
概要
本综述重点介绍了常见 综合征性 和非综合征性遗传性听力损失的临床特征和分子遗传学。本综述关于遗传性听力损失和耳聋的目标如下:
目标 1.
描述遗传性听力损失和耳聋的 临床特征.
目标 2.
回顾遗传性听力损失和耳聋的 病因.
目标 4.
对遗传性听力损失和耳聋患者家属进行 遗传咨询.
目标 5.
回顾遗传性听力损失和耳聋的 管理.
遗传性听力损失和耳聋的临床特征
类型
- 由于外耳和/或中耳小骨异常导致的传导性听力损失.
- 由于内耳结构(即, 耳蜗或听觉神经)功能障碍导致的感觉神经性听力损失.
- 传导性听力损失合并感觉神经性听力损失的混合性听力损失.
- 由于第八颅神经、听觉脑干或大脑皮层损伤或功能障碍导致的中枢性听觉功能障碍.
起病
- 语前性听力损失发生在语言发展之前。所有 先天的 (出生时存在)听力损失都是语前性的, 但不是所有的语前性听力损失都是先天性的.
- 语后性听力损失发生在正常语言发展之后.
听力损失的严重程度. 听力用分贝(dB)进行测量。每个频率的阈值或0分贝是指正常年轻人在 50% 的时间内感知到音调爆发的水平。阈值在正常阈值的15 dB之内都是正常的。听力损失严重程度如 表 1所示.
为了计算听力损失百分比,从500 Hz, 1000 Hz, 2000 Hz, 3000 Hz的纯音平均值中减去25 dB。结果乘以1.5得到耳朵特定的水平。相比于较差的听力通过给较好的听力五倍权重来确定听力障碍[JAMA 1979] (见 表 2).
注意: (1) 因为会话性语言大约在50-60 dB HL (听力水平),所以根据纯音平均值计算功能障碍可能会产生误解。例如, 45-dB 听力损失在功能上比 30% 损伤更严重。(2) 年幼的儿童适合不同的评估量表,因为即使是有限的听力损失也会对语言发展产生很大的影响 [Northern & Downs 2002]。
表 2.
听力障碍百分比
| % 障碍 | 纯音平均值(dB) 1 | % 残余听力 |
|---|---|---|
| 100% | 91 dB | 0% |
| 80% | 78 dB | 20% |
| 60% | 65 dB | 40% |
| 30% | 45 dB | 70% |
纯音平均值为500 Hz, 1000 Hz, 2000 Hz, 3000 Hz
听力损失频率指:
- 低 (<500 Hz)
- 中 (501-2000 Hz)
- 高 (>2000 Hz)
其他术语
"听力障碍" 和 "听力损失" 医疗专业人士经常交换使用,指听力测定所确定的在正常听力阈值水平以下的听力。
耳聋 (小写 "d") 是一种口语术语,指听力测定的听力阈值在重度到极重度之间。
耳聋文化(通常大写 "D"). 在美国耳聋群体中的成员都是耳聋的,他们使用美国手语。与其他文化一样,成员具有独特的社会和社会属性。耳聋群体中的成员(即,耳聋的人) 既不认为自己听力 "受损," 也不认为自己听力"丧失。"相反,他们认为自己是聋子。他们的耳聋并不被认为是一种需要治疗或治愈的病理或疾病。
“听力困难” 比听力更有效。耳聋的人用它来表示一个人有一些可用的听力 - 在轻度到重度听力损失之间的任何听力。在耳聋群体中,耳聋的人不使用口语,而听力困难的人通常会一些口语。
耳聋和听力损失的诊断
生理学测试能客观地确定听觉系统的功能,并且可在任何年龄中进行。它们包括以下内容:
- 听觉脑干反应测试(ABR, 也称为 BAER, BSER) 使用一个刺激 (点刺激)来引发电生理反应,这种反应起源于第八脑神经和听觉脑干,并且能被表面电极记录。在神经学正常个体中,ABR " V波检测阈值" 的听觉灵敏度在 1500- 到 4000-Hz 区域内最佳; ABR 不评估低频率(<1500 Hz) 灵敏度。
- 听觉稳态反应测试(ASSR) 如 ABR,它也是听觉诱发电位,并且以相似的方式测量。ASSR 使用客观的,基于统计的数学检测算法来检测和定义听力阈值。使用宽带或特定频率的刺激可获得ASSR,且在重度到极重度范围内 ASSR 可提供听力阈值差异。 ASSR 通常能给出 ABR 不能给出的特定频率信息。通常使用500, 1000, 2000, 和 4000 Hz 的测试频率。
- 诱发性耳声发射(EOAEs) 是使用带有麦克风和换能器的探头在外耳道中测量的耳蜗内发出的声音。EOAEs 主要反映了在宽频率范围内耳蜗外毛细胞的活动,并且在听觉灵敏度优于40-50 dB HL的耳朵中的出现。
- 引导测试(鼓室压测试, 声学反射阈值,声反射衰减)评估外周听觉系统,包括中耳压力,鼓膜移动性,咽鼓管功能和中耳小骨的活动性。
听力测试 主观地确定个人如何处理听觉信息(即听到)。听觉测试包括行为测试和纯音听觉测试。
- 行为测试 包括行为观察听力测试(BOA)和视觉强化听力测试(VRA)。BOA 可用于从出生到六个月的婴儿,高度依赖于测试者的技术,并且容易出错。VRA 可用于6个月到2.5岁的儿童,可提供可靠、完整的听力图,但依赖于孩子的成熟年龄和测试者的技术。
- 纯音听觉测试(空气和骨传导) 涉及确定个人“听”纯音的最低强度,作为频率(或音调)的函数。使用耳机测试从250 (接近中间 C)到 8000 Hz 的倍频程。强度或响度用分贝(dB)测量,其定义是指两声压之间的比值。0 dB HL 是正常成人听力的平均阈值; 120 dB HL 非常响亮会导致疼痛。此外还要评估语音接收阈值(SRTs)和语言辨别。
- 空气传导听力 通过耳机呈现声音; 阈值取决于外耳道,中耳和内耳的状况。
- 骨传导听力 通过位于乳突骨或前额上的振动器呈现声音,因而绕过外耳和中耳; 阈值取决于内耳的条件。
- 条件播放听力测试(CPA) 用于测试2.5岁至5岁的儿童。 孩子合作的话,可获得每只耳朵完整的特定频率听力图。
- 常规听力测试 用于测试五岁及以上的对象; 受试者需指出何时听到声音。
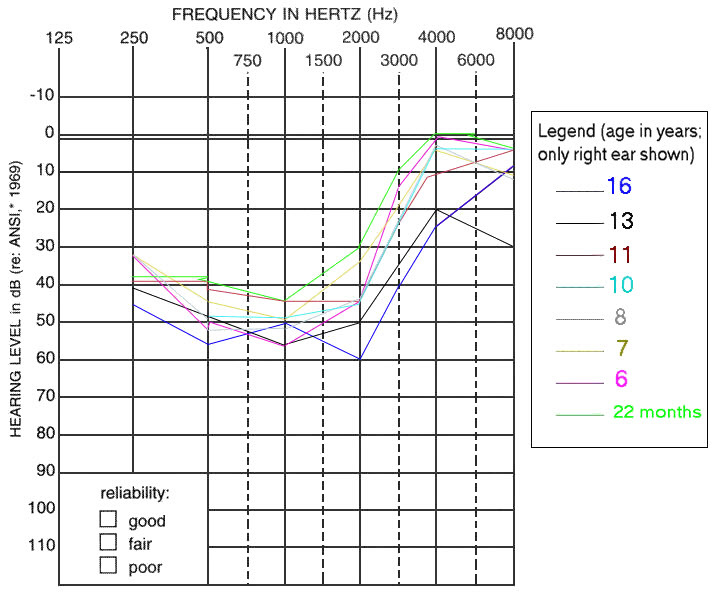
Figure 1. 情景模式
其他
- 先天性听力损失可以通过新生儿听力筛查(NBHS)来发现, 美国国立卫生研究院也倡导这项筛查。NBHS 是43个州和哥伦比亚特区的法律或法规普遍要求的。在其他州有新生儿听力筛查但不做硬性要求。因此,在美国 95% 的新生儿接受了新生儿听力筛查 [NIH RePORT]。
- 父母担心可能的听力损失或观察到的言语发育迟缓,这需要在任何儿童进行听觉筛查。
遗传性听力损失和耳聋的鉴别诊断
语言发育迟缓的儿童应该进行听觉系统评估。在伴有言语渐进性丧失和颞叶癫痫发作的正常听力测试中,应考虑 Landau-Kleffner 综合征的诊断。在患有自闭症谱系障碍或特定言语和语言障碍的幼儿中也可以发生提示可能听力损失的语言延缓。在发达国家,大约
80% 的 先天的 听力损失是由遗传因素引起的,其余为环境(获得性) 因素引起 (图 2)。获得性因素应与遗传因素相鉴别,以告知相应的评估和所需的辅助检查(即 CT, MRI, 和专家咨询) 以及预后和治疗建议。
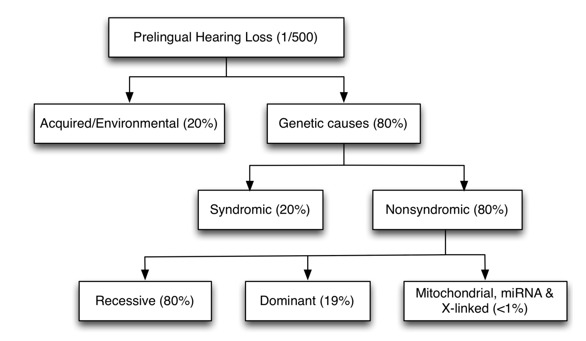
图 2.
发达国家原发性听力损失的病因
儿童获得性听力损失 通常是来自"TORCH" 生物(即弓形体病,风疹,巨细胞病毒和疱疹)的产前感染,或由脑膜炎奈瑟氏球菌,流感嗜血杆菌或肺炎链球菌引起的出生后感染,特别是细菌性脑膜炎。许多其他微生物,包括大肠杆菌、单核细胞增多性李斯特氏菌、无乳链球菌和阴沟肠杆菌等造成的脑膜炎也可导致听力损失。然而,在发达国家,非遗传性 先天的听力损失最常见的环境因素是先天性巨细胞病毒
(cCMV) 感染。其整体出生发生率大约为0.64%; 其中 10% 具有症状性 CMV, 其特征是在神经功能缺损(死亡、癫痫发作、脑性麻痹), 肝功能不全和特征性皮疹等症状中有不同数量和程度的改变。听力损失影响大约 50% 的症状性 cCMV。其余 90% 的cCMV 感染者被认为是“无症状的;” 其中高达 15% 发生单侧或双侧听力损失。因此,大多数 cCMV 感染导致的听力损失患者被分为“无症状的。”
CMV 感染性听力损失的诊断较难,往往可能无法识别,其特征是不同程度的双侧,不对称或单侧感觉神经性听力损失[Kenneson & Cannon 2007]。鉴于cCMV 在环境中普遍存在,对 cCMV 的检测需要高度怀疑,且检测应在出生后21天内完成。针对新生儿听力筛查失败的新生儿,有些州已开展了cCMV 靶向测试。鉴于新的研究显示,有症状的 CMV 患者通过抗病毒治疗可以改善听力损失,因而认识到 cCMV 听力损失越来越重要[Kimberlin et al 2015]。然而,迄今为止,使用抗病毒药物来治疗听力损失是唯一的表现 cCMV 患者仍然是实验性的。
成人获得性听力损失, 主要由环境因素导致,也最可能反映环境遗传之间的相互作用,其中最常见的是年龄相关和噪声导致的听力损失。尽管这两种类型的听力损失都反映了复杂的“环境-遗传” 听力损失,但迄今仅有少数基因的变异与这些特征有关[Yamasoba et al 2013]。在线粒体基因组 (mtDNA) 中具有特异性变异的人群中更有可能发生氨基糖苷诱导性听力损失,这也提示了与医疗相关的环境相互作用
(见非综合征性听力损失和耳聋,线粒体)。
遗传性听力损失和耳聋的病因
遗传性听力损失和耳聋可分为 综合征性 或非综合性(图 2)。综合征性听力障碍与外耳畸形,其他器官畸形或涉及其他器官系统的医疗问题有关。非综合征性听力障碍没有相关的明显外耳异常或任何相关的医疗问题; 然而,它可能与中耳和/或内耳的异常有关。
大约80% 的语前聋是遗传性的,常染色体隐性遗传 和非综合性的最多见。大多数人群中,重到极重度常染色体隐性遗传非综合性听力损失的最常见病因是 GJB2 变异。轻到中度常染色体隐性遗传听力损失的最常见病因是STRC 变异; 值得注意的是,变异都有种族差异 [Sloan-Heggen et al 2016]。
综合性听力障碍
已描述了包括听力损失在内的400多种遗传综合征[Toriello et al 2004]。尽管综合征性 听力障碍在语前聋中占比高达 30%,但与语后聋的发生和诊断相比较,其对所有耳聋的相对贡献要小得多。这里讨论的综合性听力损失按照遗传模式 分类 (表 3)。
表 3.
综合性听力障碍的病因
| MOI | 综合征 | 基因(s) | 听力障碍 | 其他临床特征 | 诊断/筛查/其他 | ||
|---|---|---|---|---|---|---|---|
| 类型 | 起病 | 严重程度 | |||||
| AD | Waardenburg 综合征 (WS) | PAX3 MITF EDNRB EDN3 SOX10 | WS1: 感觉神经的 | 先天性 | 多变的 | 最常见的 AD SHL 类型 皮肤色素异常, 头发 (白色前额发 1), & 眼睛 (异染色质虹膜) 亚型特征:
| |
| Branchiootorenal 谱系障碍 | EYA1 SIX1 SIX5 | 传导的; 感觉神经的; 混合的 | 多变的 | 多变的 | 第二常见 AD SHL BOR 3:鳃裂囊肿或瘘管,外耳耳室耳部畸形, & 肾功能异常 BOS 3: 特征同 BOR 综合征但有/无肾脏累及 高 外显率; 极度可变的表现度 | ||
| Neurofibromatosis 2 (NF2) | NF2 | 感觉神经的 | ~第三个十年 | 通常单侧 & 平缓的; 可以是双侧 & 快速的 | 听力损失继发于双侧前庭神经鞘瘤; 罕见,可能可治疗的耳聋类型 其他各种包括脑膜瘤,星形细胞瘤,室管膜瘤等肿瘤的患病风险, & 脑膜血管瘤病 | 耳蜗病变通常可以通过听力学评估来诊断,尽管明确的诊断需要MRI w /钆对比 | |
| Stickler 综合征 | COL2A1 COL11A1 COL11A2 COL9A1 COL9A2 COL9A3 | 传导的; 感觉神经的 | 多变的 | 多变的 | 结缔组织病变,包括近视,白内障, & 视网膜脱离; 面部发育不良 & 腭裂 (无论是单独还是作为Robin序列的一部分) 脊椎骨骺发育不良和/或 早发关节炎 | ||
| AR | |||||||
| Usher 综合征I型 | MYO7A USH1C CDH23 PCDH15 USH1G CIB2 | 感觉神经的 | 先天性的 | 重度到极重度 | 前庭功能异常 受累患者发现传统扩增无效& 通常需要人工沟通 因为前庭功能障碍,坐& 走的发育关键点通常比正常年龄晚 | 最常见的 AR SHL 类型 双重感觉障碍: 受累的 患者出生时是 w/SHL, 而后进展成 RP 在美国累及超过 50% 失明失聪患者. | |
| Usher 综合征II型 | ADGRV1 WHRN USH2A | 轻度到重度 | 正常前庭功能 助听器为这类患者提供有效的放大; 通常口语沟通 | ||||
| Usher 综合征III型 (OMIM 276902, 614504) | CLRN1 HARS | 进行性的 | 前庭功能进行性 | ||||
| Pendred 综合征 | SLC26A4 4, 5 | 感觉神经的 | 先天性的 | 通常 (虽然不总是) 重度到极重度 | 第二常见 AR SHL 类型 听力 &甲状腺肿 耳聋相关骨迷路异常 (Mondini 畸形或前庭导水管扩张 [扩大] ) 出生时无甲状腺肿,青春期早期 (40%) 或者成年期 (60%)出现 | Mondini 畸形或前庭导水管扩张可通过 CT 检查颞骨进行诊断 | |
| Jervell 和 Lange-Nielsen 综合征 | KCNQ1 KCNE1 | 感觉神经的 | 先天性的 | 极重度 | 第三常见的 AR HL 类型 耳聋 & ECG可检测的QT 间期延长 (异常 QTc [c=校正] >440 msec) 晕厥; 猝死 | 尽管 ECG 筛查敏感度不高,但仍可适用于聋儿筛查。高风险儿童 (即猝死家族史, SIDS, 晕厥, 或长 QT 综合征) 应进行彻底的心脏评估。 | |
| 生物素酶缺乏症 | BTD | 感觉神经的 | 多变的 | 多变的 | 如果漏诊 & 添加生物素的日常饮食不能纠正, 受累的 患者会发生神经功能 (例如, 癫痫发作,高血压,发育迟缓,共济失调) & 视觉问题 一定程度的 HL 出现在 ≥75% 的有症状的皮肤特征 (如皮疹,脱发,结膜炎)的儿童中 | 用生物素可以解决的神经 & 皮肤表现; 听力损失 & 视神经萎缩通常是不可逆转的 当儿童出现发作性或者进行性共济失调 & 进行性感觉神经性耳聋 ± 神经的 或 皮肤综合征, 考虑生物素酶缺乏症。 为阻止代谢性昏迷, 尽快进行饮食 & 治疗 ASAP. | |
| Refsum 病 | PHYH PEX7 | 感觉神经的 | 多变的 | 重度; 进行性的 | 厌食症 & 早期发作性视网膜色素变性– 神经病变,耳聋,共济失调& 鱼鳞病的不同组合的普遍发现 | 尽管非常罕见,但当耳聋患者可被日常饮食修正时考虑 Refsum 病 & 血浆麻痹症. 通过测定血清中植烷酸的浓度可确定诊断6. | |
| Alport 综合征 7 | COL4A5 COL4A3 7 COL4A4 7 | 感觉神经的 | 通常在10岁以后 | 不同程度; 进行性的 | 累及肾脏、耳蜗和眼睛 如未治疗, 肾脏疾病从微量血尿到蛋白尿, 进行性肾功能不全, ESRD进展 | ||
| XL | |||||||
| 耳聋-肌张力障碍-视神经病变综合征 (Mohr-Tranebjaerg 综合征) | TIMM8A | 感觉神经的 | 儿童早期 | 进行性的; 语前聋- 或 语后聋 | 视力障碍, 肌张力障碍, 骨折, 智力障碍 | ||
MOI = 遗传模式
AD = 常染色体显性遗传
AR = 常染色体隐性遗传
XL = X-linked
SHL = 综合征性 听力损失
ESRD =终末期肾病
RP = 色素性视网膜炎
1.
因为 受累的 患者可能会染发,所以询问病史和体格检查时应特别注意前额白色的症状。
2.
内眦外移: 眼内眦向外侧移。
3.
腮-耳-肾疾病谱包括腮-耳-肾 综合征 (BOR)和腮-耳综合征 (BOS)。
4.
在Pendre综合征中存在双基因遗传,即 受累的 患者有 SLC26A4 和 FOXI1致病性变异 的双重杂合子 [Yang et al 2007] 或者有 SLC26A4 和 KCNJ10 致病性变异的双重杂合子。
5.
SLC26A4 也与非综合征性听力损失有关 (DFNB4)。
6.
7.
X-连锁 遗传大约占Alport 综合征 85%; 常染色体隐性遗传 大约占病例的15%; 常染色体显性遗传 偶有报道。
非综合征性听力障碍
命名. 因为历史原因, 非综合征性听力障碍可用涉及的 基因 (例如, OTOF-相关的耳聋) 或用遗传位点 (例如, DFNB9)来命名。非综合征性位点被命名为 DFN (DeaFNess) ,进一步通过 遗传模式 分类(DFNA: 常染色体显性遗传; DFNB: 常染色体隐性遗传; DFNX: X-连锁) 数字表示基因做图和/或发现的顺序)。
遗传. 语前性非综合征性听力损失的遗传模式中 80% 为常染色体隐性遗传, 20% 常染色体显性遗传, 和 1%-1.5% X-连锁, 线粒体或其他遗传模式 (图 2) [Smith et al 2005]. 尽管尚无语前性非综合征性听力损失的类似数据, 但大多数已报道的家族均显示常染色体显性遗传。
遗传异质性. 非综合征性遗传性听力损失以明显的遗传异质性为特征: 迄今, 在超过110个基因中已发现超过 6,000 致病性变异。在迄今为止最大的一项使用表型靶向检测 进行全面遗传学检测的研究中,在440个确定了遗传诊断的患者中发现了超过 40 个致病性基因 [Sloan-Heggen et al 2016]。这种明显的遗传异质性强调了使用多基因测序包进行遗传诊断的重要性(见 评估策略)。
常染色体显性非综合征性听力障碍
超过 25 个基因与 常染色体显性遗传 非综合征性听力损失有关。这些基因都涉及常染色体显性非综合性听力障碍,其临床表现总结见 表 4。注意情景模式可以是独特的,因此可以指导 基因型-表型 关系 [Taylor et al 2013] (见 评估策略)。
- GJB2 和 GJB6 (DFNA3), TECTA (DFNA8/12), 和 DFNA19是语前性听力损失。
- WFS1 (DFNA6/14/38) 值得注意,因为其导致的听力损失主要影响低频。
表 4.
| 基因 | 位点 | 起病/时间 | 情景模式 |
|---|---|---|---|
| ACTG1 | DFNA20/26 | 语后性 | 高频; 进行性的 |
| CCDC50 | DFNA440 | 语后性 | 低到中等频率; 进行性的 |
| CD164 | DFNA66 | 语后性 | 平缓 或 中等-频率; 进行性的 |
| CEACAM16 | DFNA4B | 语后性 | 平缓; 进行性的 |
| COCH | DFNA9 | 语后性/2nd | 高频; 进行性的 |
| COL11A2 | DFNA13 | 语后性/2nd | 中频缺失 |
| GSDME | DFNA5 | 语后性/1st | 高频; 进行性的 |
| DIAPH1 | DFNA1 | 语后性/1st | 低频; 进行性的 |
| DMXL2 | - | 语后性/2nd | 平缓; 进行性的 |
| DSPP | DFNA39 | 语后性 | 高频; 进行性的 |
| EYA4 | DFNA10 | 语后性/3rd, 4th | 平缓/轻轻下降 |
| GJB2 1 | DFNA3 | 语前性 | 高频; 进行性的 |
| GJB3 | DFNA2B | 语后性/4th | 高频; 进行性的 |
| GJB6 1 | DFNA3 | 语前性 | 高频; 进行性的 |
| GRHL2 | DFNA28 | 语后性 | 平缓/轻轻下降 |
| HOMER2 | DFNA68 | 语后性/1st | 高频; 进行性的 |
| KCNQ4 | DFNA2 | 语后性/2nd | 高频; 进行性的 |
| MIR96 | DFNA50 | 语后性/2nd | 平缓; 进行性的 |
| MCM2 | DFNA70 | 语后性 | 高频; 进行性的 |
| MYH14 | DFNA4 | 语后性 | 平缓/轻轻下降 |
| MYH9 | DFNA17 | 语后性 | 高频; 进行性的 |
| MYO1A | DFNA48 | 语后性 | 进行性的 |
| MYO6 | DFNA22 | 语后性 | 高频; 进行性的 |
| MYO7A 2 | DFNA11 | 语后性/1st | 平缓/轻轻下降 |
| OSBPL2 | DFNA67 | 语后性 | 高频; 进行性的 |
| P2RX2 | DFNA41 | 语后性 | 平缓; 进行性的 |
| POU4F3 | DFNA15 | 语后性 | 高频; 进行性的 |
| SIX1 | DFNA23 | 语前性 | 下降 |
| SLC17A8 | DFNA25 | 语后性/2nd-6th | 高频; 进行性的 |
| TBC1D24 | DFNA65 | 语后性 | 高频; 进行性的 |
| TECTA 3, 4 | DFNA8/12 | 语前性 | 中等频率缺失 |
| TJP2 & FAM189A2 | DFNA51 | 语后性/4th | 高频; 进行性的 |
| TMC1 | DFNA36 | 语后性 | 平缓/轻轻下降 |
| WFS1 5 | DFNA6/14/38 | 语前性 | 低频; 进行性的 |
常染色体隐性非综合征性听力障碍
大型研究已强调 常染色体隐性遗传 非综合征性耳聋显著的遗传异质性。常染色体隐性遗传非综合征性听力障碍有关的70个基因及其临床表现总结见 表 5。
虽然在一些世界人群中, 高达 50% 重度到极重度 常染色体隐性遗传 非综合性听力损失的患者具有 GJB2 致病性变异(见 DFNB1), 但是最近的研究表明,GJB2 致病性变异对耳聋的贡献因种族而异 [Sloan-Heggen et al 2016]。例如,在非洲人后裔中,GJB2 的致病性变异非常罕见 [Rudman et al 2017]。
Table 5.
| 基因 | 位点 | 起病 | 类型 |
|---|---|---|---|
| ADCY1 | DFNB44 | 语前的 | 轻到中度; 稳定 |
| BDP1 | DFNB49 | 语后的 | 高频; 稳定 |
| BSND | DFNB73 | 语前的 | 重到极重度; 稳定 |
| CABP2 | DFNB93 | 语前的 | 中到重度; 稳定 |
| CDC14A | DFNB105 | 语前的 | 重到极重度 |
| CDH23 1 | DFNB12 | 语前的 | 重到极重度; 稳定 |
| CIB2 | DFNB48 | 语前的 | 重到极重度 |
| CLDN14 | DFNB29 | 语前的 | 重到极重度; 稳定d |
| CLIC5 | DFNB103 | 语前的 | 高频; 进行性的 |
| COL11A2 | DFNB53 | 语前的 | 重到极重度; 稳定的 |
| DCDC2 | DFNB66 | 语前的 | 重到极重度 |
| PJVK | DFNB59 | 语前的 | 重到极重度; 稳定的 |
| ELMOD3 | DFNB88 | 语前的 | 重到极重度; 混合的 |
| EPS8 | DFNB102 | 语前的 | 重到极重度 |
| EPS8L2 | - | 语后的 | 高频; 进行性的 |
| ESPN | DFNB36 | 语前的 | — |
| ESRRB | DFNB35 | 未知 | 重到极重度 |
| GIPC3 2 | DFNB15/72/95 | 语前的 | 重到极重度 |
| GJB2 3 | DFNB1 | 语前的 4 | 通常 稳定的 |
| GJB6 3 | DFNB1 | 语前的 4 | 通常 稳定的 |
| GPSM2 | DFNB32/82 | 语前的 | 重到极重度; 稳定的 |
| GRXCR1 | DFNB25 | 语前的 | 中到极重度; 进行性的 |
| GRXCR2 | DFNB101 | 语前的 | 高频; 进行性的 |
| HGF | DFNB39 | 语前的 | 重到极重度; 下降 |
| ILDR1 | DFNB42 | 语前的 | 中到重度的 |
| KARS | DFNB89 | 语前的 | 中到重度的; 稳定的 |
| LHFPL5 | DFNB67 | 语前的 | 重到极重度; 稳定的 |
| LOXHD1 | DFNB77 | 语后的 | 中到极重度; 进行性的 |
| LRTOMT | DFNB63 | 语前的 | 重到极重度; 稳定的 |
| MARVELD2 | DFNB49 | 语前的 | 中到极重度; 稳定的 |
| MET | DFNB97 | 语前的 | 重到极重度 |
| MSRB3 | DFNB74 | 语前的 | 重到极重度 |
| MYO15A | DFNB3 | 语前的 | 重到极重度; 稳定的 |
| MYO3A | DFNB30 | 语前的 | 重到极重度; 稳定的 |
| MYO6 | DFNB37 | 语前的 | — |
| MYO7A 5 | DFNB2 | 语前的, 语后的 | 不明 |
| NARS2 6 | DFNB94 | 语前的 | 重到极重度; 稳定的 |
| OTOG | DFNB18B | 语前的 | 轻到中度的; 稳定的 |
| OTOGL | DFNB84 | 语前的 | 高频; 稳定的 |
| OTOA | DFNB22 | 语前的 | 重到极重度; 稳定的 |
| OTOF | DFNB9 | 语前的 | 通常重到极中度; 稳定的 |
| PCDH15 | DFNB23 | 语前的 | 重到极重度; 稳定的 |
| PNPT1 | DFNB70 | 语前的 | 重到极重度; 稳定的 |
| PTPRQ | DFNB84 | 语前的 | 中到极重度; 进行性的 |
| RDX | DFNB24 | 语前的 | 重到极重度; 稳定的 |
| RIPOR2 | DFNB104 | 语前的 | 重到极重度 |
| ROR1 7 | - | 语前的 | 重到极重度 |
| S1PR2 | DFNB68 | 语前的 | 重到极重度 |
| SERPINB6 | DFNB91 | 语前的 | 中到中度 |
| SLC22A4 | DFNB60 | 语前的 | 重到极重度 |
| SLC26A4 8 | DFNB4 | 语前的, 语后的 | 稳定的; 进行性的 |
| SLC26A5 | DFNB61 | 语前的 | 重到极重度; 稳定的 |
| STRC | DFNB16 | 语前的 | 重到极重度; 稳定的 |
| SYNE4 | DFNB76 | 语前的 | 高频; 进行性的 |
| TECTA 9, 10 | DFNB21 | 语前的 | 重到极重度; 稳定的 |
| TBC1D24 | DFNB86 | 语前的 | 重到极重度 |
| TMC1 | DFNB7/11 | 语前的 | 重到极重度; 稳定的 |
| TMEM132E | DFNB99 | 语前的 | 重到极重度 |
| TMIE | DFNB6 | 语前的 | 重到极重度; 稳定的 |
| TMPRSS3 | DFNB8/10 | 语后的 11, 语前的 | 进行性的; 稳定的 |
| TPRN | DFNB79 | 语前的 | 重到极重度; 稳定的 |
| TRIOBP | DFNB28 | 语前的 | 重到极重度; 稳定的 |
| TSPEAR | DFNB98 | 语前的 | 重到极重度 |
| USH1C 12 | DFNB18 | 语前的 | 重到极重度; 稳定的 |
| WBP2 | - | 语前的 | 高频; 进行性的 |
| WHRN | DFNB31 | 语前的 | — |
1.
CDH23致病性变异也与 Usher 综合征1D型有关。
2.
GIPC3致病性变异与听源性惊厥有关。
3.
4.
语前聋也包括 先天的 耳聋。
5.
MYO7A致病性变异也与 DFNA11 (常染色体显性遗传 非综合征性听力障碍) 和 Usher 综合征 1B有关。
6.
NARS2致病性变异也能导致 Leigh 综合征。
7.
8.
SLC26A4致病性变异也与
Pendred 综合征有关。
9.
10.
11.
DFNB8 听力损失的起病是语后的 (10-12 岁), 但 DFNB10 听力损失的起病是语前的(先天的)。这种表型差异反映了基因型差异: DFNB8-致病性变异 是 剪接位点 变异, 提示无效 剪接 与正常蛋白质量的减少有关,这种蛋白的减少足以阻止语前聋,但不足以阻止最终的耳聋。
12.
USH1C致病性变异也与 Usher 综合征 1C型有关。
X-连锁非综合征性听力障碍
涉及 X-连锁 非综合征性听力障碍及其临床表现的基因总结在 表 6.
非综合征性听力损失和耳聋, 线粒体
线粒体基因中的大多数致病性变异都导致广泛的母系遗传多系统疾病; 但是一些基因中的变异, 主要是 MT-RNR1 和 MT-TS1, 通过目前未知的机制导致非综合性听力损失 [Fischel-Ghodsian 1998] (见 表 7 和 非综合征性听力损失和耳聋, 线粒体)。
MT-RNR1 编码 12S 核糖体 RNA。该基因中的变异, 1555G>A, 是母系遗传性非综合性听力损失的常见原因之一。在一些有1555G>A 变异的患者中, 服用适当剂量的氨基糖苷类会诱导听力损失; 但是, 表型变异很大,这种差异与修饰基因的作用一致[Kokotas et al 2007]。
MT-TS1 编码转运 RNASer(UCN)。已发现了在该 基因的nt7445 A-到-G 线粒体DNA异质性 的两个家系; 但是, 听力损失的 外显率 低, 这提示 MT-TS1 致病性变异在听力损失方面起着微不足道的作用。
MT-CO1 编码细胞色素 c 氧化酶亚基 1。6个具有重度到极重度耳聋患者显示MT-CO1 基因中 线粒体DNA同质的 nt7444 G-to-A 和 MT-RNR1基因 1555A>G 致病性变异的共分离[Pandya et al 1999]。六个患者中有五个显示母系遗产,且其中二人有氨基糖苷类药物使用史。与MT-RNR1 1555A>G有关的听力损失的多变性相反, 所有具有这种双重突变的患者都有重到极重度的损伤,并且完全 外显率。
Table 7.
线粒体非综合征性听力障碍: 基因及其临床表现
| 基因 | 致病性变异 | 严重度 | 外显率 |
|---|---|---|---|
| MT-RNR1 | 961 不同变异 | 多变的 | 高度多变的, 氨基糖苷类诱导 |
| 1494C>T | |||
| 1555A>G | |||
| MT-TS1 | 7445A>G | 高度多变的 | |
| 7472insC | |||
| 7510T>C | |||
| 7511T | |||
| MT-CO1 | 7444G>A | 重到极重度 | 完全, 氨基糖苷类相关的; 与 MT-RNR1 1555A>G有关 |
评估策略
最近的指南强调了遗传检测对评估听力损失患者的重要性。见Alford et al [2014] (全文), Liming et al [2016] (全文)和图3。
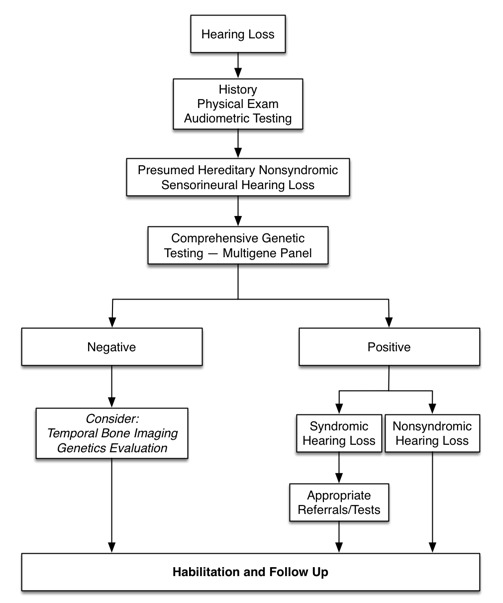
图3. 遗传性非综合性感觉神经性听力损失的诊断流程
因为分子遗传学检测是具有诊断率最高的单一类型的检测,所以除非既往史,体格检查和/或听力测试提示某种听力损失的特异性综合征类型,否则在评估遗传性感觉神经性听力损失和耳聋的待诊患者时都应首先进行分子遗传学检测(图3和表3).
初步评估 应始终包括以下内容:
- 家族史。 应尽量获得三代家族史,且注意其他患有听力丧失或具有相关发现的亲属。通过直接对这些亲属进行检查或查阅他们的医疗记录,包括听力图、耳科检查和分子遗传学检测,来完成亲属的相关记录。
- 听力学测试。 听觉状态可以在任何年龄确定(见诊断耳聋和听力损失)。
- 进行性听力损失的患者应评估Alport 综合征,Pendred 综合征,和Stickler 综合征。另外,还应考虑进行颞骨计算机断层扫描来评估前庭导水管的扩大。
进一步评估 应该基于上述检查结果进行。在明显的非综合性感觉神经性听力损失的情况下,应该在其他(辅助)检查之前先行基因测试(图3)。
分子遗传检测。 导致遗传性听力损失和耳聋常见综合征的已知基因包含在表3中。因为常染色体显性遗传综合征耳聋常有可变的表现度,因此准确的诊断可能依赖于分子遗传学检测。非特异性遗传性听力损失和耳聋的诊断是建立于已确定先证者特定基因的致病性变异(见表 4, 5, 6, 和 7)。
遗传性听力损失和耳聋的分子遗传检测过去常依赖于单基因检测;但是这种测试方法在很大程度上已被多基因组所取代,多基因组较全面(即包括已知导致耳聋的所有基因或已知导致常染色体隐性遗传或常染色体显性遗传听力损失和耳聋的所有基因) 无论是哪种遗传或种族,多基因检测包大大提高了诊断率[Shearer & Smith 2015]。在挑选的实例中,外显子组测序已被用于诊断,并且可以提供全面的遗传性听力损失和耳聋遗传检查手段,尽管这种方法容易漏掉小拷贝数变异[Zazo Seco et al 2017].
听力损失和耳聋的多基因检测包建议使用表3,4,5,6,和7中列出的大多数基因。 注:(1)基因检测包中的基因和每个基因检测的诊断敏感性因实验室和时间而异。(2)一些多基因检测包可能包括与此GeneReview中讨论的情况无关的基因;因此临床医生需要确定哪个表型靶向检测以最合理的成本鉴定出导致疾病的基因,同时可限制识别不能解释潜在表型的基因致病变异。临床医生应评估多基因检测基因包所包含的基因,并确定患者的病史和体格检查是否与明显的非综合性听力损失一致。(3)基因检测包中使用的方法可能包括序列分析,缺失/重复分析和/或其他非测序的检测。在听力损失/耳聋的评估中,因为拷贝数变异是遗传性非综合性听力损失的常见原因,因此包括检测缺失和重复的多基因检测包是必不可少的[Shearer et al 2014]。有关多基因检测包的更多信息,请点击这里。具有综合征性听力损失特征性表现的患者(表3)可以使用基因靶向测试来进行诊断。值得注意的是,目前许多遗传性听力损失和耳聋相关的多基因检测包包含了综合性听力损失的最常见原因,包括那些在继发表现和症状出现前类似非综合性听力损失(例如,Usher综合征[失明];Pendred综合征[甲状腺甲状腺肿])。
颞骨CT或MR。 颞骨的专用薄切CT或MRI可用于检测内耳畸形(即Mondini畸形,米歇尔发育不良,前庭导水管扩大,内耳道扩张),进行性听力损失的患者应考虑行该检查。
辅助检查。 一般不常规进一步评估心脏、肾脏或眼科,除非有相关临床表现或遗传检测结果提示有其他器官系统受累。
遗传咨询
遗传咨询是向患者及家属提供有关遗传疾病的性质、遗传和影响的信息,以帮助他们做出明智的医疗和个人选择。以下部分涉及遗传风险评估和家族史及基因检测的使用,以明确家庭成员的遗传状态。本部分不解决患者可能面对的所有个人的、文化的或伦理的问题,也不能替代遗传学专家的咨询。—ED.
家庭成员的患病风险 – 常染色体显性遗传性听力损失
先证者父母
- 大多数诊断为常染色体显性遗传听力损失的患者父母患有耳聋;家族史很少是阴性的。
先证者同胞
- 根据具体诊断,具有相同突变的不同患者之间临床严重程度和表型可能不同;因此,发病年龄和/或进展可能无法预测。
先证者后代
- 根据具体诊断,具有相同突变的不同患者之间临床严重程度和表型可能不同;因此,发病年龄和/或进展可能无法预测。
其他家庭成员 其他家庭成员的患病风险取决于先证者父母的情况:如果父母之中有致病性变异,他或她的亲戚都有患病风险。
携有明显新发致病性变异家庭的注意事项 患有常染色体显性遗传性耳聋的先证者其父母若没有先证者具有的致病突变或疾病的临床证据时,该突变可能是新发的。但也可能包括其他亲子关系或代孕(例如辅助生育)的非医学解释和不公开的收养。
家庭成员的风险 – X-连锁遗传性听力损失
先证者父母
先证者同胞
- 女性先证者. 同胞的患病风险取决于父母的遗传类型:
先证者后代
家庭成员的患病风险 – 经验风险
如果不能确定特定的诊断(和/或不能确定具有耳聋或听力损失阳性家族史的人的遗传模式),可以使用以下经验数据。
曾生育一个耳聋患儿且耳聋家族史阴性的正常听力夫妇再生育小孩,其小孩有18%的耳聋患病的经验概率[Green et al 1999].
耳聋和正常听力的人的后代有10%耳聋患病的经验风险[Green et al 1999]。
排除常染色体显性遗传耳聋的非近亲婚配的耳聋夫妇的孩子有大约15%的耳聋经验风险[Green et al 1999]。
- 如果父母双方都有GJB2相关的耳聋,他们后代的患病风险是100%。
耳聋先证者(疑有常染色体隐性遗传非综合征性耳聋)和耳聋患者的听力正常同胞的小孩有1/200(0.5%)的耳聋患病经验风险,是普通人群风险的五倍。
GJB2和GJB6的分子遗传学检测可以确定风险是否更高。如果听力正常的同胞是GJB2耳聋致病突变或GJB6耳聋致病突变的携带者,并且其生育伴侣患有DFNB1耳聋,那么他们生育耳聋患儿的几率是50%。
相关的遗传咨询问题
参见“风险亲属的管理和评估”,了解有关评估亲属风险的信息以早期诊断和治疗。以下几点值得注意:
- 与耳聋群体成员和打手语的人交流,需要熟练的口译员。
- 耳聋群体成员可能认为耳聋是一个显著的特征,而不是一种需要“治疗”、“治愈”或“被预防”的障碍、残疾、或医疗状态。
- 许多耳聋患者有兴趣获得关于自己耳聋原因的信息,包括有关医疗、教育和社会服务的信息,而不是关于预防、生殖或计划生育的信息。因此,确定和解决家庭或者个人的问题和担忧很重要。
- 某些术语优先使用:概率或机会与风险;耳聋和听力障碍vs听力受损。 应该避免诸如“异常”之类的词语。
家庭计划
- 确定遗传状态的最佳时间和讨论产前检查有效性都是在孕前。
- 向耳聋年轻人提供遗传咨询(包括子女耳聋的可能性和生殖选择的讨论)是适当的。
DNA银行 是储存DNA(通常从白血细胞中提取)以备将来使用。因为未来我们对基因、突变位点和遗传性听力损失的的检测技术和理解都可能有所提高,因此应该建议受累的患者储存DNA。
产前检查和植入前基因诊断
一旦在家族中确定了致病性变异,就可能增加产前诊断和为耳聋或听力损失患者使用植入前遗传诊断。
医学专业人员和患病家族内部可能存在关于产前检查使用方面的差异,特别是如果检测是用于终止妊娠而不是早期诊断。虽然大多数中心会考虑将产前检查的决定交给父母来选择,但这些问题的讨论是合适的。
资源
GeneReviews的工作人员已经选择了以下疾病特定和/或综合支持组织和/或登记处,以帮助患有此病的人及其家属。GeneReviews不负责其他组织提供的信息。有关选择标准的信息,请点击这里。
- American Society for Deaf Children (ASDC)800 Florida Avenue NortheastSuite 2047Washington DC 20002-3695Phone: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)Fax: 410-795-0965Email: info@deafchildren.org; asdc@deafchildren.org
- My46 Trait Profile
- National Association of the Deaf (NAD)8630 Fenton StreetSuite 820Silver Spring MD 20910Phone: 301-587-1788; 301-587-1789 (TTY)Fax: 301-587-1791Email: nad.info@nad.org
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- Alexander Graham Bell Association for the Deaf and Hard of Hearing3417 Volta Place NorthwestWashington DC 20007Phone: 866-337-5220 (toll-free); 202-337-5220; 202-337-5221 (TTY)Fax: 202-337-8314Email: info@agbell.org
- my baby's hearingThis site, developed with support from the National Institute on Deafness and Other Communication Disorders, provides information about newborn hearing screening and hearing loss.
管理
临床表现的治疗
理想情况下,评估和治疗耳聋患者的团队应该包括具有幼儿耳科疾病管理专业知识的耳鼻喉科医师,评估儿童听力损失经验丰富的听力学家,临床遗传学家和儿科医生。耳聋教育者、神经科医生和儿科眼科医生的专业知识也是需要的。评估的一个重要部分是确定适当的适应方案。可能情况包括助听器,振动触觉装置和人工耳蜗植入。12个月以上重度到极重度听力损失的儿童可以考虑人工耳蜗植入。
遗传性听力丧失和耳聋儿童评估和治疗的最终目标是主流教育。研究表明,对于轻到中度听力损失的儿童,三个月内诊断且六个月内进行小儿康复治疗可以实现这个目标。人工耳蜗植入的重度到极重度耳聋患儿是主流教育中的一部分,人工耳蜗的植入使得他们的社会功能和教育程度与听力正常的同龄人一样[Loy et al 2010, Langereis & Vermeulen 2015]。最近的研究主要是基于基因的人工耳蜗性能。因为耳聋的遗传异质性,每个基因的性能很难获得大样本量。但是,数据清楚地显示,与GJB2J相关的听力损失患者(见非听觉性听力丧失和耳聋, DFNB1)具有显著的人工耳蜗植入结局,其明显优于那些环境因素导致耳聋的患者[Yoshida et al 2013, Abdurehim et al 2017]。
在成人中,当遗传缺陷影响听觉神经本身时,人工耳蜗的性能可能会受到影响;但这个假设仍需要进一步的研究[Shearer et al 2017].
DFNX3是以混合的导电性感觉神经性听力损失为特征,其传导成分是由镫骨的固定引起的。与其他类型的传导性听力损失相反,外科矫正DFNX3相关听力损失可能损害听力。脑脊髓液和外淋巴之间的异常通信可能导致术中液体渗漏(“外淋巴液溢”),并在开窗术或镫骨底板移除时完全丧失听力。
主要临床表现的预防
无论何时儿童出现进行性感觉神经性听力损失和进行性共济失调,伴或不伴神经或皮肤症状时,应考虑生物素酶缺乏症,并应尽早开始治疗以防止不可逆转的后遗症。
预防继发性并发症
无论是哪种病因,未纠正的听力损失都有一致的后遗症。两岁时听觉剥夺与阅读表现差、沟通能力差以及语言能力缺乏有关。教育干预不足以完全弥补这些缺陷。相反,早期听觉干预是有效的——无论是通过放大、耳科手术还是人工耳蜗植入[Smith et al 2005]。
虽然数学和阅读的认知能力和表现下降与耳聋有关,但对遗传性听力损失患者的检查表明,这些缺陷本质上并非耳聋导致。例如,对GJB2相关听力损失患者的认知能力评估显示,人工耳蜗植入术后患者具有正常的Hiskey IQ和正常的阅读表现[Bauer et al 2003]。因此,早期诊断和及时干预对语言发展前的聋儿童的最佳认知发展至关重要。
监控
连续的听觉检查必不可少对于:
- 记录听力损失的稳定性或进展程度;
- 诊断和治疗叠加的听力损失,如中耳积液。
由SLC26A4的致病突变导致的常染色体隐性遗传的非综合征性听力损失患者,听力损失可加重且可能需要每年都进行听力测试。另外,如果诊断符合Pendred 综合征,还应追踪甲状腺功能。
Agents/避免情况
噪音暴露是公认的导致听力损失的环境因素。因为这种风险可以通过避免噪音暴露来达到最小化,因此应对记录有听力损失的人进行适当的咨询。
参考文献
出版的指南 / 共识
- Alford RL, Arnos KS, Fox M, Lin JW, Palmer CG, Pandya A, Rehm HL, Robin NH, Scott DA, Yoshinaga-Itano C; ACMG Working Group on Update of Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss; Professional Practice and Guidelines Committee. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Available online. 2014. Accessed 7-27-17. [PubMed: 24651602]
引用文献
- Abdurehim Y, Lehmann A, Zeitouni AG. Predictive value of GJB2 mutation status for hearing outcomes of pediatric cochlear implantation. Otolaryngol Head Neck Surg. 2017;157:16-24. [PubMed: 28322114]
- Bauer PW, Geers AE, Brenner C, Moog JS, Smith RJ. The effect of GJB2 allele variants on performance after cochlear implantation. Laryngoscope. 2003;113:2135-40. [PubMed: 14660916]
- Fischel-Ghodsian N. Mitochondrial mutations and hearing loss: paradigm for mitochondrial genetics. Am J Hum Genet. 1998;62:15-9. [PMC free article: PMC1376819] [PubMed: 9443888]
- Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the Midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211-6. [PubMed: 10376574]
- JAMA. Guide for the evaluation of hearing handicap. JAMA. 1979;241:2055-9. [PubMed: 430800]
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-76. [PubMed: 17579921]
- Kimberlin DW, Jester PM, Sánchez PJ, Ahmed A, Arav-Boger R, Michaels MG, Ashouri N, Englund JA, Estrada B, Jacobs RF, Romero JR, Sood SK, Whitworth MS, Abzug MJ, Caserta MT, Fowler S, Lujan-Zilbermann J, Storch GA, DeBiasi RL, Han JY, Palmer A, Weiner LB, Bocchini JA, Dennehy PH, Finn A, Griffiths PD, Luck S, Gutierrez K, Halasa N, Homans J, Shane AL, Sharland M, Simonsen K, Vanchiere JA, Woods CR, Sabo DL, Aban I, Kuo H, James SH, Prichard MN, Griffin J, Giles D, Acosta EP, Whitley RJ, et al. Valganciclovir for symptomatic congenital cytomegalovirus disease. NEJM. 2015;372:933-43. [PMC free article: PMC4401811] [PubMed: 25738669]
- Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clin Genet. 2007;71:379-91. [PubMed: 17489842]
- Langereis M, Vermeulen A. School performance and wellbeing of children with CI in different communicative–educational environments. Int J Pediatr Otorhinolaryngol. 2015;79:834-9. [PubMed: 25840945]
- Liming BJ, Carter J, Cheng A, Choo D, Curotta J, Carvalho D, Germiller JA, Hone S, Kenna MA, Loundon N, Preciado D, Schilder A, Reilly BJ, Roman S, Strychowsky J, Triglia JM, Young N, Smith RJ. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: Hearing loss in the pediatric patient. Int J Pediatr Otorhinolaryngol. 2016;90:251-8. [PubMed: 27729144]
- Loy B, Warner-Czyz AD, Tong L, Tobey EA, Roland PS. The children speak: an examination of the quality of life of pediatric cochlear implant users. Otolaryngol Head Neck Surg. 2010;142:247-53. [PMC free article: PMC2852181] [PubMed: 20115983]
- Northern JL, Downs M. Hearing in Children. Baltimore, MD: Lippincott, Williams, and Wilkins; 2002.
- Pandya A, Xia X-J, Erdenetungalag R, Amendola M, Landa B, Radnaabazar J, Dangaasuren B, Van Tuyle G, Nance WE. Heterozygous point mutations in the mitochondrial tRNA Ser(UCN) precursor coexisting with the A1555G mutation in deaf students from Mongolia. Am J Hum Genet. 1999;65:1803-6. [PMC free article: PMC1288397] [PubMed: 10577941]
- Rudman JR, Kabahuma RI, Bressler SE, Feng Y, Blanton SH, Yan D, Liu XZ. The genetic basis of deafness in populations of African descent. J Genet Genomics. 2017;44:285-94. [PubMed: 28642064]
- Zazo Seco C, Wesdorp M, Feenstra I, Pfundt R, Hehir-Kwa JY, Lelieveld SH, Castelein S, Gilissen C, de Wijs IJ, Admiraal RJ, Pennings RJ, Kunst HP, van de Kamp JM, Tamminga S, Houweling AC, Plomp AS, Maas SM, de Koning Gans PA, Kant SG, de Geus CM, Frints SG, Vanhoutte EK, van Dooren MF, van den Boogaard MH, Scheffer H, Nelen M, Kremer H, Hoefsloot L, Schraders M, Yntema HG. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet. 2017;25:308-14. [PMC free article: PMC5315517] [PubMed: 28000701]
- Shearer AE, Eppsteiner RW, Frees K, Tejani V, Sloan-Heggen CM, Brown C, Abbas P, Dunn C, Hansen MR, Gantz BJ, Smith RJH. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear Res. 2017;348:138-42. [PMC free article: PMC5527292] [PubMed: 28213135]
- Shearer AE, Kolbe DL, Azaiez H, Sloan CM, Frees KL, Weaver AE, Clark ET, Nishimura CJ, Black-Ziegelbein EA, Smith RJ. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6:37. [PMC free article: PMC4067994] [PubMed: 24963352]
- Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg. 2015;153:175-82. [PMC free article: PMC4743024] [PubMed: 26084827]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441-50. [PMC free article: PMC4796320] [PubMed: 26969326]
- Smith RJH, Bale JF, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879-90. [PubMed: 15752533]
- Taylor KR, Deluca AP, Shearer AE, Hildebrand MS, Black-Ziegelbein EA, Anand VN, Sloan CM, Eppsteiner RW, Scheetz TE, Huygen PL, Smith RJ, Braun TA, Casavant TL. AudioGene: predicting hearing loss genotypes from phenotypes to guide genetic screening. Hum Mutat. 2013;34:539-45. [PMC free article: PMC3753227] [PubMed: 23280582]
- Toriello HV, Reardon W, Gorlin RJ, eds. Hereditary Hearing Loss and Its Syndromes. New York: Oxford University Press; 2004.
- Van Camp G, Smith RJH. The Hereditary Hearing Loss Homepage. Available online. 2017. Accessed 7-25-17.
- Yamasoba T, Lin FR, Someya S, Kashio A, Sakamoto T, Kondo K. Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear Res. 2013;303:30-8. [PMC free article: PMC3723756] [PubMed: 23422312]
- Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet. 2007;80:1055-63. [PMC free article: PMC1867094] [PubMed: 17503324]
- Yoshida H, Takahashi H, Kanda Y, Usami S. Long term speech perception after cochlear implant in pediatric patients with GJB2 mutations. Auris Nasus Larynx. 2013;40:435-9. [PubMed: 23477838]
Chapter Notes
Author History
Glenn Edward Green, MD; University of Arizona (1999-2005)
Michael S Hildebrand, PhD (2010-present)
A Eliot Shearer (2012-present)
Richard JH Smith, MD (1999-present)
Guy Van Camp, PhD; University of Antwerp (1999-2017)
Revision History
- 27 July 2017 (bp) Comprehensive update posted live
- 9 January 2014 (rjhs) Revision: DFNA41 and DFNB76 added
- 3 January 2013 (cd) Revision: clinical testing available for DFNB79 and DFNX4 (DFN6)
- 5 January 2012 (cd) Revision: clinical testing for mutations in MT-CO1 associated with hearing loss and multi-基因 hearing loss/deafness panels now listed in the GeneTests™ Laboratory Directory
- 14 October 2010 (me) Comprehensive update posted live
- 2 December 2008 (rjs) Revision: DFNB23 added
- 28 October 2008 (me) Comprehensive update posted live
- 30 January 2007 (rjs) Revision: clinical testing and 产前诊断 available for DFNB9
- 4 December 2006 (rjs) Revision: clinical testing available for DFNB21 and DFNA8/12
- 22 August 2006 (rjs) Revision: to incorporate concerns of reader regarding hearing impairment scales
- 30 December 2005 (me) Comprehensive update posted to live Web site
- 18 February 2005 (rjs) Revision: clinical availability of testing, KCNQ4-related DFNA2
- 15 July 2004 (rjs) Revision: use of an interpreter
- 18 December 2003 (cd,rjs) Revision: change in test availability
- 3 November 2003 (me) Comprehensive update posted to live Web site
- 13 January 2003 (cd) Revision: test availability
- 24 April 2001 (me) Comprehensive update posted to live Web site
- 14 February 1999 (pb) Overview posted to live Web site
- 30 October 1998 (rjs) Original overview submission [Supported in part by grants 1RO1DC02842 and 1RO1DC03544 (RJHS) and Belgian National Fonds voor Wetenschappelijk Onderzoek (GVC).]