
概要
临床特点.
孤立性促性腺激素释放激素(GnRH)缺陷症(IGD)的特点是在低循环浓度类固醇激素存在的状态下出现的血清促性腺激素LH(黄体生成素)和FSH(卵泡刺激 素)的异常降低。约40%的IGD患者有正常的嗅觉(normosmic IGD),约60%的IGD患者嗅觉受损(卡尔曼综合征)。IGD可在婴儿期、青春期或成年后首次显现。先天性IGD男婴常有阴茎短小和隐睾。青少年和成人IGD,体检时可发现性腺机能减退和性成熟不全的临床表现。成年IGD男性往往有青春期前睾丸体积减小(即< 4ml),第二性征(如面部、腋窝毛发生长、声音低沉)的缺失,肌肉减少,性欲减退,勃起功能障碍和不育。成年女性乳房很少发育或没有发育,原发闭经。 虽然骨骼成熟延迟,线性增长的速度通常是正常的,除了没有明显的快速青春期生长。
诊断/检测.
青春期缺失或有部分青春期表现的青少年IGD通常经生化检测来诊断,检测揭示低血清浓度的睾酮或 雌二醇(性腺机能减退),这是由其他的正常垂体前叶解剖和功能并排除其他导致HH的继发原因的情况下GnRH介导的LH和FSH(促性腺激素分泌不足的性腺功能减退 症[HH])完全或部分缺失所致。约半数的IDG是与超过25个基因的致病变异有关;其余IGD病例的遗传原因不明。.
管理.
对症治疗: 对于第二性征的诱导和维持,可逐渐增加男性患者睾酮或人绒毛膜促性腺激素(HCG)的注射,或增加女性患者雌激素和黄体酮注射;对于刺激精子形成或卵泡发育,可联合促性腺激素(HCG和人绝经期促性腺激素治疗[hMG]或重组FSH),或脉冲GnRH疗法。不管是男性的精子发生还是女性的排卵诱导问题,如果受孕失败,体外受精可能是一种选择。
并发症的预防: 应提倡最佳的钙和维生素D摄入量和根据需要采取特异的治疗来应对骨量减少。
监管: 对于有IGD迹象的男童和女童,11岁后定期监测:性成熟(通过Tanner分期体检);促性腺激素和性激素水平;骨龄。在IGD确诊个体,定期监测:血清性激素水平(指导最佳激素替代治疗);骨密度。
风险评估: 如果家族中有已知的致病变异,青春期前的高危亲属基因检测可明确其遗传状况。由于 可变的表现度,一个带有已知致病性变异的青春期前的孩子可以有一个正常的或延迟青春期的进展,或根本没有青春期;因此,随着时间的推移,临床再评估是必要的。
GeneReview 范围
| 孤立性促性腺激素释放激素缺陷症: 包括的表型 |
|---|
|
For synonyms and outdated names see Nomenclature.
诊断
孤立性促性腺激素释放激素(GnRH)缺陷(IGD)可与嗅觉正常(normosmic IGD)或嗅觉受损(卡尔曼综合征[KS])相关联。
提示性发现
有以下表现的个体应怀疑其为孤立性促性腺激素释放激素(GnRH)缺陷症(IGD)患者:- 青少年期没有或有部分青春期表现;生化检测显示低水平的血清睾酮或雌二醇
- Tanner分期确定体格检查有不完全性成熟的结果 (见 Table 1):
- IGD男性通常有Tanner I-II期生殖器(青春期前睾丸体积<4毫升);然而,一些男性有部分青春期的成熟表现 [Pitteloud et al 2001].
- IGD女性通常有Tanner I期乳腺发育和闭经;然而,有些会有自发性乳腺发育和偶尔的月经 [Shaw et al 2011].
- IGD男性和女性通常有Tanner II-III期的阴毛,因为阴毛发育部分受控于肾上腺雄激素。
在很少的男性患者中,IGD可能发生在成年之后(即成年起病的IGD)。然而,在这些患者中,青春期没有中断,性成熟是完全的,第二性征可能得到充分发展。成年起病的IGD诊断要依靠促性腺激素分泌不足的性腺功能减退(HH)并排除其他可继发导致HH的原因。
- 男性总睾酮 (T) <100 ng/dL,女性雌二醇 (E2) <50 pg/mL
- 在低循环浓度的性类固醇激素的存在下,有异常低或正常血清LH(黄体生成素)和FSH(促卵泡刺激素)。其他垂体前叶激素水平通常是正常的。
- IGD的影像学检查结果
- IGD患者的下丘脑和垂体MRI检查通常表现正常。
- 卡尔曼综合征患者通常显示不发育或发育不全的嗅球/沟/束。
- 嗅觉检查结果. 嗅觉功能通过病史和正规的诊断性嗅觉测试来评估,如宾夕法尼亚大学的嗅觉识别测试(UPSIT)、“一刮即嗅”测试,识别40微囊化气味的能力评估,这些检测易于操作可在多数临床检测中应用 [Doty 2007]。嗅觉缺失,嗅觉减退,或使用UPSIT手册正常识别确定的无嗅觉异常,结合测试年龄和性别,获得个人评分。IGD患者或者是自述完全的嗅觉丧失,或者是经UPSIT测试发现嗅觉减退/嗅觉缺失而被诊断为KS的个体,而那些嗅觉功能正常的则被诊断为normosmic IGD(nIGD) [Lewkowitz-Shpuntoff et al 2012].
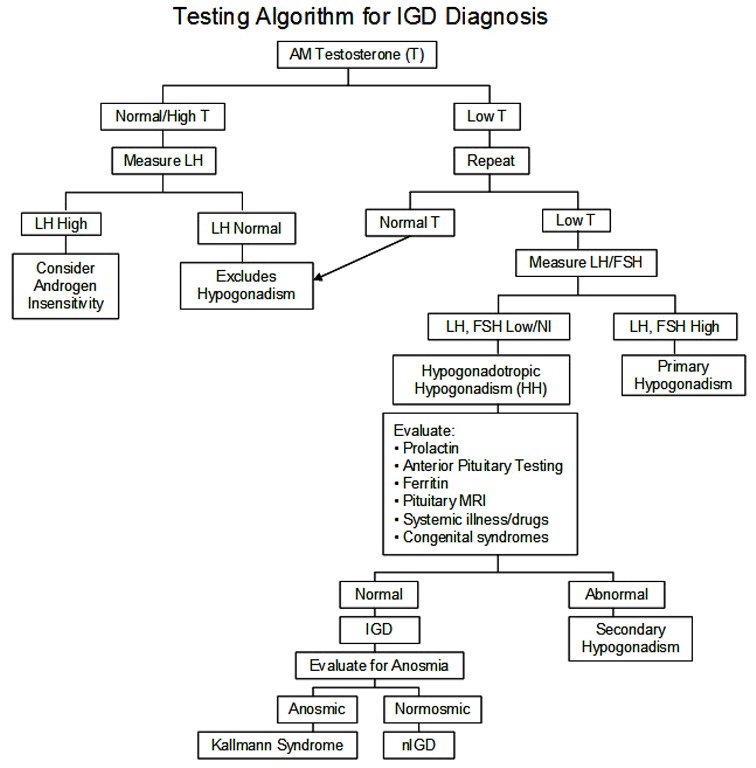
Figure 1.
诊断男性孤立性促性腺激素缺乏症(IGD)的检测程序
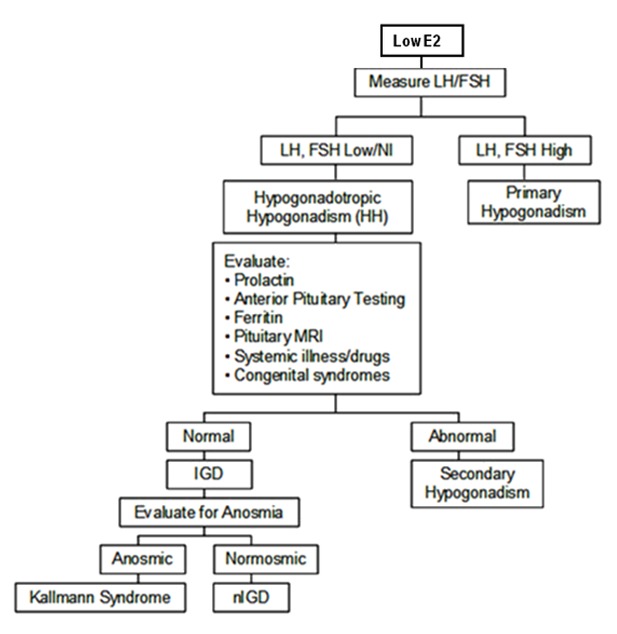
Figure 2.
诊断女性孤立性促性腺激素缺乏症(IGD)的检测程序
Table 1.
Tanner 分期
| 分期 | 正常表现 | ||
|---|---|---|---|
| 阴毛 | 男性生殖器 | 女性乳房发育 | |
| I | 无 | 幼儿型睾丸、阴囊、阴茎 (睾丸容积 <4 mL) | 没有乳芽,乳晕小,乳头略增 |
| II | 少许稀疏色浅直毛 | 睾丸增大;阴囊皮肤变红 | 乳芽形成; 乳晕增大 |
| III | 色深、粗糙、卷曲毛 | 睾丸持续增大,阴茎增长 | 乳芽和乳晕持续增长,乳房的乳晕汇合 |
| IV | 覆盖耻骨成人毛发 | 睾丸持续增大,龟头发育阴茎扩大;阴囊皮肤变暗 | 持续增长,乳晕和乳头形成二次丘投射于乳房轮廓 |
| V | 横向分布的成人型毛发 | 成熟的成人生殖器 (睾丸容积 >15 mL) | 成熟 (乳晕再次与乳房轮廓汇合;只有乳头投射) |
建立诊断
IGD先证者的诊断是根据上面描述的临床和生化研究建立的; Table 2a 和Table 2b列出了用基因诊断可识别致病变异的基因。
Table 2a为最常见的遗传原因(即> 2% IGD是由包含在该表中的任一基因的致病变异所致)和Table 2b为不常见的遗传原因(即表中任一基因的致病变异仅在几个家庭中被报道)。
分子检测方法包括用一个多基因组合进行的 系列单基因检测和更为广泛的基因检测。
系列单基因检测被认为是基于遗传方式和临床所见,尤其是非生殖表型特征表明一个特定基因的致病变异是最有可能的。如果没有发现 致病变异,首先对感兴趣的基因进行序列分析,其次是基因靶向缺失/重复分析。
下图可以帮助优选系列单基因检测的顺序 (见Figure 3 和 Figure 4):
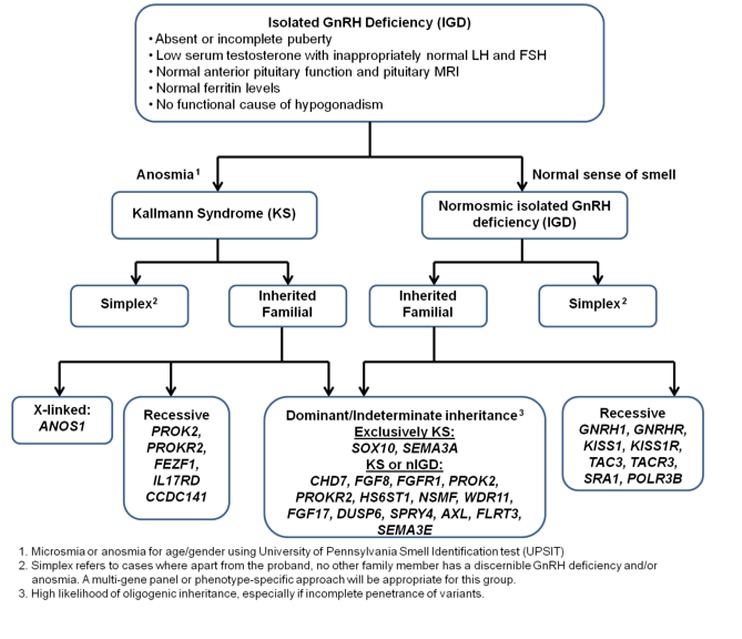
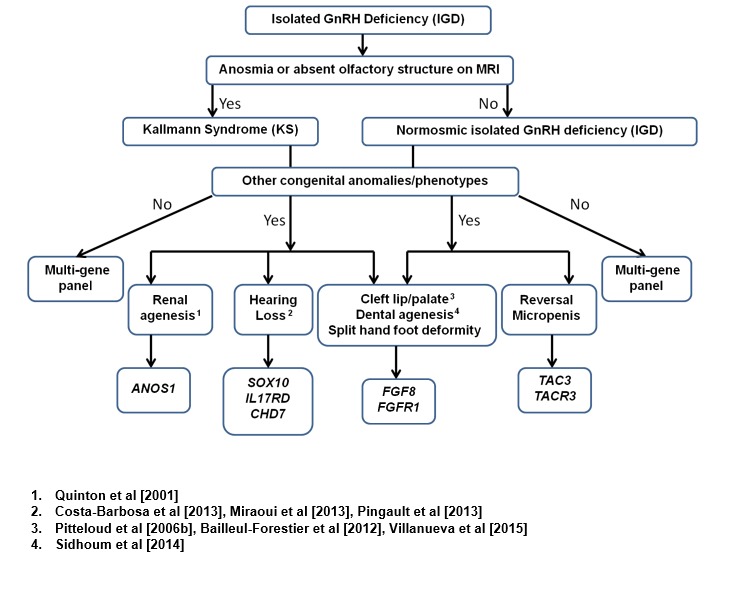
- 嗅觉
- 遗传方式
一个多基因组合,可以考虑包括表 2a和 表 2b所列的基因和其他感兴趣基因(见鉴别诊断)。当 先证者没有明显受累的家庭成员和/或没有相关的表型特征时,多基因面板应该作为第一步检测方法。 注:(1)组合中包含的基因和每个基因检测的诊断敏感性因实验室和时间的推移而有所不同。 (2)某些多基因组合可能包含与genereview讨论状态不相关的基 因;因此,临床医生需要确定哪一种多基因组合能为确定遗传原因提供最佳机会,以最合理的成本同时限制继发结果。(3)在组合中使用的方法可包括 序列分析,缺失/重复分析 和/或其他非测序为基础的检测。
更为综合的基因检测(当可用时)包括 外显子组测序和基因组测序可以考虑。这种测试可以提供或提示一个以前没有考虑的诊断(例如,不同基因突变的或导致相似临床表现的基因)。更多关于综合的基因组测序请点击 这里。
Table 2a.
孤立性促性腺激素释放激素(GnRH)缺陷症的分子遗传学检测概述:最常见的遗传原因
| 基因 1, 2 | %的IGD归结于该基因的致病变异 3 | 检测方法见得到致病变异 4 的比例 | |
|---|---|---|---|
| 序列分析 5 | 基因靶向缺失/重复分析 6 | ||
| ANOS1 ( KAL1) | 5%-10% (KS) | ~88%-99% | 在一项研究中≤12%(4/33 persons w/KS) 7 |
| CHD7 | 5%-10% (KS or nIGD) | ~100% | 未知 8 |
| FGFR1 | ~10% (KS or nIGD) | ~99% | 罕见 9 |
| GNRHR | 5%-10% (nIGD) | ~100% | 未知 8 |
| IL17RD | 2%-5% (KS or nIGD) | ~100% | 未知 8 |
| PROKR2 | ~5% (KS or nIGD) | ~100% | 未知 8 |
| SOX10 | 2%-5% (KS) | ~100% | 未知8 |
| TACR3 | ~5% (nIGD) | ~100% | 未知 8 |
Table 2b.
孤立性促性腺激素释放激素(GnRH)缺陷症的分子遗传学:不常见的遗传原因
| 基因 1, 2, 3 | 注释 | 参考文献 |
|---|---|---|
| AXL | Described in 1 report: 4/104 persons w/KS or nIGD | Salian-Mehta et al [2014] |
| CCDC141 | Described in 1 report: 1/20 persons w/KS | Hutchins et al [2016] |
| DUSP6 | Described in 1 report: 5/386 persons w/KS or nIGD | Miraoui et al [2013] |
| FEZF1 | Described in 1 report: 2/30 persons w/KS | Kotan et al [2014] |
| FGF8 | <2% 4 of persons w/KS or nIGD | |
| FGF17 | Described in 1 report: 3/386 persons w/KS or nIGD | Miraoui et al [2013] |
| FLRT3 | Described in 1 report: 3/386 persons w/KS or nIGD | Miraoui et al [2013] |
| GNRH1 | Typically AR; <2% 4 of persons w/nIGD | |
| HS6ST1 | <2% of persons w/KS or nIGD 4, 5 | |
| KISS1 | Typically AR; <2% of persons w/nIGD 4 | |
| KISS1R | Typically AR; <2% of persons w/nIGD 4 | |
| POLR3B | Described in 1 report: 3/565 persons w/KS or nIGD | Richards et al [2017] |
| PROK2 | Typically AR; <2% 4 of persons w/KS or nIGD 4 | |
| SEMA3A | <2% of persons w/KS or nIGD 4, 5 | |
| SEMA3E | Described in 1 report: 1/121 persons w/KS or nIGD | Cariboni et al [2015] |
| SPRY4 | Described in 1 report: 14/386 persons w/KS or nIGD | Miraoui et al [2013] |
| SRA1 | Described in 1 report: 3/136 persons w/nIGD | Kotan et al [2016] |
| TAC3 | Typically AR; <2% of persons w/nIGD 4 | |
| WDR11 | Described in 1 report: 1 person w/balanced 易位; 6/201 persons w/KS or nIGD | Kim et al [2010] |
临床表现
临床描述
孤立性促性腺激素释放激素缺陷症(IGD)的临床表现取决于生殖轴缺陷首先发生的阶段–婴儿期,青春期还是成年期(很少)。大多数IGD个体在青春期确诊;然而,提示性临床特征可能会表现于婴儿期。
生殖表型
婴儿期. 小阴茎(足月新生儿男性阴茎伸展长度<1.9 cm)和隐睾(睾丸未下降)代表可能在男性婴幼儿IGD中的两个临床早期所见,尽管这些发现直到青春期才能被认可。这两个临床表现反映男婴 先天性促性腺激素缺乏,出生后6个月的男婴(新生儿窗口)睾酮,LH(黄体生成素),FSH(卵泡刺激素)浓度都很低。[ Grumbach 2005 ].
虽然小阴茎、隐睾可在两种形式的IGD出现(KS和嗅觉正常的IGD),这些特征在KS男性中比在嗅觉正常IGD男性中更常见 [Pitteloud et al 2002a].
女婴通常不表现任何可能提示IGD的临床特征。
青春期. 在青春期,大多数IGD个体有性成熟异常,通常有第二性征发育不全。然而,性成熟 受影响程度是可以有所不同的(见IGD男性的生育性腺功能减退)。
男性IGD通常青春期前睾丸体积(即<4毫升),第二性特征的缺失(如面部、腋窝毛发生长和声音低沉),肌肉质量减少。
女性IGD通常很少或没有乳房发育,原发闭经,但轻度临床表现与自主月经是公认的 [Shaw et al 2011]。
由于肾上腺成熟进展正常,在肾上腺产生的低水平雄激素可足够使两性的阴毛生长正常启动(肾上腺功能初现)。
由于性激素缺乏导致骨生长板融合失败,大多数IGD男性和女性可有与身高不成比例的臂展(臂展通常超过身高5cm)。骨骼成熟延迟,线性增长率通常是正常的(除了缺乏一个明显的的青春期生长突增) [Van Dop et al 1987].
IGD男性的生育性腺功能减退. 一些IGD个体可发生一定程度的青春期发育。青春期发育异常的相对温和形式见于有性腺功能低下临床迹象的男性,低血请浓度的睾酮、有正常或近似正常的睾丸体积的部分青春期发育的迹象、正常水平的抑制素B(生精小管分泌蛋白)、精液中常有精子。
IGD的逆转,定义为性激素或GnRH短暂治疗停止后血清睾酮浓度恢复正常,占所有IGD患者的10%,包括那些KS个体 [Raivio et al 2007, Sidhoum et al 2014]。这种处理后下丘脑-垂体-性腺(HPG)轴的“觉醒”显示下丘脑的GnRH的神经元不能在青春期发挥功能,可能需要迄今未明确的刺激(潜在的环境/性激素暴露)才能启动正常活动。逆转现象的精确生理基础尚未完全理解。
嗅觉表型
嗅觉缺失. 卡尔曼综合征的嗅觉功能受损可能是嗅觉减退或嗅觉完全缺失) [Bianco & Kaiser 2009]。嗅觉减退和嗅觉缺失之间的区别是定量而非定性的(即,在嗅觉减退的人中,气味会受到不确定的 影响)。多数嗅觉受损的个体没有任何物理或社交障碍,表现常被忽视直到被诊断为IGD。
生殖和非生殖表型基因
Table 3 生殖缺陷,嗅觉功能,非生殖问题范围的基因总结
Table 3.
孤立性促性腺激素释放激素缺陷症(IGD)表型基因
| 基因 | 表型特征 | References | ||
|---|---|---|---|---|
| 生殖的 | 嗅觉的 | 其他非生殖的 | ||
| ANOS1 ( KAL1) 1, 2 | IGD (男性) | 嗅觉缺失 或 嗅觉减退 | 联带运动 (~80%男性 ) 单侧肾脏发育不全 (i ~30% 男性) 3 高足弓 | Oliveira et al [2001] Quinton et al [2001] Pitteloud et al [2002a] Massin et al [2003] Costa-Barbosa et al [2013] |
| AXL 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Salian-Mehta et al [2014] |
| CCDC141 | IGD | 嗅觉缺失 | 无 | Hutchins et al [2016] |
| CHD7 1, 2 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 高足弓or 腭裂 牙齿发育不全 耳发育不良 感音性耳聋 &半规管发育不全 眼缺损 身材矮小 智力障碍 | Kim et al [2008] Jongmans et al [2009] Costa-Barbosa et al [2013] |
| DUSP6 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Miraoui et al [2013] |
| FEZF1 1 | IGD | 嗅觉缺失 | 无 | Kotan et al [2014] |
| FGF8 1, 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 唇裂和/或腭裂 听觉缺失 眼距宽 屈曲指过度松弛 | Falardeau et al [2008] Trarbach et al [2010a] |
| FGF17 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Miraoui et al [2013] |
| FGFR1 1, 2 | 完全IGD-正常青春期 IGD 男性 > 女性 | 嗅觉缺失 或 嗅觉正常 4 | 联带运动 (~10%) 唇裂和/或腭裂 1+ 牙齿发育不全 指/趾 畸形 (指/趾短,并指/趾) | Dodé et al [2003] Pitteloud et al [2006b] Costa-Barbosa et al [2013] |
| FLRT3 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Miraoui et al [2013] |
| GNRH1 1, 2 | IGD (隐性); GD to n 4 (杂合的 致病变异) | 嗅觉正常 | 无 | Bouligand et al [2009] Chan et al [2009] |
| GNRHR 1, 2 | IGD (隐性); IGD-正常青春期 4 (杂合的致病变异) | 嗅觉正常 | 无 | de Roux et al [1997] Cerrato et al [2006] Bédécarrats & Kaiser [2007] |
| HS6ST1 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Tornberg et al [2011] |
| IL17RD 1, 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 | 听力缺失 | Miraoui et al [2013] |
| KISS1 1, 2 | IGD (隐性); IGD-正常青春期 4 (杂合的 致病变异) | 嗅觉正常 | 无 | Topaloglu et al [2012] |
| KISS1R 1, 2 | IGD (隐性); IGD-正常青春期 4 (杂合的 致病变异) | 嗅觉正常 | 无 | de Roux et al [2003] Seminara et al [2003] Semple et al [2005] |
| NSMF 2 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Xu et al [2011] |
| POLR3B | IGD (隐性) | 嗅觉正常 | 无 | Richards et al [2017] |
| PROKR2 1, 2, 4 and PROK2 1, 2, 4 | IGD (隐性); IGD-正常青春期 4 (杂合的 致病变异) | 嗅觉缺失 或 嗅觉正常 4 | 无 | Costa-Barbosa et al [2013] |
| SEMA3A 2, 4 | IGD-正常青春期 | 嗅觉缺失 或 嗅觉减退 | 无 | Cariboni et al [2011] Hanchate et al [2012] |
| SEMA3E 2, 4 | IGD | 嗅觉缺失 | 无 | Cariboni et al [2015] |
| SOX10 1 | IGD | 嗅觉缺失 | 听力缺失 虹膜色素减退 | Pingault et al [2013] Suzuki et al [2015] |
| SPRY4 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Miraoui et al [2013] |
| SRA1 | IGD | 嗅觉正常 | 无 | Kotan et al [2016] |
| TAC3 1, 2, 4 | IGD (隐性); IGD-正常青春期 4 (杂合的 致病变异) | 嗅觉正常 | 无 | Topaloglu et al [2009] |
| TACR3 1, 2, 4 | IGD (隐性); IGD-正常青春期 4 (杂合的 致病变异) | 嗅觉正常 | 无 | Guran et al [2009] Topaloglu et al [2009] Gianetti et al [2010] |
| WDR11 2, 4 | IGD-正常青春期 4 | 嗅觉缺失 或 嗅觉正常 4 | 无 | Kim et al [2010] |
病理生理学
促性腺激素释放激素脉冲式分泌进入垂体门脉循环标志着两性神经内分泌初始步骤在下丘脑-垂体-性腺(HPG)轴中的调控。因此,这个专门的GnRH 神经网络在这种生物层次中起着决定性作用,控制GnRH的分泌,调节性腺类固醇激素的反馈,并最终决定青春期发育的启动或抑制及整个生命周期的生育能力 [ Hoffman & Crowley 1982, Crowley et al 1985]。
在正常情况下,GnRH神经网络经历了一系列从胎儿到成年的动态变化。GnRH分泌的启动是在胎儿生命早期发起并持续直到婴儿期头几个月(表现为“小青春期”),然后在几年的儿童“静息”期内受到显著抑制 [Waldhauser et al 1981]。在青春期,未知的生物触发重新激动GnRH的分泌,导致完全性成熟。因此,生殖轴的控制是在动态变化中,启动和关闭取决于对生殖生命周期的各个点上未知生物信号的应答。
在IGD个体中,促性腺激素的脉冲模式的分析证明了异常发育模式相当广泛,从GnRH诱导的LH脉冲完全缺失到与青春早期别无二致的睡眠产生的GnRH释放 [Spratt et al 1987, Nachtigall et al 1997, Raivio et al 2007]. 这种广泛的神经内分泌活动说明了在IGD患者中观察到的多变的生殖表型。
外显率
潜在的遗传病因通常决定着生殖和非生殖表型的外显度。
KS表型(包括IGD和嗅觉缺失症)通常是在一个有 ANOS1(KAL1)致病变异的男性中是完全外显的。然而,其他非生殖表型即使在有相同 ANOS1(KAL1)基因缺陷的情况下也可能有不同的外显率。ANOS1 (KAL1)基因存在一个小 缺失的一组同卵双胎男性已被证实存在不一致的神经内分泌和非生殖表型:一对有室间隔 缺损和对一系列LH-RH刺激试验有更大的LH和FSH反应,而其他的有外斜视并对LH-RH刺激试验有低反应。[ Matsuo et al 2000]。
当双等位基因状态的致病变异发生时(即隐性变异)(FEZF1, GNRH1, GNRHR, IL17RD, KISS1, KISS1R, TAC3, TACR3),IGD外显率也相当高。然而,对于那些在几乎所有已知的IGD相关基因的 杂合致病变异,其生殖表型的外显率不完全,如同有正常性腺功能的人是已记载基因的致病变异杂合子。KS同卵双胞胎的不一致中也被证明,提示额外的改变可能在表型表现中发挥作用 [Hipkin et al 1990]。
ANOS1 (KAL1)致病变异男性的嗅觉缺失的 外显率通常是完全的,然而,IGD个体和一些基因(CHD7, FGFR1, FGF8, FGF17, HS6ST1, NSMF, PROK2, PROKR2)的杂合致病变异个体的异常嗅觉功能的外显率是不完全的,可见嗅觉正常、嗅觉减退或嗅觉缺失 [Pitteloud et al 2006a, Cole et al 2008, Falardeau et al 2008, Miraoui et al 2013, Balasubramanian et al 2014]。
术语
生化术语“促性腺激素分泌不足的性腺功能减退症”随着生殖生理的认识的增加演变而来。
“性腺功能低下”一词是指性发育障碍,基于个人的临床病史(如闭经、潮热、勃起功能障碍)以及体格检查(例如,小睾丸,阴道苍白)的研究结果。
随着对下丘脑-垂体-性腺(HPG)轴(见Pathophysiology)的了解越来越多和尿促性腺激素测量的介绍,“ 高促性腺激素的”性腺功能低下用来识别那些具有原发性腺缺陷,而“ 低促性腺激素的”性腺机能减退用来鉴别那些中枢性(即垂体或下丘脑)缺陷。
当中枢性性腺功能减退的解剖学因素被确定,“特发性”或“孤立性”促性腺激素分泌不足的性腺功能低下(IHH)被用来表示那些继发导致HH原因被排除的个体。
随后检测外源性GnRH治疗的效果表明,绝大多数的“特发性”HH个体有GnRH功能缺陷,由GnRH生物合成、分泌和/或作用(即“孤立性 GnRH缺陷症”[ IGD ])的缺陷导致。除了下丘脑GnRH缺陷,IGD个体通常垂体功能试验正常,且他们的性腺机能减退通常会对生外源性GnRH的生理治疗有反应 [Hoffman & Crowley 1982]。
在这一点上,“孤立性GnRH缺陷症”(IGD)更恰当地反映当前临床本质的理解而非之前的IHH的生化描述,因此是一个比IHH更好的词。
患病率
最近在芬兰的一项流行病学研究表明,KS男性和女性中的发病率最小值分别为1:30,000和1:125,000 [Laitinen et al 2011]。
在作者的250例IGD的队列研究中,男性占主导地位,男女比例近4:1[Seminara et al 1998]。
孤立性促性腺激素释放激素缺陷症(IGD)患者中近三分之二是KS。
基因相关(等位基因)疾病
CHD7.CHD7基因致病性变异杂合或微缺失可引起CHARGE综合征。该综合征的特征有眼缺损,心脏畸形,先天性后鼻孔闭锁、生长和发育迟缓,生殖器发育不全,耳朵异常[ Vissers et al 2004]。CHARGE综合征的生殖器异常是由性腺功能减退所致,常伴随有嗅觉缺陷和唇腭裂 [Pinto et al 2005]。
FGFR1. KS除了有高度可变的表现度,FGFR1致病变异相关的其他表型包括Pfeiffer综合征1型和osteoglophonic侏儒症。
- Pfeiffer综合征的致病变异,95%发生在FGFR2基因,只有5%发生在FGFR1基因(见 FGFR-相关的颅缝早闭综合征)。Pfeiffer综合征1型 的特点是冠状面颅缝早闭合并中重度面中部发育不全,通常智力正常,拇指宽并内偏,大脚趾有不同程度的短指。偶尔会出现听力受损和脑积水。
- Osteoglophonic侏儒症包括四肢近端发育不良,面部特征畸形,骨纤维发育不良和三叶草样的头骨。
SOX10.SOX10基因致病性变异杂合或微缺失可引起Waardenburg综合征,无IGD。该综合征的特点是耳聋、皮肤/头发/虹膜色素减退、先天性巨结肠病、神经系统的缺陷 [Pingault et al 2013]。
鉴别诊断
促性腺激素分泌不足的性腺功能减退症的其他原因
促性腺激素分泌不足的性腺功能减退症指一组不同临床状况,特点是性腺功能减退发生时,LH(促黄体生成激素)和FSH(卵泡刺激素)血清浓度异常降低。
鉴别孤立性GnRH缺陷症(IGD),导致促性腺激素分泌不足的性腺功能减退的继发原因,促性腺激素分泌不足的性腺功能减退的 综合征/ 遗传原因,通常需要更多的临床、实验室和影像学评估。这些评估可能包括用于发现其他系统表现的体格检查,家族史,其他垂体激素血清浓度的测量,血清铁的研究,下丘脑 /垂体成像。值得注意的是,尽管进行了彻底的评估,IGD有时是很难与其他原因导致的促性腺激素分泌下降区分的。因此,已知的IGD相关基因( 表 2)的 分子遗传学检测可能有助于IGD的诊断。
获得原因. 多种疾病的过程,从全身性疾病到大脑和垂体肿瘤可导致促性腺激素分泌障碍。比较常见的,经常引起其他垂体激素分泌障碍的情况如下:
- 中枢神经系统或垂体肿瘤
- 垂体卒中
- 大脑/垂体辐射
- 头部创伤
- 药物: GnRH 激动剂/拮抗剂, 糖皮质激素,麻醉剂,化疗药物, 致高泌乳素血症的药物
- 高泌乳素血症导致的功能缺陷,慢性系统性疾病,进食障碍,营养不良,甲状腺功能减退,糖尿病,库兴氏病
- 全身性疾病如结节病和组织细胞增生症
综合征Table 4 中列出的综合征可与促性腺激素分泌不足的性腺功能减退以及其他重要的临床表现和/或其他垂体激素缺陷有关。
Table 4.
促性腺激素分泌不足的性腺功能减退相关综合征
| 综合征 | 遗传机制 / 相关基因 | 表型 | 参考文献 | |
|---|---|---|---|---|
| Bardet-Biedl 综合征 | 19个基因中任何一个的致病变异 1 | 发育迟缓,视觉障碍,轴后多指趾畸形,肥胖,肾功能损害 | ||
| CHARGE 综合征 | CHD7 | 眼缺损,心脏缺陷,鼻后孔闭锁、生长迟缓、耳畸形 | Pinto et al [2005], Lalani & Belmont [2009] | |
| 合并垂体激素缺乏 | HESX1 | 不同程度的垂体机能减退 | PROP1-相关联合垂体激素缺乏症, 鉴别诊断联合垂体激素缺乏 | |
| LHX3 | ||||
| LHX4 | ||||
| POU1F1 | ||||
| PROP1 | ||||
| Gordon-Holmes 综合征 | OTUD4 PNPLA6 RNF216 STUB1 | 小脑共济失调,痴呆 | Seminara et al [2002], Margolin et al [2013] | |
| HFE-相关的 遗传性血色沉着病 | HFE | 肝硬化,糖尿病心肌病,关节炎,皮肤色素沉着过度 | ||
| 肥胖综合征 | PCSK1 ( PC1) | 病态肥胖,肾上腺皮质功能减退,低胰岛素血症 | Jackson et al [1997], Jackson et al [2003] | |
| LEP | 病态肥胖 | Strobel et al [1998] | ||
| LEPR | Clément et al [1998] | |||
| Prader-Willi 综合征 | 父源15q11.2缺失 | 婴儿期张力减退,发育迟缓,男性隐睾/小阴茎,异常饱腹感,智力障碍 | Cassidy et al [2012] | |
| TUBB3 E410K 综合征 (见 Tubulinopathies Overview) | TUBB3 | 先天性眼外肌纤维化,面神经无力, 发育迟缓,感觉运动神经多发性神经病,刻板的面中部发育不良,智力障碍和声带麻痹 ,气管软化和周期性呕吐。 | Chew et al [2013], Balasubramanian et al [2015] | |
| X-连锁先天性肾上腺发育不良 adrenal hypoplasia congenita | NR0B1 | 肾上腺皮质功能减退 | ||
| Xp22.3 邻近基因缺失综合征 | Xp22.3 微缺失 | KS,鱼鳞病,身材矮小,智力障碍, 点状软骨发育不良,手指联带运动 | Bick et al [1989], Hou [2005] |
至少有19个基因与BBS相关: ARL6, BBIP1, BBS1, BBS2, BBS4, BBS5, BBS7, BBS9, BBS10, BBS12, CEP290, IFT27, LZTFL1, MKKS, MKS1, SDCCAG8, TRIM32, TTC8和WDPCP.
特定发育阶段的IGD鉴别诊断
婴儿期. 虽然IGD男性出生时可有隐睾和/或小阴茎,但这些特点并非是IGD特异的。许多疾病可以引起这些生殖缺陷,从 孤立的研究结果到Prader-Willi 综合征或垂体发育异常(见 PROP1-相关联合垂体激素缺乏症)。隐睾是尤为准确的、男性生殖器最常见的出生缺陷。
青春期. 也许最难的区别是IGD和原发性的青春期延迟(CDP)。时间是鉴别这两个状况的关键因素。在CDP中,自发的和其他正常的青春期最终发生,而在IGD中 任何时间都不会发生自发的性成熟。有证据表明,CDP和IGD不是离散的临床存在,而是一个表型谱的一部分。在IGD家庭,青春期延迟发生在其他的“正 常”家庭成员的频率要高于一般人群的发生率,这表明CDP可以代表一个IGD 表型温和的临床变异 [ Waldstreicher et al 1996, Pitteloud et al 2006a]。
虽然在任何年龄短CDP和IGD的差异不能被可靠地区分,但习惯上认为18岁是IGD的可确诊年龄;然而,最近一些人IGD“逆转”的描述发生在20多岁以后,提示这些人可能是的CDP严重形式 [Raivio et al 2007]。相比之下,与IGD相关的其他临床特征(例如嗅觉丧失,联带运动)( Table 3)可能会导致IGD在18岁之前被确诊。
目前,临床上尚无测试能可靠区分CDP与IGD。数据分析表明,在GnRH或hCG(人绒毛膜促性腺激素)刺激后,CDP个体的LH和性激素的平均 血清浓度与IGD个体有显著差异。然而,在GnRH或hCG刺激后检测血清LH和性激素浓度的的临床效果受限于个体血清LH和性激素浓度的显著变化,导致 CDP与IGD群体间有相当大的重叠 [Degros et al 2003]。
GnRH治疗后,游离α亚基峰基比(FAS)有助于区分CDP和IGD,通过95%范围内的 敏感性与特异性以及在10%范围内的重叠率 [Mainieri & Elnecave 2003]。最近,一个19天的HCG测试与传统的GnRH试验相结合改进了IGD和CDP的区分 [ Segal et al 2009]。然而,鉴于在这两项研究中相对较少的研究个体和有限的随访,仍需要前瞻性验证来确定以上检测真正的诊断可靠性。管理
初步诊断后评估
建议使用以下对诊断为IGD的个体建立疾病程度与需求的评估:
- 低促性腺素性功能减退症实验室检查*的评估,是否未作为诊断建立的一部分来执行对性腺功能实验室检查评估是否已经进行的诊断工作的一部分* 血清LH(黄体生成素)和FSH(卵泡刺激素)浓度,男性总睾酮(T) <100 ng/dL和女性雌二醇(E 2) <50 pg/mL
- 可能的非生殖特征表现的评估包括:肾超声检查(检测单侧肾缺如),听力测试(检测感音神经性耳聋),骨骼测量(检测肢体/脊柱骨性畸形),牙科检查(检测牙发育不全),视力检查(检测虹膜和脉络膜视网膜缺损)和发育评估(是否有发育延迟的证据)
- 除了评估性腺功能减退/促性腺激素释放激素缺乏的程度,在骨骼健康的潜在恶化可能由低循环性激素导致这个问题需要解决。根据青春期的时间,GnRH缺乏的持续时间,和其他骨质疏松症的危险因素(如糖皮质激素过量,吸烟),患病个体应该考虑接受骨矿物质密度检测(见 继发并发症的预防)
- 咨询临床遗传学家和/或遗传顾问
对症治疗
一个IGD管理的欧洲专家共识已于最近出版 [Boehm et al 2015] (全文)。
通常,一个明确的IGD诊断大约在18岁左右确立。然而,偶尔会有一个临床高度怀疑的IGD可能在青春期出现,表现为嗅觉丧失和青春期延迟或在婴儿期出现小阴茎、隐睾。
年龄≥18岁的IGD男性
治疗选择包括性激素、促性腺激素和脉冲GnRH治疗。成人治疗的选择取决于治疗的目的(即诱导和维持第二性征和/或诱导和维持生育)。激素替代疗法 的选择也基于个体治疗的偏好;然而,当生育需要不是非常迫切时,睾酮替代疗法是最实际的选择。由于多数IGD个体在诊断时尚未进入青春期,初始治疗应从低 剂量开始逐渐增加至成人剂量,达到第二性征的发育。
无生育要求男性的激素替代治疗
- 睾酮注射和经皮给药途径睾酮治疗通常用于诱导青春期和维持成年睾酮水平。最近鼻睾酮已可使用,但尚无IGD患者使用的报道。注射睾酮制剂有一个“过山车”样药代动力学的效应,有高峰和低谷的水平,可达到生理学外水 平;因此,透皮制剂提供一个更有利的药代动力学特性,有增益效应。一个典型的成人睾酮替代剂量是每2周注射200毫克睾酮酯或每天注射5克1%的睾酮凝 胶。剂量与新的睾酮制剂不同;个人睾酮制剂应遵循制造商的说明。使用局部雄激素替代物的男性必须注意避免将皮肤上暴露于其他个体。有传闻表明,患者与其他家庭成员(包括妇女和儿童)之间的临床效应水平的睾酮水平传输可能有不良副作用。一旦青春期开始,睾酮替代疗法通常要无限期需要,以确保正常的性功能和维持适当的肌肉,骨骼和红细胞的量。然 而,约10%的男性IGD可发生逆转;因此,如果临床证据表明下丘脑-垂体轴的内源性活性(例如,睾酮可使睾丸生长,睾酮水平可保持尽管没有治疗/保守治 疗),应同时性一个简短的睾酮治疗冲刷应并监测睾酮水平。如果睾酮水平下降,治疗应重新开始。如果睾酮水平正常,不需要进一步的睾酮治疗;由于有些个体可 能需要重新开始治疗,应行系列的水平动态监测。
- 人绒毛膜促性腺激素(hCG)睾酮治疗的一种替代方法,HCG注射促进睾丸生长,使血清睾酮浓度正常化,并诱导第二性征的发展。在成人,hCG治疗通常在1500 IU肌注或隔日皮下注射直至血清睾酮浓度达到正常。如果血清睾酮水平持续低水平,剂量应增加250 IU。
- hCG治疗必须增加体重以对抗乳房女性化(hCG刺激男性的睾丸产生的雌激素导致)的风险。在一定程度上对男性乳房女性化的风险可以通过逐步降低hCG的剂量,达到在中正常范围内维持血清睾酮浓度最小需要剂量(~ 500 ng/dL)。
疑似IGD的男性婴儿/青少年
如果临床怀疑是IGD(例如,低睾酮水平较低并有低/正常促性腺激素)低剂量睾酮或hCG治疗可在婴儿早期给小阴茎男孩应用来增加阴茎长度 [Bin-Abbas et al 1999, Young 2012]。
由于IGD确切诊断可能到18岁才能确定,婴儿期后这些孩子一般不需要治疗到青春期的时间。在这个时候,如果高度怀疑有IGD(例如,相关的嗅觉缺 失和青春期延迟),这些患者可能会从早期开始的激素替代治疗并青春期早期睾酮或hCG治疗中受益。暗示诱导疗法可在在青少期开始,25-50mg剂量的长 效睾酮酯隔周肌肉注射。剂量可每两至三个月增加25-50mg直至完全实现男性化。一旦达到成人剂量(~200mg/ 2周),进一步的调整要基于血清睾酮水平。
有生育需求男性的激素替代治疗(男性患者的生育诱导). 当睾酮替代疗法抑制睾丸精子发生,通常需要促性腺激素或脉冲式GnRH治疗来实现男性生育潜能。
- 促性腺激素治疗。在 大多数IGD男性,促性腺激素联合使用(hCG和人绝经期促性腺激素[hMG]或重组FSH)来刺激精子发生。睾丸体积很小(≤~ 8ml)的男性,HCG的起始剂量一般为1500 IU肌注或隔日皮下注射;FSH添加剂量范围从37.5到75 IU作为hMG或重组配方。波谷血清睾酮浓度(目标:中度至正常范围[500 ng/dL]),波谷血清FSH水平(目标:中度至正常范围),精子计数监测以评估治疗反应。最近的试验表明,对于睾丸体积较低的个体,FSH先于联合治 疗前应用,可能会改善生精结局 [Dwyer et al 2013].在睾丸体积高于基线的男性中,单独的hCG治疗可能足以使精子发生和怀孕 [Burris et al 1988] 。但是,如果六到九个月后,精液分析显示持续性无精子症或明显的少精子症,应增加FSH剂量,范围从37.5到75 IU作为HMG或重组制剂添加到治疗方案中。在任何一种治疗中,必须追踪睾丸体积的变化,因为这是成功精子发生的主要决定因素之一。事实上,直到睾丸体积达到8毫升时精液分析才可见精子的存在惠特科姆和克劳利 [Whitcomb & Crowley 1990]。在大多数无隐睾病史的男性中,精子功能通常是正常的,甚至在相对低精子数的也可怀孕。注: Liu et al [2009]指出,先前的促性腺激素治疗可缩短精子发生起始所需的后续促性腺激素的治疗时间。
- 如果存在垂体缺陷,促性腺激素治疗成为治疗的选择。
- 脉冲GnRH刺激 vs. 促性腺激素治疗. 而无论是促性腺激素治疗或脉冲GnRHIGD女性的刺激都可在大约90%-95%的IGD男性中诱发精子生成,一些人对脉冲GnRH刺激的反应要好于促性腺激素治疗的反应。通过一个便携式泵送素丸每两小时GnRH脉冲方式皮下给药是一种诱导睾丸发育和精子发生的有效方法 [Pitteloud et al 2002b]。由于IGD的原发缺陷通常位于下丘脑,垂体可对生理剂量的GnRH适当反应。注:在美国,用于男性不育的脉冲GnRH治疗目前尚未得到FDA的批准,因此只能在专门的研究中心进行这种治疗。
IGD女性
对没有生育要求女性的激素替代治疗. 虽然女性IGD的确诊通常是18岁左右,偶尔临床高度怀疑IGD可能表现为青春期出现嗅觉丧失和青春期延迟,治疗可能需要早一点开始(~14岁)
- 为了保证理想的乳房发育,初始治疗应包括口服或外用制剂的雌激素替代。多种雌激素可用;建议倍力美口服剂每日0.3毫克,逐渐增加至成人替代剂量每日1-1.25mg。
- 一旦乳腺发育达到最佳,应添加孕激素来保护子宫内膜(例如,周期性口服孕酮制剂每日200mg,持续10-12天)。
- 虽然个人偏好在治疗方案的选择中有重要作用,凝血功能异常女性应考虑给予低雌激素制剂(见 Factor V Leiden Thrombophilia and Prothrombin Thrombophilia)。
有生育需求女性的激素替代治疗(女性生育诱导). 脉冲GnRH刺激和外源性促性腺激素是FDA批准用于IGD女性卵泡发育的疗法。这两种疗法都应该在专门从事排卵诱导的医生密切监督下进行。在整个月经周期中,GnRH不同频率的静脉给药,都能很好地模拟正常的月经周期,从而使单个卵泡排卵 [Santoro et al 1986]。该疗法比传统的外源性促性腺激素治疗能明显增加多胎妊娠和卵巢过度刺激综合征的发生率。这两种方法在每个排卵周期的受孕率约为30%[ Martin et al 1990]。
排卵诱导失败IGD患者的生育选择
体外受精. 虽然大多数IGD男性通过脉冲GnRH治疗或联合促性腺激素治疗可获得成功的精子发生,一些由 ANOS1(KAL1)致病变异导致的KS男性患者可对治疗有非典型的反应[ Sykiotis et al 2010a]。对于那些对治疗有反应的人来说,低精子数往往会导致受孕;然而,尽管精子发生成功,如果不育仍继续存在,体外受精(IVF)是一种选择。
同样,如果自然妊娠失败的IGD女性通过诱导排卵,IVF可以作为一种选择。
二级并发症的预防
鼓励摄取最佳剂量的钙和维生素D,根据骨矿化程度考虑双膦酸盐用于骨量减少的特异性治疗(见 初步诊断后评估)。
监督
有IGD提示性检查发现的两种性别儿童(如, 小阴茎, 嗅觉缺失) 应该从11岁开始定期监测以下情况:
- 通过Tanner分期对性成熟的评价(Table 1)
- 男性血清LH、FSH、总睾酮(T)的测定,女性雌二醇(E2)的测定
- 骨龄测定
IGD确诊个体, 血清类固醇水平 (用以指导最佳激素替代治疗) 和骨矿物质密度应该定期检测。
亲属风险评估
评估受累个体无明显临床症状的老年人和年轻人高危亲属,以期尽早明确那些受益于监管的人,促使其尽快开始治疗。
评估包括:
- 如果家庭中存在已知的致病变异,则进行分子遗传检测。然而,一个带有已知致病性变异的青春期孩子可以正常方式度过青春期,或者青春期延迟,或者完全没有延迟。因此,随着时间的推移,对这些人进行重新评估是非常重要的,只有IGD伴有青春期发育缺陷时才应开始激素治疗。
- 如果家庭中存在未知的致病变异,需要对处于青春期的高危亲属进行临床检查,以评估青春期发育的临床起始表现,如果有延迟,需要启动适当的青春期诱导治疗。
以遗传咨询为目的的高危亲属检测问题详见 Genetic Counseling 。
在研究中的治疗
搜索ClinicalTrials.gov获取各种疾病的临床研究信息取。注意:有些疾病可能没有临床试验
遗传咨询
遗传咨询是向个人和家庭提供关于遗传病的性质,传递和影响信息的过程,以帮助他们做出明智的医疗和个人决定。 以下部分涉及遗传风险评估和家族史和遗传检测的应用,以明确家庭成员的遗传状况。 本节不能涉及个人所面临的所有的个人,文化或伦理问题,也不能替代遗传学专业人士的咨询。—ED.
遗传方式
孤立性促性腺激素释放激素(GnRH) 缺陷 (IGD) 可以 X-连锁, 常染色体显性, 或常染色体隐性方式遗传(见 Figure 3).
几乎所有的IGD相关基因也与不确定的或寡基因遗传相关(特别是一个IGD相关 致病变异的杂合状态时)[Sykiotis et al 2010b, Hanchate et al 2012, Miraoui et al 2013])。
三代家族史的获取可以明确IGD的遗传模式并有助于基因检测和 遗传咨询。所有家族成员的详细的家族史都应该获取,包括血缘的 相关问题,生殖特征(例如,小阴茎和隐睾、青春期发育、生育/不孕)、嗅觉功能(正常嗅觉、嗅觉减退、嗅觉丧失),和非生殖功能(例如,颅面畸形,包括唇 腭裂/缺牙,听力缺失,联带运动,肾发育不全)。如果其他IGD个体或这些相关的结果在家系中确定,遗传方式可能会变得明显。然而,在大多数人中,没有这 样的家族史存在。家庭成员的风险 – X-连锁遗传
男性先证者的父母
男性先证者的同胞。同胞的罹患风险取决于母亲的遗传状态:
男性先证者的后代
其他家系成员.先证者 的舅父可能有 受累风险,其姨母有成为ANOS1致病变异携带者的风险。姨母的后代, 根据他们的性别, 可有成为携带者或受累的风险。
杂合子 (携带者) 检测. 如果先证者已确定有ANOS1致病变异,其高危女性亲属的分子遗传学检测是明确他们遗传状况最翔实的方法。
注: (1) 女性 ANOS1致病变异的杂合子 偶尔会有诊断为IGD的临床表现[Shaw et al 2011]. (2) 女性杂合子鉴定需要 (a) 家系中预先明确的 ANOS1致病变异,或 (b) 如果受累 男性不可检测,首先通过序列分析进行 分子遗传学检测,如果没有确定致病性变异, 再行靶向基因的 缺失/重复分析。
家系成员风险 – 常染色体显性遗传
先证者的父母
- 父母双方详细的青春期发育史;和
先证者的后代.常染色体显性遗传IGD患者的后代有50%的几率遗传 致病变异;然而,由于可变的表现度和降低的 外显率,实际风险可能会低于50%。
其他家庭成员. 其他家庭成员的风险取决于先证者父母的遗传状态:如果父母之一 受累或者有一个已知致病变异,他或她的家庭成员可能存在风险。
其他家庭成员的风险 – 常染色体隐性遗传
先证者的父母
- 杂合子通常不会发病。
- 偶尔, 杂合子会有IGD的临床表现[Cole et al 2008, Gianetti et al 2010, Sykiotis et al 2010b, Chan et al 2011, Miraoui et al 2013];这可能是由于未知的环境、表观或寡基因因素。
先证者的同胞
- 偶尔, 杂合子会有IGD的临床表现 [ Cole et al 2008, Gianetti et al 2010, Sykiotis et al 2010b, Chan et al 2011, Miraoui et al 2013];这可能是由于未知的环境、表观或寡基因因素。
先证者的后代.患者的后代是IGD相关的 致病变异的肯定杂合子(携带者)。
相关的遗传咨询问题
见风险亲属评估的管理,评估风险亲属的信息,以便及早诊断和治疗。
生育计划
- 怀孕前是确定遗传风险,明确携带者的状态,讨论产前检查有效性的最佳时机。
DNA存储是对将来可能用到的DNA(通常从白细胞提取)进行存储。因为将来我们可能对检测方式和对基因、等位基因变异和疾病的理解有所改进,应考虑存储患者的DNA。
资源
GeneReviews的工作人员选了以下疾病特异的和/或伞支持组织和/或为患此疾病的个人及其家人的利益提供登记。 GeneReviews不对其他组织提供的信息负责。有关选择标准的信息,请点击此处。
- Pituitary Network AssociationPO Box 1958Thousand Oaks CA 91358Phone: 805-499-9973Fax: 805-480-0633Email: info@pituitary.org
- My46 Trait Profile
- Pituitary FoundationPO Box 1944Bristol BS99 2UBUnited KingdomPhone: 0845 450 0375 (Helpline); 0845 450 0376Fax: 0117 933 0910Email: helpline@pituitary.org.uk
- The Pituitary SocietyVA Medical Center423 East 23rd StreetRoom 16048aWNew York NY 10010Phone: 212-951-7035Fax: 212-951-7050
分子遗传学
分子遗传学和OMIM表格中的信息可能与GeneReview中的其他信息有不同:表中可能包含了更能多最近的信息 —ED.
Table A.
孤立性促性腺激素释放激素(GnRH) 缺陷:基因和数据库
Table B.
孤立性促性腺激素释放激素(GnRH) 缺陷的OMIM条目 ( View All in OMIM)
| 109135 | AXL RECEPTOR TYROSINE KINASE; AXL |
| 136350 | FIBROBLAST GROWTH FACTOR RECEPTOR 1; FGFR1 |
| 138850 | GONADOTROPIN-RELEASING HORMONE RECEPTOR; GNRHR |
| 146110 | HYPOGONADOTROPIC HYPOGONADISM 7 WITH OR WITHOUT ANOSMIA; HH7 |
| 147950 | HYPOGONADOTROPIC HYPOGONADISM 2 WITH OR WITHOUT ANOSMIA; HH2 |
| 152760 | GONADOTROPIN-RELEASING HORMONE 1; GNRH1 |
| 162330 | TACHYKININ 3; TAC3 |
| 162332 | TACHYKININ RECEPTOR 3; TACR3 |
| 244200 | HYPOGONADOTROPIC HYPOGONADISM 3 WITH OR WITHOUT ANOSMIA; HH3 |
| 300836 | KAL1 GENE; KAL1 |
| 308700 | HYPOGONADOTROPIC HYPOGONADISM 1 WITH OR WITHOUT ANOSMIA; HH1 |
| 600483 | FIBROBLAST GROWTH FACTOR 8; FGF8 |
| 602229 | SRY-BOX 10; SOX10 |
| 602748 | DUAL-SPECIFICITY PHOSPHATASE 6; DUSP6 |
| 603286 | KISS1 METASTASIS SUPPRESSOR; KISS1 |
| 603725 | FIBROBLAST GROWTH FACTOR 17; FGF17 |
| 603819 | STEROID RECEPTOR RNA ACTIVATOR 1; SRA1 |
| 603961 | SEMAPHORIN 3A; SEMA3A |
| 604161 | KISS1 RECEPTOR; KISS1R |
| 604808 | FIBRONECTIN-LIKE DOMAIN-CONTAINING LEUCINE-RICH TRANSMEMBRANE PROTEIN 3; FLRT3 |
| 604846 | HEPARAN SULFATE 6-O-SULFOTRANSFERASE 1; HS6ST1 |
| 606417 | WD REPEAT-CONTAINING PROTEIN 11; WDR11 |
| 606807 | INTERLEUKIN 17 RECEPTOR D; IL17RD |
| 607002 | PROKINETICIN 2; PROK2 |
| 607123 | PROKINETICIN RECEPTOR 2; PROKR2 |
| 607984 | SPROUTY, DROSOPHILA, HOMOLOG OF, 4; SPRY4 |
| 608137 | NMDA RECEPTOR SYNAPTONUCLEAR SIGNALING AND NEURONAL MIGRATION FACTOR; NSMF |
| 608166 | SEMAPHORIN 3E; SEMA3E |
| 608892 | CHROMODOMAIN HELICASE DNA-BINDING PROTEIN 7; CHD7 |
| 610628 | HYPOGONADOTROPIC HYPOGONADISM 4 WITH OR WITHOUT ANOSMIA; HH4 |
| 612370 | HYPOGONADOTROPIC HYPOGONADISM 5 WITH OR WITHOUT ANOSMIA; HH5 |
| 612702 | HYPOGONADOTROPIC HYPOGONADISM 6 WITH OR WITHOUT ANOSMIA; HH6 |
| 613301 | FEZ FAMILY ZINC FINGER PROTEIN 1; FEZF1 |
| 614366 | POLYMERASE III, RNA, SUBUNIT B; POLR3B |
| 614381 | LEUKODYSTROPHY, HYPOMYELINATING, 8, WITH OR WITHOUT OLIGODONTIA AND/OR HYPOGONADOTROPIC HYPOGONADISM; HLD8 |
| 614837 | HYPOGONADOTROPIC HYPOGONADISM 8 WITH OR WITHOUT ANOSMIA; HH8 |
| 614838 | HYPOGONADOTROPIC HYPOGONADISM 9 WITH OR WITHOUT ANOSMIA; HH9 |
| 614839 | HYPOGONADOTROPIC HYPOGONADISM 10 WITH OR WITHOUT ANOSMIA; HH10 |
| 614840 | HYPOGONADOTROPIC HYPOGONADISM 11 WITH OR WITHOUT ANOSMIA; HH11 |
| 614841 | HYPOGONADOTROPIC HYPOGONADISM 12 WITH OR WITHOUT ANOSMIA; HH12 |
| 614842 | HYPOGONADOTROPIC HYPOGONADISM 13 WITH OR WITHOUT ANOSMIA; HH13 |
| 614858 | HYPOGONADOTROPIC HYPOGONADISM 14 WITH OR WITHOUT ANOSMIA; HH14 |
| 614880 | HYPOGONADOTROPIC HYPOGONADISM 15 WITH OR WITHOUT ANOSMIA; HH15 |
| 614897 | HYPOGONADOTROPIC HYPOGONADISM 16 WITH OR WITHOUT ANOSMIA; HH16 |
| 615266 | HYPOGONADOTROPIC HYPOGONADISM 17 WITH OR WITHOUT ANOSMIA; HH17 |
| 615267 | HYPOGONADOTROPIC HYPOGONADISM 18 WITH OR WITHOUT ANOSMIA; HH18 |
| 615269 | HYPOGONADOTROPIC HYPOGONADISM 19 WITH OR WITHOUT ANOSMIA; HH19 |
| 615270 | HYPOGONADOTROPIC HYPOGONADISM 20 WITH OR WITHOUT ANOSMIA; HH20 |
| 615271 | HYPOGONADOTROPIC HYPOGONADISM 21 WITH OR WITHOUT ANOSMIA; HH21 |
| 616030 | HYPOGONADOTROPIC HYPOGONADISM 22 WITH OR WITHOUT ANOSMIA; HH22 |
| 616031 | COILED-COIL DOMAIN-CONTAINING PROTEIN 141; CCDC141 |
分子遗传发病机理
在过去的三十年里,临床研究策略与现代遗传学研究方法的结合已揭示了超过25个致病/促成非综合征型IGD的基因,具有不同的遗传方式。大致有两种 遗传途径与IGD有关:(i) 神经发育基因支配的GnRH神经元的起源,通常导致卡尔曼综合征型IGD(KS)。(ii) 一组神经内分泌基因控制GnRH的分泌或作用导致的嗅觉正常的 孤立性促性腺激素释放激素缺乏型IGD(nIGD)。一小部分的基因可引起 KS和nIGD型IGD,这表明这些基因可以控制GnRH迁移和GnRH分泌/作用(见 Table 2a and Table 2b)。
除了孟德尔遗传方式,IGD复杂遗传结构(发生在10% - 15%的病例)现已有记录,其中两个或两个以上IGD相关基因的致病性变异在一个单一个体存在。这些致病性变异通常是 杂合子,本身不足以导致IGD,但是在另外一个 基因存在额外致病性变异的情况下,可导致IGD [Sykiotis et al 2010]。几乎所有已知IGD相关基因与 寡基因遗传有关。这些寡基因致病变异可能具有协同作用的方式,可能导致IGD的一些 可变表现度和外显不全。
ANOS1 (KAL1)
基因结构.ANOS1 (KAL1) 包含14个外显子,没有选择性剪接变异体。
致病性变异.ANOS1中报告的致病性变异包括全基因的 缺失,单个或多个外显子的缺失,几个碱基的缺失,致病性错义变异, 致病性无义变异和剪接变异。 更多信息, 见 Table A。
正常基因产物.ANOS1基因的编码蛋白是anosmin 1,有680个氨基酸,与神经发育相关分子的功能相似 [ Rugarli et al 1993]. N末端包含与乳清酸蛋白家族序列同源的结构域和蛋白酶抑制剂的模体。C末端含有一系列与神经细胞粘附分子相似的纤连蛋白III型重复序列。
异常基因产物. Anosmin功能不全会导致嗅觉和GnRH神经元在发育过程中从嗅板的迁移缺陷[ Cariboni et al 2004]。这些神经元的迁移受阻会出现KS、IGD和嗅觉缺失迹象,导致的嗅球畸形可通过MRI在大多数个体检测到。
CHD7
致病性变异.CHD7基因的IGD致病性变异主要是错义突变,他们或是 亚效等位基因或是显性等位基因;相反,功能丧失(即截短的)的CHD7致病性变异导致更为广泛的表型表现,如 CHARGE综合征患者的表现[Balasubramanian et al 2014]。
正常基因产物. 正常的基因产物是染色质解旋酶DNA结合蛋白7。它属于一个被认为改变核小体结构和介导染色质相互作用的蛋白质家族。
异常基因产物. 通过与CHD7同源基因的比较发现KS或nIGD患者CHD7基因致病性变异可导致截短蛋白或氨基酸替换[ Kim et al 2008]。
FGFR1
基因结构.FGFR1 包含18个外显子,有一个终止于外显子10 的剪接变异体。
致病性变异.FGFR1基因的致病性变异包括致病性的缺失突变、错义突变、 无义突变和剪接突变。更多信息,见 Table A。
正常基因产物.FGFR1 编码一个跨膜受体蛋白,包括三个胞外免疫球蛋白样结构域和一个胞内酪氨酸激酶 结构域[Lee et al 1989]。结合配体后导致受体二聚化和细胞内信号蛋白的募集。
异常基因产物. 异常FGFR1基因产物导致信号转导受损。 Anosmin的基因剂量效应和它与FGFR1的相互作用以介导GnRH神经元迁移的假说,可用来解释为何IGD男性的 表型比女性更明显[Dodé et al 2003]。
GNRHR
基因结构.GNRHR 包含三个编码外显子,有一个选择性剪接变异体。
致病性变异.GNRHR基因的致病性变异通常是错义突变, 导致嗅觉正常的IGD,常染色体隐性遗传方式。其他GNRHR基因致病性变异包括 无义突变和移码突变,可导致常染色体隐性遗传的IGD。杂合子致病突变(错义, 移码和无义) 也出现在有不同临促航表型的IGD患者中,提示寡基因的遗传模式[ ianetti et al 2012]。
IL17RD
基因结构.IL17RD 包含17个编码外显子,有7个选择性剪接变异体。
致病性变异.IL17RD基因纯合和杂合的 错义突变可导致KS型IGD,包括寡基因遗传。
正常基因产物. 该基因编码一种膜蛋白,属于白介素-17受体(IL-17R)蛋白家族,是白介素-17受体信号复合体的组成部分。该基因产物通过MAPK/ERK信号通路影响成纤维细胞生长因子信号转导,抑制或刺激细胞的生长。
PROKR2
致病性变异.PROKR2基因的致病性变异包括错义和 无义突变。
正常基因产物. 正常基因编码前动力蛋白受体-2,G蛋白偶联跨膜受体。
异常基因产物. 在KS/nIGD型患者中明确的PROKR2基因的致病变异导致受体功能减弱和信号受损[Cole et al 2008, Monnier et al 2009, Martin et al 2011]。选定的PROKR2致病变异的功能研究未能证明其有显性负效应。 PROKR2基因敲除小鼠嗅球缺失,并有因下丘脑GNRH合成神经元缺失导致的严重生殖系统萎缩[ Matsumoto et al 2006, Martin et al 2011]。
参考文献
发布的准则/共识声明
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Available online. 2015. Accessed 2-27-17. [PubMed: 26194704]
引用文献
- Au MG, Crowley WF Jr, Buck CL. Genetic counseling for isolated GnRH deficiency. Mol Cell Endocrinol. 2011;346:102–9. [PMC free article: PMC3185214] [PubMed: 21664415]
- Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, Stankovic KM, Plummer L, Buck CL, Quinton R, Nebesio TD, Mericq V, Merino PM, Meyer BF, Monies D, Gusella JF, Al Tassan N, Katsanis N, Crowley WF Jr. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci U S A. 2014;111:17953–8. [PMC free article: PMC4273325] [PubMed: 25472840]
- Balasubramanian R, Chew S, MacKinnon SE, Kang PB, Andrews C, Chan WM, Engle EC. Expanding the phenotypic spectrum and variability of endocrine abnormalities associated with TUBB3 E410K syndrome. J Clin Endocrinol Metab. 2015;100:E473–7. [PMC free article: PMC4333039] [PubMed: 25559402]
- Bédécarrats GY, Kaiser UB. Mutations in the human gonadotropin-releasing hormone receptor: insights into receptor biology and function. Semin Reprod Med. 2007;25:368–78. [PubMed: 17710733]
- Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569–76. [PMC free article: PMC2864719] [PubMed: 19707180]
- Bick D, Curry CJ, McGill JR, Schorderet DF, Bux RC, Moore CM. Male infant with ichthyosis, Kallmann syndrome, chondrodysplasia punctata, and an Xp chromosome deletion. Am J Med Genet. 1989;33:100–7. [PubMed: 2750777]
- Bin-Abbas B, Conte FA, Grumbach MM, Kaplan SL. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J Pediatr. 1999; 134:579–83. [PubMed: 10228293]
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11:547–64. [PubMed: 26194704]
- Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–8. [PubMed: 19535795]
- Burris AS, Rodbard HW, Winters SJ, Sherins RJ. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 1988;66:1144–51. [PubMed: 3372679]
- Cariboni A, Davidson K, Rakic S, Maggi R, Parnavelas JG, Ruhrberg C. Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum Mol Genet. 2011;20:336–44. [PubMed: 21059704]
- Cariboni A, Pimpinelli F, Colamarino S, Zaninetti R, Piccolella M, Rumio C, Piva F, Rugarli EI, Maggi R. The product of X-linked Kallmann's syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet. 2004;13:2781–91. [PubMed: 15471890]
- Cariboni A, André V, Chauvet S, Cassatella D, Davidson K, Caramello A, Fantin A, Bouloux P, Mann F, Ruhrberg C. Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J Clin Invest. 2015; 125:2413–28. [PMC free article: PMC4497752] [PubMed: 25985275]
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi Syndrome. Genet Med. 2012;14:10–26. [PubMed: 22237428]
- Cerrato F, Shagoury J, Kralickova M, Dwyer A, Falardeau J, Ozata M, Van Vliet G, Bouloux P, Hall JE, Hayes FJ, Pitteloud N, Martin KA, Welt C, Seminara SB. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism. Eur J Endocrinol. 2006 Nov;155 Suppl 1:S3–S10. [PubMed: 17074994]
- Chan YM, Broder-Fingert S, Paraschos S, Lapatto R, Au M, Hughes V, Bianco SD, Min L, Plummer L, Cerrato F, De Guillebon A, Wu IH, Wahab F, Dwyer A, Kirsch S, Quinton R, Cheetham T, Ozata M, Ten S, Chanoine JP, Pitteloud N, Martin KA, Schiffmann R, Van der Kamp HJ, Nader S, Hall JE, Kaiser UB, Seminara SB. GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J Clin Endocrinol Metab. 2011;96:E1771–81. [PMC free article: PMC3205899] [PubMed: 21880801]
- Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, Gaspert A, Lavoie HB, Wu CH, Crowley WF Jr, Amory JK, Pitteloud N, Seminara SB. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2009; 106:11703–8. [PMC free article: PMC2710623] [PubMed: 19567835]
- Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD, MacKinnon SE, Oystreck DT, Rankin J, Crawford TO, Geraghty M, Pomeroy SL, Crowley WF Jr, Jabs EW, Hunter DG, Grant PE, Engle EC. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013;136:522–35. [PMC free article: PMC3572929] [PubMed: 23378218]
- Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougnères P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. [PubMed: 9537324]
- Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Ravio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley WF Jr, Zhou QY, Pitteloud N. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotropin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab. 2008;93:3551–9. [PMC free article: PMC2567850] [PubMed: 18559922]
- Costa-Barbosa FA, Balasubramanian R, Keefe KW, Shaw ND, Al-Tassan N, Plummer L, Dwyer A, Buck CL, Choi J-H, Seminara SB, Quinton R, Monies D, Meyer B, Hall JE, Pitteloud N, Crowley WF. Prioritizing Genetic Testing in Patients With Kallmann Syndrome Using Clinical Phenotypes. J Clin Endocrinol Metab. 2013;98:E943–53. [PMC free article: PMC3644607] [PubMed: 23533228]
- Crowley WF Jr, Filicori M, Spratt DI, Santoro NF. The physiology of gonadotropin-releasing hormone (GnRH) secretion in men and women. Recent Prog Horm Res. 1985;41:473–531. [PubMed: 3931190]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003; 100:10972–6. [PMC free article: PMC196911] [PubMed: 12944565]
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–602. [PubMed: 9371856]
- Degros V, Cortet-Rudelli C, Soudan B, Dewailly D. The human chorionic gonadotropin test is more powerful than the gonadotropin-releasing hormone agonist test to discriminate male isolated hypogonadotropic hypogonadism from constitutional delayed puberty. Eur J Endocrinol. 2003;149:23–9. [PubMed: 12824862]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003; 33:463–5. [PubMed: 12627230]
- Doty RL. Office procedures for quantitative assessment of olfactory function. Am J Rhinol. 2007;21:460–73. [PubMed: 17882917]
- Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, Dym M, Sluss PM, Crowley WF Jr, Pitteloud N. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013;98:E1790–5. [PMC free article: PMC3816270] [PubMed: 24037890]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–31. [PMC free article: PMC2441855] [PubMed: 18596921]
- Gianetti E, Hall JE, Au MG, Kaiser UB, Quinton R, Stewart JA, Metzger DL, Pitteloud N, Mericq V, Merino PM, Levitsky LL, Izatt L, Lang-Muritano M, Fujimoto VY, Dluhy RG, Chase ML, Crowley WF Jr, Plummer L, Seminara SB. When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR). J Clin Endocrinol Metab. 2012;97:E1798–807. [PMC free article: PMC3431570] [PubMed: 22745237]
- Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LF, Costa EM, de Mendonça BB, de Castro M, Lofrano A, Hall JE, Bolu E, Ozata M, Quinton R, Amory JK, Stewart SE, Arlt W, Cole TR, Crowley WF, Kaiser UB, Latronico AC, Seminara SB. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–67. [PMC free article: PMC2902066] [PubMed: 20332248]
- Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab. 2005;90:3122–7. [PubMed: 15728198]
- Guran T, Tolhurst G, Bereket A, Rocha N, Porter K, Turan S, Gribble FM, Kotan LD, Akcay T, Atay Z, Canan H, Serin A, O'Rahilly S, Reimann F, Semple RK, Topaloglu AK. Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J Clin Endocrinol Metab. 2009;94:3633–9. [PMC free article: PMC4306717] [PubMed: 19755480]
- Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, Leroy C, Baron S, Campagne C, Vanacker C, Collier F, Cruaud C, Meyer V, García-Piñero A, Dewailly D, Cortet-Rudelli C, Gersak K, Metz C, Chabrier G, Pugeat M, Young J, Hardelin JP, Prevot V, Dodé C. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012;8:e1002896. [PMC free article: PMC3426548] [PubMed: 22927827]
- Hipkin LJ, Casson IF, Davis JC. Identical twins discordant for Kallmann's syndrome. J Med Genet. 1990;27:198–9. [PMC free article: PMC1017005] [PubMed: 2325096]
- Hoffman AR, Crowley WF Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307:1237–41. [PubMed: 6813732]
- Hou JW. Detection of gene deletions in children with chondrodysplasia punctata, ichthyosis, Kallmann syndrome, and ocular albinism by FISH studies. Chang Gung Med J. 2005;28:643–50. [PubMed: 16323556]
- Hutchins BI, Kotan LD, Taylor-Burds C, Ozkan Y, Cheng PJ, Gurbuz F, Tiong JD, Mengen E, Yuksel B, Topaloglu AK, Wray S. CCDC141 Mutation Identified in Anosmic Hypogonadotropic Hypogonadism (Kallmann Syndrome) Alters GnRH Neuronal Migration. Endocrinology. 2016;157:1956–66. [PMC free article: PMC4870868] [PubMed: 27014940]
- Jackson RS, Creemers JW, Farooqi IS, Raffin-Sanson ML, Varro A, Dockray GJ, Holst JJ, Brubaker PL, Corvol P, Polonsky KS, Ostrega D, Becker KL, Bertagna X, Hutton JC, White A, Dattani MT, Hussain K, Middleton SJ, Nicole TM, Milla PJ, Lindley KJ, O'Rahilly S. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–60. [PMC free article: PMC259128] [PubMed: 14617756]
- Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O'Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. [PubMed: 9207799]
- Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, Crowley WF Jr, Hoefsloot LH. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71. [PMC free article: PMC2854009] [PubMed: 19021638]
- Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010;87:465–79. [PMC free article: PMC2948809] [PubMed: 20887964]
- Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–9. [PMC free article: PMC2561938] [PubMed: 18834967]
- Kotan LD, Hutchins BI, Ozkan Y, Demirel F, Stoner H, Cheng PJ, Esen I, Gurbuz F, Bicakci YK, Mengen E, Yuksel B, Wray S, Topaloglu AK. Mutations in FEZF1 cause Kallmann syndrome. Am J Hum Genet. 2014;95:326–31. [PMC free article: PMC4157145] [PubMed: 25192046]
- Kotan LD, Cooper C, Darcan Ş, Carr IM, Özen S, Yan Y, Hamedani MK, Gürbüz F, Mengen E, Turan İ, Ulubay A, Akkuş G, Yüksel B, Topaloğlu AK, Leygue E. Idiopathic Hypogonadotropic Hypogonadism Caused by Inactivating Mutations in SRA1. J Clin Res Pediatr Endocrinol. 2016;8:125–34. [PMC free article: PMC5096466] [PubMed: 27086651]
- Laitinen EM, Vaaralahti K, Tommiska J, Eklund E, Tervaniemi M, Valanne L, Raivio T. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. 2011; 6:41. [PMC free article: PMC3143089] [PubMed: 21682876]
- Lalani SR, Belmont JW. CHARGE syndrome. In: Lang F, ed. Encyclopedia of Molecular Mechanisms of Disease. Springer Publishing House. 2009.
- Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245:57–60. [PubMed: 2544996]
- Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, Chan YM, Pitteloud N, Crowley WF Jr, Balasubramanian R. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. 2012;97:E136–44. [PMC free article: PMC3251934] [PubMed: 22072740]
- Liu PY, Baker HW, Jayadev V, Zacharin M, Conway AJ, Handelsman DJ. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 2009;94:801–8. [PubMed: 19066302]
- Mainieri AS, Elnecave RH. Usefulness of the free alpha-subunit to diagnose hypogonadotropic hypogonadism. Clin Endocrinol (Oxf) 2003;59:307–13. [PubMed: 12919153]
- Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O'Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N, Seminara SB. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. [PMC free article: PMC3738065] [PubMed: 23656588]
- Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, Seminara SB, Pitteloud N, Zhou QY, Crowley WF Jr. The Role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev. 2011;32:225–46. [PMC free article: PMC3365793] [PubMed: 21037178]
- Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF Jr (1990) Clinical review 15: Management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 71:1081A-G. [PubMed: 2229271]
- Massin N, Pecheux C, Eloit C, Bensimon JL, Galey J, Kuttenn F, Hardelin JP, Dode C, Touraine P. X chromosome-linked Kallmann syndrome: clinical heterogeneity in three siblings carrying an intragenic deletion of the KAL-1 gene. J Clin Endocrinol Metab. 2003; 88:2003–8. [PubMed: 12727945]
- Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci U S A. 2006;103:4140–5. [PMC free article: PMC1449660] [PubMed: 16537498]
- Matsuo T, Okamoto S, Izumi Y, Hosokawa A, Takegawa T, Fukui H, Tun Z, Honda K, Matoba R, Tasumi K, Amino N. A novel mutation of the KAL1 gene in monozygotic twins with Kallmann syndrome. Eur J Endocrinol. 2000;143:783–7. [PubMed: 11124862]
- Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, Keefe K, Niedziela M, Raivio T, Crowley WF Jr, Seminara SB, Quinton R, Hughes VA, Kumanov P, Young J, Yialamas MA, Hall JE, Van Vliet G, Chanoine JP, Rubenstein J, Mohammadi M, Tsai PS, Sidis Y, Lage K, Pitteloud N. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 2013;92:725–43. [PMC free article: PMC3644636] [PubMed: 23643382]
- Monnier C, Dode C, Fabre L, Teixeira L, Labesse G, Pin JP, Hardelin JP, Rondard P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009; 18:75–81. [PMC free article: PMC3298864] [PubMed: 18826963]
- Nachtigall LB, Boepple PA, Pralong FP, Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism--a treatable form of male infertility. N Engl J Med. 1997;336:410–5. [PubMed: 9010147]
- Oliveira LM, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, Schipani E, Costa EM, Latronico AC, Crowley WF Jr, Vallejo M. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–8. [PubMed: 11297579]
- Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. 2008;14:367–70. [PMC free article: PMC2434956] [PubMed: 18463157]
- Pingault V, Bodereau V, Baral V, Marcos S, Watanabe Y, Chaoui A, Fouveaut C, Leroy C, Vérier-Mine O, Francannet C, Dupin-Deguine D, Archambeaud F, Kurtz FJ, Young J, Bertherat J, Marlin S, Goossens M, Hardelin JP, Dodé C, Bondurand N. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet. 2013;92:707–24. [PMC free article: PMC3644631] [PubMed: 23643381]
- Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, Hertz-Pannier L, Bertrand AM, Lyonnet S, Rappaport R, Netchine I. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005; 90:5621–6. [PubMed: 16030162]
- Pitteloud N, Acierno JS Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF Jr. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006a;103:6281–6. [PMC free article: PMC1458869] [PubMed: 16606836]
- Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF Jr, Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–5. [PubMed: 11397842]
- Pitteloud N, Hayes FJ, Boepple PA, DeCruz S, Seminara SB, MacLaughlin DT, Crowley WF Jr. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002a;87:152–60. [PubMed: 11788640]
- Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002b; 87:4128–36. [PubMed: 12213860]
- Pitteloud N, Meysing A, Quinton R, Acierno JS Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, Bouloux P, Mohammadi M, Crowley WF Jr. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006b;254-255:60–9. [PubMed: 16764984]
- Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, Azcona C, MacColl GS, Jacobs HS, Conway GS, Besser M, Stanhope RG, Bouloux PM. Idiopathic gonadotropin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001;55:163–74. [PubMed: 11531922]
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF Jr, Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–73. [PubMed: 17761590]
- Richards MR, Plummer L, Chan YM, Lippincott MF, Quinton R, Kumanov P, Seminara SB. Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J Med Genet. 2017;54:19–25. [PMC free article: PMC5189673] [PubMed: 27512013]
- Rugarli EI, Lutz B, Kuratani SC, Wawersik S, Borsani G, Ballabio A, Eichele G. Expression pattern of the Kallmann syndrome gene in the olfactory system suggests a role in neuronal targeting. Nat Genet. 1993; 4:19–26. [PubMed: 8513320]
- Salian-Mehta S, Xu M, Knox AJ, Plummer L, Slavov D, Taylor M, Bevers S, Hodges RS, Crowley WF Jr, Wierman ME. Functional consequences of AXL sequence variants in hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2014;99:1452–60. [PMC free article: PMC3973777] [PubMed: 24476074]
- Santoro N, Filicori M, Crowley WF Jr. Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev. 1986;7:11–23. [PubMed: 3082615]
- Sato N, Ogata T. Kallmann syndrome. Nihon Rinsho. 2006 Suppl 2:220–4. [PubMed: 16817388]
- Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Clin Endocrinol Metab. 2009;94:780–5. [PubMed: 19017752]
- Seminara SB, Hayes FJ, Crowley WF Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–39. [PubMed: 9793755]
- Seminara SB, Acierno JS Jr, Abdulwahid NA, Crowley WF Jr, Margolin DH. Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J Clin Endocrinol Metab. 2002;87:1607–12. [PubMed: 11932290]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–27. [PubMed: 14573733]
- Semple RK, Achermann JC, Ellery J, Farooqi IS, Karet FE, Stanhope RG, O'rahilly S, Aparicio SA. Two novel missense mutations in g protein-coupled receptor 54 in a patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2005;90:1849–55. [PubMed: 15598687]
- Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Meriq V, Merino PM, Gusella JF, Crowley WF Jr, Pitteloud N, Hall JE. Expanding the phenotype and genotype of female GnRH deficiency. J. Clin. Endocrinol. Metab. 2011;96:E566–76. [PMC free article: PMC3047229] [PubMed: 21209029]
- Sidhoum VF, Chan YM, Lippincott MF, Balasubramanian R, Quinton R, Plummer L, Dwyer A, Pitteloud N, Hayes FJ, Hall JE, Martin KA, Boepple PA, Seminara SB. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab. 2014;99:861–70. [PMC free article: PMC3942233] [PubMed: 24423288]
- Spratt DI, Carr DB, Merriam GR, Scully RE, Rao PN, Crowley WF Jr. The spectrum of abnormal patterns of gonadotropin-releasing hormone secretion in men with idiopathic hypogonadotropic hypogonadism: clinical and laboratory correlations. J Clin Endocrinol Metab. 1987;64:283–91. [PubMed: 3098771]
- Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–5. [PubMed: 9500540]
- Suzuki E, Izumi Y, Chiba Y, Horikawa R, Matsubara Y, Tanaka M, Ogata T, Fukami M, Naiki Y. Loss-of-function SOX10 mutation in a patient with Kallmann syndrome, hearing loss, and iris hypopigmentation. Horm Res Paediatr. 2015;84:212–6. [PubMed: 26228106]
- Sykiotis GP, Hoang XH, Avbelj M, Hayes FJ, Thambundit A, Dwyer A, Au M, Plummer L, Crowley WF Jr, Pitteloud N. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J Clin Endocrinol Metab. 2010a;95:3019–27. [PMC free article: PMC2902061] [PubMed: 20382682]
- Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, Seminara SB, Crowley WF Jr, Pitteloud N. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A. 2010b; 107:15140–4. [PMC free article: PMC2930591] [PubMed: 20696889]
- Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–8. [PMC free article: PMC4312696] [PubMed: 19079066]
- Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, Gurbuz F, Temiz F, Millar R, Yuksel B. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. NEJM. 2012;366:629–35. [PubMed: 22335740]
- Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Hughes V, Seminara SB, Van Uum S, Crowley WF, Habuchi H, Kimataf K, Piteloud N, Bülow HE. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2011;108:11524–9. [PMC free article: PMC3136273] [PubMed: 21700882]
- Trarbach EB, Abreu AP, Silveira LF, Garmes HM, Baptista MT, Teles MG, Costa EM, Mohammadi M, Pitteloud N, de Mendonca BB, Latronico AC. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J Clin Endocrinol Metab. 2010a;95:3491–6. [PMC free article: PMC3213864] [PubMed: 20463092]
- Trarbach EB, Teles MG, Costa EM, Abreu AP, Garmes HM, Guerra G Jr, Baptista MT, de Castro M, Mendonca BB, Latronico AC. Screening of autosomal gene deletions in patients with hypogonadotropic hypogonadism using multiplex ligation-dependent probe amplification: detection of a hemizygosis for the fibroblast growth factor receptor 1. Clin Endocrinol (Oxf) 2010b;72:371–6. [PubMed: 19489874]
- Van Dop C, Burstein S, Conte FA, Grumbach MM. Isolated gonadotropin deficiency in boys: clinical characteristics and growth. J Pediatr. 1987;111:684–92. [PubMed: 2889818]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7. [PubMed: 15300250]
- Waldhauser F, Weissenbacher G, Frisch H, Pollak A. Pulsatile secretion of gonadotropins in early infancy. Eur J Pediatr. 1981;137:71–4. [PubMed: 6791927]
- Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, Boepple PA, Holmes LB, Crowley WF Jr. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388–95. [PubMed: 8954047]
- Whitcomb RW, Crowley WF Jr. Clinical review 4: Diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990;70:3–7. [PubMed: 2403572]
- Xu N, Kim HG, Bhagavath B, Cho SG, Lee JH, Ha K, Meliciani I, Wenzel W, Podolsky RH, Chorich LP, Stackhouse KA, Grove AM, Odom LN, Ozata M, Bick DP, Sherins RJ, Kim SH, Cameron RS, Layman LC. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril. 2011;95:1613–20.e1. [PMC free article: PMC3888818] [PubMed: 21300340]
- Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2012;97:707–18. [PubMed: 22392951]
推荐阅读
- Ballabio A, Rugarli EI. Kallmann syndrome. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). New York: McGraw-Hill. Chap 225. Available online.
- Crowley WF, Balasubramanian R, eds. Special Issue: Genetics of GnRH Deficiency. Available online. 2011.
- Semple RK, Kemal Topaloglu A. The recent genetics of hypogonadotropic hypogonadism - novel insights and new questions. Clin Endocrinol (Oxf) 2010;72:427–35. [PubMed: 19719764]
章注
作者历史
Margaret Au, MBE, MS, CGC; Massachusetts General Hospital (2010-2013)
Ravikumar Balasubramanian, MD, PhD (2013-present)
Cassandra Buck, MS, CGC; Massachusetts General Hospital (2013-2017)
Marissa Caudill; University of Connecticut Health Center (2007-2010)
William F Crowley Jr, MD (2007-present)
J Carl Pallais, MD, MPH; Massachusetts General Hospital (2007-2013)
Nelly Pitteloud, MD; Massachusetts General Hospital (2007-2013)
Stephanie Seminara, MD; Massachusetts General Hospital (2007-2013)
修订历史
- 2 March 2017 (ha) Comprehensive update posted live
- 18 July 2013 (me) Comprehensive update posted live
- 4 January 2011 (cd) Revision: changes in nomenclature, Tanner staging, and test availability; references added
- 8 April 2010 (me) Comprehensive update posted live
- 23 May 2007 (me) Review posted to live Web site
- 1 June 2006 (jcp) Original submission