
简介
临床特征。 半乳糖血症是指半乳糖代谢紊乱,包括典型半乳糖血症,临床变异半乳糖血症和生化变异半乳糖血症。 这篇GeneReview专注于:
- 经典半乳糖血症,可导致危及生命的并发症,包括喂养问题,生长障碍,肝细胞损伤,出血和大肠杆菌败血症。 如果在出生后的前十天提供乳糖限制性饮食,新生儿体征通常会很快消退,并且可以预防肝功能衰竭,败血症和新生儿死亡的并发症; 然而,尽管从幼年时期接受了充分的治疗,患有典型半乳糖血症的儿童仍然存在发育迟缓,言语问题(称为儿童时期的言语失调和构音障碍)以及运动功能异常的风险 。 几乎所有患有典型半乳糖血症的女性都表现出卵巢功能不全(POI)。
- 临床变异半乳糖血症,可导致危及生命的并发症,包括喂养问题,生长障碍,肝细胞损伤,包括肝硬化和出血。 在非洲裔美国人和南非本土非洲人易发生。 新生儿筛查(NBS)可能会遗漏患有临床变异半乳糖血症的人,因为高半乳糖血症并不像经典半乳糖血症那样明显,并且呼气测试是正常的。 如果在生命的前十天提供乳糖限制饮食,通常会预防严重的急性新生儿并发症。 患有临床变异半乳糖血症和充分早期治疗的非裔美国人似乎没有患POI等长期并发症的风险。
诊断/检测。通过检测GALT中升高的红细胞半乳糖-1-磷酸浓度,降低的红细胞半乳糖-1-磷酸尿苷酰酶(GALT)酶活性和/或 双等位基因的的致病变异来确定经典半乳糖血症和临床变异半乳糖血症的诊断。
在典型半乳糖血症中,红细胞半乳糖-1-磷酸通常高于10mg / dL,并且红细胞GALT酶活性不存在或几乎检测不到。在临床变异半乳糖血症中,红细胞GALT酶活性(可能不存在或几乎不可检测,如在非洲裔美国人中)但在脑和肠组织中高得多(例如,对照值的10%)。其他具有临床变异半乳糖血症的个体可能具有接近或高于对照值的1%的红细胞GALT酶活性,但可能从未超过10%-15%。
几乎100%的患有典型半乳糖血症或临床变异半乳糖血症的婴儿可以在新生儿筛查计划中检测到,其中包括在他们的筛查套餐中测试半乳糖血症。然而,如果仅测量血液总半乳糖水平而不测量红细胞GALT酶活性,则可能错过具有临床变异半乳糖血症的婴儿。
管理。预防主要表现:对于半乳糖血症“筛查阳性”的任何新生儿的护理标准是在进行诊断测试时立即进行饮食干预。如果红细胞半乳糖-1-磷酸浓度> 10 mg / dL且红细胞GALT酶活性≤对照活性的10%(即,儿童患有典型半乳糖血症或临床变异半乳糖血症),则继续限制半乳糖摄入并且所有乳制品用含有非半乳糖碳水化合物的无乳糖配方(如Isomil®或Prosobee®)代替;在婴儿期和幼儿期后,饮食管理变得不那么重要了。
表型的治疗:在极少数情况下,可能需要在出生后第一年进行白内障手术。儿童时期的言语失调和构音障碍需要专家言语治疗。建议由心理学家和/或发育儿科医生在一岁时进行发育评估,以便与言语治疗师和治疗医师一起制定治疗计划。适合学龄儿童,个人教育计划和/或专业帮助学习技能和特殊教室。激素替代疗法是青春期发育迟缓和/或原发性或继发性闭经所需的。
预防继发性并发症:建议摄入钙,维生素D和维生素K,以防止骨质矿化减少。
监测:常规监测:有毒分析物(如红细胞半乳糖-1-磷酸和尿液半乳糖醇)的积累;白内障;语言发展; POI;和骨质疏松症。
需要避免的药物/情况:母乳,含有乳糖,牛奶,乳制品和含酪蛋白或乳清的食品的专有婴儿配方奶粉;用乳糖和半乳糖制成的药物。
妊娠管理:患有典型半乳糖血症的女性应在怀孕期间保持乳糖限制饮食。
对有风险的亲属的评估:尽早诊断和治疗有风险的同胞:
- 当家族中的GALT致病变异已知时,执行产前诊断;要么
- 如果尚未进行产前检测,则测试新生儿是否存在家族特异性GALT致病变异或红细胞GALT酶活性。
遗传咨询。 经典的半乳糖血症和临床变异的半乳糖血症以 常染色体隐性遗传方式遗传。有一个 受累的孩子的夫妻每次怀孕后有25%的几率患有受影响的孩子。如果家族中的GALT致病变异已知,则分子遗传 携带者检测风险同胞和产前诊断是一种选择。如果家族中的GALT致病变异未知,则产前检测可依赖于培养的羊水细胞中GALT酶活性的测定。
诊断
经典半乳糖血症和临床变异半乳糖血症是本GeneReview所涵盖的主题。患有这些形式的半乳糖血症的个体将会或可能表现出临床疾病。已发表了关于经典半乳糖血症的诊断,治疗,治疗和随访的国际临床指南[Welling et al 2017]。
半乳糖血症生化变异形式以Duarte Variant Galactosemia为例,并被许多人认为不是真正的疾病(参见Genetically Related Disorders) [Berry 2012]。
可疑发现
对于具有以下新生儿筛查结果,临床特征,家族史和支持性实验室检查结果的个体,应怀疑经典半乳糖血症和临床变异半乳糖血症:
新生儿筛查
- 半乳糖血症阳性新生儿筛查 (National Newborn Screening Status Report [pdf])
- 新生儿筛查使用从足跟刺中获得的少量血液来量化:
- 红细胞半乳糖-1-磷酸和血液半乳糖浓度的总含量;和/或
- 红细胞GALT酶活性
- 州新生儿筛查(NBS)计划在执行哪些测试方面有所不同 - 或者,如果两者都执行,则按照顺序进行。
临床表现
- 未治疗婴儿:
- 喂养困难
- 发育障碍
- 肝功能衰竭
- 出血
- 大肠杆菌脓毒血症
- 未治疗大龄儿:
- 发育迟缓
- 语言障碍
- 运动功能异常,包括伴有共济失调的锥体外系表现
- 白内障
- 肝功能衰竭/肝硬化
- 女性卵巢早衰
受累的兄弟姐妹的家族史。 注意:缺乏半乳糖血症家族史并不排除诊断。
支持性实验室发现
- 在经典半乳糖血症中:
- 红细胞半乳糖-1-磷酸可高达120mg / dL,但在新生儿期通常> 10mg / dL。 当受累的个体接受无乳糖饮食时,水平≥1.0mg/ dL。 红细胞半乳糖-1-磷酸的正常水平<1mg / dL。
- 血浆半乳糖游离半乳糖通常> 10 mg / dL,但可能高达90-360 mg / dL(5-20 mmol / L)。
- 半乳糖-1-磷酸尿苷酰酶(GALT)酶活性不存在或几乎检测不到。
- 在临床变异半乳糖血症中:
建立诊断
通过检测 先证者红细胞半乳糖-1-磷酸盐浓度升高,红细胞半乳糖-1-磷酸尿苷酶(GALT)酶活性降低,和/或 双等位基因的GALT中的致病变异的确定,确定经典半乳糖血症和临床变异链球血症的诊断。(见 Table 1)。
分子遗传测试方法可包括单基因测试:
- 首先进行GALT的序列分析,然后如果仅发现一个或没有 致病性变异,则进行 基因靶向deletion/duplication analysis。
- 可以首先在欧洲或非洲血统的个体中进行针对常见致病变异的靶向分析(参见Table 1)。 当测试大量样品时(例如, 携带者筛选或 新生儿筛查),该方法最有效。
用于经典半乳糖血症和临床变异半乳糖血症的分子遗传学检测
| 基因1 | 测试方法 | 先症者致病性变异占比 2 |
|---|---|---|
| GALT | Targeted analysis 3, 4 | ~90% 5, 6 |
| Sequence analysis 7 | >95% 8 | |
| Gene-targeted deletion/duplication analysis 9 | See footnotes 4, 10 |
- 1.
染色体 位点和蛋白质信息见 Table A. Genes and Database
- 2.
有关在该基因中检测到的等位基因变异的信息见 Molecular Genetics
- 3.
与典型的半乳糖血症相关的常见致病变异包括p.Gln188Arg, p.Lys285Asn, p.Leu195Pro, p.Tyr209Cys, p.Phe171Ser和 c.253-2A>G(在西班牙裔常见) [Elsas & Lai 1998],以及与临床变异半乳糖血症和D2 Duarte(c.[940A>G;c.-119_116delGTCA])致病变异相关的p.Ser135Leu致病性变异,几乎总是与生化变异半乳糖血症有关(见Genetically Related Disorders).
- 4.
在Ashkenazim中,5.2-kb缺失是常见的(参见 Molecular Genetics, 病理变异).
- 5.
靶向变异套餐中包含的致病变异可能因实验室而异; 检测率会相应变化。
- 6.
在具有生物化学证实的经典半乳糖血症和临床变异半乳糖血症的个体中 [Elsas & Lai 1998]
- 7.
序列分析检测分为良性,可能是良性, 意义不确定,可能是致病性或致病性的变异。 致病变异可包括小的基因内缺失/插入和 错义, nonsense, and 剪接位点; 通常,无法检测到外显子或全基因缺失/重复。 对于序列分析结果解释中需要考虑的问题. 对于序列分析结果解释时要考虑的问题点击 here.
- 8.
- 9. 基因靶向deletion/duplication analysis检测基因内缺失或重复。 使用的方法可以包括: quantitative PCR,长程PCR和多重连接依赖性探针扩增(MLPA)和设
- 计用于检测单 外显子缺失或重复的基因靶向微阵列。
- 10.没有关于基因靶向deletion/duplication analysis的数据。
临床特征
临床表现
由半乳糖-1-磷酸尿苷酰酶(GALT)缺乏引起的半乳糖血症可分为三种临床/生化表型:(1)经典半乳糖血症; (2)临床变异半乳糖血症; (3)生化变异半乳糖血症。该分类基于:残留的红细胞GALT酶活性;在乳糖限制饮食中观察到的半乳糖代谢物(例如,红细胞半乳糖-1-磷酸和尿液半乳糖醇)的水平;最重要的是,受累的个体可能会发生急性和慢性长期并发症。这种分类允许对患有半乳糖血症的婴儿的父母进行适当的咨询,特别是关于所谓的与饮食无关的并发症。
经典半乳糖血症
在摄取母乳或含乳糖配方奶粉的几天内,患有典型半乳糖血症的婴儿会出现危及生命的并发症,包括喂养困难,生长障碍,低血糖,肝细胞损伤,出血素质和黄疸(见 Table 2)。如果不治疗经典半乳糖血症,可能会发生大肠埃希菌败血症,休克和死亡[Levy et al 1977]。在新生儿期存活并继续摄入乳糖的婴儿可能会出现严重的脑损伤 [Otaduy et al 2006]
Table 2.
经典半乳糖血症新生儿事件的发生频率
| 事件 | 患病新生儿发生比例 | 伴发症状 |
|---|---|---|
| 肝细胞损伤 | 89% | 黄疸 (74%) 肝大(43%) 肝功能异常(10%) 凝血障碍 (9%) 腹水 (4%) |
| 食物不耐受 | 76% | 呕吐 (47%) 腹泻 (12%) 喂养困难 (23%) |
| 生长障碍 | 29% | |
| 昏睡 | 16% | |
| 癫痫发作 | 1% | |
| 脓血症 | 10% | 大肠杆菌 (26 cases) 克雷伯菌 (3) 肠杆菌 (2) 葡萄球菌 (1) β-链球菌 (1) 粪链球菌 (1) |
来自270名有症状新生儿的调查报告[Waggoner et al 1990]
如果在生命的前三到十天内提供无乳糖饮食,则症状迅速消退,预防肝衰竭,大肠杆菌败血症和新生儿死亡,预后良好。未能实施有效的 新生儿筛查可能会导致肝功能衰竭等灾难性后果 [Malone et al 2011]。
如果未确定经典半乳糖血症的诊断,婴儿表现出随静脉内抗生素和自限性乳糖治疗的反复性和间断性黄疸以及由于乳糖的引入伴随改变的出血。如果延迟治疗,可能会出现生长迟缓和进行性肝病等并发症。罕见受累的 个体可能会出现可能导致失明的玻璃体出血 [Levy et al 1996, Takci et al 2012]。
即使早期和充分的治疗,老年儿童和患有典型半乳糖血症的成人的长期结果可能包括白内障,言语缺陷,生长不良,智力缺陷,神经功能缺损(主要是伴有共济失调的锥体外系症状)和卵巢功能不全(POI) )[Schweitzer-Krantz 2003]。
经典半乳糖血症慢性和/或长期并发症变异很大。即使是在新生儿期没有发病并且从出生开始接受无乳糖饮食开始的个体(例如,家庭中有先前受累的同胞),也可能表现出语言延迟,言语缺陷,学习障碍,认知障碍,女性卵巢功能不全。这些问题可能早在一到两岁就会出现,并且在几乎所有情况下,婴儿早期无法预测最终脑和卵巢功能障碍。少数个体可能表现出有记录的神经系统异常,包括震颤 ,小脑性共济失调和肌张力障碍。在疾病进程早期没有发现这些长期并发症的良好预测因素。总体而言,患有典型半乳糖血症的成年人的生活质量降低,与phenylketonuria (PKU)患者相比更是如此[Gubbels et al 2011, ten Hoedt et al 2011, Hoffmann et al 2012]。
预后和“疾病负担”可以基于红细胞GALT酶活性水平,GALT 基因型,达到成功治疗控制的年龄和遵守乳糖限制来预测。 POI和语言性运动障碍 分析发现13CO2呼气试验(仅在研究基础上可用)是最敏感和特异性的预后参数[Guerrero et al 2000, Webb et al 2003, Barbouth et al 2006]。
Waggoner et al [1990]报道了长期预后的详细信息,这是对270名经典半乳糖血症患者的回顾性横断面调查结果。总之,长期结果的数据表明,涉及神经系统和卵巢的并发症与任何众所周知的生化变量(例如,红细胞半乳糖-1-磷酸水平)无关;此外,这些并发症中的一种或多种的表现甚至在具有与经典半乳糖血症相关的基因型相同的个体中也不同(参见 Table 3) [Doyle et al 2010, Schadewaldt et al 2010, Hoffmann et al 2011, Krabbi et al 2011, Coss et al 2012, Waisbren et al 2012].
智力发展。 177名年龄在6岁或以上的人除了半乳糖血症外没有明显的发育迟缓的医学原因,45%被描述为发育迟缓。随着年龄的增长,个体作为一组的平均IQ分数略有下降(4-7分)。使用生活质量调查问卷对不同年龄的荷兰人进行的研究表明认知结果不正常 [Bosch et al 2004b]。
语言 56%(136/243)三岁或以上的个体报告了语言问题。
超过90%的有语言障碍的人被描述为词汇障碍和发音问题。语言问题仅解决了24%。更正式的分析发现44%的人有语言问题; 38%的患者具有特定的诊断,包括儿童时期的失语症[Robertson & Singh 2000, Webb et al 2003]。语言缺陷是异质的,涉及中枢缺陷和运动异常,并且随着时间的推移而发展[Potter et al 2013]。
在语言障碍患者中观察到的发育商和智商分数显著低于言语正常的个体;然而,一些有语言障碍的人测试在平均范围内。
运动功能。在5岁以上的人群中,18%的人有精细运动性震颤和协调、步态和平衡问题。两名青少年观察到严重的共济失调。成人表现出震颤,构音障碍,小脑性共济失调和肌张力障碍 [Waisbren et al 2012, Rubio-Agusti et al 2013]。
性腺功能。在47名女孩和妇女中,81%有卵巢功能不全(POI)的迹象。月经初潮的平均年龄为14岁,范围为10至18岁。年龄超过17岁的34名女性中有8名(包括2名“性腺条纹”)患有原发性闭经。大多数女性在月经初潮的几年内出现月经稀发和继发性闭经。年龄大于22岁的17名女性中只有5名月经正常。其中两人在18岁和26岁时分娩,从未经历过正常的月经期。
Guerrero et al [2000]认为如果以下情况属实,则患有半乳糖血症的女性POI的发展更为可能:
- 个体是纯合性p.Gln188Arg;
- 治疗期间平均红细胞半乳糖-1-磷酸浓度大于3.5 mg / dL;和
- 从全身13C半乳糖氧化中回收13CO2降低至对照的13C半乳糖的5%。
据报道,男性患者的睾酮和/或卵泡刺激素(FSH)和黄体生成素(LH)的血清浓度正常。然而,文献中很少有关于患有儿童的经典半乳糖血症的男性的报道[Panis et al 2006a, Waisbren et al 2012, Gubbels et al 2013]。目前还没有数据支持男性生殖道结构异常导致不孕;初步数据表明隐睾症患病率增加,精液量减少[Gubbels et al 2013]。
生长。在许多人中,儿童期和青春期早期的生长严重延迟;当青春期延迟并且青少年后期持续增长时,最终成人身高在正常范围内。低于父母平均身高与低胰岛素样生长因子-I(IGF-I)有关[Panis et al 2007]。
白内障 报告314例患者中有30%白内障。近一半的白内障被描述为“轻度”,“短暂的”或“新生儿”,并通过饮食治疗解决;只有8人接受了手术治疗。对于患有白内障的人,饮食控制平均年龄为77天,而没有白内障的人则为20天。然而,需要白内障手术的八个人中有一个是从出生开始接受治疗的婴儿。
治疗与结果之间的关系。在治疗和结果之间没有发现显著关联,除了出生后2个月后才治疗的个体发育迟缓的发生率更高。然而,IQ分数与治疗开始的年龄并不高度相关。在27个同胞中研究了早期治疗对结果的影响,其中三个有三个受累的同胞。在出现临床症状或报告了新生儿筛查结果后,对同胞进行了诊断和治疗,而年轻的同胞则在出生后两天内接受治疗。尽管年轻的同胞早期治疗且只有一个出现新生儿症状,但同胞之间智商得分的差异没有统计学意义,并且年轻同胞的语言和卵巢功能并不比他们的同胞更好。
据报道,38名从出生开始接受治疗的婴儿中,有21名婴儿在怀孕期间限制了母亲的饮食。这21人的长期结果并不比17名在怀孕期间母乳摄入量不受限制的人更好。
在具有残留酶活性的个体和没有可测量的酶活性的个体之间没有观察到显著差异,除了具有一些酶活性的个体倾向于更高的年龄。
有/无神经系统并发症的个体。在具有正常智力,言语和运动功能的56个个体和具有发育迟缓和言语和运动问题的25个个体之间在治疗或生化因子方面没有观察到差异。
并发症的关系。发育迟缓和低IQ分数与言语问题,运动问题和延迟生长相关,但与卵巢功能异常无关。
性别差异。 10岁以后女性的平均智商低于男性(p <0.05),5至12岁年龄段的平均身高较低(p <0.05),但言语或运动问题或治疗频率没有差异,包括治疗开始年龄,新生儿疾病或半乳糖-1-磷酸红细胞浓度。然而,智力发育,言语和运动功能的问题可能表明在一些半乳糖血症病例中存在特定的神经异常 [Schadewaldt et al 2010]。
临床变异型半乳糖血症 具有变异形式的半乳糖血症的个体可能具有经典半乳糖血症的一些方面,包括早期白内障,肝病,轻度智力障碍伴共济失调和生长迟缓 [Fridovich-Keil et al 2011]。临床变异型半乳糖血症可导致未经治疗的婴儿出现危及生命的并发症,包括喂养问题,生长障碍,肝细胞损伤(包括肝硬化)和出血。
临床变异半乳糖血症的例子是在非洲裔美国人和南非的非洲本地人患有p.Ser135Leu/Ser135Leu 基因型的疾病。 新生儿筛查 (NBS)可能遗漏具有临床变异半乳糖血症的新生儿,因为高半乳糖血症并不像经典半乳糖血症那样明显,呼吸试验也是正常的[Crushell et al 2009]。
如果在生命的前十天提供乳糖限制饮食,通常会预防严重的急性新生儿并发症。
据目前所知,患有临床变异半乳糖血症和充分早期治疗的非裔美国人不会出现包括POI在内的长期并发症。
病理生理学
GALT酶催化半乳糖-1-磷酸和UDP葡萄糖转化为UDP半乳糖和Glu-1-P的两步过程称为乒乓球或双 - 双分子反应(图1): (Figure 1):
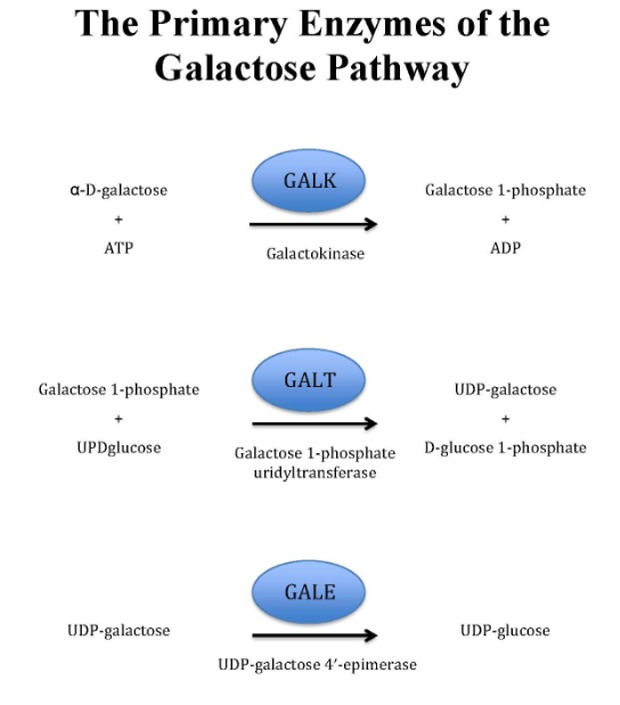
Figure 1.
半乳糖代谢,Leloir途径
- 1.
UDP葡萄糖与活性位点结合并释放葡萄糖-1-磷酸,使UMP与酶共价连接。
- 2.
然后,半乳糖-1-磷酸盐在活性位点着陆,与UMP结合,并且在磷酸键断裂后,UDP半乳糖被释放。
当GALT酶活性不足时,半乳糖-1-磷酸,半乳糖和半乳糖积累(Figure 2)。 半乳糖在细胞中转化为半乳糖醇并产生渗透作用,包括可能导致白内障的晶状体纤维肿胀。 已经假设相同的过程产生脑细胞肿胀,随后产生假瘤。
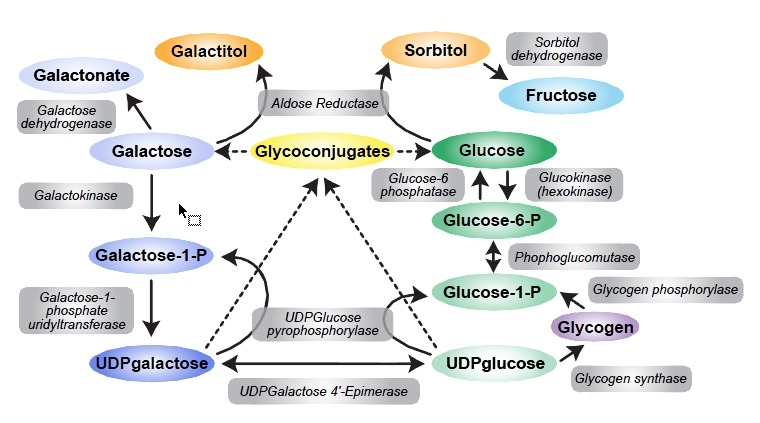
Figure 2.
半乳糖代谢, 半乳糖-1-磷酸尿苷酰酶缺陷
经典半乳糖血症患者可能表现出无关于饮食的慢性并发症,并且由于受累的胎儿的羊水含有高水平的半乳糖醇,受影响新生儿的脐带血含有较高水平的红细胞半乳糖 - 1-磷酸盐,必须考虑GALT酶缺乏的长期并发症是否是由于产前毒性 [Komrower 1982]。 一个假设是产前CNS损伤继发于肌醇缺乏[Berry 2011].
基因型 - 表型相关性
已经注意到显著的 基因型-表型相关性[Shield et al 2000, Tyfield 2000]。 虽然GALT基因型可以预测预后[Guerrero et al 2000, Webb et al 2003],但是关于基因型 - 表型相关性的一些模糊似乎是由于经典半乳糖血症慢性并发症的表现和严重程度的变异所致。
使用Table 3 中的半乳糖血症分类系统有助于消除混淆。 导致三种半乳糖血症表型的最常见致病变异- 经典,临床变异和生化变异 - 显示在Table 3中。(有关定义,请参见Diagnosis和Genetically Related Disorders)。
Table 3.
GALT基因型和生化/临床表型
| 经典半乳糖血症 (Alias 1) | 临床变异型半乳糖血症 (Alias 1) | 生化变异型半乳糖血症 (Alias 1) |
|---|---|---|
| p.[Gln188Arg]+[p.Gln188Arg] (Q188R/Q188R) | p.[Ser135Leu]+[Ser135Leu] (S135L/S135L) 2 | c.[940A>G; c.-16_119delGTCA] (4bp 5' del + N314D/Q188R) 3 |
| p.[Lys285Asn]+[Lys285Asn] (K285N/K285N) | ||
| p.[Leu195Pro]+[Leu195Pro] (L195P/L195P) | ||
| (Δ5.2 kb del/ Δ5.2 kb del) 4 |
- 1.
变异名称不符合当前的命名约定
- 2.
p.Ser135Leu致病性变异的原始鉴定仅限于非裔美国人; 然而,它偶尔出现在非裔美国人婴儿身上
- 3.
被称为“Duarte variant galactosemia”或“Duarte D2变异”
- 4.
见 Table 5, 注脚 4.
p.Gln188Arg。来自北欧背景的白人群体中具有GALT缺乏的人中大约70%的等位基因在氨基酸位置188(p.Gln188Arg)处用精氨酸取代谷氨酰胺。
在纯合性状态下,致病性变异干扰催化反应。它与早产卵巢功能不全(POI)和儿童语言失用的风险增加有关[Robertson & Singh 2000]。
在一项横断面回顾性研究中,将基因型与经典半乳糖血症患者的预后相关联,结果较差的个体p.Gln188Arg纯合性 致病性变异比例较高,预后较好的为非纯合型 p.Gln188Arg变异。然而,在3岁时开始正常乳糖摄入的p.Gln188Arg致病变异的一个成年雌性和一个成年雄性纯合子没有表现典型半乳糖血症 表型 [Lee et al 2003, Panis et al 2006a]。
p.Ser135Leu。 p.Ser135Leu 等位基因,其中亮氨酸取代氨基酸135处的丝氨酸,在非洲很普遍。
如果在新生儿期开始治疗,那么在 纯合性状态下具有这种 等位基因的患有半乳糖血症的非洲裔美国人具有良好的预后。通常,这些个体在新生儿期不易患大肠杆菌败血症或从婴儿期治疗时不易患慢性并发症(即语言障碍,POI和智力障碍) [Lai et al 1996]。
复合杂合的人数据有限(p.[Ser135Leu];[Gln188Arg]);然而,它们似乎比基因型p.[Gln188Arg]+[p.Gln188Arg]的典型半乳糖血症相关的个体具有更少的并发症。
p.Asn314Asp。 是Duarte(D2)等位基因变异,其中天冬氨酸取代残基314处的天冬酰胺(p.Asn314Asp)和 cis构型的第二种变异 - 启动子区中的4-bp缺失(c.-119_116delGTCA) ) - 导致红细胞GALT酶活性降低。 D2等位基因名称为c.[940A>G; c.-119_116delGTCA]。
在纯合性状态下,D2红细胞GALT酶活性降低了50%。
具有D2和与经典半乳糖血症相关的致病性变异的复合杂合子具有良好的预后[Langley et al 1997, Lai et al 1998]。
然而,已有报道D2(Duarte)和D1(LA变异)的复合杂合性发生。 D1等位基因具有cis构型的p.Leu218Leu,p.Asn314Asp致病性变异 p.[Leu218Leu;Asn314Asp],其赋予“超活性”(即,杂合子具有~117%的红细胞GALT活性,而纯合子显示〜 134%的活动)。
其他。 在285位(p.Lys285Asn)用天冬酰胺取代赖氨酸,在德国南部,奥地利和克罗地亚很普遍; p.Gln188Arg的纯合性状态或复合杂合状态与的神经和认知功能预后不良有关,被认为是典型半乳糖血症。
其他复杂杂合子(例如,p.[Gln188Arg]+[p.Arg333Gly])具有良好的长期结果 [Ng et al 2003]。
- 当具有基因型如p.[Gln188Arg]+[p. Gln188Arg]的经典半乳糖血症时,可以看到清晰的基因型-表型相关性。 例如, 将Gln188Arg]与由 p.[Ser135Leu]+[p.Ser135Leu]基因型引起的临床变异半乳糖血症进行比较,几乎所有具有p.[Gln188Arg]+[p.Gln188Arg]基因型的女性都表现出POI,而具有p.[Ser135Leu]+[p.Ser135Leu]基因型的非洲裔美国女性的POI几乎闻所未闻。 一个关键的未解决的问题是,必须有多少残留的GALT酶活性才能消除慢性饮食无关的并发症。 隐性残留GALT酶活性可能是学龄儿童预后的潜在影响者[Ryan et al 2013]。
- 在乳糖限制性饮食中,患有典型半乳糖血症的人显示红细胞半乳糖-1-磷酸盐水平在1至5 mg / dL之间,尿液半乳糖醇水平在100至400μmol/ mmol肌酐之间,而患有p.[Ser135Leu]+[p.Ser135Leu] 基因型通常具有低于1 mg / dL的红细胞半乳糖-1-磷酸盐水平,以及低于100μmol/ mmol肌酐的尿液半乳糖醇,通常在正常范围内 [Saudubray et al 2012, Walter & Fridovich-Keil 2014].
- 具有生化变异半乳糖血症的人 - 例如,c.563A> G(p.Gln188Arg)和D2 c.[940A>G;的复合杂合子。c.-119_116delGTCA] 基因型 - 与经典半乳糖血症或临床变异半乳糖血症不同:它们通常没有疾病的迹象和症状,只有生化异常。
注意:新生儿筛查后发现300多种GALT致病变异中的许多变异,很少或没有长期随访数据。 在这些情况下,术语经典的半乳糖血症应谨慎使用或根本不应用。 如果没有支持性数据,如果向新父母提出建议,即他们的婴儿会在半乳糖血症中发生一种或多种慢性并发症,这是不合适的。
命名法
遗传性高半乳糖血症
- GALK中继发于致病变异的半乳糖激酶缺乏症
- 在GALE中继发于致病变异的Epimerase deficiency galactosemia
- 在GALT中继发于致病变异的半乳糖-1-磷酸尿苷酰基酶短缺
- 经典的半乳糖血症
- 严重的GALT酶缺乏,在红细胞和肝脏中缺乏或几乎检测不到活性
- 也称为G / G,携带者为G / N.
- 临床变异型半乳糖血症
- 红细胞和/或肝脏中1%-10%的残留GALT酶活性
- 生化变异半乳糖血症
- 红细胞中15%-33%的残留GALT酶活性
- 包括D2 Duarte生化变异状态,也称为G / D.
流行
根据新生儿筛查计划的结果,经典半乳糖血症的患病率为1:48,000 [National Newborn Screening and Genetics Resource Center 2014]。然而,当红细胞GALT酶活性<5%的对照活性和红细胞半乳糖-1-磷酸浓度> 2 mg / dL用作诊断标准时,一些新生儿筛查程序记录的患病率为1:10,000[Bosch et al 2005]。
爱尔兰经典半乳糖血症的发病率为1:16,476 [Coss et al 2013]。
虽然不可能提供临床变异型半乳糖血症的患病率数据,但p.Ser135Leu/Ser135Leu 基因型的估计患病率为1:20,000 [Henderson et al 2002]。
遗传相关(等位)疾病
Duarte variant galactosemia是生化变异半乳糖血症的一个例子,与GALT中的特定致病变异有关。
Duarte变异(D2)在启动子区 (c.-119-116delGTCA中具有 cis构型(即,在相同的 等位基因中)致病性 错义变异p.Asn314Asp和GTCA 缺失,其损害正调节结构域。它被命名为c.[940A>G; c.-119_116delGTCA](见 Table 3)。
注意:洛杉矶(LA)变异(D1)具有与Duarte变异相同的p.Asn314Asp致病性错义变异,但不具有 缺失的GTCA启动子。相反,它是带有错义变异p.Leu218Leu的 cis配置。该变异不会引起半乳糖血症并且与红细胞GALT酶活性增加有关[Langley et al 1997, Elsas et al 2002]。
在生化变异半乳糖血症:
- 红细胞半乳糖-1-磷酸通常> 1mg / dL,但可高达35mg / dL。当个体进行无乳糖饮食时,水平<1 mg / dL。
- 残留的红细胞GALT酶活性通常> 15%,平均为对照值的25%。
专家意见和信息表明,Duarte variant galactosemia是最常见的生化变异半乳糖血症形式,不会导致临床表现。没有前瞻性的长期循证医学研究来证明,例如,患有D2c.[940A>G; c.-119_116delGTCA] 基因型没有临床表现,无论有或没有饮食干预。这仍然存在争议,因为三分之二的相对较小的队列中发现语言异常和/或发育问题的患病率较高,这表明中枢神经系统功能障碍可能存在于Duarte变异半乳糖血症患者的一部分中[Ficicioglu et al 2008, Powell et al 2009, Lynch et al 2015]。
关于Duarte变异半乳糖血症患者的残留红细胞GALT酶活性是否在对照活性的13%-33%范围内,是否在婴儿期和幼儿期应限制摄入半乳糖尚未达成协议。摄入乳糖可能会出现持续的半乳糖-1-磷酸积累,但通常没有后遗症。
鉴别诊断
新生儿肝毒性的鉴别诊断包括:感染性疾病;阻塞性胆道疾病包括Alagille syndrome, severe ATP8B1 deficiency (进行性家族性肝内胆汁淤积)和 citrin deficiency; hereditary fructose intolerance; tyrosinemia type;和其他代谢疾病,包括Neimann-Pick disease type C。
注意:建立脓毒症的诊断并不排除半乳糖血症的可能性, 特别是大肠杆菌败血症脓毒症,常见于患有典型半乳糖血症的婴儿。
半乳糖激酶(GALK)缺乏症(OMIM 230200)患有白内障,血浆半乳糖浓度增加和半乳糖醇尿液排泄增加的,但其他方面都是健康的个体应考虑。这些个体具有正常的红细胞GALT酶活性,并且不会积累红细胞半乳糖-1-磷酸。白内障是由晶状体纤维中半乳糖的积累及其还原为半乳糖醇引起的,半乳糖醇是一种不渗透的酒精,导致细胞内渗透压和水吸收增加。其他患有GALK缺陷的个体会发展为CNS疾病。检测GALK酶活性降低可以诊断。 GALK1中的双等位基因致病变异是致病原因[Kolosha et al 2000, Hunter et al 2001]。 GALK缺乏症的患病率尚不清楚,但可能不到1:100,000。
Epimerase deficiency galactosemia(UDP-半乳糖4'-表异构酶[GALE]缺乏症)患有肝病,生长障碍和红细胞半乳糖-1-磷酸浓度升高,但红细胞GALT酶活性正常的个体应考虑。迄今为止,仅报告了8例严重GALE缺乏症患者。相比之下,大多数患有GALE缺陷的个体无临床表现:它们是具有新生儿筛查阳性,红细胞半乳糖-1-磷酸增加和正常红细胞GALT酶活性的健康新生儿。检测GALE酶活性降低可以诊断。 GALE中的双等位基因致病变异是致病因素。 GALE缺乏症在日本的估计患病率为1:23,000,其他人群患病率不明。
管理
新生儿期初步诊断后的评估
为确定典型半乳糖血症或临床变异型半乳糖血症的新生儿的疾病和需求程度,建议进行以下评估 [Walter et al 1999]:
- 咨询生化遗传疾病专家
- 测量红细胞半乳糖-1-磷酸浓度和尿液半乳糖醇作为监测治疗效果的基线(见
Prevention of Primary Manifestations). - 根据需要进行神经系统检查和脑MRI检查
- 眼科检查,包括白内障裂隙灯检查
- 特别是在患有晚期治疗疾病且可能有肝硬化风险的受累的个体中评估肝细胞疾病。
治疗表型和并发症
已发布了一份针对管理的国际临床指南 [Welling et al 2017] (full text)。
有关饮食干预的信息,请参见Prevention of Primary Manifestations。乳糖限制可逆转已患有肝细胞疾病的新生儿的肝脏疾病。
眼科治疗。白内障手术可能需要在出生后的第一年进行,尤其是在未能进行NBS导致诊断延迟的罕见个体中。
语言治疗。患有言语和构音障碍的儿童失语的受影响个体需要由语言专家进行治疗,直到并发症得到控制。这可能需要多年的强化治疗。
发育治疗应包括心理学家和/或发育儿科医生在一岁时进行的发育检查,然后心理学家和/或发育儿科医生与语言治疗师和治疗医师密切合作,制定适合每个孩子的治疗计划和评估方案。 。根据临床表现的时间和并发症的性质,可能需要个人教育计划和/或具有学习技能和特殊教室的专业帮助,即使对于具有相同 基因型的儿童,也可能变化很大。
卵巢功能不全(POI)治疗。虽然生化/内分泌测试可能表明婴儿早期的POI,但在青春期发育迟缓或原发性或继发性闭经之前,女性通常不会出现POI征象。因此,在青春期或成人女性转诊给儿科内分泌科医生进行咨询。在适当的时候,应该由专门从事青少年服务和/或不孕症的妇产科医生看到受累的女性。
在少数情况下可能需要给予雌激素和孕酮以促进青春期发育,或者在发生继发性闭经时。
关于女性是否应该在青春期早期服用雌激素和黄体酮以延缓“卵母细胞的浪费”以及是否应采用更积极的治疗计划(类似于肿瘤学中使用的计划)存在争议。在这方面,必须尊重受累的女性的自主权,如果是青春期前的个体,则应考虑她做出明智决定的能力。医院伦理委员会应参与任何决策,每个案例都要单独处理[van Erven et al 2013]。
对于患有典型半乳糖血症和POI的女性,考虑卵巢活检保存卵母细胞以备将来是有争议的。如果正在考虑这种类型的手术(对于正在接受癌症化疗的女性而言越来越常见),建议咨询医院伦理委员会。
对于某些患有典型半乳糖血症的人来说,生育能力是可能的。因此,应该给予成年人这个机会,向专门研究不孕症妇产科医生进行咨询, 确保促进受孕。
不孕不育。由于卵巢功能障碍,FSH刺激可能对某些女性产生排卵有用。
一名患有卵巢功能不全(POI)的女性在接受FSH治疗后受孕,随后分娩为正常儿童[Menezo et al 2004]。其他人已经发现,经典半乳糖血症中的POI可能是由卵巢卵泡数量减少或成熟引起的,并且在某些情况下可能通过促性腺激素的外源药物刺激而治疗[Rubio-Gozalbo et al 2010]。
预防主要表现
饮食干预。婴儿的红细胞GALT酶活性≤对照活性的10%且红细胞半乳糖-1-磷酸浓度> 10 mg / dL时,即刻进行饮食干预。
因为90%的新生儿碳水化合物来源是乳糖,人乳含有6%-8%的乳糖,牛奶含有3%-4%的乳糖,而大多数专有的婴儿配方奶含有7%的乳糖,所有这些乳制品必须立即更换为不含乳糖的配方(如Isomil®或Prosobee®)。这种大豆配方含有蔗糖,果糖和含半乳糖的低聚糖,它们不能在小肠中水解。
含有少量半乳糖的元素配方,例如由酪蛋白水解产物制成的Alimentum®,Nutramigen®和Pregestimil®,过去已经使用,没有明显的副作用。既不含游离也不结合半乳糖的配方(Neocate®)没有任何副作用[Zlatunich & Packman 2005]。
所有含乳糖食品(包括牛奶和其他乳制品)的饮食限制应该持续一生;然而,在婴儿期和幼儿期,当牛奶和乳制品不再是主要的能量来源时,管理饮食变得不那么重要了。关于婴儿期饮食是否应该严格是有争论的 [Berry et al 2004, Bosch et al 2004a, Schadewaldt et al 2004],因为内源性半乳糖的产生比从牛奶以外的食物摄取的产量高一个数量级。在对大量受试者进行更多前瞻性循证医学研究之前,应教育父母终身需要限制牛奶和乳制品的饮食。
预防继发性并发症
由于患有典型半乳糖血症和临床变异型半乳糖血症的儿童和成人的骨矿物质密度可能会减少,因此提倡补充超过1000 IU /天的维生素D和维生素K [Panis et al 2006b, Batey et al 2013]。
对于经典和临床变异型半乳糖血症患者的钙和维生素D摄入量,请参阅Health and Medicine Division of the National Academies of Sciences, Engineering, and Medicine推荐的 dietary allowances and adequate intakes(pdf)。点击此处查看其他膳食参考摄入量表。
如果常规随访有代谢紊乱知识的营养师,则证实钙和维生素D摄入量以适应年龄,如果血浆25-羟基维生素D在正常范围内,但骨矿物质密度降低,请咨询儿科和/或或成人内分泌学家可能是有保证的。
监控
另Welling et al [2017] (full text)发布的管理指南。
生化测试 应定期监测具有典型半乳糖血症和临床变异型半乳糖血症的个体[Walter et al 1999] ,以分析物质的积累:
- 在每次门诊就诊时根据需要(例如,在引入新食物期间)测量红细胞半乳糖-1-磷酸水平,浓度<5mg / dL被认为在治疗范围内。
注意:红细胞半乳糖-1-磷酸是评估急性摄入半乳糖的好方法。
- 可以进行尿液半乳糖醇(半乳糖代谢的替代途径的产物)水平但不是长期监测所必需的。
- 注意,红细胞半乳糖-1-磷酸和尿液半乳糖醇可以提供关于乳糖限制饮食的依从性的信息。
- 尿液半乳糖醇分析在受累的红细胞输血的受累个体中特别有价值; > 78 mmol / mol肌酐为异常。
- 尿液半乳糖醇通常不受急性饮食摄入半乳糖的影响。
如果检测到红细胞半乳糖-1-磷酸或尿液半乳糖醇的突然增加,应寻求过量半乳糖的膳食来源或对其他原因进行评估。
具有经典或临床变异型半乳糖血症的个体的时间表 生化遗传诊所就诊
- 根据潜在急性并发症的性质,第一年每三个月或根据需要
- 生命第二年每六个月一次
- 每年一次
代谢性营养师门诊就诊。如上所述,加上临时访问和电话咨询
眼科评估。随访检查的时间取决于新生儿白内障的存在与否,否则,可以在一岁,五岁和青春期进行检查。注意:经典半乳糖血症的人婴儿早期患有白内障非常罕见,因为白内障发展可能需要大量摄入牛奶和乳制品。
语言评估
- 在18个月时由语言专家评估(推荐所有受累的儿童)
- 如果初步评估未显示语言或语言障碍的诊断,则在婴儿期和幼儿期每3至12个月重新评估一次,具体取决于发育专家和医生进行的评估
发育评估。受影响的个体应在一年后由心理学家和/或发育儿科医生进行发育检查,此后每一至三年进行一次,这取决于一个或多个发育领域的延迟程度。
评估卵巢早衰(POI)。如果年龄为12岁且第二性征不足或年龄14岁且没有正常月经,则建议测量女性血浆中的17-β-雌二醇和FSH [Welling et al 2017]。
评估骨质减少
- 每年根据需要测量血浆钙,磷和25-羟基维生素D.
- 由于经典半乳糖血症患者的骨密度降低普遍存在,因此建议在六岁时,青春期,青春期以及成年后每五年进行一次DEXA扫描。
- 常规代谢性饮食检查应验证钙和维生素D的摄入量是否适合年龄,血浆25-羟基维生素D是否在正常范围内。即便如此,某些受累个体的骨矿物质密度可能会降低,在这种情况下,可能需要咨询儿科和/或成人内分泌科医生。
要避免的药物/情况
应避免以下情况:
- 母乳,含有乳糖,牛奶,乳制品和酪蛋白或含乳清食品的专有婴儿配方奶粉
- 含乳糖或半乳糖的药物制剂
- 含有乳糖(片剂,胶囊,甜味酏剂)的药物,特别是在婴儿期
风险亲属的评估
当已知在家族中引起经典半乳糖血症或临床变异型半乳糖血症的致病变异时,可以通过羊膜穿刺术或CVS进行有危险胎儿的 产前诊断,以允许在出生时进行治疗。
如果没有进行产前检查,应使用红细胞GALT酶检测和/或基因检测(如果已知家族中的 家族性病原体变异)从出生开始对每个有风险的新生儿同胞进行治疗,并筛选出典型的半乳糖血症或临床变异型半乳糖血症, 尽早诊断。注意:如果担心产前检测的可靠性,可能会在进行诊断检测时给予大豆配方奶粉。
有关为Genetic Counseling目的测试有风险亲属的相关问题,请参阅遗传咨询。
孕期管理 具有典型半乳糖血症和临床变异性半乳糖血症的女性在怀孕期间应该使用乳糖限制饮食。
没有证据表明,当他们的母亲(携带者)在怀孕期间接受乳糖限制饮食时,患有典型半乳糖血症或临床变异型半乳糖血症的儿童的结果会得到改善。因此,未受影响的孕妇在GALT(携带者女性)具有杂合的致病性变异,在怀孕期间不需要限制乳糖的饮食。
正在调查的疗法
研究表明,尽管限制外源性半乳糖,内源性半乳糖产量可能接近1.0-2.0克/天 [Berry et al 2004, Schadewaldt et al 2004]。如果这是真的,那么半乳糖的“自我中毒”可能比管理年龄较大的儿童和不再依赖牛奶作为其主要能量来源的成年人对外源性半乳糖的限制更具问题。
使用GALK酶的小抑制剂正在研究降低半乳糖-1-磷酸的内源性产生的方法[Tang et al 2010]。虽然GALT酶缺陷型人成纤维细胞的体外研究证明了证据,但尚未在动物模型中进行。由Leslie et al [1992]生成的GALT敲除小鼠不表达半乳糖血症的人表型,除了由于高半乳糖血症和高半乳糖尿症导致的多尿症在很大程度上没有表现。由于小鼠在进化过程中失去了ARHI(DIRAS3)[Lai et al 2008, Rubio-Gozalbo et al 2010, Tang et al 2010],需要一种表达ARHI(DIRAS3)信号的GALT酶缺陷小鼠模型来检验该假设。 ARHI基因表达在疾病的表型表达中发挥作用,并确定抑制半乳糖-1-磷酸的产生是否限制或消除“人类表型”。
在ClinicalTrials.gov中搜索有关各种疾病和病症的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质,遗传和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况。 本节不是为了解决个人可能面临的所有个人,文化和伦理问题,也不是用遗传专业人员代替咨询。-编者.
遗传方式
经典的半乳糖血症和临床变异的半乳糖血症以常染色体隐性遗传方式遗传。
家庭成员的风险
先证者的父母
先证者的同胞
- 在受孕时,具有经典半乳糖血症或临床变异性半乳糖血症的 先证者的每个同胞有25%的机会成为 受累的,有50%的几率成为致病 等位基因的携带者 (杂合子),25%的机会不受影响也不是携带者。
- 杂合子(携带者)是无症状的,不会发展为半乳糖血症。
先证者的后代
- 具有经典半乳糖血症或临床变异半乳糖血症的个体的后代是GALT中 致病性变异的肯定杂合子(携带者)。
- 如果父母一方患有典型的半乳糖血症或临床变异性半乳糖血症而另一方是携带者,则每个孩子有50%的机会成为杂合子,并有50%的几率患有典型的半乳糖血症或临床变异性半乳糖血症。
其他家庭成员。先证者父母的每个同胞都有50%的风险成为GALT致病性变异的携带者。
携带者(杂合子)检测
携带者是这种常染色体隐性遗传 病的杂合子,并且没有患上这种疾病的风险。
分子遗传学测试。如果已在家族中鉴定出GALT致病变异,则可以对有风险的家庭成员进行携带者检测。
生化基因检测
- 通过测量红细胞GALT酶活性进行携带者测试,当 致病性变异为p.Ser135Leu时,其为经典半乳糖血症或临床变异半乳糖血症的携带者几率约50%。
- 这将与其他临床变异引起的半乳糖血症致病变异不同,其在纯合性状态下导致1%-10%的残留红细胞GALT酶活性。
相关的遗传咨询问题
有关为早期诊断和治疗目的评估高危亲属的信息,请参阅管理,Evaluation of Relatives at Risk。
家庭计划
- 确定遗传风险的最佳时间, 携带者状态的澄清以及产前检测可用性的讨论是在怀孕前进行的。
- 向年轻人提供遗传咨询(包括对后代和生殖选择的潜在风险的讨论)是适当的,这些年轻人是受累的,携带者,或者有成为携带者的风险。
DNA库是DNA的存储(通常从白细胞中提取),以备将来使用。因为测试方法和我们对基因,等位基因变体和疾病的理解将来可能会有所改善,所以应该考虑 受累的个体的银行DNA。
产前检查和植入前遗传学诊断
分子遗传学测试。一旦在受累的家庭成员中鉴定出两种GALT致病变异,就可以进行经典半乳糖血症或临床变异型半乳糖血症产前检测和植入前遗传诊断(PGD)。
使用分子遗传学检测的产前检测(或 植入前遗传诊断)优于酶分析。
生化检测。 GALT酶活性和分子诊断的分析依赖于在妊娠约10至12周通过绒毛膜绒毛取样(CVS)获得的细胞或通常在妊娠约15至18周进行的羊膜穿刺术。
注意:(1)当胎儿患有典型的半乳糖血症或临床变异型半乳糖血症时,半乳糖醇的羊水浓度在妊娠晚期晚期升高,并且过去曾用于产前检测。 (2)妊娠年龄表示为从最后一次正常月经的第一天或通过超声测量计算的月经周。
医疗专业人员和家庭内部关于使用产前检查的观点可能存在差异,特别是如果考虑将检测用于终止妊娠而不是早期诊断。虽然大多数中心会认为这是父母的选择,但讨论这些问题是恰当的。
资源
GeneReviews的工作人员选择了以下疾病特异性和/或orumbrella支持组织和/或登记处,以使这种疾病患者及其家人受益。 GeneReviews不对其他组织提供的信息负责。 有关selectioncriteria的信息,请单击 here.
- Galactosemia Support Group (GSG)31 Cotysmore RoadSutton Coldfield West Midlands B75 6BJUnited KingdomPhone: +44 0121 378 5143Email: sue@galactosaemia.org
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- Save Babies Through Screening Foundation, Inc.P. O. Box 42197Cincinnati OH 45242Phone: 888-454-3383Email: email@savebabies.org
- The Galactosemia FoundationP.O. Box 2401Mandeville LA 70471Phone: 866-900-7421Email: president@galactosemia.org
- Association for Neuro-Metabolic Disorders (ANMD)5223 Brookfield LaneSylvania OH 43560-1809Phone: 419-885-1809; 419-885-1497Email: volk4olks@aol.com
- Children Living with Inherited Metabolic Diseases (CLIMB)United KingdomPhone: 0800-652-3181Email: info.svcs@climb.org.uk
分子遗传
Molecular Genetics和OMIM表中的信息可能与GeneReview中的其他信息不同:表格可能包含更多最新信息。 - 编者。
Table A.
经典的半乳糖血症和临床变异型半乳糖血症:基因和数据库
Table B.
经典半乳糖血症和临床变异型半乳糖血症OMIM条目 (View All in OMIM)
基因结构。 基因长约4kb,有11个外显子和10个内含子。 启动子富含GC,就像“管家基因”一样。 有关基因和蛋白质信息的详细摘要,请参阅Table A,基因。
致病变异。 已知超过300种GALT致病变异 [Elsas & Lai 1998, Tyfield et al 1999, Bosch et al 2005, Calderon et al 2007]。
Table 4中显示了在美国最普遍的致病变异,Suzuki et al [2001]报道了不同种族群体中五种最常见的GALT致病变异的频率。
在Ashkenazi Jewish背景下,人们常见GALT 5.2-kb缺失[Coffee et al 2006]。 5.2-kb缺失等位基因是一个复杂的缺失,涉及GALT启动子的3163个核苷酸缺失和5' 基因区以及3'基因的2295-bp缺失; 只保留 外显子8和 内含子8的片段 [Coffee et al 2006]。 MLPA可检测到5.2kb的复合物缺失。
Table 4.
美国284例经典半乳糖血症患者GALT突变等位基因的患病率
| Pathogenic Variant | Number of Alleles | Percent of Total |
|---|---|---|
| p.Gln188Arg | 280 | 49% |
| p.Ser135Leu | 40 | 7% |
| p.Lys285Asn | 20 | 4% |
| p.Leu195Pro | 11 | 2% |
| p.Tyr209Cys | 5 | 1% |
| 5.2-kb 缺失 1 | 7 | 1% |
| p.Asn314Asp | 141 | 25% |
| Other | 64 | 11% |
| TOTAL | 568 | 100% |
- 1.
见 Table 5,注脚 4.
Table 5.
在此GeneReview中讨论GALT致病变异
| DNA Nucleotide Change (Alias 1) | Predicted Protein Change | Reference Sequences |
|---|---|---|
| c.404C>T | p.Ser135Leu | NM_000155 NP_000146 |
| c.512T>C | p.Phe171Ser | |
| c.563A>G | p.Gln188Arg | |
| c.584T>C | p.Leu195Pro | |
| c.607G>A | p.Glu203Lys | |
| c.626A>G | p.Tyr209Cys | |
| c.855G>T | p.Lys285Asn | |
| c.[940A>G; c.-119_116delGTCA] 2 | p.Asn314Asp; effect on promoter variant | |
| c.997C>G | p.Arg333Gly | |
| c.253-2A>G (IVS2-2A>G) 3 | -- | |
| (Δ5.2kb) or (5.2kbdel) 4, 5 | -- |
关于变异分类的注意事项:表中列出的变异由作者提供。 GeneReviews工作人员尚未独立验证变异的分类。
关于命名法的注释:GeneReviews遵循人类基因组变异学会(varnomen- .hgvs.org )的标准命名惯例。 有关术语的解释,请参见Quick Reference 。- 1.
变异名称不符合当前的命名约定
- 2.
- 3.
见于西班牙裔的个人
- 4.
一种复杂的缺失,涉及GALT启动子的3163-bp缺失和5' 基因区以及该基因3'末端的2295-bp缺失; 仅保留 外显子8和 内含子8的片段[Barbouth et al 2006, Coffee et al 2006]。 该缺失的标准HGVS命名法同样复杂,可以最好地描述为 c.[-1039_753del; 820+50_*789delinsGAATAGACCCCA].
- 5.
见于 Ashkenazi Jewish人
正常基因产物。 GALT蛋白起二聚体的作用,表现出独特的双分子乒乓动力学。 GALT酶首先结合UDP-葡萄糖,然后释放葡萄糖-1-磷酸。第二次置换反应需要稳定的GALT-UMP复合物,其涉及半乳糖-1-磷酸与UDP-半乳糖和游离GALT酶的释放和结合。
异常基因产物
- 致病性变异p.Gln188Arg在很大程度上阻止了GALT-UMP中间体的形成[McCorvie et al 2016]。
- LA变异 (D1)涉及翻译速率加快,至少部分是由于残基218中的亮氨酸密码子从共有到稀有(c.652C> T)的核苷酸变化,从而稳定p.Asn314Asp的致病性变异。已经提出了“密码子偏好”和由GALT启动子 良性变异引起的增加的 基因表达的组合以解释LA变异增加活性[Langley et al 1997]。密码子218LA变异(D1)编码是p.Leu218 =(c.652C> T)。
- Duarte D2变异是E-box中的 缺失,这是一种降低GALT 基因表达的碳水化合物反应元件 [Elsas et al 2001]。
参考文献
发布的准则/共识声明
- 国家新生儿筛查和遗传资源中心。 全国新生儿筛查状况报告。 在 online可查. 2014. Accessed 3-6-17.
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar SC, Waisbren SE, Bosch AM; Galactosemia Network (GalNet). International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. Available online. 2017. Accessed 3-6-17. [PMC free article: PMC5306419] [PubMed: 27858262]
Literature Cited
- Barbouth D, Slepak T, Klapper H, Lai K, Elsas LJ. Prevention of a molecular misdiagnosis in galactosemia. Genet Med. 2006;8:178 - 82. [PubMed: 16540753]
- Batey LA, Welt CK, Rohr F, Wessel A, Anastasoaie V, Feldman HA, Guo CY, Rubio-Gozalbo E, Berry G, Gordon CM. Skeletal health in adult patients with classic galactosemia. Osteoporos Int. 2013;24:501 - 9. [PubMed: 22525982]
- Berry GT. Galactosemia: when is it a newborn screening emergency? Mol Genet Metab. 2012 May;106(1):7 - 11. [PubMed: 22483615]
- Berry GT. Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J Inherit Metab Dis. 2011;34:345 - 55. [PubMed: 21246399]
- Berry GT, Moate PJ, Reynolds RA, Yager CT, Ning C, Boston RC, Segal S. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81:22 - 30. [PubMed: 14728988]
- Bosch AM, Bakker HD, Wenniger-Prick LJ, Wanders RJ, Wijburg FA. High tolerance for oral galactose in classical galactosaemia: dietary implications. Arch Dis Child. 2004a;89:1034 - 6. [PMC free article: PMC1719730] [PubMed: 15499058]
- Bosch AM, Grootenhuis MA, Bakker HD, Heijmans HS, Wijburg FA, Last BF. Living with classical galactosemia: health-related quality of life consequences. Pediatrics. 2004b;113:e423 - 8. [PubMed: 15121984]
- Bosch AM, Ijlst L, Oostheim W, Mulders J, Bakker HD, Wijburg FA, Wanders RJ, Waterham HR. Identification of novel mutations in classical galactosemia. Hum Mutat. 2005;25:502. [PubMed: 15841485]
- Calderon FR, Phansalker A, Crockett D, Miller M, Mao R. Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat. 2007;28:939 - 43. [PubMed: 17486650]
- Coffee B, Hjelm LN, DeLorenzo A, Courtney EM, Yu C, Muralidharan K. Characterization of an unusual deletion of the galactose-1-phosphate uridyl transferase (GALT) gene. Genet Med. 2006;8:635 - 40. [PubMed: 17079880]
- Coss KP, Byrne JC, Coman DJ, Adamczyk B, Abrahams JL, Saldova R, Brown AY, Walsh O, Hendroff U, Carolan C, Rudd PM, Treacy EP. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol Genet Metab. 2012;105:212 - 20. [PubMed: 22133299]
- Coss KP, Doran PP, Owoeye C, Codd MB, Hamid N, Mayne PD, Crushell E, Knerr I, Monavari AA, Treacy EP. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis. 2013;36:21 - 7. [PubMed: 22870861]
- Crushell E, Chukwu J, Mayne P, Blatny J, Treacy EP. Negative screening tests in classical galactosaemia caused by S135L homozygosity. J Inherit Metab Dis. 2009;32:412 - 5. [PubMed: 19418241]
- Doyle CM, Channon S, Orlowska D, Lee PJ. The neuropsychological profile of galactosaemia. J Inherit Metab Dis. 2010;33:603 - 9. [PubMed: 20607611]
- Elsas LJ 2nd, Lai K. The molecular biology of galactosemia. Genet Med. 1998;1:40 - 8. [PubMed: 11261429]
- Elsas LJ, Lai K, Saunders CJ, Langley SD. Functional analysis of the human galactose-1-phosphate uridyltransferase promoter in Duarte and LA variant galactosemia. Mol Genet Metab. 2001;72:297 - 305. [PubMed: 11286503]
- Elsas LJ, Webb AL, Langley SD. Characterization of a carbohydrate response element regulating the gene for human galactose-1-phosphate uridyltransferase. Mol Genet Metab. 2002;76:287 - 96. [PubMed: 12208133]
- Ficicioglu C, Thomas N, Yager C, Gallagher PR, Hussa C, Mattie A, Day-Salvatore DL, Forbes BJ. Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab. 2008;95:206 - 12. [PubMed: 18976948]
- Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011;34:357 - 66. [PMC free article: PMC3063539] [PubMed: 20978943]
- Gubbels CS, Thomas CM, Wodzig WK, Olthaar AJ, Jaeken J, Sweep FC, Rubio-Gozalbo ME. FSH isoform pattern in classic galactosemia. J Inherit Metab Dis. 2011;34:387 - 90. [PMC free article: PMC3063565] [PubMed: 20814826]
- Gubbels CS, Welt CK, Dumoulin JC, Robben SG, Gordon CM, Dunselman GA, Rubio-Gozalbo ME, Berry GT. The male reproductive system in classic galactosemia: cryptorchidism and low semen volume. J Inherit Metab Dis. 2013;36:779 - 86. [PubMed: 23053469]
- Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ. Risk factors for premature ovarian failure in females with galactosemia. J Pediatr. 2000;137:833 - 41. [PubMed: 11113841]
- Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr. 2002;2:7. [PMC free article: PMC126267] [PubMed: 12350230]
- Hoffmann B, Dragano N, Schweitzer-Krantz S. Living situation, occupation and health-related quality of life in adult patients with classic galactosemia. J Inherit Metab Dis. 2012;35:1051 - 8. [PubMed: 22447152]
- Hoffmann B, Wendel U, Schweitzer-Krantz S. Cross-sectional analysis of speech and cognitive performance in 32 patients with classic galactosemia. J Inherit Metab Dis. 2011;34:421 - 7. [PubMed: 21347587]
- Hunter M, Angelicheva D, Levy HL, Pueschel SM, Kalaydjieva L. Novel mutations in the GALK1 gene in patients with galactokinase deficiency. Hum Mutat. 2001;17:77 - 8. [PubMed: 11139256]
- Kolosha V, Anoia E, de Cespedes C, Gitzelmann R, Shih L, Casco T, Saborio M, Trejos R, Buist N, Tedesco T, Skach W, Mitelmann O, Ledee D, Huang K, Stambolian D. Novel mutations in 13 probands with galactokinase deficiency. Hum Mutat. 2000;15:447 - 53. [PubMed: 10790206]
- Komrower GM. Galactosemia – thirty years on. The experience of a generation. J Inherit Metab Dis. 1982;5:96 - 104.
- Krabbi K, Uudelepp ML, Joost K, Zordania R, Õunap K. Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab. 2011;103:249 - 53. [PubMed: 21501963]
- Lai K, Langley SD, Dembure PP, Hjelm LN, Elsas LJ 2nd. Duarte allele impairs biostability of galactose-1-phosphate uridyltransferase in human lymphoblasts. Hum Mutat. 1998;11:28 - 38. [PubMed: 9450900]
- Lai K, Langley SD, Singh RH, Dembure PP, Hjelm LN, Elsas LJ 2nd. A prevalent mutation for galactosemia among black Americans. J Pediatr. 1996;128:89 - 95. [PubMed: 8551426]
- Lai K, Tang M, Yin X, Klapper H, Wierenga K, Elsas L. ARHI: A new target of galactose toxicity in classic galactosemia. Biosci Hypotheses. 2008;1:263 - 71. [PMC free article: PMC2613282] [PubMed: 19122833]
- Langley SD, Lai K, Dembure PP, Hjelm LN, Elsas LJ. Molecular basis for Duarte and Los Angeles variant galactosemia. Am J Hum Genet. 1997;60:366 - 72. [PMC free article: PMC1712399] [PubMed: 9012409]
- Lee PJ, Lilburn M, Wendel U, Schadewaldt P. A woman with untreated galactosaemia. Lancet. 2003;362:446. [PubMed: 12927432]
- Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, Elsas LJ. The human galactose-1-phosphate uridyltransferase gene. Genomics. 1992;14:474 - 80. [PubMed: 1427861]
- Levy HL, Sepe SJ, Shih VE, Vawter GF, Klein JO. Sepsis due to Escherichia coli in neonates with galactosemia. N Engl J Med. 1977;297:823 - 5. [PubMed: 331112]
- Levy HL, Brown AE, Williams SE, de Juan E Jr. Vitreous hemorrhage as an ophthalmic complication of galactosemia. J Pediatr. 1996;129:922 - 5. [PubMed: 8969739]
- Lynch ME, Potter NL, Coles CD, Fridovich-Keil JL. Developmental Outcomes of School-Age Children with Duarte Galactosemia: A Pilot Study. JIMD Rep. 2015;19:75 - 84. [PMC free article: PMC4501238] [PubMed: 25681083]
- Malone JI, Diaz-Thomas A, Swan K. Problems with the new born screen for galactosaemia. BMJ Case Rep. 2011 Jun 3;2011. [PMC free article: PMC3109760] [PubMed: 22693313]
- McCorvie TJ, Kopec J, Pey AL, Fitzpatrick F, Patel D, Chalk R, Shrestha L, Yue WW. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum Mol Genet. 2016;25:2234 - 44. [PMC free article: PMC5081055] [PubMed: 27005423]
- Menezo YJ, Lescaille M, Nicollet B, Servy EJ. Pregnancy and delivery after stimulation with rFSH of a galatosemia patient suffering hypergonadotropic hypogonadism: case report. J Assist Reprod Genet. 2004;21:89 - 90. [PMC free article: PMC3455405] [PubMed: 15202737]
- National Newborn Screening and Genetics Resource Center. National newborn screening status report. Available online. 2014. Accessed 3-6-17.
- Ng WG, Xu YK, Wong LJ, Kaufman FR, Buist NR, Donnell GN. Two adult galactosaemia females with normal ovarian function and identical GALT mutations (Q188R/R333G). J Inherit Metab Dis. 2003;26:75 - 9. [PubMed: 12872845]
- Otaduy MC, Leite CC, Lacerda MT, Costa MO, Arita F, Prado E, Rosemberg S. Proton MR spectroscopy and imaging of a galactosemic patient before and after dietary treatment. Am J Neuroradiol. 2006;27:204 - 7. [PubMed: 16418384]
- Panis B, Bakker JA, Sels JP, Spaapen LJ, van Loon LJ, Rubio-Gozalbo ME. Untreated classical galactosemia patient with mild phenotype. Mol Genet Metab. 2006a;89:277 - 9. [PubMed: 16621642]
- Panis B, Gerver WJ, Rubio-Gozalbo ME. Growth in treated classical galactosemia patients. Eur J Pediatr. 2007;166:443 - 6. [PubMed: 17024348]
- Panis B, Vermeer C, van Kroonenburgh MJ, Nieman FH, Menheere PP, Spaapen LJ, Rubio-Gozalbo ME. Effect of calcium, vitamins K1 and D3 on bone in galactosemia. Bone. 2006b;39:1123 - 9. [PubMed: 16782422]
- Potter NL, Nievergelt Y, Shriberg LD. Motor and speech disorders in classic galactosemia. JIMD Rep. 2013;11:31 - 41. [PMC free article: PMC3755563] [PubMed: 23546812]
- Powell KK, Van Naarden Braun K, Singh RH, Shapira SK, Olney RS, Yeargin-Allsopp M. Long-term speech and language developmental issues among children with Duarte galactosemia. Genet Med. 2009;11:874 - 9. [PubMed: 19904210]
- Robertson A, Singh RH. Outcomes analysis of verbal dyspraxia in classic galactosemia. Genet Med. 2000;2:142 - 8. [PubMed: 11397328]
- Rubio-Agusti I, Carecchio M, Bhatia KP, Kojovic M, Parees I, Chandrashekar HS, Footitt EJ, Burke D, Edwards MJ, Lachmann RH, Murphy E. Movement disorders in adult patients with classical galactosemia. Mov Disord. 2013;28:804 - 10. [PubMed: 23400815]
- Rubio-Gozalbo ME, Gubbels C, Bakker J, Menherre P, Wodzig W, Land J. Gonadal function in male and female patients with classic galactosemia. Hum Reprod Update. 2010;16:177 - 88. [PubMed: 19793842]
- Ryan EL, Lynch ME, Taddeo E, Gleason TJ, Epstein MP, Fridovich-Keil JL. Cryptic residual GALT activity is a potential modifier of scholastic outcome in school age children with classic galactosemia. J Inherit Metab Dis. 2013;36:1049 - 61. [PMC free article: PMC3657299] [PubMed: 23319291]
- Saudubray JM, van den Berghe G, Walter JH, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. 5 ed. New York, NY: Springer. 2012.
- Schadewaldt P, Hoffmann B, Hammen HW, Kamp G, Schweitzer-Krantz S, Wendel U. Longitudinal assessment of intellectual achievement in patients with classical galactosemia. Pediatrics. 2010;125:e374 - 81. [PubMed: 20100763]
- Schadewaldt P, Kamalanathan L, Hammen HW, Wendel U. Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab. 2004;81:31 - 44. [PubMed: 14728989]
- Schweitzer-Krantz S. Early diagnosis of inherited metabolic disorders towards improving outcome: the controversial issue of galactosaemia. Eur J Pediatr. 2003;162 Suppl 1:S50 - 3. [PubMed: 14614623]
- Shield JP, Wadsworth EJ, MacDonald A, Stephenson A, Tyfield L, Holton JB, Marlow N. The relationship of genotype to cognitive outcome in galactosaemia. Arch Dis Child. 2000;83:248 - 50. [PMC free article: PMC1718484] [PubMed: 10952646]
- Suzuki M, West C, Beutler E. Large-scale molecular screening for galactosemia alleles in a pan- ethnic population. Hum Genet. 2001;109:210 - 5. [PubMed: 11511927]
- Takci S, Kadayifcilar S, Coskun T, Yigit S, Hismi B. A rare galactosemia complication: vitreous hemorrhage. JIMD Rep. 2012;5:89 - 93. [PMC free article: PMC3509908] [PubMed: 23430922]
- Tang M, Wierenga K, Elsas LJ, Lai K. Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem Biol Interact. 2010;188:376 - 85. [PMC free article: PMC2980576] [PubMed: 20696150]
- ten Hoedt AE, Maurice-Stam H, Boelen CC, Rubio-Gozalbo ME, van Spronsen FJ, Wijburg FA, Bosch AM, Grootenhuis MA. Parenting a child with phenylketonuria or galactosemia: implications for health-related quality of life. J Inherit Metab Dis. 2011;34:391 - 8. [PMC free article: PMC3063540] [PubMed: 21290186]
- Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, Elsas LJ 2nd, Strobl W, Kozak L, Coskun T, Novelli G, Okano Y, Zekanowski C, Shin Y, Boleda MD. Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum Mutat. 1999;13:417 - 30. [PubMed: 10408771]
- Tyfield LA. Galactosemia and allelic variation at the galactose-1-phosphate uridyltransferase gene: a complex relationship between genotype and phenotype. Eur J Pediatr. 2000;159:S204 - 7. [PubMed: 11216901]
- van Erven B, Gubbels CS, van Golde RJ, Dunselman GA, Derhaag JG, de Wert G, Geraedts JP, Bosch AM, Treacy EP, Welt CK, Berry GT, Rubio-Gozalbo ME. Fertility preservation in female classic galactosemia patients. Orphanet J Rare Dis. 2013;8:107. [PMC free article: PMC3718676] [PubMed: 23866841]
- Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13:802 - 18. [PubMed: 1706789]
- Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D’Anna K, Gentile J, Guo C-Y, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu B-L, Levy H, Elsas L, Berry GT. The adult galactosemic phenotype. J Inherit Metab Dis. 2012;35:279 - 86. [PMC free article: PMC3641771] [PubMed: 21779791]
- Walter JH, Collins JE, Leonard JV. Recommendations for the management of galactosaemia. UK Galactosaemia Steering Group. Arch Dis Child. 1999;80:93 - 6. [PMC free article: PMC1717786] [PubMed: 10325771]
- Walter JH, Fridovich-Keil JL. Galactosemia. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 72. New York, NY: McGraw-Hill. 2014. Available online.
- Webb AL, Singh RH, Kennedy MJ, Elsas LJ. Verbal dyspraxia and galactosemia. Pediatr Res. 2003;53:396 - 402. [PubMed: 12595586]
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar SC, Waisbren SE, Bosch AM. Galactosemia Network (GalNet). International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40:171 - 6. [PMC free article: PMC5306419] [PubMed: 27858262]
- Zlatunich CO, Packman S. Galactosaemia: early treatment with an elemental formula. J Inherit Metab Dis. 2005;28:163 - 8. [PubMed: 15877205]
Suggested Reading
- Berry GT, Elsas LJ. Introduction to the Maastricht workshop on galactosemia: lessons from the past and new directions in galactosemia. J Inherit Metab Dis. 2011;34:249 - 55. [PubMed: 21116719]
- Berry GT, Walter JH and Friedovich-Keil J. Disorders of galactose metabolism. In: Saudubray JM, Baumgartner MR, Walter JH, eds. Inborn Metabolic Diseases – Diagnosis and Treatment. 6 ed. New York: Springer Inc; 2016:139-47.
- Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29:516 - 25. [PubMed: 16838075]
本章节的注解
作者历史
Gerard T Berry, MD, FFACMG (2014-present)
Louis J Elsas II, MD, FFACMG; University of Miami (1999-2014)
更新历史
- 9 March 2017 (ma) 系统性更新发布到公开网页上
- 3 April 2014 (me) 系统性更新发布到公开网页上
- 26 October 2010 (me) 系统性更新发布到公开网页上
- 27 September 2007 (me) 系统性更新发布到公开网页上
- 2 May 2005 (me) 系统性更新发布到公开网页上
- 27 March 2003 (me) 系统性更新发布到公开网页上
- 4 February 2000 (me) 内容发布到公开网页上
- 31 August 1999 (le) 最早稿件