table A). 很多基因几乎没有致病突变被报道. 通过透射电子显微镜分析受累的 个体的纤毛特征 (见 Table 2), 免疫荧光分析, 和/或 高速视频显微镜分析可以帮助更进一步解释等位基因突变的临床重要性或者分析 意义不确定的突变.注意
: 通常的突变基因的信息 (也就是说, 那些 >1% PCD的基因突变; 见 Table 1A) 在下方列出来, 按 位点的顺序.点
这 (pdf) 可获得少发的突变基因的信息 (也就是说, 那些 <1% PCD的基因突变; 见 Table 1B).
获得基因 总结的信息和以下基因的蛋白的信息, 见 Table A, 基因.
DNAI1 / CILD1
致病突变. 见 Table 3. 基于大量研究发现, 15% 的致病突变被鉴定为错义; 其余的是插入, 缺失, 和 无义 以及 剪接位点 突变 [Zariwala et al 2006]. 大约三分之一的致病突变发生在外显子13, 16, 和 17上, 保守的WD (Trp-Asp 氨基酸)基因重复区域.出现在两个或更多无关家庭中的致病基因包括致病突变在外显子
13,16, 17上和内含子 c.48+2_3insT (也叫做 IVS1+2_3insT) 上. c.48+2_3insT 致病性变异 占等位基因突变的大约55%; mRNA研究表明它废除cDNA的剪切供体位点,导致内含子1序列多了一个,预测会使得翻译提前终止 [Pennarun et al 1999]. 微卫星分析说明c.48+2_3insT起源单一 [Zariwala et al 2006]. 这种突变在阿米什人群中被观察到[Ferkol et al 2013].
剪接位点致病性变异c.1490G>A 在外显子16起始部引起外显子15和16 非移码缺失.
致病性变异c.2001+1G>A (也叫做IVS19+1G>A)引起cDNA外显子19非移码缺失 [Zariwala et al 2006].
Table 3.
这篇GeneReview讨论的DNAI1 突变
| DNA核苷酸改变 (别名) 1 | 预测的蛋白改变 | 参考序列 |
|---|---|---|
| c.48+2dupT (c.48+2_3insT) (IVS1+2_3insT) (219+3insT) | p.Ser17ValfsTer12 | NM_012144-.2 NP_036276-.1 |
| c.1490G>A r.1402_1569del | p.Arg468_Lys523del | |
| c.2001+1G>A (IVS19+1G>A) r.1819_2001del | p.Ala607_Lys667del |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
1.
不遵从现在的命名标准的突变命名
正常 基因产物.DNAI1 编码699-氨基酸蛋白纤毛动力蛋白轴丝中间链 1, 属于马达蛋白大家庭 [Pennarun et al 1999]. 它包括五个保守的WD重复区域(富含色氨酸-天冬氨酸) 在基因的碳端. 在人类和小鼠中, DNAI1 只在含有动纤毛或鞭毛的组织中表达(包括小鼠胚胎节点在E7.5 dpc).
DNAH5 / CILD3
致病突变. 见 Table 4. 所有42个致病性变异等位基因中,大约47% 集中在五个外显子中(34,50,63,76,和77) [Hornef et al 2006]. 百分之十五的致病突变是错义其余的是无义 和 剪接位点 突变, 插入,和缺失[Hornef et al 2006].
一个致病性变异 (c.10815delT) 在外显子 63上在欧洲人中出现通过单倍体分析发现可能是共同起源[Hornef et al 2006].突变
p.Gln1450Ter在来自阿米什和门诺派教徒的PCD患者中被发现[Ferkol et al 2013].两个患有PCD和猫叫综合症的个体有
DNAH5缺失由于染色体5p 缺失并且一个 DNAH5致病性变异在残留的等位基因上 [Shapiro et al 2014b].在一个个体上
,致病 剪接位点 突变p.Arg577Thr 引起外显子 13跳过缺失预测会引起翻译终止信号提前终止[Hornef et al 2006].
Table 4.
DNAH5突变在这篇GeneReview讨论
| DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
| c.10815delT | p.Pro3606HisfsTer23 | NM_001369-.2 NP_001360-.1 |
| c.4348C>T | p.Gln1450Ter | |
| c.1730G>C | p.Arg577Thr |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews 工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.DNAH5 编码纤毛动力蛋白轴丝重链 5, 一个4624氨基酸蛋白. N端结构域是外动力蛋白臂复合体干结构域并与其他重链,中间链,和轻链有作用. C端结构域形成球状头包含6个保守的6p-环结构域和一个保守的微管结构位点(MTB).第一个p环结构域可结合并水解ATP[Olbrich et al 2002].
异常 基因产物. 致病突变在DNAH5 引起有缺陷的外动力蛋白臂.
DNAH11 (CILD7)
致病突变. 见 Table 5. 描述了移码, 剪接位点, 和 错义 致病突变 [Knowles et al 2012].一个个体有父亲遗传的单亲同二体
染色体 7 既有PCD又有全内脏反转 (也就是说, 卡塔格内综合症) 同时有囊胞性纤维症是二倍的纯合性 p.Arg2852Ter致病性变异 在DNAH11上,和在 CFTR 上p.Phe508del 致病突变[Bartoloni et al 2002].
在一个德国起源的有PCD并且纤毛动力蛋白臂正常患者的家庭中, 5个 受累的 个体内脏正位一个内脏全反转. 所有六个患者有 复合杂合DNAH11 致病突变 (p.Tyr4128Ter 和 p.Ala4518_Ala4523delinsGln) [Schwabe et al 2008].
Table 5.
此篇 GeneReview讨论的DNAH11突变
DNA核甘酸改变 | 预测的蛋白改变 | 参考序列 |
|---|---|---|
c.12384C>G | p.Tyr4128Ter | |
c.13552_13608del57 | p.Ala4518_Ala4523delinsGln | |
c.8554C>T | p.Arg2852Ter |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物. 纤毛动力蛋白轴丝重链 11 是一个 4,516-氨基酸蛋白行使有关纤毛运动依靠微管马达ATP酶的功能. C端构成球状头包含6个保守的p-环结构域和一个保守的微管结构位点[Bartoloni et al 2002].
在老鼠中, 直系同源 基因 (lrd 或 Dnah11)参与左右轴的决定.
异常 基因产物. 纤毛动力蛋白轴丝重链11 位于外动力蛋白臂; 然而, DNAH11 致病突变引起的PCD个体的呼吸性纤毛动力蛋白臂结构是正常的 [Schwabe et al 2008].
DNAI2 (CILD9)
致病突变. 见 Table 6. 纯合子 剪接位点 和 无义DNAI2 致病突变在患有PCD的无关家庭中被发现 [Loges et al 2008, Knowles et al 2013b, Kim et al 2014].在大的近亲婚配的伊朗犹太家族所有的
受累的 个体是 纯合性 剪接位点致病性变异c.1494+1G>C (IVS11+1G>C) 引起 非移码 跳过 外显子 11 预测会失去49氨基酸残基. [Loges et al 2008]. 另一个剪切位点致病突变, c.346-3T>G, 导致外显子4跳过引起外显子3,5融合跳过预测形成异常蛋白 (p.Ile116GlyfsTer54).
致病性变异 p.Trp453Ter 犹太人 群体中被鉴定 [Knowles et al 2013b].
Table 6.
挑选的 DNAI2 突变
DNA 核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.346-3T>G | p.Ile116GlyfsTer54 | |
c.787C>T | p.Arg263Ter | |
c.1494+1G>C | p.Val450_Ser498del |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
1.
不遵从现在的命名标准的突变命名
正常 基因产物. 纤毛动力蛋白轴丝中间链 2 (DNAI2)是一个605氨基酸蛋白和DNAI1平行同源属于马达蛋白大家庭. 它含有5个保守的WD (Trp-Asp)重复域在基因的C端.
异常基因产物.DNAI2 上的致病突变引起外动力蛋白臂缺陷 [Loges et al 2008].
DNAAF2 (C14orf104, KTU, PF13) (CILD10)
基因结构.DNAAF2 (以前叫做 C14orf104, KTU, 或 PF13)有3个外显子 .
致病突变. 见 Table 7. 三个 纯合性 截断致病突变在PCD患者中被发现: p.Ser8Ter, c.31delG, 和 c.1199_1216dup16 (p.Gly406ArgfsTer90) [Omran et al 2008, Kim et al 2014].
Table 7.
此篇GeneReview讨论DNAAF2 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.23C>A | p.Ser8Ter | |
c.31delG | p.Glu11ArgfsTer5 | |
c.1199_1214dup16 | p.Gly406ArgfsTer90 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物. Kintoun (之前叫做ktu)是一个837氨基酸胞浆蛋白被认为与动力蛋白臂复合物在细胞质内在转移到纤毛部位之前提前组装有关[Omran et al 2008]. 两个作为选择的剪切异型体 被发现, 一个是全长另一个缺少非移码外显子 2.
异常 基因产物.DNAAF2 上的致病突变导致内外动力蛋白臂的缺失 [Omran et al 2008].
RSPH4A (CILD11)
致病突变. 见 Table 8. 大约20个 RSPH4A 致病突变, 大部分观察到的是 无义 和移码的变体. 一个普遍的 致病性变异 (p.Gln154Ter) 在来自美国祖先是巴基斯坦的 受累的 群体中被发现 [Castleman et al 2009]. 大量研究发现 双等位基因的 致病突变出现在184 个东欧血统家庭中的四个(~2%) [Ziętkiewicz et al 2012]. 一个截断 (c.166dupC)突变在伊朗流浪者祖先的一对同胞中发现 [Casey et al 2015].另外
,一个普遍的剪接位点突变(c.921+3_6delAAGT) 在来自波多黎各西班牙祖先的群体中被发现[Daniels et al 2013]. 剪切位点突变记录分析发现失帧缺失 外显子 2 导致终止信号提前终止翻译(p.Tyr230GlnfsTer8).
Table 8.
此篇GeneReview讨论RSPH4A 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.166dupC | p.Arg56ProfsTer11 | |
c.460C>T | p.Gln154Ter | |
c.921+3_+6delAAGT | p.Tyr230GlnfsTer8 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.RSPH4A 编码径向轴条头蛋白4同系物A, 一个有716个氨基酸的蛋白,调控动力蛋白诱导的移动并控制轴丝波浪样运动 [Castleman et al 2009].
异常 基因产物.RSPH4A 上的致病突变导致纤毛有9+0 或 8+1 微管结构的移动缺陷 [Castleman et al 2009].
DNAAF1 (CILD13)
致病突变. 见 Table 9. DNAAFI 失去功能致病突变引起疾病 [Loges et al 2009]. Duquesnoy et al [2009] 发现了7个 致病性变异 等位基因在4个 受累的 个体身上包括5个 无义 或移码突变; 一个 缺失外显子2和3 (p.Glu42_Lys117del)和一个 错义 致病突变.
错义致病性变异, p.Leu175Arg, 表现 纯合性 的状态, 位于LRR 结构域 可能和蛋白-蛋白相互作用相关. 使用 Chlamymodonas 进行功能分析说明p.Leu175Arg 是一个 可能的致病性变异 [Duquesnoy et al 2009].
Loges et al [2009] 观察到5个致病DNAAF1 等位基因在6个群体中: 一个是纯合性 移码 致病性变异, c.1349dupC (p.Pro451AlafsTer5); 一个是 杂合的nonsense, p.Arg271Ter, 致病突变; 和三个大的多基因缺失包括 DNAAF1.
Table 9.
挑选的DNAAF1 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.524T>G | p.Leu175Arg | |
c.811C>T | p.Arg271Ter | |
c.1349dupC | p.Pro451AlafsTer5 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物. DNAAF1 是一个725氨基酸蛋白编码富含亮氨酸重复序列(LRR)蛋白超家族中的一部分并对在胞浆内提前组装和/或动力蛋白臂复合物的匹配起到重要作用 [Loges et al 2009].
异常 基因产物.DNAAF1 上的致病突变导致纤毛微结构分析上发现内外动力蛋白臂都有缺陷. 免疫荧光分析发现蛋白 DNAH5, DNAH9, DNAI2 (外动力蛋白臂), 和 DNALI1 (内动力蛋白臂)缺失 [Loges et al 2009].
CCDC39 (CILD14)
致病突变. 见 Table 10. 无义, 移码, 剪接位点, 和 错义 致病突变被观察到. 三个致病突变 (p.Thr358GlnfsTer3, p.Ser786IlefsTer33, 和 c.357+1G>C)每个都有个共享的单体型, 暗示每个都有共同的来源. 一个 致病性变异 (p.Glu731AsnfsTer31) 在三个家庭中有共享的单体型, 但是在一个家庭中有一个独特的单体型暗示可能有反复性的突变[Merveille et al 2011].
Table 10.
此篇GeneReview讨论的CCDC39 突变
DNA核苷酸改变 | 预测的蛋白改变 | 参考序列 |
|---|---|---|
c.357+1G>C | -- | |
c.1072delA | p.Thr358GlnfsTer3 | |
c.2190delA | p.Glu731AsnfsTer31 | |
c.2357_2359delinsT | p.Ser786IlefsTer33 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.CCDC39 编码941个氨基酸轴丝蛋白CCDC39包含弯曲螺旋结构域. 免疫荧光分析来自健康个体的呼吸上皮细胞在顶端细胞质中沿着轴丝全长定位CCDC39.
异常 基因产物.CCDC39 上的致病突变引起内动力蛋白臂缺陷和轴丝解体 (包括异常的径向轴条蛋白和微管连接蛋白, 减少内动力蛋白臂的数量, 和外周二联体的取代) (见 Table 2) [Merveille et al 2011].
CCDC40 (CILD15)
致病突变. 见 Table 11. 在 CCDC40 上的致病突变在24个有轴丝解体的无关家庭中的14个 被发现. 这14个家庭, 13个有 双等位基因的 致病突变. 有 14不同的等位基因, 包括插入/删除, 无义 和 剪接位点 致病突变, 和大的缺失. 一个 致病性变异 (p.Ala83ValfsTer82) 在7个无关的人群中至少一个等位基因 上表达在 ; 其他的致病突变 (p.Arg942MetinsTrp) 在两个无关人群中被观察 [Becker-Heck et al 2011].
Table 11.
此篇GeneReview讨论CCDC40 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.248delC | p.Ala83ValfsTer82 | |
c.2824_2825insTGT | p.Arg942MetinsTrp |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.CCDC40 编码1142个氨基酸的包含弯曲螺旋结构域的轴丝蛋白. 在小鼠中, 蛋白定位到来自多纤毛气管细胞的 9+2 呼吸轴丝结构上并且9+0 节点纤毛不是很容易被探测到.
异常 基因产物. 与 CCDC39相似, CCDC40 上的致病突变引起内动力蛋白臂缺陷和轴丝解体(包括异常的径向轴条蛋白和微管连接蛋白, 内动力蛋白臂数量减少, 和外周二联体被取代) (见 Table 2) [Merveille et al 2011].
另外, CCDC39在所有有 CCDC40 致病突变的人群中缺失暗示说CCDC39轴丝更新可能需要CCDC40 [Becker-Heck et al 2011].
CCDC103 (CILD17)
基因结构.CCDC103 有4个外显子并编码242氨基酸的蛋白.
致病突变. 见 Table 12. 2个 致病性变异 等位基因被发现. 一个截断致病突变在3个无关的近亲婚配的 巴基斯坦祖先家庭中被发现并且一个 错义 致病突变在2个巴基斯坦祖先的无关系家庭和一个德国家庭中被发现 [Panizzi et al 2012].
Table 12.
此篇GeneReview讨论CCDC103 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.383dupG | p.Gly128fsTer25 | |
c.461A>C | p.His154Pro |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.CCDC103 编码含弯曲螺旋 结构域蛋白 103, 一个有242个氨基酸的蛋白并且N端有弯曲螺旋结构域. 这个蛋白在呼吸上皮细胞的细胞质中被表达 [Panizzi et al 2012].
异常 基因产物.CCDC103 上的致病突变导致纤毛微结构分析可观察到的内外动力蛋白臂缺陷. 另外, 免疫荧光分析发现 DNAH5, DNAI2, 和 DNAH9 (ODA的标记) 在有双等位基因的CCDC103 致病突变的个体的纤毛远端缺失, 与ODA异常组装一致 [Panizzi et al 2012].
LRRC6 (CILD19)
致病突变.Table 13. 超过10个 LRRC6 致病突变, 观察到很多预测是截断突变 [Kott et al 2012, Zariwala et al 2013]. 尤其是, 突变 c.630delG 在巴基斯坦祖先 受累的 群体中被观察到 [Zariwala et al 2013, Watson et al 2014].
Table 13.
此篇GeneReview讨论LRRC6 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.598_599delAA | p.Lys200GlufsTer3 | |
c.574C>T | p.Gln192Ter | |
c.576dupA | p.Glu193ArgfsTer4 | |
c.220G>C | p.Ala74Pro | |
c.436G>C | p.Asp146His | |
c.630delG | p.Trp210CysfsTer12 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.LRRC6 编码富含亮氨酸重复蛋白 6 (也是TILB 蛋白同系物), 一个有466个氨基酸的蛋白,N端有5个LRR结构具有决定含LRR蛋白的SDS22类似亚纲的共同序列. LRRC6也包含LRR帽,是一个弯曲螺旋 结构域, 一个多聚赖氨酸结构,和一个C端alpha-晶体-p23-类似 结构域 .另外,来自一个健康人的免疫荧光分析说明 LRRC6 定位在线毛细胞的胞浆中[Kott et al 2012].
异常 基因产物. LRRC6 上的致病突变通过纤毛微结构分析观察到可以引起内外动力蛋白臂的缺损 [Kott et al 2012].
CCDC114 (CILD20)
致病突变. 见 Table 14. CCDC114 上的致病突变包括移位和 剪接位点 突变[Knowles et al 2013b]. 所有4个剪切位点突变等位基因引起异常转录 [Knowles et al 2013b, Onoufriadis et al 2013].
相同祖先的 致病性变异, c.742G>A, 在8个来自北荷兰的福伦丹渔村的基因隔离的家庭中被观察到 [Onoufriadis et al 2013]. c.742G>A 剪切位点等位基因 与许多 受累的 家庭有共同的单体型说明一个可能的 建立者效应 [Knowles et al 2013b]; 实际上, 相同的等位基因在所有福伦丹村的受累个体中都被发现, 说明人群中的 携带者 的频率为1:10 [Onoufriadis et al 2013].
Table 14.
这篇GeneReview讨论CCDC114 突变
DNA核苷酸改变 | 预测的蛋白改变 | 参考序列 |
|---|---|---|
c.742G>A | p.Ala248SerfsTer52 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.CCDC114 编码670个氨基酸的含弯曲螺旋结构域的轴丝蛋白. 这个蛋白是纤毛细胞中外动力蛋白臂对接复合物的一部分. 人类呼吸道上皮细胞的研究中表明纤毛细胞分化的时候会诱导 CCDC114 全部翻译 [Knowles et al 2013b]. CCDC114 在人类呼吸上皮细胞的纤毛中被发现表达 [Hjeij et al 2013, Onoufriadis et al 2013].
异常 基因产物.CCDC114 上的致病突变导致外动力蛋白缺陷和纤毛活动异常 [Knowles et al 2013b, Onoufriadis et al 2013].
ZMYND10 (CILD22)
致病突变. 见 Table 15. 13种不同的在 ZMYND10 上的致病等位基因包括截断突变, 错义 突变, 和一个大的 缺失被报道. 有 双等位基因的 错义突变 c.47T>G (p.Val16Gly)的个体表现出动力蛋白臂部分保留伴随着活动功能部分保留 (但是震动模式僵硬并且震动幅度异常) 说明这是一个 功能下降的等位基因 [Moore et al 2013].
Table 15.
这篇GeneReview讨论ZMYND10 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.47T>G | p.Val16Gly | |
c.797T>C | p.Leu66Pro |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.ZMYND10 编码440个氨基酸的胞浆蛋白此蛋白包含一个C端的MYND 锌指 结构域. ZMYND10 与LRRC6相互作用, LRRC6是另一个胞浆蛋白与原发性纤毛不动症相关.
异常 基因产物. Pathogenic variants in ZMYND10 上的致病突变引起内外动力蛋白臂缺损和纤毛静止 [Moore et al 2013, Zariwala et al 2013].
ARMC4 (CILD23)
致病突变. 见 Table 16. 8个不相关的在ARMC4上的致病等位基因被发现包括一个大的 缺失 和 无义, 剪切位点, 移码, 和 错义 突变 [Hjeij et al 2013].
一个 无义 突变, p.Glu557Ter, 在多个无关的家庭中被观察到. 功能研究说明错义突变 p.Leu927Trp 是 功能下降的 [Hjeij et al 2013].
Table 16.
这篇GeneReview讨论ARMC4 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.1669G>T | p.Glu557Ter | |
c.2780T>G | p.Leu927Trp |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.ARMC4 编码含1044个氨基酸的轴丝蛋白并有十个armadillo重复结构(ARMs) 和一个HEAT重复并参与各种各样的细胞. ARMC4在纤毛轴丝中出现 [Hjeij et al 2013].
异常 基因产物.ARMC4 上的致病突变导致外动力蛋白臂缺损和纤毛震动频率下降以及纤毛不动 [Hjeij et al 2013]. ARMC4 好像不是外动力蛋白臂结构的一部分 [Hjeij et al 2013].
RSPH1 (CILD24)
致病突变. 见 Table 17. RSPH1 上的致病突变包括截断和剪切位点突变和一个 错义 突变 [Kott et al 2013, Knowles et al 2014]. 8个被报道的致病突变中, 3个 (p.Glu29Ter, c.407_410delAGTA, 和 c.275-2A>C) 有共同的单倍体在2个或更多的无关系家庭中 [Knowles et al 2014].分析4个剪切位点突变
(c.275-2A>C, c.727+5G>A, c.366-3C>A, 和 c.366G>A) 终止信号提前终止翻译表现异常的转录.
Table 17.
这篇 GeneReview讨论RSPH1 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.85G>T | p.Glu29Ter | |
c.275-2A>C | p.Gly92AlafsTer10 | |
c.366G>A | p.Arg122SerfsTer22 | |
c.366-3C>A | Not determined. | |
c.407_410delAGTA | p.Lys136MetfsTer6 | |
c.727+5G>A | p.Ala244ValfsTer22 |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.RSPH1 编码309个氨基酸的轴丝蛋白,该蛋白N端有5个MORN (膜占据和识别连结)重复随后是一个连接和6个MORN重复. RSPH1 在人类呼吸上皮细胞的纤毛细胞分化的时候表达. RSPH1 在人类鼻上皮细胞的纤毛轴丝中表达 [Kott et al 2013, Knowles et al 2014].
异常 基因产物.RSPH1 上的致病突变导致大多数但不是所有的中央微管二联体缺损,以及异常的震动模式 [Kott et al 2013, Knowles et al 2014]. 纤毛全长上的RSPH1的缺失导致RSPH4A缺失, 另一个径向轴条头蛋白, 来自纤毛远端 [Kott et al 2013].
SPAG1 (CILD28)
致病突变. 见 Table 18. 总共8个独立的致病等位基因被检测到包括截断等位基因, 一个等位基因起始密码子失效和一个大的 缺失 (11,971-bp) 包括5’ 端的 SPAG1. 8个独立的致病突变中, 3个等位基因(p.Gln672Ter, c.902_906delAAGTA, 和 11,971-bp 缺失)有共同的单体型说明每个等位基因都有相同的来源[Knowles et al 2013c].
Table 18.
此篇GeneReview讨论SPAG1 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.2014C>T | p.Gln672Ter | |
c.902_906delAAGTA | p.Lys301ThrfsTer4 | |
11,971 bp del |
|
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.SPAG1 编码含926个氨基酸的胞浆蛋白包含一个三角形四肽(TPR)九重复结构参与蛋白-蛋白之间的相互作用. 人类呼吸道上皮细胞的研究说明纤毛细胞分化诱导SPAG1 全部转录. SPAG1 在人工培养的呼吸上皮细胞溶菌产物中表达.
异常 基因产物.SPAG1 上的基因突变导致内外动力蛋白臂缺陷. 另外, 视频显微镜分析表现出几乎不动性 [Knowles et al 2013c].
CCDC151 (CILD30)
致病突变. 见 Table 19. p.Glu309Ter致病性变异 在三个阿拉伯血缘的家庭中被发现 以及 p.Ser419Ter 在巴基斯坦血缘的个体中被发现. 内脏位置异常在5个受累的 个体中的4个被发现, 说明CCDC151 对确定左右轴有重要作用 [Alsaadi et al 2014, Hjeij et al 2014].
Table 19.
这篇GeneReview讨论CCDC151 突变
DNA核苷酸改变 | 预测蛋白改变 | 参考序列 |
|---|---|---|
c.925G>T | p.Glu309Ter | |
c.1256C>A | p.Ser419Ter |
突变种类上的注意事项: 此表所列突变由作者提供. GeneReviews工作人员没有独立证实突变的种类.
命名法上的注意事项: GeneReviews 依从Human Genome Variation Society的标准命名约定 (varnomen-.hgvs.org). 见 Quick Reference 可知对命名法的解释.
正常 基因产物.CCDC151 编码含595个氨基酸的轴丝蛋白有3个高度保守的弯曲螺旋结构域. 通过免疫荧光分析CCDC151表现位于呼吸道纤毛. 共-免疫沉淀反应研究表明CCDC151 和CCDC114共沉淀,但是不和其他的外动力蛋白臂蛋白DNAI1, DNAI2, 和 DNAL1共沉淀 [Hjeij et al 2014].
异常 基因产物.CCDC151 上的致病突变引起外动力蛋白臂缺陷和纤毛不动症. CCDC114 和 ARMC4 (外动力蛋白臂蛋白) 在CCDC151上有致病突变的个体的呼吸上皮上探测不到, 说明它们的位置依靠彼此 [Hjeij et al 2014].
参考文献
引用文献
- Afzelius BA. Cilia-related diseases. J Pathol. 2004;204:470-7. [PubMed: 15495266]
- Afzelius BA, Mossberg B, Bergstrom SE. Immotile cilia syndrome (primary ciliary dyskinesia), including Kartagener syndrome. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Basis of Inherited Disease. New York, NY: McGraw-Hill; 2001:4817-27.
- Alsaadi MM, Erzurumluoglu AM, Rodriguez S, Guthrie PA, Gaunt TR, Omar HZ, Mubarak M, Alharbi KK, Al-Rikabi AC, Day IN. Nonsense mutation in coiled-coil domain containing 151 gene (CCDC151) causes primary ciliary dyskinesia. Hum Mutat. 2014;35:1446-48. [PMC free article: PMC4489323] [PubMed: 25224326]
- Austin-Tse C, Halbritter J, Zariwala MA, Gilberti RM, Gee HY, Hellman N, Pathak N, Liu Y, Panizzi JR, Patel-King RS, Tritschler D, Bower R, O’Toole E, Porath JD, Hurd TW, Chaki M, Diaz KA, Kohl S, Lovric S, Hwang DY, Braun DA, Schueler M, Airik R, Otto EA, Leigh MW, Noone PG, Carson JL, Davis SD, Pittman JE, Ferkol TW, Atkinson JJ, Olivier KN, Sagel SD, Dell SD, Rosenfeld M, Milla CE, Loges NT, Omran H, Porter ME, King SM, Knowles MR, Drummond IA, Hildebrandt F. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am J Hum Genet. 2013;93:672-86. [PMC free article: PMC3791264] [PubMed: 24094744]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125-48. [PubMed: 16722803]
- Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N, Rossier C, Jorissen M, Armengot M, Meeks M, Mitchison HM, Chung EM, Delozier-Blanchet CD, Craigen WJ, Antonarakis SE. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A. 2002;99:10282-6. [PMC free article: PMC124905] [PubMed: 12142464]
- Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J, McSheene J, Loges NT, Olbrich H, Haeffner K, Fliegauf M, Horvath J, Reinhardt R, Nielsen KG, Marthin JK, Baktai G, Anderson KV, Geisler R, Niswander L, Omran H, Burdine RD. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet. 2011;43:79-84. [PMC free article: PMC3132183] [PubMed: 21131974]
- Ben Khelifa M, Coutton C, Zouari R, Karaouzène T, Rendu J, Bidart M, Yassine S, Pierre V, Delaroche J, Hennebicq S, Grunwald D, Escalier D, Pernet-Gallay K, Jouk PS, Thierry-Mieg N, Touré A, Arnoult C, Ray PF. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2014;94:95-104. [PMC free article: PMC3882734] [PubMed: 24360805]
- Blanchon S, Legendre M, Copin B, Duquesnoy P, Montantin G, Kott E, Dastot F, Jeanson L, Cachanado M, Rousseau A, Papon JF, Beydon N, Brouard J, Crestani B, Deschildre A, Désir J, Dollfus H, Leheup B, Tamalet A, Thumerelle C, Vojtek AM, Escalier D, Coste A, de Blic J, Clément A, Escudier E, Amselem S. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J Med Genet. 2012;49:410-6. [PubMed: 22693285]
- Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, Dougherty GW, Raidt J, Werner C, Amirav I, Hevroni A, Abitbul R, Avital A, Soferman R, Wessels M, O’Callaghan C, Chung EM, Rutman A, Hirst RA, Moya E, Mitchison HM, Van Daele S, De Boeck K, Jorissen M, Kintner C, Cuppens H, Omran H. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 2014;5:4418. [PubMed: 25048963]
- Brown DE, Pittman JE, Leigh MW, Fordham L, Davis SD. Early lung disease in young children with primary ciliary dyskinesia. Pediatr Pulmonol. 2008;43:514-6. [PubMed: 18383332]
- Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura M, Lenzner S, Latos-Bielenska A, Ropers HH. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171-8. [PubMed: 16783569]
- Bukowy-Bieryłło Z, Ziętkiewicz E, Loges NT, Wittmer M, Geremek M, Olbrich H, Fliegauf M, Voelkel K, Rutkiewicz E, Rutland J, Morgan L, Pogorzelski A, Martin J, Haan E, Berger W, Omran H, Witt M. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr Pulmonol. 2013;48:352-63. [PubMed: 22888088]
- Casey JP, McGettigan PA, Healy F, Hogg C, Reynolds A, Kennedy BN, Ennis S, Slattery D, Lynch SA. Unexpected genetic heterogeneity for primary ciliary dyskinesia in the Irish Traveller population. Eur J Hum Genet. 2015;23:210-7. [PMC free article: PMC4297907] [PubMed: 24824133]
- Castleman VH, Romio L, Chodhari R, Hirst RA, de Castro SC, Parker KA, Ybot-Gonzalez P, Emes RD, Wilson SW, Wallis C, Johnson CA, Herrera RJ, Rutman A, Dixon M, Shoemark A, Bush A, Hogg C, Gardiner RM, Reish O, Greene ND, O’Callaghan C, Purton S, Chung EM, Mitchison HM. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet. 2009;84:197-209. [PMC free article: PMC2668031] [PubMed: 19200523]
- Chilvers MA, Rutman A, O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol. 2003;112:518-24. [PubMed: 13679810]
- Daniels ML, Leigh MW, Davis SD, Armstrong MC, Carson JL, Hazucha M, Dell SD, Eriksson M, Collins FS, Knowles MR, Zariwala MA. Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum Mutat. 2013;34:1352-6. [PMC free article: PMC3906677] [PubMed: 23798057]
- Davis SD, Ferkol TW, Rosenfeld M, Lee H-S, Dell SD, Sagel SD, Milla C, Zariwala MA, Pittman JE, Shapiro AJ, Carson JL, Krischer J, Hazucha MJ, Cooper ML, Knowles MR, Leigh MW. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med. 2015;191:316-24. [PMC free article: PMC4351577] [PubMed: 25493340]
- DiBella LM, King SM. Dynein motors of the Chlamydomonas flagellum. Int Rev Cytol. 2001;210:227-68. [PubMed: 11580207]
- Diggle CP, Moore DJ, Mali G, zur Lage P, Ait-Lounis A, Schmidts M, Shoemark A, Garcia Munoz A, Halachev MR, Gautier P, Yeyati PL, Bonthron DT, Carr IM, Hayward B, Markham AF, Hope JE, von Kriegsheim A, Mitchison HM, Jackson IJ, Durand B, Reith W, Sheridan E, Jarman AP, Mill P. HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet. 2014;10:e1004577. [PMC free article: PMC4168999] [PubMed: 25232951]
- Djakow J, Svobodová T, Hrach K, Uhlík J, Cinek O, Pohunek P. Effectiveness of sequencing selected exons of DNAH5 and DNAI1 in diagnosis of primary ciliary dyskinesia. Pediatr Pulmonol. 2012;47:864-75. [PubMed: 22416021]
- Duquesnoy P, Escudier E, Vincensini L, Freshour J, Bridoux AM, Coste A, Deschildre A, de Blic J, Legendre M, Montantin G, Tenreiro H, Vojtek AM, Loussert C, Clément A, Escalier D, Bastin P, Mitchell DR, Amselem S. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am J Hum Genet. 2009;85:890-6. [PMC free article: PMC2790569] [PubMed: 19944405]
- Duriez B, Duquesnoy P, Escudier E, Bridoux AM, Escalier D, Rayet I, Marcos E, Vojtek AM, Bercher JF, Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc Natl Acad Sci U S A. 2007;104:3336-41. [PMC free article: PMC1805560] [PubMed: 17360648]
- Failly M, Bartoloni L, Letourneau A, Munoz A, Falconnet E, Rossier C, De Santi MM, Santamaria F, Sacco O, Delozier-Blanchet CD, Lazor R, Blouin JL. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J Med Genet. 2009;46:281-6. [PubMed: 19357118]
- Failly M, Saitta A, Muñoz A, Falconnet E, Rossier C, Santamaria F, de Santi MM, Lazor R, DeLozier-Blanchet CD, Bartoloni L, Blouin JL. DNAI1 mutations explain only 2% of primary ciliary dykinesia. Respiration. 2008;76:198-204. [PubMed: 18434704]
- Ferkol T, Leigh M. Primary ciliary dyskinesia and newborn respiratory distress. Semin Perinatol. 2006;30:335-40. [PubMed: 17142159]
- Ferkol TW, Puffenberger EG, Lie H, Helms C, Strauss KA, Bowcock A, Carson JL, Hazucha M, Morton DH, Patel AC, Leigh MW, Knowles MR, Zariwala MA. Primary ciliary dyskinesia-causing mutations in Amish and Mennonite communities. J Pediatr. 2013;163:383-7. [PMC free article: PMC3725203] [PubMed: 23477994]
- Hadfield PJ, Rowe-Jones JM, Bush A, Mackay IS. Treatment of otitis media with effusion in children with primary ciliary dyskinesia. Clin Otolaryngol Allied Sci. 1997;22:302-6. [PubMed: 9298603]
- Hjeij R, Lindstrand A, Francis R, Zariwala MA, Liu X, Li Y, Damerla R, Dougherty GW, Abouhamed M, Olbrich H, Loges NT, Pennekamp P, Davis EE, Carvalho CM, Pehlivan D, Werner C, Raidt J, Köhler G, Häffner K, Reyes-Mugica M, Lupski JR, Leigh MW, Rosenfeld M, Morgan LC, Knowles MR, Lo CW, Katsanis N, Omran H. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am J Hum Genet. 2013;93:357-67. [PMC free article: PMC3738828] [PubMed: 23849778]
- Hjeij R, Onoufriadis A, Watson CM, Slagle CE, Klena NT, Dougherty GW, Kurkowiak M, Loges NT, Diggle CP, Morante NF, Gabriel GC, Lemke KL, Li Y, Pennekamp P, Menchen T, Konert F, Marthin JK, Mans DA, Letteboer SJ, Werner C, Burgoyne T, Westermann C, Rutman A, Carr IM, O'Callaghan C, Moya E, Chung EM. UK10K Consortium, Sheridan E, Nielsen KG, Roepman R, Bartscherer K, Burdine RD, Lo CW, Omran H, Mitchison HM. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am J Hum Genet. 2014;95:257-74. [PMC free article: PMC4157146] [PubMed: 25192045]
- Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, Thornton KC, Giacalone JC, Albee AJ, Wilson KS, Turner EH, Nickerson DA, Shendure J, Bayly PV, Leigh MW, Knowles MR, Brody SL, Dutcher SK, Ferkol TW. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet. 2012;91:685-93. [PMC free article: PMC3484505] [PubMed: 23040496]
- Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M, Antonarakis SE, Blouin JL, Bartoloni L, Nusslein T, Ahrens P, Griese M, Kuhl H, Sudbrak R, Knowles MR, Reinhardt R, Omran H. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120-6. [PMC free article: PMC2662904] [PubMed: 16627867]
- Imtiaz F, Allam R, Ramzan K, Al-Sayed M. Variation in DNAH1 may contribute to primary ciliary dyskinesia. BMC Med Genet. 2015;16:14. [PMC free article: PMC4422061] [PubMed: 25927852]
- Jeanson L, Copin B, Papon JF, Dastot-Le Moal F, Duquesnoy P, Montantin G, Cadranel J, Corvol H, Coste A, Désir J, Souayah A, Kott E, Collot N, Tissier S, Louis B, Tamalet A, de Blic J, Clement A, Escudier E, Amselem S, Legendre M. RSPH3 mutations cause primary ciliary dyskinesia with central-complex and near absence of radial spokes. Am J Hum Genet. 2015;97:153-62. [PMC free article: PMC4571005] [PubMed: 26073779]
- Katsuhara K, Kawamoto S, Wakabayashi T, Belsky JL. Situs inversus totalis and Kartagener’s syndrome in a Japanese population. Chest. 1972;61:56-61. [PubMed: 4538074]
- Kennedy MP, Leigh MW, Dell S, Morgan L, Molina PL, Zariwala M, Minnix S, Noone PG, Knowles MR. Primary ciliary dyskinesia and situs ambiguus/heterotaxy: Organ laterality defects other than situs inversus totalis. Proc Am Thorac Soc. 2006;3:A399.
- Kim RH, A, Hall D, Cutz E, Knowles MR, Nelligan KA, Nykamp K, Zariwala MA, Dell SD. The role of molecular genetic analysis in the diagnosis of primary ciliary dyskinesia. Ann Am Thorac Soc. 2014;11:351-9. [PMC free article: PMC4028737] [PubMed: 24498942]
- Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia: recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013a;188:913-22. [PMC free article: PMC3826280] [PubMed: 23796196]
- Knowles MR, Leigh MW, Carson JL, Davis SD, Dell SD, Ferkol TW, Olivier KN, Sagel SD, Rosenfeld M, Burns KA, Minnix SL, Armstrong MC, Lori A, Hazucha MJ, Loges NT, Olbrich H, Becker-Heck A, Schmidts M, Werner C, Omran H, Zariwala MA. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax. 2012;67:433-41. [PMC free article: PMC3739700] [PubMed: 22184204]
- Knowles MR, Leigh MW, Ostrowski LE, Huang L, Carson JL, Hazucha MJ, Yin W, Berg JS, Davis SD, Dell SD, Ferkol TW, Rosenfeld M, Sagel SD, Milla CE, Olivier KN, Turner EH, Lewis AP, Bamshad MJ, Nickerson DA, Shendure J, Zariwala MA., Genetic Disorders of Mucociliary Clearance Consortium. Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am J Hum Genet. 2013b;92:99-106. [PMC free article: PMC3542458] [PubMed: 23261302]
- Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, Hazucha MJ, Carson JL, Olivier KN, Sagel SD, Rosenfeld M, Ferkol TW, Dell SD, Milla CE, Randell SH, Yin W, Sannuti A, Metjian HM, Noone PG, Noone PJ, Olson CA, Patrone MV, Dang H, Lee HS, Hurd TW, Gee HY, Otto EA, Halbritter J, Kohl S, Kircher M, Krischer J, Bamshad MJ, Nickerson DA, Hildebrandt F, Shendure J, Zariwala MA. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med. 2014;189:707-17. [PMC free article: PMC3983840] [PubMed: 24568568]
- Knowles MR, Ostrowski LE, Loges NT, Hurd T, Leigh MW, Huang L, Wolf WE, Carson JL, Hazucha MJ, Yin W, Davis SD, Dell SD, Ferkol TW, Sagel SD, Olivier KN, Jahnke C, Olbrich H, Werner C, Raidt J, Wallmeier J, Pennekamp P, Dougherty GW, Hjeij R, Gee HY, Otto EA, Halbritter J, Chaki M, Diaz KA, Braun DA, Porath JD, Schueler M, Baktai G, Griese M, Turner EH, Lewis AP, Bamshad MJ, Nickerson DA, Hildebrandt F, Shendure J, Omran H, Zariwala MA. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am J Hum Genet. 2013c;93:711-20. [PMC free article: PMC3791252] [PubMed: 24055112]
- Kosaki K, Ikeda K, Miyakoshi K, Ueno M, Kosaki R, Takahashi D, Tanaka M, Torikata C, Yoshimura Y, Takahashi T. Absent inner dynein arms in a fetus with familial hydrocephalus-situs abnormality. Am J Med Genet A. 2004;129A:308-11. [PubMed: 15326634]
- Kott E, Duquesnoy P, Copin B, Legendre M, Moal FD, Montantin G, Jeanson L, Tamalet A, Papon J, Siffroi J, Rives N, Mitchell V, de Blic J, Coste A, Clement A, Escalier D, Touré A, Escudier E, Amselem S. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet. 2012;91:958-64. [PMC free article: PMC3487148] [PubMed: 23122589]
- Kott E, Legendre M, Copin B, Papon JF, Dastot-Le Moal F, Montantin G, Duquesnoy P, Piterboth W, Amram D, Bassinet L, Beucher J, Beydon N, Deneuville E, Houdouin V, Journel H, Just J, Nathan N, Tamalet A, Collot N, Jeanson L, Le Gouez M, Vallette B, Vojtek AM, Epaud R, Coste A, Clement A, Housset B, Louis B, Escudier E, Amselem S. Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am J Hum Genet. 2013;93:561-70. [PMC free article: PMC3769924] [PubMed: 23993197]
- Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, Davis SD, Knowles MR, Zariwala MA. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11:473-87. [PMC free article: PMC3739704] [PubMed: 19606528]
- Loges NT, Olbrich H, Becker-Heck A, Häffner K, Heer A, Reinhard C, Schmidts M, Kispert A, Zariwala MA, Leigh MW, Knowles MR, Zentgraf H, Seithe H, Nürnberg G, Nürnberg P, Reinhardt R, Omran H. Deletion and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arms defects. Am J Hum Genet. 2009;85:883-9. [PMC free article: PMC2795801] [PubMed: 19944400]
- Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, Kuhl H, Baktai G, Peterffy E, Chodhari R, Chung EM, Rutman A, O’Callaghan C, Blau H, Tiszlavicz L, Voelkel K, Witt M, Zietkiewicz E, Neesen J, Reinhardt R, Mitchison HM, Omran H. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet. 2008;83:547-58. [PMC free article: PMC2668028] [PubMed: 18950741]
- Lucas JS, Leigh MW. Diagnosis of primary ciliary dyskinesia: searching for a gold standard. Eur Respir J. 2014;44:1418-22. [PubMed: 25435529]
- MacCormick J, Robb I, Kovesi T, Carpenter B. Optimal biopsy techniques in the diagnosis of primary ciliary dyskinesia. J Otolaryngol. 2002;31:13-7. [PubMed: 11881766]
- Majithia A, Fong J, Hariri M, Harcourt J. Hearing outcomes in children with primary ciliary dyskinesia--a longitudinal study. Int J Pediatr Otorhinolaryngol. 2005;69:1061-4. [PubMed: 16005347]
- Marthin JK, Petersen N, Skovgaard LT, Nielsen KG. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med. 2010;181:1262-8. [PubMed: 20167855]
- Mazor M, Alkrinawi S, Chalifa-Caspi V, Manor E, Sheffield VC, Aviram M, Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet. 2011;88:599-607. [PMC free article: PMC3146731] [PubMed: 21496787]
- Merveille AC, Davis EE, Becker-Heck A, Legendre M, Amirav I, Bataille G, Belmont J, Beydon N, Billen F, Clément A, Clercx C, Coste A, Crosbie R, de Blic J, Deleuze S, Duquesnoy P, Escalier D, Escudier E, Fliegauf M, Horvath J, Hill K, Jorissen M, Just J, Kispert A, Lathrop M, Loges NT, Marthin JK, Momozawa Y, Montantin G, Nielsen KG, Olbrich H, Papon JF, Rayet I, Roger G, Schmidts M, Tenreiro H, Towbin JA, Zelenika D, Zentgraf H, Georges M, Lequarré AS, Katsanis N, Omran H, Amselem S. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet. 2011;43:72-8. [PMC free article: PMC3509786] [PubMed: 21131972]
- Mitchison HM, Schmidts M, Loges NT, Freshour J, Dritsoula A, Hirst RA, O’Callaghan C, Blau H, Al Dabbagh M, Olbrich H, Beales PL, Yagi T, Mussaffi H, Chung EM, Omran H, Mitchell DR. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat Genet. 2012;44:381-9. [PMC free article: PMC3315610] [PubMed: 22387996]
- Moore A, Escudier E, Roger G, Tamalet A, Pelosse B, Marlin S, Clement A, Geremek M, Delaisi B, Bridoux AM, Coste A, Witt M, Duriez B, Amselem S. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet. 2006;43:326-33. [PMC free article: PMC2563225] [PubMed: 16055928]
- Moore DJ, Onoufriadis A, Shoemark A, Simpson MA, zur Lage PI, de Castro SC, Bartoloni L, Gallone G, Petridi S, Woollard WJ, Antony D, Schmidts M, Didonna T, Makrythanasis P, Bevillard J, Mongan NP, Djakow J, Pals G, Lucas JS, Marthin JK, Nielsen KG, Santoni F, Guipponi M, Hogg C, Antonarakis SE, Emes RD, Chung EM, Greene ND, Blouin JL, Jarman AP, Mitchison HM. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am J Hum Genet. 2013;93:346-56. [PMC free article: PMC3738835] [PubMed: 23891471]
- Mullowney T, Manson D, Kim R, Stephens D, Shah V, Dell S. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics. 2014;134:1160-6. [PMC free article: PMC4243067] [PubMed: 25422025]
- Narayan D, Krishnan SN, Upender M, Ravikumar TS, Mahoney MJ, Dolan TF Jr, Teebi AS, Haddad GG. Unusual inheritance of primary ciliary dyskinesia (Kartagener’s syndrome). J Med Genet. 1994;31:493-6. [PMC free article: PMC1049931] [PubMed: 8071978]
- Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459-67. [PubMed: 14656747]
- O’Callaghan C, Chetcuti P, Moya E. High prevalence of primary ciliary dyskinesia in a British Asian population. Arch Dis Child. 2010;95:51-52. [PubMed: 19720631]
- Olbrich H, Häffner K, Kispert A, Völkel A, Volz A, Sasmaz G, Reinhardt R, Hennig S, Lehrach H, Konietzko N, Zariwala M, Noone PG, Knowles M, Mitchison HM, Meeks M, Chung EM, Hildebrandt F, Sudbrak R, Omran H. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet. 2002;30:143-4. [PubMed: 11788826]
- Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J, Banki NF, Shoemark A, Burgoyne T, Al Turki S, Hurles ME. UK10K Consortium, Köhler G, Schroeder J, Nürnberg G, Nürnberg P, Chung EM, Reinhardt R, Marthin JK, Nielsen KG, Mitchison HM, Omran H. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet. 2012;91:672-84. [PMC free article: PMC3484652] [PubMed: 23022101]
- Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H, Zhang Q, Leblond G, O’Toole E, Hara C, Mizuno H, Kawano H, Fliegauf M, Yagi T, Koshida S, Miyawaki A, Zentgraf H, Seithe H, Reinhardt R, Watanabe Y, Kamiya R, Mitchell DR, Takeda H. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611-6. [PMC free article: PMC3279746] [PubMed: 19052621]
- Onoufriadis A, Paff T, Antony D, Shoemark A, Micha D, Kuyt B, Schmidts M, Petridi S, Dankert-Roelse JE, Haarman EG, Daniels JM, Emes RD, Wilson R, Hogg C, Scambler PJ, Chung EM. UK10K, Pals G, Mitchison HM. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet. 2013;92:88-98. [PMC free article: PMC3542455] [PubMed: 23261303]
- Panizzi JR, Becker-Heck A, Castleman VH, Al-Mutairi DA, Liu Y, Loges NT, Pathak N, Austin-Tse C, Sheridan E, Schmidts M, Olbrich H, Werner C, Häffner K, Hellman N, Chodhari R, Gupta A, Kramer-Zucker A, Olale F, Burdine RD, Schier AF, O’Callaghan C, Chung EM, Reinhardt R, Mitchison HM, King SM, Omran H, Drummond IA. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat Genet. 2012;44:714-9. [PMC free article: PMC3371652] [PubMed: 22581229]
- Pennarun G, Escudier E, Chapelin C, Bridoux AM, Cacheux V, Roger G, Clement A, Goossens M, Amselem S, Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet. 1999;65:1508-19. [PMC free article: PMC1288361] [PubMed: 10577904]
- Perrone CA, Myster SH, Bower R, O’Toole ET, Porter ME. Insights into the structural organization of the I1 inner arm dynein from a domain analysis of the 1beta dynein heavy chain. Mol Biol Cell. 2000;11:2297-313. [PMC free article: PMC14920] [PubMed: 10888669]
- Satir P. The cilium as a biological nanomachine. FASEB J. 1999;13 Suppl 2:S235-7. [PubMed: 10619134]
- Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, Olbrich H, Fliegauf M, Failly M, Liebers U, Collura M, Gaedicke G, Mundlos S, Wahn U, Blouin JL, Niggemann B, Omran H, Antonarakis SE, Bartoloni L. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat. 2008;29:289-98. [PubMed: 18022865]
- Sha YW, Ding L, Li P. Management of primary ciliary dyskinesia/Kartagener’s syndrome in infertile male patients and current progress in defining the underlying genetic mechanism. Asian J Androl. 2014;16:101-6. [PMC free article: PMC3901865] [PubMed: 24369140]
- Shapiro AJ, Davis SD, Ferkol TF, Dell SD, Rosenfeld M, Olivier KN, Sagel SD, Milla C, Zariwala MA, Wolf W, Carson JL, Hazucha MJ, Burns K, Robinson B, Knowles MR, Leigh MW. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest. 2014a;146:1176-86. [PMC free article: PMC4219335] [PubMed: 24577564]
- Shapiro AJ, Weck KE, Chao KC, Rosenfeld M, Nygren AO, Knowles MR, Leigh MW, Zariwala MA. Cri du chat syndrome and primary ciliary dyskinesia: a common genetic cause on chromosome 5p. J Pediatr. 2014b;165:858-61. [PMC free article: PMC4177261] [PubMed: 25066065]
- Tarkar A, Loges NT, Slagle CE, Francis R, Dougherty GW, Tamayo JV, Shook B, Cantino M, Schwartz D, Jahnke C, Olbrich H, Werner C, Raidt J, Pennekamp P, Abouhamed M, Hjeij R, Köhler G, Griese M, Li Y, Lemke K, Klena N, Liu X, Gabriel G, Tobita K, Jaspers M, Morgan LC, Shapiro AJ, Letteboer SJ, Mans DA, Carson JL, Leigh MW, Wolf WE, Chen S, Lucas JS, Onoufriadis A, Plagnol V, Schmidts M, Boldt K. UK10K, Roepman R, Zariwala MA, Lo CW, Mitchison HM, Knowles MR, Burdine RD, Loturco JJ, Omran H. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet. 2013;45:995-1003. [PMC free article: PMC4000444] [PubMed: 23872636]
- Tobin JL, Beales PL. The nonmotile ciliopathies. Genet Med. 2009;11:386-402. [PubMed: 19421068]
- Torgersen J. Situs inversus, asymmetry, and twinning. Am J Hum Genet. 1950;2:361-70. [PMC free article: PMC1716380] [PubMed: 14837905]
- Wallmeier J, Al-Mutairi DA, Chen CT, Loges NT, Pennekamp P, Menchen T, Ma L, Shamseldin HE, Olbrich H, Dougherty GW, Werner C, Alsabah BH, Köhler G, Jaspers M, Boon M, Griese M, Schmitt-Grohé S, Zimmermann T, Koerner-Rettberg C, Horak E, Kintner C, Alkuraya FS, Omran H. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46:646-51. [PubMed: 24747639]
- Watson CM, Crinnion LA, Morgan JE, Harrison SM, Diggle CP, Adlard J, Lindsay HA, Camm N, Charlton R, Sheridan E, Bonthron DT, Taylor GR, Carr IM. Robust diagnostic genetic testing using solution capture enrichment and a novel variant-filtering interface. Hum Mutat. 2014;35:434-41. [PMC free article: PMC4285299] [PubMed: 24307375]
- Wessels MW, den Hollander NS, Willems PJ. Mild fetal cerebral ventriculomegaly as a prenatal sonographic marker for Kartagener syndrome. Prenat Diagn. 2003;23:239-42. [PubMed: 12627427]
- Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, Loges NT, Pennekamp P, Lindberg S, Stenram U, Carlén B, Horak E, Köhler G, Nürnberg P, Nürnberg G, Porter ME, Omran H. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nature Genet. 2013;45:262-68. [PMC free article: PMC3818796] [PubMed: 23354437]
- Zariwala MA, Gee HY, Kurkowiak M, Al-Mutairi DA, Leigh MW, Hurd TW, Hjeij R, Dell SD, Chaki M, Dougherty GW, Adan M, Spear PC, Esteve-Rudd J, Loges NT, Rosenfeld M, Diaz KA, Olbrich H, Wolf WE, Sheridan E, Batten TF, Halbritter J, Porath JD, Kohl S, Lovric S, Hwang DY, Pittman JE, Burns KA, Ferkol TW, Sagel SD, Olivier KN, Morgan LC, Werner C, Raidt J, Pennekamp P, Sun Z, Zhou W, Airik R, Natarajan S, Allen SJ, Amirav I, Wieczorek D, Landwehr K, Nielsen K, Schwerk N, Sertic J, Köhler G, Washburn J, Levy S, Fan S, Koerner-Rettberg C, Amselem S, Williams DS, Mitchell BJ, Drummond IA, Otto EA, Omran H, Knowles MR, Hildebrandt F. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am J Hum Genet. 2013;93:336-45. [PMC free article: PMC3738827] [PubMed: 23891469]
- Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423-50. [PubMed: 17059358]
- Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noone PG, Carson JL, Hazucha MJ, Lori A, Horvath J, Olbrich H, Loges NT, Bridoux AM, Pennarun G, Duriez B, Escudier E, Mitchison HM, Chodhari R, Chung EM, Morgan LC, de Iongh RU, Rutland J, Pradal U, Omran H, Amselem S, Knowles MR. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am J Respir Crit Care Med. 2006;174:858-66. [PMC free article: PMC2648054] [PubMed: 16858015]
- Zhu L, Belmont JW, Ware SM. Genetics of human heterotaxias. Eur J Hum Genet. 2006;14:17-25. [PubMed: 16251896]
- Ziętkiewicz E, Bukowy-Bieryłło Z, Voelkel K, Klimek B, Dmeńska H, Pogorzelski A, Sulikowska-Rowińska A, Rutkiewicz E, Witt M. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS One. 2012;7:e33667. [PMC free article: PMC3308995] [PubMed: 22448264]
建议阅读
- Afzelius BA, Mossberg B, Bergström SE. Immotile cilia syndrome (primary ciliary dyskinesia), including Kartagener syndrome. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 187. McGraw-Hill. Available online.
- Berdon WE, Willi U. Situs inversus, bronchiectasis, and sinusitis and its relation to immotile cilia: history of the diseases and their discoverers-Manes Kartagener and Bjorn Afzelius. Pediatr Radiol. 2004;34:38-42. [PubMed: 14551758]
- Boon M, Vermeulen FL, Gysemans W, Proesmans M, Jorissen M, De Boeck K. Lung structure-function correlation in patients with primary ciliary dyskinesia. Thorax. 2015;70:339-45. [PubMed: 25673230]
- Eley L, Yates LM, Goodship JA. Cilia and disease. Curr Opin Genet Dev. 2005;15:308-14. [PubMed: 15917207]
- Funkhouser WK 3rd, Niethammer M, Carson JL, Burns KA, Knowles MR, Leigh MW, Zariwala MA, Funkhouser WK Jr. A new tool improves diagnostic test performance for transmission em evaluation of axonemal dynein arms. Ultrastruct Pathol. 2014;38:248-55. [PMC free article: PMC3990650] [PubMed: 23957500]
- Gerdes JM, Katsanis N. Microtubule transport defects in neurological and ciliary disease. Cell Mol Life Sci. 2005;62:1556-70. [PubMed: 15924265]
- Horani A, Brody SL, Ferkol TW. Picking up speed: advances in the genetics of primary ciliary dyskinesia. Pediatr Res. 2014;75:158-64. [PMC free article: PMC3946436] [PubMed: 24192704]
- Hosie P, Fitzgerald DA, Jaffe A, Birman CS, Morgan L. Primary ciliary dyskinesia: overlooked and undertreated in children. J Paediatr Child Health. 2014;50:952-8. [PubMed: 24943508]
- Hosie PH, Fitzgerald DA, Jaffe A, Birman CS, Rutland J, Morgan LC. Presentation of primary ciliary dyskinesia in children: 30 years’ experience. J Paediatr Child Health. 2015;51:722-6. [PubMed: 25510893]
- Kennedy MP, Noone PG, Carson J, Molina PL, Ghio A, Zariwala MA, Minnix SL, Knowles MR. Calcium stone lithoptysis in primary ciliary dyskinesia. Respir Med. 2007;101:76-83. [PubMed: 16757159]
- Leigh MW, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: improving the diagnostic approach. Curr Opin Pediatr. 2009;21:320-5. [PMC free article: PMC3665363] [PubMed: 19300264]
- Lobo J, Zariwala MA, Noone PG. Primary ciliary dyskinesia. Semin Respir Crit Care Med. 2015;36:169-79. [PMC free article: PMC4873960] [PubMed: 25826585]
- Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, National PCD. Service, UK. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child. 2014;99:850-6. [PMC free article: PMC4145427] [PubMed: 24771309]
- Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, Yagi H, Khalifa O, Kureshi S, Chatterjee B, Sabol SL, Swisher M, Connelly PS, Daniels MP, Srinivasan A, Kuehl K, Kravitz N, Burns K, Sami I, Omran H, Barmada M, Olivier K, Chawla KK, Leigh M, Jonas R, Knowles M, Leatherbury L, Lo CW. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125:2232-42. [PMC free article: PMC3770728] [PubMed: 22499950]
- Noone PG, Zariwala M, Knowles MR. Primary ciliary dyskinesia. In: MS Runge, C Patterson, eds. Principles of Molecular Medicine. Totowa, New Jersey: Humana Press; 2006:239-50.
- Omran H, Haffner K, Volkel A, Kuehr J, Ketelsen UP, Ross UH, Konietzko N, Wienker T, Brandis M, Hildebrandt F. Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am J Respir Cell Mol Biol. 2000;23:696-702. [PubMed: 11062149]
- Popatia R, Haver K, Casey A. Primary ciliary dyskinesia: an update on new diagnostic modalities and review of the literature. Pediatr Allergy Immunol Pulmonol. 2014;27:51-9. [PMC free article: PMC4062113] [PubMed: 24963453]
- Quinlan RJ, Tobin JL, Beales PL. Modeling ciliopathies: Primary cilia in development and disease. Curr Top Dev Biol. 2008;84:249-310. [PubMed: 19186246]
- Shapiro AJ, Tolleson-Rinehart S, Zariwala MA, Knowles MR, Leigh MW. The prevalence of clinical features associated with primary ciliary dyskinesia in a heterotaxy population: results of a web-based survey. Cardiol Young. 2015;25:752-9. [PMC free article: PMC4369774] [PubMed: 24905662]
- Werner C, Onnebrink JG, Omran H. Diagnosis and management of primary ciliary dyskinesia. Cilia. 2015;4:2. [PMC free article: PMC4300728] [PubMed: 25610612]
- Ziętkiewicz E, Nitka B, Voelkel K, Skrzypczak U, Bukowy Z, Rutkiewicz E, Humińska K, Przystałowska H, Pogorzelski A, Witt M. Population specificity of the DNAI1 gene mutation spectrum in primary ciliary dyskinesia (PCD). Respir Res. 2010;11:174. [PMC free article: PMC3014902] [PubMed: 21143860]
章注释
作者注释
网站:
UNC - Department of Pathology and Laboratory Medicine
UNC - Pulmonary Diseases and Critical Care Medicine - PCD
UNC - Department of Pediatrics
感谢
我们非常感谢所有参与的患者和他们的家庭,感谢US PCD基金会,感谢Dr. Johnny Carson为本节作图。
此研究获得下列基金的资助:
- NIH/ORD/NCRR U54 HL096458
- NIH/NHLBI 1 R01 HL071798
- NHLBI/NIH 5 R01 HL117836
本章节的内容由参与的作者负责,并不一定代表NIH的官方观点。
修订历史
- 2015.9.3 (me) 全面更新
- 2012.6.7 (cd) 更新:命名改变:TXNDC3 →NME8;已经载入 Molecular Genetics
- 2011.9.15 (me) 全面更新
- 2009.10.6 (me) 全面更新
- 2007.1.24 (me) 更新,添加网址
- 2006.7.19 (mbz) 初版上传