
摘要
临床特征.
9q22.3微缺失,包括 PTCH1缺失, 该基因是Gorlin综合征(痣基底细胞癌综合征)中突变的基因,该疾病已详细的描述,可根据临床发现进行断定,以及发育迟缓和/或智力障碍,特应性颅缝 管狭窄,阻塞性脑积水,出生前和出生后的巨大儿以及癫痫。受累个体Wilms肿瘤的风险增加。 Gorlin综合征的常见表现包括:20岁以前的大脑镰 钙化、皮肤基底细胞癌(BCC)、下颌角膜囊肿、手掌/脚底皮肤凹痕 、儿童成神经管细胞瘤以及心脏和卵巢纤维瘤的风险增加。9q22.3微缺失的临床谱是可变的,临床发现在某种程度上取决于微缺失的大小。
诊断/检查.
9q22.3微缺失的诊断通过染色体9q22.3上的杂合缺失来证实。受影响个体(但不在对照组)中通常缺失的最小临界区域为352 kb,包括 PTCH1和 FANCC。通过常规G带染色体分析或其他常规细胞遗传学条带技术不能鉴定出9q22.3微缺失,除非是极大的缺失。
管理.
对症治疗:常规治疗和管理是根据心脏,神经和皮肤表现由适当的专家进行。需要全面的体格、职业和言语治疗服务。切除或治疗下颌角膜囊肿、基底细胞癌或其他肿瘤需要术干预。
预防主要表现:限制暴露于电离辐射,如计算机断层扫描和X射线。
监护:通过神经病学家和/或神经外科医生随时监测整个儿童的头围和 神经状态,及时评估脑部大小、行为变化或意识改变,这是障碍性脑积水、成神经管细胞瘤和/或其他脑肿瘤的证据。与Beckwith-Wiedemann综 合征的监测相似,推荐用常规腹部超声波诊断Wilms肿瘤。八岁以上,至少每年皮肤检查,每12-18个月用曲面断层照相片确定颌骨角化囊肿。
因子/环境避免:阳光照射过度;使用放射治疗,因为在治疗区域有发展多个BCC的风险。
妊娠管理:对于具有大头畸形的受累胎儿,可能需要进行剖腹产。
遗传咨询.
9q22.3微缺失是以常染色体显性方式遗传的。在大多数个体 中,微缺失似乎是由新发事件或由平衡重排的父母遗传来的不平衡染色体重排引起的;然而,缺失的遗传自有症状的嵌合体母亲的遗传模式也有报道。当父母双方没 有平衡染色体重排或缺失时,未来怀孕的复发风险很低(大概<5%),但大于一般人群,因为父母可能具有生殖腺嵌合或包括生殖腺的低水平体细胞镶嵌。 基于父母中鉴定的平衡染色体重排或缺失,以及关生殖腺嵌合的可能性,可以对高风险的怀孕进行产前检测。
诊断
临床诊断
9q22.3微缺失的临床谱是可变的,临床发现在某种程度上取决于微缺失的大小。
所有报告的 9q22.3 微缺失都包括 PTCH1,该基因突变导致格林综合征 (痣样基底细胞癌综合征);因此,9q22.3 基因微缺失的个体都有这个已经充分描述的疾病的临床表现 [ Kimonis et al 2004]。
格林综合征的主要特征包括 :
- 20岁以前的大脑镰 钙化
- 一生有五个以上的基底细胞癌或三十岁以前有一个基底细胞癌
- 下颌角膜囊肿
- 手掌/脚底皮肤凹痕
- 一级亲属有格林综合征
格林综合征的次要特征包括 [ Kimonis et al 2004]:
- 唇裂和/或腭裂
- 近端或远端多指趾畸形
- 大头畸形 (枕-额周长>97th百分位)
- 眼睛异常(包括小眼睛,白内障,视网膜异常,发育缺陷)
- 肋骨和/或椎骨异常
- 心脏和卵巢纤维瘤
- 儿童成神经管细胞瘤(也称为原始神经外胚层肿瘤[PNET])
- 淋巴肉瘤或胸膜囊肿
其他9q22.3 微缺失的常见表现包括 [ Muller et al 2012]:
- 发育迟缓和/或智力障碍
- 短鼻子和长人中
- 额侧的颅缝早闭
- 阻塞性脑积水
- 产前/产后身高体重大于95th百分位
- 癫痫
9q22.3微缺失的罕见异常可能包括肾脏异常,Wilms肿瘤,Chiari畸形和胼胝体发育不良。
检查
细胞遗传学检查.通过常规G带染色体分析或其他常规细胞遗传学条带技术不能鉴定出9q22.3微缺失,除非是极大的缺失。
分子遗传学检查
关键区域.9q22.3微缺失的诊断通过染色体9q22.3上的杂合缺失来证实。受影响个体(但不在对照组)中通常缺失的最小临界区域为352 kb,包括 PTCH1(human homolog 1 of Drosophila Patched) 和 FANCC1(Fanconi anemia complementation group C) [ Muller et al 2012]。
基因.PTCH1的删除是导致9q22.3微缺失的大部分特征的唯一的基因;然而,该基因的缺失似乎不足以引起与Gorlin综合征中通常观察到的特征不同的特征。在连续 基因缺失的间隔内包括的两个至273个基因中是编码微小RNA的,转录因子,未识别的开放阅读框和未知功能蛋白的基因 [ Muller et al 2012]。 这些基因中的许多基因,其单个基因缺失或突变表型仍然没有明确。
注意:被删除的基因随着微缺失的大小和断点变化而变化。
临床检查
表 1.
9q22.3微缺失所用分子诊断总结
| 染色体区域 | 实验方法 | 检测到的突变 1 | 该方法的突变监测频率 2 |
|---|---|---|---|
| 9q22.3 | 染色体微阵列 (CMA) 3 | 352 kb到20.5 Mb的缺失 | 有合适的BACs, SNPs, 或寡核苷酸:>99% |
| 缺失/重复分析 4, 5 | 有合适的探针:>99% |
检测结果解释
诊断策略
在先证者确认/建立诊断 要求检测到 最小临界352 kb 缺失,该缺失常见于 9q22.3 微缺失或任何重叠的较大缺失。
- 如果临床上怀疑有 9q22.3 微缺失,可以采用有针对性的技术检测(如FISH、 MLPA)。
临床特点
临床描述
许多有 9q22.3 微缺失的患者在婴儿期表现出张力减退,所有的患者都表现出大动作延迟。有较大缺失的个体张力减退可能甚至持续到童年晚期和青春期 [ Shimojima et al 2009, Yamamoto et al 2009, Muller et al 2012]. 有最小缺失的个人可能有大动作延迟但没有其他残疾或延迟。
拥有大约 2 Mb 或更大的缺失的个体表现出运动、 语言和行为/社会里程碑的持续延迟 [ Shimojima et al 2009, Muller et al 2012]. 有这些大型的缺失个体通常上学年龄就会显现出来的智力障碍,需要特殊教育。缺失大小增加预计将带来更严重的残疾;有报告称智商或发育商 (DQ) 分数在 40 多到 30 多,或者更低 [ Kroes et al 1994, Redon et al 2006, Fujii et al 2007, Nowakowska et al 2007, Yamamoto et al 2009, Muller et al 2012].
一小部分有9q22.3 基因微缺失的个体发展出癫痫 [ Shimojima et al 2009, Yamamoto et al 2009, Muller et al 2012].
报告称有 9q22.3 基因微缺失的许多 (16/37) 个体有严重到轻度的脑心室扩张,或者无症状,可以与脑萎缩或占位性病变 (例如,髓母细胞瘤)有关 [ Muller et al 2012]. 其中的一小部分 (7/16) 将会有病因不明的严重梗阻性脑积水,需要脑室分流 [ Muller et al 2012].
大约 20%有 9q22.3 基因微缺失的个体有产前发病的巨大儿,出生的长度和重量在第 95 百分位以上并持续到产后 [ Muller et al 2012].
少数病例报告描述9q22.3 微缺失患者的巨大儿或偏侧增生 [ Cajaiba et al 2006, Chen et al 2006, Redon et al 2006, Shimojima et al 2009, Yamamoto et al 2009, Muller et al 2012, Isidor et al 2013].
Eight of the 37 受累的individuals reported and reviewed by Muller et al [2012]描述并回顾的37个病例中的8个表现出额缝提前融合,导致颅缝早闭和三角头畸形。
Cajaiba et al [2006]描述了单独一个人患有肾母细胞瘤和盆腔横纹肌肉瘤并得出结论,虽然肾母细胞瘤与格林综合征是不相关的,但横纹肌肉瘤可能是相关的 [ Cajaiba et al 2006]. 然而,最近报告了四个额外个体有生殖系 9q22.3 基因微缺失与肾母细胞瘤 [ Garavelli et al 2013, Isidor et al 2013]. Isidor et al [2013]从四个受影响的个人中的一个的肿瘤里测序了 PTCH1并证实了一个不是该患者的正常肾组织或血液中存在的非缺失等位基因的体细胞突变。因而,12%(5/42) 的文献报道的有9q22.3 基因微缺失的个体发展出肾母细胞瘤。
受累的个体可能有面部异常,包括宽阔隆起的前额,垂直的额头皱,要么向上或向下的成角的睑裂,一个短鼻子和长人中 [ Ying et al 1982, Farrell et al 1991, Olivieri et al 2003, Midro et al 2004, Redon et al 2006, Nowakowska et al 2007, Yamamoto et al 2009, Muller et al 2012]. 有的个体面部特征随时间愈发明显,然而有的有大缺失的患者一出生特征就很明显 [ Ying et al 1982, Muller et al 2012].
基因型-表型的相关性
许多 9q22.3 微缺失患者中出现的特征源于 PTCH1单倍体剂量不足,并且符合格林综合征。然而,由于迄今为止大多数报道的个体在其缺失内不止PTCH1基因,因此预期这些其他基因中的一种或多种的缺失导致不属于Gorlin综合征的附加表型特征。
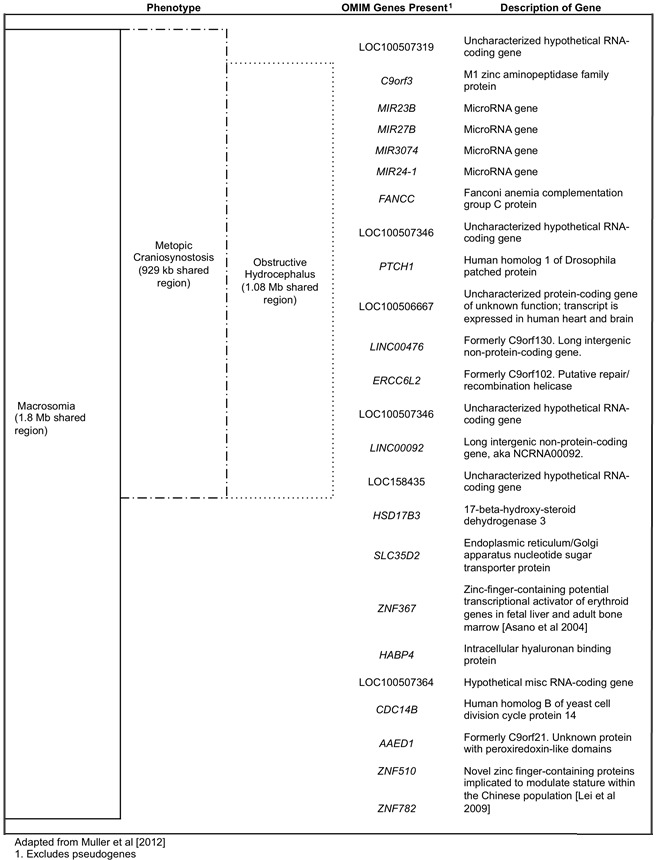
Muller et al [2012]确定了10个9q22.3微缺失患者中最小的常见缺失间隔和相关断点,为以下发现:
- 特应性颅缝管狭窄:含有16个基因的929-kb区域
- 严重阻塞性脑积水:含有18个基因的1.08-Mb区域
- 巨大胎儿:一个含有31个基因的1.8Mb区域
注意:这些间隔间的一些基因尚未得到充分验证,没有鉴定出特异性候选基因。
多位作者提出存在于具有9q22.3微缺失突变的个体亚群中的巨噬细胞特异性是丧失父系等位基因的结果 [ Redon et al 2006, Shimojima et al 2009]. 然而,到目前为止没鉴定出缺失区间内有印迹基因。
外显率
预计9q22.3 基因微缺失是完全外显的表型,但表现多样。到目前为止已经有此微缺失但是没有表性的人报道。
患病率
9q22.3微缺陷是罕见的。 迄今为止已有42名受累个体在文献中被报道,其中一名患有体细胞嵌合 [ Yamamoto et al 2009, Muller et al 2012, Garavelli et al 2013, Isidor et al 2013]. 可能在有发育延迟和/或智力障碍且没有其他特征的人群中,9q22.3 微缺失只占很小一部分。
遗传相关(等位基因)疾病
有描述过一个母亲和她的孩子包含含有 PTCH1和 FANCC1号外显子的360kb区域组成的9q22.3重复, 两者均有小头畸形和轻度发育迟缓 [ Derwinska et al 2009].
包括基因内或全基因缺失在内的 PTCH1的显性功能缺失性突变已知导致Gorlin综合征[ Hahn et al 1996].
已经在少数个体中描述了 PTCH1中的显性功能获得性突变与脑膜发育不全7型(OMIM 610828)相关的,表现为与胚胎发生sonic hedgehog蛋白表达降低 [ Ming et al 2002, Ribeiro et al 2006].
在不存在该综合征的任何其他发现的情况下作为单一肿瘤发生的散发性肿瘤(包括成神经管细胞瘤,牙源性角膜囊肿,心脏纤维瘤,卵巢纤维瘤和基底细胞癌)可以是 PTCH1中存在体细胞突变; 因此,这些肿瘤不是遗传性的。 详情请参阅 Cancer and Benign Tumors.
鉴别诊断
一个男性具有轻度异常面部特征,发音困难,漏斗胸和单侧肾发育不全的,鉴定出不包括 PTCH1的5.3Mb的9q22.2-q22.3缺失。 他的两个女儿也出现智力障碍,以及他的异常特征,但没有任何畸形 [ Siggberg et al 2011].
在9q22.3微缺失有多种共同特征的综合征中,Gorlin综合征(痣基底细胞癌综合征)是最常见的。
Beckwith-Wiedemann综合征(BWS)特征为巨大儿症,巨舌,内脏肿大,胚胎性肿瘤(如Wilms肿瘤,肝母细胞瘤,成神经细胞瘤 和横纹肌肉瘤),卵母细胞瘤,新生儿低血糖症,耳朵皱纹/凹陷,肾上腺皮质细胞瘤和肾脏的生长障碍异常(例如,髓质发育不良,肾钙质沉着,髓质海绵肾和肾 脏病)。早期死亡可能发生于早产儿,低血糖症,心肌病,巨大症或肿瘤的并发症。然而,以前报告的20%的死亡率可能是高估的,因为对该疾病更好地认识以及 治疗选择的增加。大黄疸症和巨大儿症通常在出生时出现,但可能出生后发病。七至八岁的增长速度减慢。血红素增生可能会影响身体的部分区域或特定的器官和组 织。分子遗传学检测可以鉴定BWS个体染色体11p15的表观遗传和基因组变化:(1)50%受影响个体的印记中心2(IC2)上母体染色体上甲基化的丧 失; (2)20%的个体染色体11p15的父系单亲二倍体;和(3)5%的个体在印记中心1(IC1)的母体染色体上获得甲基化。 CDKN1C的序列分析鉴定了大约40%的家族性病例和5%-10%无BWS家族史的病例的突变。
Sotos综合征的特点是典型的面部外观,过度生长(身高和/或头围高于平均值≥2SD),学习障碍范围从轻度(儿童上正常学校,成年后可独立) 到严重(可能需要终身护理和支持)。Sotos综合征有行为问题,先天性心脏异常,新生儿黄疸,肾脏异常,脊柱侧凸和癫痫发作的主要特征相关。 约80%-90%的Sotos综合征患者具有 NSD1异常。
许多其他基因组微缺失或微缺失综合征导致9q22.3微缺失相同的表型如发育迟缓或智力障碍和/或一些个体非特异性表型特征。
管理
初次诊断后的后续评估
为了确定诊断为微缺失9q22.3的个体的疾病严重程度,建议进行以下评估:
- 脑成像(不使用CT)和神经系统评估
- 完整体检,包括Gorlin综合征表现的皮肤病学评估
- 综合发育评估
- 肾脏和盆腔超声检查评估可能的肾异常和卵巢纤维瘤
- 超声心动图
- 眼科评估
- 仔细考虑相关骨骼异常和/或牙科成像观察异常
- 家庭遗传咨询
孕期管理
许多具有9q22.3微缺失的个体存在产前发病的巨细胞病和/或大头症。 正如一些人所报道的那样,这可能需要剖腹产,包括紧急情况[ Isidor et al 2013].
在研治疗
搜索ClinicalTrials.gov获取关于各种疾病和病症的临床研究的信息。注意:这种疾病可能没有临床试验
遗传咨询
遗 传咨询是向个人和家庭提供关于遗 传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和个人决定的过程。 以下部分涉及遗传风险评估和使用家族史及遗传检测来明确家庭成员的遗传状况的内容。 本节并不意味着能解决患者面临的所有个人,文化或伦理问题,或替代遗传学专业人士的咨询。-ED。家庭成员风险
先证者双亲
- 大多数病例是由明显的新发事件或具有平衡异位的父母引起的不平衡染色体重排的遗传所致。
先证者兄弟姐妹
- 先证者同胞的风险取决于父母的遗传状况。
先证者后代. 诊断为典型的9q22.3微缺失的患者中仅有一个人已知可以生殖; 然而,这个人的缺失是嵌合性的 [ Isidor et al 2013].
其他家系成员.其他家庭成员的风险取决于先证者父母的遗传状况。 如果父母具有平衡的染色体重排或缺失,则其家庭成员可能也具有重排或缺失的风险。
相关遗传咨询事项
计划生育
- 确定遗传风险,澄清携带者状态的和产前检测的可行性的最佳时间是在怀孕之前。
DNA银行是存储DNA的(通常从白血细胞提取),以备日后使用。因为测试方法和我们对基因、等位基因变异和疾病的理解在将来会有所提升,所以受累个体可以考虑保存DNA。
产前诊断
产前检测在技术上是可行的。在妊娠的约15至18周通过羊膜穿刺术或妊娠地约10至12周通过绒毛获得的胎儿细胞获得染色体,可以使用特异性FISH探针分析或染色体微阵列(CMA)以分子遗传学测试。
由于与生殖腺嵌合的可能性会导致一定的复发风险(可能<5%),向具有9q22.3微缺失儿童的父母可提供产前检测。 产前检测也适用于携带平衡染色体重排或具有微缺失的家长。
注意:妊娠年龄表示为从上次正常月经的第一天或超声测量计算的月经周。
植入前遗传学诊断(PGD)可能是9q22.3微缺失的怀孕风险增加的家庭的良好选择。
资源
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Chromosome Disorder Outreach (CDO)PO Box 724Boca Raton FL 33429-0724Phone:561-395-4252 (Family Helpline)Email:info@chromodisorder.org
- Unique: The Rare Chromosome Disorder Support GroupG1 The StablesStation Road WestOxted Surrey RH8 9EEUnited KingdomPhone:+44 (0) 1883 723356Email:info@rarechromo.org; rarechromo@aol.com
分子诊断
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
表A.
分子遗传病理学
使9q22.3区域易于缺失的机制尚不清楚。 位于 PTCH1和 该区域相邻基因之间有众多SINE,大型LINE和LTR,潜在地可能易于导致这些基因的缺失或重复的重组事件。 类似地,不清楚在9q22.3微缺失内的基因(PTCH1除外)删除如何影响表型,因为在最大报告的20.5Mb缺失的273个基因中的许多基因的功能和 单倍体不足表型仍未得到鉴定 [ Muller et al 2012].
报 告过的受累个体的微缺失大小似乎并不复发。 最早的9q22.3缺失报告先于CMA技术; 在此时只描述了常规细胞遗传条带技术可见的大的缺失,使得与过去几年的报道断点相比较比较困难。 随着CMA技术的发展,报道的最小的9q22.3微缺失只包含两个基因,PTCH1和FANCC [ Muller et al 2012].
PTCH1 (Drosophila Patched的人类同系物1)编码肿瘤抑制蛋白,是 sonic hedgehog (SHH)蛋白的受体,其以未结合的形式通常用于抑制SHH信号传导。 FANCC(范科尼贫血症互补组C)编码一种蛋白质,是具有E3泛素连接酶活性的核心FA核蛋白复合物的一部分,其在DNA损伤和S期中起作用。 该基因中的纯合或复合杂合突变导致范科尼贫血。
Muller et al [2012]在10名9q22.3微缺失个体中,试图定义三种特征性额侧颅缝早闭,阻塞性脑积水和巨大儿症的关键区域和基因; 使用这种方法,他们能够将重叠区域缩小到0.929至1.8 Mb。 这些间隔内的公共基因总结在图1中。
癌症和良性肿瘤
PTCH1中的功能丧失性体细胞突变已经在Gorlin综合征中的散发性癌症中被描述,包括成神经管细胞瘤,牙源性角膜囊肿,心脏和卵巢纤维瘤以及基底细胞癌 [ Kimonis et al 2004].
在具有胚系9q22.3微缺失的个体的Wilms肿瘤组织中,在未删除的等位基因上鉴定了 PTCH1中的体细胞无义突变。 正常肾组织或血液中不存在体细胞无义突变 [ Isidor et al 2013].
参考文献
Literature Cited
- Asano H, Murate T, Naoe T, Saito H, Stamatoyannopoulos G. Molecular cloning and characterization of ZFF29: a protein containing a unique Cys2His2 zinc-finger motif. Biochem J.2004; 384:647–53.[ PMC free article : PMC1134151] [ PubMed : 15344908]
- Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol.2006; 3:575–80.[ PubMed : 17019435]
- Chen CP, Lin SP, Wang TH, Chen YJ, Chen M, Wang W. Perinatal findings and molecular cytogenetic analyses of de novo interstitial deletion of 9q (9q22.3-->q31.3) associated with Gorlin syndrome. Prenat Diagn.2006; 26:725–9.[ PubMed : 16927391]
- Derwinska K, Smyk M, Cooper ML, Bader P, Cheung SW, Stankiewicz P. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur J Hum Genet.2009; 17:267–71.[ PMC free article : PMC2986050] [ PubMed : 18830227]
- Farrell SA, Siegel-Bartelt J, Teshima I. Patients with deletions of 9q22q34 do not define a syndrome: three case reports and a literature review. Clin Genet.1991; 40:207–14.[ PubMed : 1773536]
- Fujii K, Ishikawa S, Uchikawa H, Komura D, Shapero MH, Shen F, Hung J, Arai H, Tanaka Y, Sasaki K, Kohno Y, Yamada M, Jones KW, Aburatani H, Miyashita T. High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum Genet.2007; 122:459–66.[ PubMed : 17703323]
- Garavelli L, Piemontese MR, Cavazza A, Rosato S, Wischmeijer A, Gelmini C, Albertini E, Albertini G, Forzano F, Franchi F, Carella M, Zelante L, Superti-Furga A. Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A.2013; 161A(11):2894–901.[ PubMed : 24124115]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, Bale AE. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell.1996; 85:841–51.[ PubMed : 8681379]
- Isidor B BF, Lafon D, Plessis G, Lacaze E, Kannengiesser C, Rossignol S, Pichon O, Briand A, Martin-Coignard D, Piccione M, David A, Delattre O, Jeanpierre C, Sévenet N, Le Caignec C. WIlms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1in nephroblastomas. Eur J Hum Genet.2013; 21:784–7.[ PMC free article : PMC3722950] [ PubMed : 23169491]
- Kimonis VE, Mehta SG, Digiovanna JJ, Bale SJ, Pastakia B. Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med.2004; 6:495–502.[ PubMed : 15545745]
- Kroes HY, Tuerlings JH, Hordijk R, Folkers NR, ten Kate LP. Another patient with an interstitial deletion of chromosome 9: case report and a review of six cases with del(9)(q22q32). J Med Genet.1994; 31:156–8.[ PMC free article : PMC1049682] [ PubMed : 8182726]
- Lei SF, Yang TL, Tan LJ, Chen XD, Guo Y, Guo YF, Zhang L, Liu XG, Yan H, Pan F, Zhang ZX, Peng YM, Zhou Q, He LN, Zhu XZ, Cheng J, Liu YZ, Papasian CJ, Deng HW. Genome-wide association scan for stature in Chinese: evidence for ethnic specific loci. Hum Genet.2009; 125:1–9.[ PMC free article : PMC2730511] [ PubMed : 19030899]
- Midro AT, Panasiuk B, Tumer Z, Stankiewicz P, Silahtaroglu A, Lupski JR, Zemanova Z, Stasiewicz-Jarocka B, Hubert E, Tarasow E, Famulski W, Zadrozna-Tolwinska B, Wasilewska E, Kirchhoff M, Kalscheuer V, Michalova K, Tommerup N. Interstitial deletion 9q22.32-q33.2 associated with additional familial translocation t(9;17)(q34.11;p11.2) in a patient with Gorlin-Goltz syndrome and features of Nail-Patella syndrome. Am J Med Genet A.2004; 124A:179–91.[ PubMed : 14699618]
- Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet.2002; 110:297–301.[ PubMed : 11941477]
- Muller EA, Aradhya S, Atkin JF, Carmany EP, Elliott AM, Chudley AE, Clark RD, Everman DB, Garner S, Hall BD, Herman GE, Kivuva E, Ramanathan S, Stevenson DA, Stockton DW, Hudgins L. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia and developmental delay. Am J Med Genet A.2012; 158A:391–9.[ PubMed : 22190277]
- Nowakowska B, Kutkowska-Kazmierczak A, Stankiewicz P, Bocian E, Obersztyn E, Ou Z, Cheung SW, Cai WW. A girl with deletion 9q22.1-q22.32 including the PTCH and ROR2 genes identified by genome-wide array-CGH. Am J Med Genet A.2007; 143A:1885–9.[ PubMed : 17632781]
- Olivieri C, Maraschio P, Caselli D, Martini C, Beluffi G, Maserati E, Danesino C. Interstitial deletion of chromosome 9, int del(9)(9q22.31-q31.2), including the genes causing multiple basal cell nevus syndrome and Robinow/brachydactyly 1 syndrome. Eur J Pediatr.2003; 162:100–3.[ PubMed : 12548386]
- Redon R, Baujat G, Sanlaville D, Le Merrer M, Vekemans M, Munnich A, Carter NP, Cormier-Daire V, Colleaux L. Interstitial 9q22.3 microdeletion: clinical and molecular characterisation of a newly recognised overgrowth syndrome. Eur J Hum Genet.2006; 14:759–67.[ PubMed : 16570072]
- Ribeiro LA, Murray JC, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A.2006; 140:2584–6.[ PubMed : 17001668]
- Shimkets R, Gailani MR, Siu VM, Yang-Feng T, Pressman CL, Levanat S, Goldstein A, Dean M, Bale AE. Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am J Hum Genet.1996; 59(2):417–22.[ PMC free article : PMC1914731] [ PubMed : 8755929]
- Shimojima K, Adachi M, Tanaka M, Tanaka Y, Kurosawa K, Yamamoto T. Clinical features of microdeletion 9q22.3 (pat). Clin Genet.2009; 75:384–93.[ PubMed : 19320658]
- Siggberg L, Peippo M, Sipponen M, Miikkulainen T, Shimojima K, Yamamoto T, Ignatius J, Knuutila S. 9q22 deletion - first familial case. Orphanet J Rare Dis.2011; 6:45.[ PMC free article : PMC3135502] [ PubMed : 21693067]
- Yamamoto K, Yoshihashi H, Furuya N, Adachi M, Ito S, Tanaka Y, Masuno M, Chiyo H, Kurosawa K. Further delineation of 9q22 deletion syndrome associated with basal cell nevus (Gorlin) syndrome: report of two cases and review of the literature. Congenit Anom (Kyoto)2009; 49:8–14.[ PubMed : 19243411]
- Ying KL, Curry CJ, Rajani KB, Kassel SH, Sparkes RS. De novo interstitial deletion in the long arm of chromosome 9: a new chromosome syndrome. J Med Genet.1982; 19:68–70.[ PMC free article : PMC1048822] [ PubMed : 7069749]
Chapter Notes
Revision History
- 20 February 2014 (me) Comprehensive update posted live
- 18 August 2011 (me) Review posted live
- 25 April 2011 (em) Original submission