
摘要
临床特征.
未经治疗的酪氨酸血症I型通常在小婴儿期表现为严重肝脏损害,或在随后第一年表现为肝功能障碍和肾小管功能障碍伴生长障碍及佝偻病。未治疗的儿童可能出现重复、经常不易识别的神经学危象,持续1-7天,包括精神改变、腹痛、周围神经病变和/或需要机械通气的呼吸衰竭。 未治疗的儿童通常在10岁前死亡,通常死于肝衰竭,神经系统危象或肝细胞癌。用尼替西农和低酪氨酸饮食联合治疗可达到大于90%的存活率、正常生长,肝功能改善、预防肝硬化,纠正肾小管性酸中毒和改善继发性佝偻病.
诊断/试验.
I型酪氨酸血症由FAH编码的延胡索酰乙酰乙酸水解酶(FAH)缺乏所致。 典型的生化表现包括:血液和尿液中琥珀酰丙酮浓度增加; 血浆酪氨酸,甲硫氨酸和苯丙氨酸浓度增高; 及尿液酪氨酸代谢产物和化合物δ-ALA增高。皮肤成纤维细胞FAH酶活性测定是可能的但不一定成功获得。通过靶向分子遗传学检测可分析4种常见FAH致病变异的和整个编码区的序列分析可检出超过95%患者中致病变异.
治疗.
临床表现的治疗:尼替西农(Orfadin®),即2-(2-硝基-4-三氟甲基苯甲酰基)-1,3-环己二酮(NTBC)阻断酪氨酸降解通路中第二步即对羟基苯丙酮酸双氧化酶(p-HPPD),预防延胡索酰乙酰乙酸的堆积及其向琥珀酰丙酮转化。一旦诊断为酪氨酸血症I型就应开始给予尼替西农治疗。由于尼替西农可增加血酪氨酸浓度,因此诊断后立即开始限制饮食苯丙氨酸和酪氨酸摄入以防止角膜中酪氨酸晶体的形成。如果血苯丙氨酸浓度太低(<20μmol/L),饮食中应添加天然蛋白质。在获得尼替西农之前,对于酪氨酸血症I型的唯一有效治疗是肝移植,肝移植用于治疗那些对尼替西农无效、或已证实肝组织恶变的具有严重肝功能衰竭症状的儿童.
原发症状的预防:诊断一旦明确后就应开始尼替西农治疗.
预防继发性并发症:肉碱缺乏、骨质疏松和继发肾小管型范可尼综合征的佝偻病的早期体征的治疗.
监测:已经建立了对酪氨酸血症I型患者的常规监测指南.
高危亲属的评估:I型酪氨酸血症儿童的父母之后所生育的孩子应在出生后尽快接受尿液和血液琥珀酰丙酮分析,以便尽早的诊断和开始治疗.
妊娠管理:人类怀孕期间使用尼替西农数据很少; 然而,至少有两名妇女在接受治疗剂量的尼替西农治疗时分娩了健康婴儿.
遗传咨询.
I型酪氨酸血症属常染色体隐性遗传。 在受孕时,每个患者的同胞有25%的机率患病,50%的机率为无症状的携带者,25%的机率即不是患者也不是携带者。如果已知家系中的两种致病性等位基因变异,则对于高危孕妇进行携带者检测和产前诊断是可能的.
诊断
酪氨酸血症I型,一种酪氨酸代谢紊乱,通常在新生儿筛查时被检出。 在未治疗情况下,酪氨酸血症I型在小婴儿期表现为为严重的肝病。6个月以上儿童可能因出现肾脏疾病,佝偻病和/或神经系统危象而引起医生注意.
病因是由于延胡索酰乙酰乙酸水解酶(FAH)(EC 3.7.1.2)缺乏所致[Lindblad et al 1977]。(见图1 和病理生理)
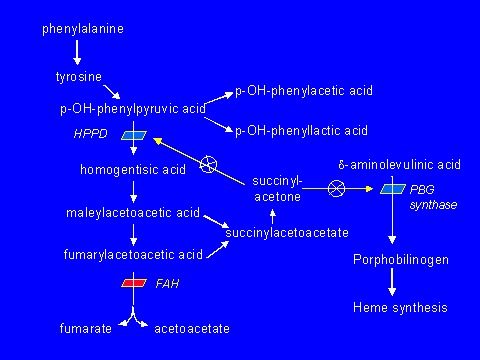
图1.
酪氨酸分解代谢途径
I型酪氨酸血症有以下特征
- 血和尿中琥珀酰丙酮浓度增高
注意:(1)肝衰竭或严重肾脏疾病的儿童尿液中琥珀酰丙酮的排泄增加是酪氨酸血症I型的病理特征。(2)许多实验室要求在测定尿有机酸时特别需要检测琥珀酰丙酮. - 血浆酪氨酸,甲硫氨酸和苯丙氨酸的浓度升高
注意:(1)受影响婴儿脐带血和新生儿期血浆酪氨酸浓度在可以是正常的。(2)升高的血浆酪氨酸浓度也可以是肝损伤或不成熟的非特异性指标; 例如,食用高蛋白配方[Techakittiroj et al 2005]包括未稀释的山羊奶[Hendriksz&Walter 2004]的的婴儿. - 尿有机酸检测发现酪氨酸代谢物对羟基苯丙酮酸盐、对羟基苯乳酸盐和对羟基苯乙酸盐浓度升高
- 继发于肝脏和血红细胞中琥珀酰丙酮对δ-氨基乙酰丙酸(δ-ALA)脱水酶的抑制,尿中排出化合物δ-ALA增加 [Sassa & Kappas 1983]
- 延胡索酰乙酰乙酸水解酶(FAH)活性降低。 虽然可能测定皮肤成纤维细胞中酶活性,但该方法尚未建立.
未治疗的酪氨酸血症I型在肝功能方面的改变如下
血清甲胎蛋白浓度明显升高(平均160,000 ng/mL)(正常:1-3个月婴儿<1000 ng/mL,3个月至18岁儿童 <12 ng/mL)- 延长凝血酶原和部分凝血活酶时间
注意:(1)酪氨酸血症I型其甲胎蛋白血症浓度(AFP)和凝血酶原时间/部分凝血活酶时间(PT / PTT)的改变比在非特异性肝脏疾病中改变更严重,并且通常是酪氨酸血症I型的表现。(2)转氨酶和胆红素如有改变则仅为适度升高。(3)伴有肝脏疾病和正常血清AFP和PT/PTT者患酪氨酸血症I型的可能性较小.
新生儿筛查诊断酪氨酸血症I型
- 血酪氨酸或甲硫氨酸浓度升高。血中酪氨酸或甲硫氨酸浓度升高提示肝脏疾病; 通过血浆或尿琥珀酰丙酮的定量来进一步评估酪氨酸血症I型的诊断。
- 注意:(1)患有酪氨酸血症I型的婴儿在新生儿筛查原标本中血酪氨酸和甲硫氨酸浓度可能仅轻度升高或正常水平。(2)新生儿筛查发现的酪氨酸浓度升高可能是新生儿暂时性酪氨酸血症II型或III型或其他肝脏疾病。(3)升高的甲硫氨酸浓度可提示肝功能不全、甲硫氨酸代谢缺陷或同型半胱氨酸尿(见由胱硫醚β-合酶缺乏导致同型半胱氨酸尿症)
- 酪氨酸血症I型更敏感和特异性指标
- 琥珀酰基丙酮,通过串联质谱直接从新生儿血滴中测定 [Allard et al 2004, Rashed et al 2005, Al-Dirbashi et al 2008]
注意:琥珀酰丙酮是现在新生儿筛查实验室中诊断酪氨酸血症I型的常规生化标志物. - 在加拿大魁北克的新生儿筛查项目中测定Δ-ALA脱水酶(PBG合成酶)活性[Giguère et al 2005]。 然后在具有明显δ-ALA脱水酶缺乏的婴儿尿中测定琥珀酰丙酮[Schulze et al 2001].
个体具有携带编码延胡索酰乙酰乙酸水解酶的FAH基因两个致病性变异这一特征性生化表现时可确定酪氨酸血症I型. 见表1.
分子遗传学检测可开始针对某些具有创始性致病变异群体成员中的个体进行靶向分析; 否则,开始即进行测序分析。如果没有或只有一个致病变异被检出,考虑缺失/重复分析.
表 1.
酪氨酸血症I型的分子遗传学检测汇总
基因 | 检测方法 | 该方法检测到致病性变异的先症者比例 |
|---|---|---|
| FAH | 致病变异的靶向分析 2 | 见脚注 3, 4, 5 |
| 测序分析 6 | >95% | |
| 缺失/重复分析 7 | 不明确; 一个大缺失报道 8 |
- 1.
- 2.
包括在一个基因组中致病变异可因不同实验室而有所不同.
- 3.
四种常见的FAH致病变异 – c.1062+5G>A (IVS12+5 G>A), c.554-1G>T (IVS6-1 G>T), c.607-6T>G (IVS7-6 T>G), and p.Pro261Leu (P261L) –占美国人群I型酪氨酸血症致病变异中约60%[CR Scott,未发表的数据].
- 4. p.Pro261Leu(P261L)在Ashkenazi犹太人群致病变异
- 中占> 99%[Elpeleg et al 2002].
- 5.
c.1062 + 5G> A(IVS12 + 5 G> A)占法国加拿大人群致病变异中87.9% [Poudrier et al 1996].
- 6.
测序分析可检出各种变异,有良性的、可能良性的、不明意义的、可能致病的、或致病的变异。 致病变异可包括小片段基因缺失/插入和错义、无义及剪接位点变异; 通常,未检测到一个外显子或全基因缺失/重复。 对于解释测序分析结果时需要考虑的问题,请点击这里.
- 7.
用于鉴定基因组DNA编码区和侧翼内含子区域序列分析不能检测的外显子或全基因缺失/重复的试验。 可以使用的多种方法包括:定量PCR,长片段PCR,多重连接依赖性探针扩增(MLPA)和包括该基因/染色体片段的染色体微阵列(CMA)技术.
- 8.
Park et al [2009]报道了FAH的大片段的缺失.
临床特征
临床描述
未治疗的酪氨酸血症I型通常在小婴儿时表现为严重肝脏受累,或者在出生第一年后期出现肝脏受损和显着的肾脏受累,生长障碍及佝偻病。生长障碍由于伴有营养摄入不足的慢性疾病、肝脏受损和/或慢性肾脏疾病所致。 未治疗的儿童通常在十岁前死于肝衰竭,神经系统危象或肝细胞癌。
肝脏受损: 未治疗的儿童在出生6个月前出现急性肝功能衰竭,伴初期凝血因子的合成功能丧失[Croffie等1999]。 PT和PTT明显延长,并且给予维生素K治疗不能纠正; 凝血因子II,VII,IX,XI和XII水平降低; 而相反凝血因子V和VIII水平正常。血清转氨酶水平可能仅有轻度升高; 血清胆红素浓度可能正常或仅轻微升高,与大多数形式的严重肝脏疾病相反,后者转氨酶和血清胆红素浓度的显着升高伴随PT和PTT的延长。 受损肝细胞对细胞死亡的抵抗可以解释所观察到肝功能方面的不相一致[Vogel et al 2004].
疾病早期阶段可以发展为肝衰竭伴随腹水,黄疸和胃肠道出血。 儿童可能有“烂白菜”或“烂蘑菇”特异性气味。 婴儿偶尔有持续性低血糖; 一些患儿有高胰岛素血症[Baumann et al 2005]。 其他有慢性轻度酸中毒[CRS cott,未发表的数据]。 未治疗的患病婴儿可在首次出现症状的数周或数月内死于肝衰竭。
.肾小管受损: 在未治疗的慢性病症中,症状在6个月后发展; 肾小管受损是主要表现。 肾小管功能障碍包括一种Fanconi样肾综合征,伴有广泛的氨基酸尿症,磷酸盐丢失、许多患者表现为肾小管性酸中毒。 持续的肾脏磷酸盐的丢失是佝偻病的主要原因; 血清钙浓度通常正常。
.神经系统危象: 未治疗的儿童可能有反复发作的神经系统危象,类似于患有急性间歇性卟啉症的年长者。 这些危象包括精神状态的改变,腹痛,周围神经病变和/或需要机械通气的呼吸衰竭。 危象可持续1-7天。反复发作的神经系统危象往往不易识别。 Mitchell et al [1990]报道,42%未经治疗的酪氨酸血症I型法籍加拿大儿童出现过这种发作现象。 在一项国际调查中,van Spronsen et al [1994] 报道,未治疗儿童中10%死亡发生在神经系统危象期间。
.肝细胞癌。 那些没有接尼替西农和低酪氨酸饮食治疗的儿童、及那些发生急性肝衰竭后存活的儿童具有发展和死于肝细胞癌的高风险。
.未治疗儿童的存活率。 在年龄2个月前诊断而未治疗的婴儿其两年存活率为29%[van Spronsen et al 1994]。 那些年龄2〜6个月诊断的患者其两年生存率为74%;那些在年龄6个月后诊断的患者其两年生存率为96%。在年龄2至6个月之间诊断的患者其5年以上的存活率降至约30%,在年龄6个月后诊断的患者5年以上的存活率降至约60%(图2)
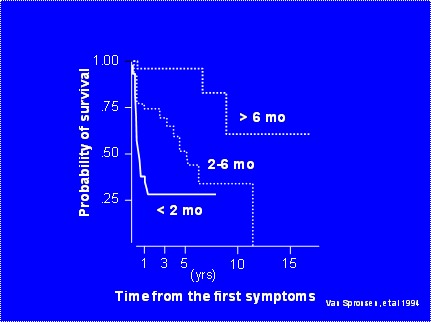
用尼替西农治疗的酪氨酸血症I型儿童的自然史与未治疗的儿童不同。年龄小于2岁的儿童患儿联用尼替西农和低酪氨酸饮食治疗,其临床症状的改善较单独用低酪氨酸饮食治疗者更显着。尼替西农和低酪氨酸饮食联合治疗者存活率大于90%,生长正常、肝功能改善、预防肝硬化、肾小管性酸中毒纠正及继发性佝偻病得到改善。[McKiernan 2006, Masurel-Paulet et al 2008].
在治疗的儿童中观察到的神经危象通常与长时间中断尼替西农治疗有关[CRScott,未发表的数据]。
急性肝功能衰竭的儿童在尼替西农治疗前及开始治疗阶段需要支持疗法。 临床改善通常发生在尼替西农开始治疗的一周内。
.
角膜晶体。 尼替西农阻断酪氨酸分解代谢途径,阻止了琥珀酰丙酮产生,但是组织中酪氨酸水平升高。血液酪氨酸浓度大于600mol/ L时,具有酪氨酸沉积风险犹如双侧、线性、分支状的上皮下角膜混浊[Ahmad et al 2002],导致眼睛畏光和瘙痒,敏感。 一旦酪氨酸水平降低,晶体消失。
肝细胞癌。 虽然Holme & Lindstedt [2000] and van Spronsen et al [2005] 报道在多年尼替西农治疗后的患者中发生肝细胞癌,估计少于5%在2岁前接受尼替西农治疗的儿童在10岁前发展成肝细胞癌 [CR Scott,未发表数据]。 在魁北克,新生儿筛查项目中包括酪氨酸血症I型,未受影响者因出现酪氨酸血症I型的表现而住院,也未报道过在生后30天之内接受尼替西农治疗的患者发生肝细胞癌。此组报告最长治疗时间为12年[Larochelle et al 2012]
.
病理生理学
延胡索酰乙酰乙酸水解酶(FAH)是酪氨酸分解代谢途径中的终末酶(图1)。 FAH缺乏(EC 3.7.1.2)导致酪氨酸血症I型[Lindblad et al 1977]。 在FAH缺乏症中,延胡索酰乙酰乙酸酯(FAA),即前质:
- 被转变为琥珀酰乙酰乙酸酯和琥珀酰丙酮。 琥珀酰丙酮会干扰以下肝酶:;:
- 对羟基苯丙酮酸双氧化酶(p-HPPD),导致血浆酪氨酸浓度升高
- PBG合成酶,导致(1)在肝脏和循环红细胞中δ-ALA脱水酶活性降低; (2)血红素合成减少; (3)增加δ-氨基乙酰丙酸(δ-ALA),其可引起急性神经症状的发作; 和(4)尿δ-ALA排出增加
基因型-表型关系
一般来说,临床表现和基因型无相关性。在同一家庭中存在急性和慢性形式,同样无关系的患者具有相同基因型 [Poudrier et al 1998].
解释这种临床变异的一种机制是基因逆转。从具有慢性形式的酪氨酸血症I型患者肝中切除的肝结节中已发现了FAH蛋白免疫阳性的细胞,并且具有FAH酶活性[Kvittingen et al 1994, Grompe 2001]。 这些看起来“正常”的细胞似乎是通过基因逆转产生的,即在体细胞分裂期间,种系致病性病变异对正常基因序列的自发性自身修复(即突变逆转)。
已报道了自发体细胞突变,这种突变能抑制致病性变异的作用,并促使这些细胞中正常或接近正常的基因表达 [Bliksrud et al 2005]。 这是一种真实的突变序列反转,而不是母体细胞克隆或母体细胞融合的结果[Bergeron et al 2004]。 “正常”(即还原)细胞具有选择性生长优势,因为它们不再因FAA堆积而具有凋亡的风险。 在慢性酪氨酸血症I型未治疗、具有轻度生化和临床表型的患者中,这些还原“正常”细胞集落形成许多肝结节[Kim et al 2000, Demers et al 2003]。 然而,通过非还原突变细胞持续产生琥珀酰丙酮和FAA使患者处于肝细胞癌的持续风险中 [Kim et al 2000].
已经报道在1例具有严重肝脏疾病的4个月龄的比利时婴儿中发现了一种罕见的及非典型的酪氨酸血症I型。肝功能检查异常伴α-甲胎蛋白明显升高,PT和PTT延长,尿中未检测到琥珀酰丙酮。延胡索酰乙酰乙酸酶(FAH)蛋白和活性降低,但未缺乏。发现了一种独特的纯合致病性变异c.103G> A(p.Ala35Thr) [Cassiman et al 2009].
命名
先前使用的酪氨酸血症I型相关术语包括酪氨酸病.
患病率
酪氨酸血症I型患病率1/100,000-120,000 [Mitchell et al 2001]。由于其临床表现的不一致和自然转归不清,估计少于50%的患者被诊断时仍活着。
在普通美国人群中,携带率约为1:150-1:100.
因为一种创始效应引起的某些致病性变异的频率增加,世界上两个地区具有高于预期的酪氨酸血症I型发病率:
在挪威[c.1062+5G>A (IVS 12+5 G>A), p.Gly337Ser (G337S),和/或
p.249HisfsTer5.5]及芬兰[p.Trp262Ter (W262X)],出生患病率估计在1/ 74,000-60,000活产儿].
法国定居者殖民化的创始者效应存在于加拿大魁北克省。在该群体中,c.1062 + 5G> A(IVS12 + 5G> A)致病性变异占等位基因变异中87%。 魁北克省的出生患病率为 1:16,000。在魁北克的Saguenay-Lac Saint-Jean地区,患病率是1:1,846活产儿。 根据新生儿筛查统计数据,魁北克地区的总体携带率1:66。在Saguenay-Lac St-Jean地区携带率为1:16-1:20。
遗传相关(等位基因)疾病
除了在此GeneReview中讨论的那些表型之外,没有其他与FAH中的致病变异相关的表型。
鉴别诊断
具有以下任何表现的儿童应进行I型酪氨酸血症的评估:
表 2.
对具有下列表现的婴儿进行酪氨酸血症I型的鉴别诊断
| 临床表现 | 鉴别诊断 |
|---|---|
| 高酪氨酸血症 |
|
| Hypermethioninemia |
|
| Liver disease |
|
| Renal syndrome |
|
| Rickets |
|
| Neurologic crisis |
|
酪氨酸血症II型由于酪氨酸氨基转移酶(TAT)缺陷引起(EC 2.6.1.5)。酪氨酸血症II型的诊断见以下几点:
- 血浆酪氨酸浓度通常大于500μmol/L,可能超过1000μmol/ L(其他氨基酸的浓度正常)。
- 增加对羟基苯丙酮酸盐,对羟基苯乳酸盐和对羟基苯乙酸盐的排出,尿有机酸分析显示尿中出现少量N-乙酰酪氨酸和4-酪胺。
患者在足底和手掌上出现疼痛、无瘙痒和角化过度的斑块。手指表面可出现明显角化过度相关的黄色增厚。眼损害表现为顽固性、假树枝状角膜炎[Macsai et al 2001]。 虽然发育迟缓似乎常见,但是否对这点解释和神经症状报道有一定偏差尚不清楚。
发现限制酪氨酸和苯丙氨酸饮食可改善症状[Ellaway et al 2001].
酪氨酸血症III型,最罕见的酪氨酸疾病,它是由于对羟基苯丙酮酸双氧化酶缺乏引起(EC.1.13.11.27)。血浆酪氨酸浓度为350〜650μmol/L。 4-羟基苯丙酮酸,4-羟基苯乳酸和4-羟基苯乙酸的排出增加。 精确定量随蛋白摄入量不同而不同.
少数该病患者已得到诊断,而临床表型仍未明确。第一个患者因为智力残疾或共济失调就医; 另一个患者通过常规筛查被发现[Mitchell et al 2001]。 这些患者如同酪氨酸血症II型者,没有肝脏受累,但有皮肤或眼睛改变。 尚不清楚III型酪氨酸血症是否与认知延迟真正相关,或者如果关联是由确定偏差导致的[Ellaway,et al 2001]
低苯丙氨酸和酪氨酸的饮食可以降低血浆酪氨酸浓度。.
治疗
为了在临床表现基础上确定I型酪氨酸血症患者的疾病程度和需求,推荐以下评估(见表3):
- CBC与血小板计数; 血清电解质水平;肝功能评估(PT,PTT,血清胆红素浓度,肝酶浓度[AST,ALT,GGT,碱性磷酸酶],血清AFP浓度); 肾功能评估(BUN,肌酐,尿蛋白/肌酸比值)
- CT或MRI基础腹部影像检查对比以评估肝腺瘤或结节(see Dubois et al [1996])和肾大小。
- 手腕X摄片以证实有无佝偻病表现。
- 医学遗传学咨询
临床表现的对症治疗
急性肝功能衰竭的治疗。儿童可能需要辅助呼吸、适当的补液和血制品以纠正出血体质。
尼替西农(Orfadin)。 美国食品和药物管理局于2002年4月批准2-(2-硝基-4-三氟甲基苄基)-1,3-环己二酮(NTBC)用于治疗酪氨酸血症I型[Schwetz 2002]。尼替西农阻断对羟基苯丙酮酸双氧化酶(p-HPPD),即酪氨酸降解途径的第二步,并阻止FAA的堆积并转化为琥珀酰丙酮(图1).
一旦酪氨酸血症I型的诊断明确,应立即给予尼替西农治疗。.
尼替西农通常治疗剂量为1.0mg/kg/d; 个体需要量有所不同。 应调整剂量以维持血中尼替西农水平在40-60μmol/L,这一水平理论上能阻断99%以上的p-HPPD活性。 很罕见,个别患者可能需要更高的血液水平的尼替西农(70μmol/ L)抑制琥珀酰丙酮排出。 只要尼替西农的血浓度在治疗范围内,就不需要测定尿琥珀酰丙酮。
尼替西农剂量通常分二次给予; 然而,由于该药具有较长的半衰期(50-60小时),年龄较大和病情稳定的患者可以每天一次服药维持适当的治疗[Schlune et al 2012]。
尼替西农罕见的副作用包括暂时性血小板减低和暂时性嗜中性粒细胞减少,其通常不需要干预即可恢复,另有畏光,其可通过严格的饮食控制及血酪氨酸浓度降低后可消失。
低酪氨酸饮食。 尼替西农可增加血酪氨酸浓度,需要低酪氨酸饮食以防止角膜中形成酪氨酸晶体。饮食管理应该在诊断后立即开始,并且应提供限制苯丙氨酸和酪氨酸的全营养饮食,可食用低蛋白素食和医疗配方如Tyrex®(Ross)或Tyros-1®(Mead Johnson)。
苯丙氨酸和酪氨酸需求是相互依存的,个体之间及在同一个体内这种需求不相同,取决于个体生长速率、能量和蛋白质足够程度以及健康状况。通过适当的饮食管理,各年龄段血浆酪氨酸浓度应为200-500μmol/L; 血浆苯丙氨酸浓度应为20-80μmol/ L(0.3-1.3 mg /dL)。 如果血苯丙氨酸浓度太低(<20μmol/L),应该从牛奶或食物中添加额外的蛋白质。
肝移植 在用于治疗酪氨酸血症I型的尼替西农获取前,唯一的肯定性治疗是肝移植。
最近临床经验表明,如今当儿童出现以下情况时可进行肝移植:(1)临床表现为严重肝功能衰竭,并对尼替西农治疗无效或(2)具有肝组织恶性变化的依据 [Mohan et al 1999, Bartlett et al 2014].
接受肝移植患者需要长期免疫抑制剂。 年幼儿童肝移植相关死亡率为10%或更高。
接受肝移植患者也可受益于低剂量(0.1mg / kg /天)尼替西农治疗,以预防由于血浆和尿中持续存在琥珀酰丙酮导致持续性肾小管和肾小球功能障碍 [Pierik et al 2005, Bartlett et al 2013].
原发症状的干预
一旦诊断确诊后,立即用尼替西农(Orfadin®)治疗。
继发并发症的干预
因为肾小管范可尼综合征继发肉碱缺乏可引起骨骼肌无力,应测定血清肉碱浓度,以确定肉碱缺乏,给予治疗[Nissenkorn et al 2001].
通过纠正酸中毒,重建钙磷盐平衡以及给予25-羟基维生素D治疗由肾小管损伤引起的骨质疏松症和佝偻病。
监测
在酪氨酸血症I型患者的治疗中需经常监测以下指标(表3)[CR Scott个人建议]。
表 3.
酪氨酸血症I型的监测指南
| 评估指标 | 治疗开始 (基础值) | 治疗头6月 | 治疗6月后 | 当 临床需要时 | |||
|---|---|---|---|---|---|---|---|
| 每月1次 | 每3月1次 | 每3月1次 | 每6月1次 | 每年一次 | |||
| 酪氨酸血症I型指标 | |||||||
| 血浆甲硫氨酸,苯丙氨酸,酪氨酸浓度 | x | x | x | ||||
| 尿琥珀酰丙酮 | x | x | x | ||||
| 血尼替西农浓度 | x | x | |||||
| CBC(全血计数) | |||||||
| 血红蛋白,血细胞容积,白,血小板计数 | x | x | x | ||||
| 肝功能评估 | |||||||
| 血清AFP浓度 | x | x | x | ||||
| 凝血酶原时间(PT) | x | x (until normal) | |||||
| 部分凝血活酶时间(PTT) | x | x (until normal) | |||||
| 胆红素 | x | + | |||||
| ALT/AST | x | x (until normal) | + | ||||
| GGT | x | x (until normal) | + | ||||
| 碱性磷酸酶 | x | x (until normal) | + | ||||
| CT or MRI 1 | x | x | |||||
| 肾脏评估 | |||||||
| 尿素氮,肌酐 | x | x | |||||
| 尿:PO4,Ca,Prot / Cr比值 | x | ||||||
| 骨骼评估 | |||||||
| 腕骨X摄片 | x | x | |||||
x =如果临床有指征
ALT/AST = 丙氨酸转氨酶/谷草转氨酶
GGT = γ-谷氨酰转肽
1.MRI对比评估肝腺瘤或结节及肾大小
更新的处理建议,见 de Laet et al [2013].
避免的因素/环境
避免不适当的蛋白摄入.
相关危险因素的评估
虽然新诊断的酪氨酸血症I型婴儿的健康年长同胞患有酪氨酸血症I型不太可能,分析他/她们尿琥珀酰丙酮需谨慎。
所有酪氨酸血症I型儿童的父母以后所生育的孩子应该在出生后尽快做血或尿琥珀酰丙酮测定以尽可能早期诊断及开始治疗。
注:如果在家系中已知FAH基因中存在致病性变异,可以进行产前诊断以在出生前明确高危同胞基因携带状况,以在出生后尽早可能给予治疗。
见高危家庭成员相关问题遗传咨询以达到遗传咨询目的。.
妊娠期处理
人妊娠期使用尼替西农相关数据极少。推测孕妇仍安全的免于不良事件;然而,发育中的胎儿可能因酪氨酸代谢异常存在风险。
至少有两名妇女在接受治疗剂量的尼替西农时生育了健康的婴儿 [Garcia Segarra et al 2010].
- 在一个例子中,患病母亲生下了一个未受影响的婴儿,并报道该婴儿在2.5岁时健康并发育正常 [Garcia Segarra et al 2010, Vanclooster et al 2012].
- 另一个例子,患病妇女生育了一名患病孩子。 据报道,该孩子在7个月时有正常的生长和发育[Garcia Segarra et al 2010]。 作者推测这名受影响的孩子在宫内因孕妇接受尼替西农治疗而预防了肝损伤。.
监测治疗
搜索ClinicalTrials.gov获取关于疾病和病症广泛的临床研究方面的信息。 注意:该病尚无临床试验。.
其他
在获取尼替西农前,唯一可用的非移植治疗方法是限制苯丙氨酸和酪氨酸的饮食。 虽然这种方法有一定帮助,但仍可发生反复的神经系统危象和肝脏疾病的进展。 平均生存年龄小于10岁。
遗传咨询
遗传咨询是向患者和家庭成员提供有关遗传疾病的自然史,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估和家族史和遗传诊断试验,以明确家庭成员的遗传状况。 本节内容不是为了解决个人可能面临的所有个体,文化或伦理问题或替代遗传专家的咨询。—ED.
遗传方式
酪氨酸血症I型属于常染色体隐性遗传。
家庭成员的风险
先证者的父母
- 患者父母是杂合子(即一种致病性等位基因变异的携带者)。
- 杂合子(携带者)无症状
先证者的同胞
- 在受孕时,患者的每个同胞有25%的机率患病,50%的机率为无症状的携带者,25%的机率即不是患者,也不是携带者。.
- 一旦已知高危同胞不受影响,他/她成为携带者的风险是2/3。
- 杂合子(携带者)无症状。
先证者的后代 酪氨酸血症I型患者的后代一定是携带一个致病性FAH变异的杂合子(携带者)。
先证者的其他家庭成员。先证者父母的每个同胞有50%的风险为携带者。
携带者检测
一旦在家庭中鉴定出基因致病变异,有可能对高危亲属进行携带者的检测。
相关遗传咨询问题
参见处理,高危亲属风险评估的信息异达到早期诊断和治疗目的。
家庭计划
- 提供遗传咨询、澄清携带者状况及讨论产前诊断的合适时间是在怀孕前。
- 对年轻成人患者、携带者或有携带风险的年轻成人提供遗传咨询(包括其子代潜在风险和生育选择的讨论)是合适的。
- .DNA库是DNA(通常从白血细胞中提取)储存库,以备可能将来使用。 因为基因检测方法和我们对基因、碱基变异及疾病的理解可能会在将来得到改善,应该考虑对基因库中患者DNA重新分析。 .
产前诊断
分子遗传检测。 如果在患者家庭成员中已检测出FAH致病变异,则可以通过能提供该基因检测或常规产前诊断的临床实验室对高危风险的孕妇进行产前诊断。
生化检测。 通过对孕15-18周、具有25%患 病风险胎儿的孕妇羊膜穿刺获取的羊水检测琥珀酰丙酮水平以进行产前诊断成为可能。 虽然羊水中琥珀酰丙酮的检测具有诊断意义,但是已有假阴性结果的报道,因此,该方法只能在采用稳定同位素检测方法始终能检出低水平琥珀酰丙酮的实验室进 行。由于这些生化试验问题,分子遗传检测是产前诊断的优选方法。
植入前遗传诊断(PGD)可能是一些已明确FAH致病变异家庭的一种选择。
资源
GeneReviews员工选择以下疾病特异和/或相关支持组织机构和/或有益于该疾病患者个人及其家人的注册系统。 GeneReviews不对其他组织提供的信息负责。 获取有关选择标准信息,请单击此处.
- 酪氨酸血症1型
- National Library of Medicine Genetics Home Reference
- Save Babies Through Screening Foundation, Inc.P. O. Box 42197Cincinnati OH 45242Phone: 888-454-3383Email: email@savebabies.org
- American Liver Foundation75 Maiden LaneSuite 603New York NY 10038Phone: 800-465-4837 (Toll-free HelpLine); 212-668-1000Fax: 212-483-8179Email: info@liverfoundation.org
- Children Living with Inherited Metabolic Diseases (CLIMB)United KingdomPhone: 0800-652-3181Email: info.svcs@climb.org.uk
分子遗传学
分子遗传学和OMIM表格中的信息可能与GeneReview中的其他地方不同:表格可能包含更新的信息—ED.
表 A.
酪氨酸血症I型:基因和数据库
| 基因 | 染色体部位 | 蛋白质 | 专用数据库 | HGMD |
|---|---|---|---|---|
| FAH | 15q25.1 | Fumarylacetoacetase | FAH database | FAH |
数据来自以下标准参考文献:来自HGNC的基因; 染色体位点,基因名称,关键部位,来自OMIM互补基团组;来自UniProt蛋白质。 有关提供链接的数据库(Locus Specific,HGMD)描述,请单击此处。
基因结构 FAH大小约为35kb,包含14个外显子。 关于基因和蛋白质的详细信息参见表A,基因。
良性等位基因变异。 单一假性缺失的等位基因变异(p.Arg341Trp [c.1021C> T])导致FAH酶活性降低及极少的免疫反应蛋白,但有适当量的FAH mRNA。
致病性等位基因变体。 参见表4.已经报道了错义,无义和剪接位点致病变体,以及FAH的小缺失和插入缺失。 Park等人[2009]报道了涉及FAH的大的缺失。
以下群体特异性致病变异来源于创始者效应或遗传漂移 [Bergman et al 1998, Bergeron et al 2001, Arranz et al 2002, Elpeleg et al 2002, Heath et al 2002]:
- Ashkenazi犹太: p.Pro261Leu (P261L)
- 芬兰人: p.Trp262Ter (W262X)
- 加拿大法国人: c.1062+5G>A (IVS 12+5 G>A)
- 巴基斯坦人: p.Gln64His (Q64H)
- 斯堪的纳维亚人: p.Gly337Ser (G337S)
- 土耳其人: p.Asp233Val (D233V)
- 北欧人: c.1062+5G>A (IVS 12+5 G>A)
- 南欧人: c.554-1G>T (IVS 6-1 G>T)
表 4.
选择的FAH等位基因变异
| 变异分类 | DNA核苷酸改变(别名 1) | 蛋白质氨基酸改变 | 参考序列 |
|---|---|---|---|
| 假性缺失 | c.1021C>T | p.Arg341Trp | NM_000137.1 NP_000128.1 |
| 致病性 | c.192G>T | p.Gln64His | |
| c.554-1G>T (IVS6-1G>T) | -- | ||
| c.607-6T>G (IVS7-6T>G) | -- | ||
| c.698A>T | p.Asp233Val | ||
| c.782C>T | p.Pro261Leu | ||
| c.786G>A | p.Trp262Ter | ||
| c.1009G>A | p.Gly337Ser | ||
| c.1062+5G>A (IVS12+5 G>A) | -- |
注意变异分类:表中列出的变异已由作者提供。genereviews人员没有独立核实变种分类。
命名的注释:Gene Review遵循人类基因组变异学会(www.hgvs.org)的标准命名方法。 有关命名的解释,请见快速参考文献。
1.不符合当前命名法的变异设定
对于在北美患者中检出的其他致病变异,参见表5(pdf)。
正常基因产物。 FAH是作为同源二聚体的细胞溶质蛋白,分子量约80kd的。 野生型FAH 有约3.5μmol/L的FAA km值。 FAH催化FAA转化为富马酸酯和乙酰乙酸酯,并将琥珀酰乙酰乙酸酯转化为琥珀酸酯和乙酰乙酸酯。
异常基因产物。 错义、无义和剪接位点致病性变异最终导致FAH酶活性消失,导致细胞内FAA,琥珀酰乙酰乙酸酯和琥珀酰丙酮的堆积造成导致细胞损伤和凋亡。
参考文献
引用文献
- Ahmad S, Teckman JH, Lueder GT. Corneal opacities associated with NTBC treatment. Am J Ophthalmol. 2002;134:266–8. [PubMed: 12140036]
- Al-Dirbashi OY, Mohamed S, Rashed MS, Jacob M, Al-Ahaideb LY, Al-Amoudi M, Rahbeeni Z, Al-Sayed MM, Al-Hassnan Z, Al-Owain M, Al-Zeidan H. Improved method to determine succinylacetone in dried blood spots for diagnosis of tyrosinemia type 1 using UPLC-MS/MS. Biomed Chromatogr. 2008;22:1181–5. [PubMed: 18651606]
- Allard P, Grenier A, Korson MS, Zytkovicz TH. Newborn screening for hepatorenal tyrosinemia by tandem mass spectrometry: analysis of succinylacetone extracted from dried blood spots. Clin Biochem. 2004;37:1010–5. [PubMed: 15498530]
- Arranz JA, Pinol F, Kozak L, Perez-Cerda C, Cormand B, Ugarte M, Riudor E. Splicing mutations, mainly IVS6-1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat. 2002;20:180–8. [PubMed: 12203990]
- Bansal S, Dhawan A. Acute liver failure. In: Walker WA, Kleinman RE, Sherman PM, Shneider BL, Sanderson IR, eds. Pediatric Gastrointestinal Disease - Pathophysiology, Diagnosis, Management. 4 ed. Ch 58. Hamilton, Ontario: BC Decker, Inc; 2004:1491-507.
- Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis. 2014 Feb 11; [Epub ahead of print] [PubMed: 24515874]
- Bartlett DC, Preece MA, Holme E, Lloyd C, Newsome PN, McKiernan PJ. Plasma succinylacetone is persistently raised after liver transplantation in tyrosinaemia type 1. J Inherit Metab Dis. 2013;36:15–20. [PubMed: 22456946]
- Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. 2005;28:131–5. [PubMed: 15877201]
- Bergeron A, D'Astous M, Timm DE, Tanguay RM. Structural and functional analysis of missense mutations in fumarylacetoacetate hydrolase, the gene deficient in hereditary tyrosinemia type 1. J Biol Chem. 2001;276:15225–31. [PubMed: 11278491]
- Bergeron A, Lettre F, Russo P, Morissette J, Tanguay RM. No evidence of maternal cell colonization in reverted liver nodules of tyrosinemia type I patients. Gastroenterology. 2004;127:1381–5. [PubMed: 15521007]
- Bergman AJ, van den Berg IE, Brink W, Poll-The BT, Ploos van Amstel JK, Berger R. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat. 1998;12:19–26. [PubMed: 9633815]
- Bliksrud YT, Brodtkorb E, Backe PH, Woldseth B, Rootwelt H. Hereditary tyrosinaemia type I in Norway: incidence and three novel small deletions in the fumarylacetoacetase gene. Scand J Clin Lab Invest. 2012;2012;72:369–73. [PubMed: 22554029]
- Bliksrud YT, Brodtkorb E, Andresen PA, van den Berg IE, Kvittingen EA. Tyrosinaemia type I--de novo mutation in liver tissue suppressing an inborn splicing defect. J Mol Med. 2005;83:406–10. [PubMed: 15759101]
- Cassiman D, Zeevaert R, Holme E, Kvittingen EA, Jaeken J. A novel mutation causing mild, atypical fumarylacetoacetase deficiency (Tyrosinemia type I): a case report. Orphanet J Rare Dis. 2009;4:28. [PMC free article: PMC2802351] [PubMed: 20003495]
- Croffie JM, Gupta SK, Chong SK, Fitzgerald JF. Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics. 1999;103:675–8. [PubMed: 10049978]
- Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34:1313–20. [PubMed: 14691918]
- Dubois J, Garel L, Patriquin H, Paradis K, Forget S, Filiatrault D, Grignon A, Russo P, St-Vil D. Imaging features of type 1 hereditary tyrosinemia: a review of 30 patients. Pediatr Radiol. 1996;26:845–51. [PubMed: 8929295]
- Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, Ugarte M, Trefz FK, Leonard JV. Outcome of tyrosinaemia type III. J Inherit Metab Dis. 2001;24:824–32. [PubMed: 11916315]
- Elpeleg ON, Shaag A, Holme E, Zughayar G, Ronen S, Fisher D, Hurvitz H. Mutation analysis of the FAH gene in Israeli patients with tyrosinemia type I. Hum Mutat. 2002;19:80–1. [PubMed: 11754109]
- Endo F, Sun MS. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells. J Inherit Metab Dis. 2002;25:227–34. [PubMed: 12137232]
- Garcia Segarra N, Roche S, Imbard A, Benoist JF, Grenèche MO, Davit-Spraul A, Ogier de Baulny H. Maternal and fetal tyrosinemia type I. J Inherit Metab Dis. 2010;33 Suppl 3:S507–10. [PubMed: 23250512]
- Giguère Y, Ruel J, Belanger N, Grenier A, Laberge C, Quebec Neonatal Blood Screening Programme, CHUL du CHUQ, Quebec, Canada. Neonatal mass screening for hereditary tyrosinemia type 1 in Quebec: a historical perspective (1970-2005). Portland, OR: Newborn Screening and Genetic Testing Symposium. 2005.
- Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21:563–71. [PubMed: 11745044]
- Heath SK, Gray RG, McKiernan P, Au KM, Walker E, Green A. Mutation screening for tyrosinaemia type I. J Inherit Metab Dis. 2002;25:523–4. [PubMed: 12555948]
- Hendriksz CJ, Walter JH. Feeding infants with undiluted goat's milk can mimic tyrosinaemia type 1. Acta Paediatr. 2004;93:552–3. [PubMed: 15188986]
- Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4:805–14. [PubMed: 11232358]
- Kim SZ, Kupke KG, Ierardi-Curto L, Holme E, Greter J, Tanguay RM, Poudrier J, D'Astous M, Lettre F, Hahn SH, Levy HL. Hepatocellular carcinoma despite long-term survival in chronic tyrosinaemia I. J Inherit Metab Dis. 2000;23:791–804. [PubMed: 11196105]
- Kvittingen EA, Rootwelt H, Berger R, Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type I. J Clin Invest. 1994;94:1657–61. [PMC free article: PMC295327] [PubMed: 7929843]
- de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, de Baulny HO, Pintos-Morell G, Spiekerkötter U. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;2013;8:8. [PMC free article: PMC3558375] [PubMed: 23311542]
- Larochelle J, Alvarez F, Bussières JF, Chevalier I, Dallaire L, Dubois J, Faucher F, Fenyves D, Goodyer P, Grenier A, Holme E, Laframboise R, Lambert M, Lindstedt S, Maranda B, Melançon S, Merouani A, Mitchell J, Parizeault G, Pelletier L, Phan V, Rinaldo P, Scott CR, Scriver C, Mitchell GA. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107:49–54. [PubMed: 22885033]
- Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci USA. 1977;74:4641–5. [PMC free article: PMC432003] [PubMed: 270706]
- Macsai MS, Schwartz TL, Hinkle D, Hummel MB, Mulhern MG, Rootman D. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522–7. [PubMed: 11589874]
- Masurel-Paulet A, Poggi-Bach J, Rolland MO, Bernard O, Guffon N, Dobbelaere D, Sarles J, de Baulny HO, Touati G. NTBC treatment in tyrosinaemia type 1: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81–7. [PubMed: 18214711]
- McKiernan PKJ. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006;66:743–50. [PubMed: 16706549]
- Mitchell G, Larochelle J, Lambert M, Michaud J, Grenier A, Ogier H, Gauthier M, Lacroix J, Vanasse M, Larbrisseau A, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432–7. [PubMed: 2153931]
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2001:1777-806.
- Mohan N, McKiernan P, Preece MA, Green A, Buckels J, Mayer AD, Kelly DA. Indications and outcome of liver transplantation in tyrosinaemia type 1. Eur J Pediatr. 1999;158 Suppl 2:S49–54. [PubMed: 10603099]
- Nissenkorn A, Korman SH, Vardi O, Levine A, Katzir Z, Ballin A, Lerman-Sagie T. Carnitine-deficient myopathy as a presentation of tyrosinemia type I. J Child Neurol. 2001;16:642–4. [PubMed: 11575602]
- Park HD, Lee DH, Choi TY, Lee YK, Kim JW, Ki CS, Lee YW. Clinical, biochemical, and genetic analysis of a Korean neonate with hereditary tyrosinemia type 1. Clin Chem Lab Med. 2009;47:930–3. [PubMed: 19569981]
- Pierik LJ, van Spronsen FJ, Bijleveld CM, van Dael CM. Renal function in tyrosinaemia type I after liver transplantation: a long-term follow-up. J Inherit Metab Dis. 2005;28:871–6. [PubMed: 16435179]
- Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab. 1998;64:119–25. [PubMed: 9705236]
- Poudrier J, St-Louis M, Lettre F, Gibson K, Prevost C, Larochelle J, Tanguay RM. Frequency of the IVS12 + 5G-->A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat Diagn. 1996;16:59–64. [PubMed: 8821854]
- Rashed MS, Al-Ahaidib LY, Al-Dirbashi OY, Al Amoudi M, Al-Sayed MM, Rahbeeni Z, Al-Hassnan Z, Al-Dbaas A, Al-Owain M, Ni Luanaigh M. Tandem mass spectrometric assay of succinylacetone in urine for the diagnosis of hepatorenal tyrosinemia. Anal Biochem. 2005;339:310–7. [PubMed: 15797572]
- Sassa S, Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71:625–34. [PMC free article: PMC436912] [PubMed: 6826727]
- Schlune A, Thimm E, Herebian D, Spiekerkoetter U. Single dose NTBC-treatment of hereditary tyrosinemia type I. J Inherit Metab Dis. 2012;2012;35:831–6. [PubMed: 22307209]
- Schulze A, Frommhold D, Hoffmann GF, Mayatepek E. Spectrophotometric microassay for delta-aminolevulinate dehydratase in dried-blood spots as confirmation for hereditary tyrosinemia type I. Clin Chem. 2001;47:1424–9. [PubMed: 11468232]
- Schwetz BA. From the Food and Drug Administration. JAMA. 2002;287:1103. [PubMed: 11879090]
- Techakittiroj C, Cunningham A, Hooper PF, Andersson HC, Thoene J. High protein diet mimics hypertyrosinemia in newborn infants. J Pediatr. 2005;146:281–2. [PubMed: 15689925]
- Vanclooster A, Devlieger R, Meersseman W, Spraul A, Kerckhove KV, Vermeersch P, Meulemans A, Allegaert K, Cassiman D. Pregnancy during nitisinone treatment for tyrosinaemia type I: first human experience. JIMD Rep. 2012;5:27–33. [PMC free article: PMC3509920] [PubMed: 23430914]
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifl). J Pediatr Gastroenterol Nutr. 2005;40:90–3. [PubMed: 15625434]
- van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, Berger R, Heymans HS. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187–91. [PubMed: 7927251]
- Vogel A, van Den Berg IE, Al-Dhalimy M, Groopman J, Ou CN, Ryabinina O, Iordanov MS, Finegold M, Grompe M. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology. 2004;39:433–43. [PubMed: 14767996]
推荐读物
- Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G. Hypertyrosinemia. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 79. New York, NY: McGraw-Hill. Available online. 2014. Accessed 12-21-15.
- Thimm E, Herebian D, Assmann B, Klee D, Mayatepek E, Speikerkoetter U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol Genet Metab. 2011;102:122–5. [PubMed: 21112803]
章节标注
致谢
由食品和药物管理局(FD-4-001445)和罕见病治疗药物项目的支持。 作者感谢加拿大蒙特利尔的Grant Mitchell博士,瑞典哥德堡的Sven Lindstedt博士和Elisabeth Holme博士的合作和讨论。
历年版本
- 17 July 2014 (me) Comprehensive update posted live
- 25 August 2011 (me) Comprehensive update posted live
- 21 October 2008 (cg) Comprehensive update posted live
- 24 July 2006 (me) Review posted to live Web site
- 29 June 2005 (crs) Original submission