
概述
临床特征.
脊髓小脑性共济失调 7型(Spinocerebellar ataxia type 7,SCA7)以进行性小脑性共济失调为特征,包括构音障碍和吞咽困难,锥杆细胞和视网膜营养不良伴进行性中心视力丧失,导致受累的成年人失明。 在儿童早期或婴儿期发病,并具有特别快速和积极的进程,往往与未能成功发育和运动技能退化有关。
诊断/检测.
绝大多数成年人根据临床表型诊断为SCA7的结果是值得怀疑的。ATXN7是唯一一个SCA7的致病变异基因。通过分子遗传学检测手段对ATXN7异常CAG三核苷酸重复扩增的检测可用于确诊成年人的SCA7以及建立对儿童的诊断。受影响的个体通常具有超过36个CAG重复,尽管重复较少的个体也可能出现症状。
处理.
对症处理:对于成年人:拐杖和助步器的使用可防止跌倒,家庭设施改造(如扶手杆、升高的马桶座和斜坡)有助于行走,加重型餐具和辅料钩有助于保持独立性,言语治疗和通 信设备有助于那些构音障碍的患者,喂养评估有助于那些吞咽困难的患者,低视力助视器和行走训练有助于那些视力受损的患者。
继发性并发症的预防:体重控制促进移动和行走。
监测:常规眼科检查。
其他:控制震颤的药物是无效的。
遗传咨询.
SCA7是常染色体显性遗传疾病。受累的个体的后代有50%几率遗传变异的基因。父母在传递基因给孩子的过程中,由于CAG重复的进一步扩大会引起疾病预期的发生。如果家族中受累成员已通过分子遗传学检测确诊,则可对家族中高危妊娠进行产前检查。
诊断
临床诊断
虽然尚未建立正式的诊断标准,但对于有以下发现的成年人,可确诊为脊髓小脑性共济失调 7型(Spinocerebellar ataxia type 7,SCA7):
- 脊髓小脑性共济失调引起的进展性不协调,包括构音障碍/吞咽困难、辨距障碍和轮替运动障碍
- 视网膜锥杆细胞营养不良伴以下情况:
- 视网膜电图测试显示视杆细胞和视锥细胞功能异常
在详细的颜色视觉检测中检出蓝/黄色轴缺陷
眼底检查发现黄斑改变(在疾病晚期)
- 家族史符合常染色体显性遗传模式
在儿童中,疾病的进展较成年人通常更快,更具侵袭性。在婴幼儿中, 临床诊断是困难的,因为共济失调和视力丧失不明显;未能茁壮成长和动作发展指标的丧失可能是最早的临床发现[Benton et al 1998]。
分子遗传学检测
基因.ATXN7是其变异会导致脊髓小脑性共济失调 7型(Spinocerebellar ataxia type 7,SCA7)的唯一基因。
等位基因大小
- 可变的正常等位基因. 28-33个CAG重复[Lebre et al 2003]。先前称之为“中间等位基因”,可变的正常等位基因是减数分裂不稳定产生的,并没有令人信服地与异常表型相关联。由于等位基因在可变正常范围内的不稳定性,具有可变正常等位基因的无症状个体其后代可能更倾向于扩大的等位基因[Mittal et al 2005]。
- 一名具有34个CAG重复的女性在65岁时表现出“非常温和的症状”[Nardacchione et al 1999]。
- Koob et al [1998]描述了具有35个CAG重复的有症状个体,相比之下David et al [1998]和Stevanin et al [1998]描述了具有35个CAG重复的无症状个体
- 36个CAG重复个体在其63岁时发展出相对温和的症状[Nardacchione et al 1999]。
- 全外显率的等位基因. 超过36个CAG重复的等位基因[Nardacchione et al 1999, Michalik et al 2004],例如460个CAG重复的极端扩增[van de Warrenburg et al 2001]被认为是完全外显。
注:外显率降低的等位基因和全外显率等位基因之间等位基因大小之间的界限仍然不清楚,有待更多家族被报道;尽管如此,无论这些等位基因如何描述,它们都应该被认为是不稳定的和致病的。
临床检测——致病变异的靶向技术
检测策略
为了对一名先证者进行确诊或建立诊断:
1.使用 2.如果单个正常大小的等位基因被确认,需判断是否有SCA7和/或常染色体显性遗传小脑共济失调的家族史伴有视网膜变性。基于先证者的家族史和发病年龄,判断使用Southern blot技术对先证者及其他家族成员进行ATXN7更大的CAG重复扩增的检测是否合适。对无症状成年家族成员的风险进行
预先筛查需要事先确认这个家族中的致病性变异是什么。对具有风险的产妇进行
临床特征
临床描述
脊髓小脑性共济失调 7型(Spinocerebellar ataxia type 7,SCA7)患者的范围从婴儿(加速病程和早期死亡)到第五十或偶然第六十个年头的老人(慢性进行性视网膜变性和小脑性共济失调)[Giunti et al 1999]。
在婴儿或幼年早期,共济失调可能并不明显但肌肉萎缩、无力和张力减退是普遍的[Enevoldson et al 1994]。两名患有严重疾病和扩增超过200和306个CAG重复的婴儿具有新生儿发育不良,发育迟缓,喂养不良,吞咽困难,充血性心力衰竭,大脑和小脑萎缩以及视网膜疾病[Babovic-Vuksanovic et al 1998, Benton et al 1998]。Ansorge et al [2004]报道一名具有180个 CAG重复的婴儿在婴儿期死亡。
在那些婴幼儿发病的患者中,小脑和脑干变性非常迅速,以至于视网膜变性和相关的视力丧失可能不明显。
当初始症状发生在青春期或青春期前时,视网膜变性的失明可能在几年内发生。在十几岁时出现症状的患者可能在十年或更短时间内失明。
在成年人中,进行性小脑共济失调(即
辨距困难,轮替运动障碍和协调差)可能会先出现,但通常会随后表现出视觉症状。虽然进展速度有所不同,但最终的结果是严重的构音障碍,吞咽困难和卧床状态,伴随着运动控制的丧失。随着疾病的进展,肌腱反射和痉挛变得明显。眼跳可能会显著减慢。
认知衰退和精神病已有报道[Benton et al 1998]。一些SCA7患者的神经精神病学检测结果揭示了这些患者对社会认知的选择性缺陷[Sokolovsky et al 2010]。
视网膜变性是导致完全失明的进行性锥体杆体营养不良[To et al 1993, Aleman et al 2002, Ahn et al 2005, Hugosson et al 2009]。视网膜变性的发作通常表现出在青少年晚期或20岁初期具有夜盲症(无法在明亮的光线下清晰地看到),畏光(对光的极度敏感)以及色觉和中央视敏度异常的表征[Miller et al 2009]。
在视网膜变性的早期阶段,年轻人可能没有症状,但可能在黄斑部有细微的颗粒变化,并且在使用法恩斯沃思二原色视者(D15)方法对详细颜色的视觉检测中检出蓝/黄色轴缺陷。视网膜电图在疾病过程早期始终不正常,最初显示明视(视锥)反应减少,随后暗视(视杆)反应减少[Miller et al 2009]。
随着视锥功能随时间减弱,中心视力下降到20/200(法定失明)范围,出现更显著的黄斑变化(见图 1),所有色彩辨别消失,最终全部视力消失。
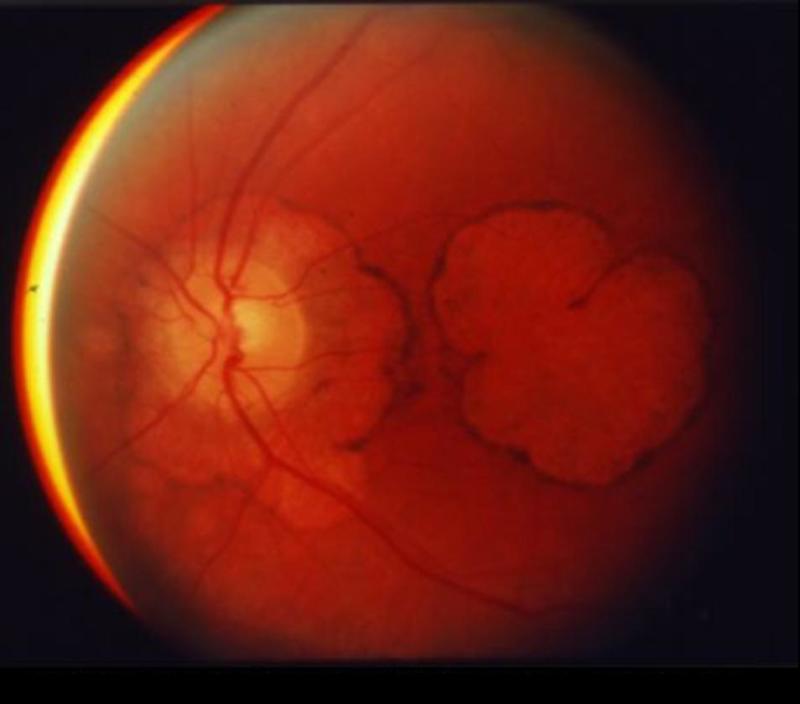
图 1. 眼底镜照片显示晚期SCA7患者严重的黄斑变性。
值得注意的是,在成人发病的疾病中,视网膜变性引起的视力丧失可能先于,伴随或随后出现共济失调[Miller et al 2009],而且最不起眼的眼底检查结果和微弱的共济失调可能伴有严重的视力丧失[Thurtell et al 2009]。
病理学。在小脑(尤其是浦肯野细胞)、下橄榄核,齿状核和脑桥核中观察到神经元丢失、有髓纤维丢失和神经胶质增生;在大脑皮层、基底神经节、丘脑和中脑中的程度较低[Rüb et al 2008, Seidel et al 2012]。
在一项详细的病理解剖研究中,Rüb et al [2008]将广泛的脑神经元变性模式与多变的临床表型相关联,比较18个不同区域的伴有各种临床表现如共济失调、锥体征、视力丧失、复视和听力受损的神经元变性。
在包括变性和非变性区域内的神经元中表达突变ataxin-7,由此产生的核内包涵体在浦肯野细胞中基本不存在[Michalik & Van Broeckhoven 2003]。在一个严重受累的婴儿中,Ansorge et al [2004]在海马体和许多非神经系统组织如肠、胰腺和心血管系统中鉴定了ataxin-7核内包涵体。 在来自对骨骼肌和肝脏的活组织检查中还观察到异常线粒体[Han et al 2010a]。
在脊髓的后柱和脊髓小脑束中观察到明显的退化[Martin et al 1994, Lebre et al 2003, Koeppen 2005]。
视网膜中光感受器、双极细胞和颗粒细胞的退化是明显的,特别是在中央凹和中央凹周围区域[Martin et al 1994]。
基因型-表型相关性
CAG重复长度与疾病严重程度之间存在相关性:CAG重复越长,发病年龄越早,疾病越严重且进展迅速。尽管观察到CAG重复长度与发病年龄、疾病严重程度和病程相关,但目前的共识是ATXN7等位基因的大小不能为临床预后提供足够的预测价值[Andrew et al 1997]。
预判
在患有致病性ATXN7等位基因的家族中,重复长度往往随着传代而逐渐扩大,在受累男性的受累的后代中观察到更显著的扩增[Gouw et al 1998]。这解释了在遗传水平上,SCA7家族中显著的遗传早现,现在被认为是CAG重复疾病中最不稳定的。对家庭的预判可能是戏剧性的,以至于在ATXN7 CAG重复扩增的父母或祖父母表现出症状前,孩子可能会被诊断为患有散发性神经退行性疾病[van de Warrenburg et al 2001, Ansorge et al 2004]。
命名
视网膜变性与小脑性共济失调的关系已经被认识了几十年[Havener 1951, Carpenter & Schumacher 1966, Weiner et al 1967, Konigsmark & Weiner 1970, Anttinen et al 1986, Gouw et al 1994]。过去用于描述SCA7的术语包括橄榄桥脑小脑性共济失调(OPCA) III型和OPCA II型。
发病率
人群发病率低于1:100,000。在几个研究中,SCA7占所有SCAs的2%[Filla et al 2000, Storey et al 2000]。在日本北海道[Sasaki et al 2000, Jardim et al 2001, Kim et al 2001]和中国大陆[Tang et al 2000]的人口研究中没有报道SCA7的个体。然而,另一些研究报道了中国北京4个SCA7家系[Gu et al 2000]和台湾的1个家系[Tsai et al 2004]。Han et al [2010b]回顾了东南亚14个SCA7家系(3个来自中国大陆,1个来自台湾,2个来自日本,8个来自韩国)的7篇报道。这些家庭的临床特征和遗传特征与西方国家SCA7的典型特征相一致。
遗传相关(等位基因)疾病
除了本GeneReview章节描述的那些以外,没有任何表型与ATXN7的致病突变相关联。
鉴别诊断
虽然许多其他脊髓小脑性共济失调(Spinocerebellar ataxias,SCAs)的临床和病理表型与SCA7相重叠,视网膜变性是SCA7的鉴别特征(见共济失调综述)。
部分SCA1的患者被报道具有渐进性的视力丧失[Illarioshkin et al 1996, Abe et al 1997]。
SCA7的表型可能与伴有其他形式的视力丧失的获得性共济失调(如糖尿病性视网膜病变,多发性硬化症或年龄相关性黄斑变性)混淆。
Leber 遗传性视神经病(Leber hereditary optic neuropathy,LHON)等线粒体脑病可表现为共济失调,有时伴有视觉退化;这些基于线粒体基因型的共济失调可以通过分子遗传学检测和遗传模式(母系遗传而不是常染色体显性遗传)以及SCA7中通常见到的遗传早现相鉴别(见线粒体疾病概述)。
婴儿和儿童发病的SCA7可能与脂质储存疾病和神经元蜡样脂褐质沉积症相混淆。
患者管理
初次诊断的评估
为了确认脊髓小脑性共济失调 7型(spinocerebellar ataxia type 7,SCA7)的疾病程度和个体确诊需求,建议如下:
- 病史
- 神经科检查
- 眼科检查包括视力,视野和色觉的评估
- 临床遗传咨询
对症治疗
对受累的个体的管理仍然是支持性的,因为没有已知的治疗方法来延缓或阻止疾病进展的存在。
小脑共济失调. 尽管运动和物理疗法都不能阻止运动失调或肌无力的进展,但脊髓小脑性共济失调 7型(spinocerebellar ataxia type 7,SCA7)的患者应保持活动。拐杖和助步器有助于防止跌倒。对家中一些可提供便利的装置进行改装,如扶手杆、升高的马桶座和适用于轮椅车的斜坡道将是必要的。言语治疗和通信设备(如书写板和基于计算机的设备)将有助于构音障碍的患者。加重型餐具和敷料钩有助于患者保持独立感。当吞咽困难变成障碍时,吞咽研究视频可以帮助患者辨认那些不易引发误吸的食物的一致性。注意:控制震颤的药物对于小脑震颤不起作用。
视网膜变性. 为了限制对视网膜的损伤,鼓励使用太阳镜和限制紫外线照射。已经为具有周边视力丧失仅保持中央视觉的患者提出了各种助视器,虽然它们都有着各自缺点。低视力辅助器如放大镜和闭路电视可以为中心视力下降和视野受限的人提供有效的阅读视野。宽视野、高强度的手电筒产生明亮的宽光束,这能改善视网膜变性患者的夜间活动能力。这些辅助器价格便宜,可以双目观看,但又大又重又显眼。Prevention of Secondary
继发性并发症的预防
没有饮食因素可以减轻症状;然而,建议补充维生素,特别是如果卡路里摄入量降低的时候。体重控制是重要的,因为肥胖可以加剧步行和移动能力的困难性。
监测
找眼科医生进行例行的常规检查有助于测量视力和视野,并帮助确定合适的视觉辅助工具。
正在研究的疗法
Scholefield et al [2009]表明等位基因-特异的RNA干扰可作为SCA7的治疗方法。搜索ClinicalTrials.gov获取关于更大范围疾病和病症临床研究的信息。注:可能没有关于该疾病的临床试验。
遗传咨询
遗传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够背景知识,以及符合个人情况的决定。 接着几个段落是涉及遗传风险评估, 根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或伦理问题,或者企图替代遗传学专业人员的咨询工作。-作者ED
家族成员的风险
先证者的父母
- 绝大多数诊断为SCA7的患者有一个受累的父母。
注:
虽然大多数被确诊为SCA7的患者都有受累的家长,但可能因为对家族中其他成员疾病的错诊、家长在症状出现前过早死亡或患病家长的发病年龄晚等因素而导致家族史成阴性。先证者的后代.受累的患者的后代理论上有50%的几率遗传CAG重复扩增。遗传了CAG重复扩增的个体发展为SCA7表型的风险取决于等位基因扩增的大小。
先证者的其他家庭成员. 其他家庭成员的风险取决于先证者父母的遗传状况:如果一个亲本具有扩增的CAG重复,他或她的家庭成员具有患病风险。
遗传咨询相关问题
风险个体. 发病年龄、病情严重程度、特殊症状和疾病进展是可变的,不能通过家族史或分子遗传学检测来进行预测。
具有患病风险的无症状个体的检测. 对高危的成年人进行SCA7的检测可用分子遗传学检测中提及的技术。这种检测不可用于预测无症状个体的发病年龄、病情严重程度、特殊症状和疾病进展速度。 在对高危人群进行SCA7检测时,首先应对受累的家庭成员进行检测,以确认其分子诊断。应该注意的是,在没有明确症状的情况下检测致病性变异是预测性检测。高危无症状的成年家庭成员可能会寻求检测,以便对生育、财务事宜和职业规划作出个人决定。其他人可能存在不同的动机,包括“需要知道”。对高危无症状的成年家庭成员检测通常涉及检测前的访谈,其中需对要求检测的动机、个体对SCA7的认识,阳性和阴性检测结果的可能影响以及神经系统状态进行评估。对于那些正在寻求检测的人,应该就他们在健康、生命、残疾保险覆盖、就业和教育歧视以及社会和家庭互动改变中可能遇到的问题进行咨询。其他要考虑的问题是对其他家庭成员的危险状态的影响。应当征得知情同意,并将记录进行保密。 测试结果为阳性的个体需要安排长期跟踪和评估。
对年龄小于18岁的高危个体的检测. 共识认为年龄小于18岁且有成年发病性疾病风险的人不应在没有症状的情况下接受检测。反对检测年龄小于18岁的无症状个体的主要观点是,这会剥夺了他们知情权或不知情权,提高了其在家庭和其他社会环境中被歧视的可能性,并且可能会对教育和职业产生严重影响[Bloch & Hayden 1990, Harper & Clarke 1990]。年龄小于18岁的有症状的个人通常从确诊的特异诊断中受益。另见国家遗传咨询师协会关于对未成年人进行成年发病的遗传检测的立场声明,以及美国儿科学会和美国医学遗传学和基因组学协会的政策声明:儿童基因检测和筛查中的伦理和政策问题。
家庭计划
DNA银行用以储存DNA(通常从白细胞中提取),以备将来使用。因为测试方法和我们对基因、等位基因变体和疾病的理解可能会在将来得到完善,所以应考虑对受累的个体的DNA进行存储。
人力资源
GeneReviews员工选择以下疾病特定和/或伞状组织和/或登记处,为患有此疾病的个人及其家人提供帮助。GeneReviews不对其他组织提供的信息负责。有关选择标准的信息,请单击此处。
- NCBI Genes and Disease
- Spinocerebellar Ataxia: Making an Informed Choice about Genetic TestingBooklet providing information about Spinocerebellar Ataxia
- Ataxia UKLincoln House1-3 Brixton RoadLondon SW9 6DEUnited KingdomPhone: 0845 644 0606 (helpline); 020 7582 1444 (office); +44 (0) 20 7582 1444 (from abroad)Email: helpline@ataxia.org.uk; office@ataxia.org.uk
- euro-ATAXIA (European Federation of Hereditary Ataxias)Ataxia UKLincoln House, Kennington Park, 1-3 Brixton RoadLondon SW9 6DEUnited KingdomPhone: +44 (0) 207 582 1444Email: smillman@ataxia.org.uk
- National Ataxia Foundation2600 Fernbrook LaneSuite 119Minneapolis MN 55447Phone: 763-553-0020Email: naf@ataxia.org
- Spanish Ataxia Federation (FEDAES)SpainPhone: 34 983 278 029; 34 985 097 152; 34 634 597 503Email: sede.valladolid@fedaes.org; sede.gijon@fedaes.org; sede.bilbao@fedaes.org
- CoRDS RegistrySanford Research2301 East 60th Street NorthSioux Falls SD 57104Phone: 605-312-6423Email: sanfordresearch@sanfordhealth.org
分子遗传学
分子遗传学章节和OMIM表中的信息可能与GeneReview中的其他信息不同:表格中可能包含更多最新信息。-作者ED
表 A.
脊髓小脑性共济失调 7型:基因和数据库
表 B.
OMIM 脊髓小脑性共济失调 7型(在OMIM中查看全部)
View in own window
基因结构. ATXN7全长为136,094 bp,编码892个氨基酸的蛋白。关于基因和蛋白信息的详细总结见表 A, 基因.
良性变异体. 在第一个外显子中存在CAG重复的多态性;正常等位基因的CAG重复数目在4-19个。在不受影响的,无关的参考人群中,(CAG)10 是最常见的等位基因,并且大于19个重复的正常等位基因还没有见过[Del-Favero et al 1998, Gouw et al 1998]。剪切变异体似乎是存在的,虽然其意义不明。
致病性变异体.ATXN7的致病性变异是蛋白编码区中(CAG)n 三核苷酸重复的异常扩增[David et al 1997]。致病性等位基因CAG重复数目从36到450个[Michalik et al 2004]。28-36个CAG重复的等位基因意义不确定(见分子遗传学检测)。
正常的基因产物.ATXN7编码的ataxin-7蛋白大小约为95 kd。蛋白在细胞核中显著表达,但穿梭于细胞核和细胞质之间。Ataxin-7其中一项功能是作为转录共激活复合体STAGA的核心组件[Garden & LaSpada 2008]. Ataxin-7在细胞质中也发挥稳定微管的作用[Nakamura et al 2012]。已有报道表明ataxin-7正常分布于人的大脑和视网膜中[Cancel et al 2000]。
异常的基因产物.ATXN7中的CAG重复编码一串谷氨酰胺。在未受影响的个体中,多聚谷氨酰胺链长达4-19个氨基酸。异常的蛋白具有37个或者更多的多聚谷氨酰胺扩增。来自受累的个体的蛋白已在细胞核中检出,其大小约为130 kd[Trottier et al 1995]。ATXN7的CAG重复扩增抑制了反义非编码RNA(可促进对ataxin-7启动子的抑制性染色质修饰)的转录[Sopher et al 2011],从而促进ataxin-7变异蛋白的表达水平的升高。
变异的ataxin-7引起神经变性的确切分子机制尚不清楚。在细胞培养模型中,Ajayi et al [2012]报道变异的ataxin-7导致活性氧的产量增加,这导致了细胞毒性。Mookerjee et al [2009]和Janer et al [2010]指出,ataxin-7中第257位的赖氨酸的翻译后修饰在发病机制中是重要的。
在SCA7的转基因小鼠模型中,扩增的多聚谷氨酰胺链可诱导小脑和视网膜中的神经变性和跨神经变性[Yvert et al 2000]。 对STAGA复合物中的酶组分Gcn5的敲除会使小脑和视网膜病变恶化[Chen et al 2012]。小脑中的异常Bergmann神经胶质可能通过谷氨酸转运通路的受损引起变性,进而导致小脑浦肯野细胞的非细胞自主变性[Garden et al 2002, Custer et al 2006]。那些将神经纤维爬升到小脑下橄榄体的神经元似乎在SCA7发病机制中也发挥作用。从三种细胞类型——Bergmann神经胶质细胞、下橄榄体神经元细胞和浦肯野细胞中——敲除变异的基因,使转基因小鼠的前症状期增加一倍[Furrer et al 2011]。在转基因小鼠中抑制突变蛋白50%的表达可逆转小鼠SCA7表型的几个方面,提示了潜在治疗的途径[Furrer et al 2013]。
参考文献
发布的指导方针/共识性声明
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 2-7-17. [PubMed: 23428972]
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset disorders. Available online. 2012. Accessed 2-7-17.
文献引用
- Abe T, Abe K, Aoki M, Itoyama Y, Tamai M. Ocular changes in patients with spinocerebellar degeneration and repeated trinucleotide expansion of spinocerebellar ataxia type 1 gene. Arch Ophthalmol. 1997;115:231-6. [PubMed: 9046258]
- Ahn JK, Seo JM, Chung H, Yu HG. Anatomical and functional characteristics in atrophic maculopathy associated with spinocerebellar ataxia type 7. Am J Ophthalmol. 2005;139:923-5. [PubMed: 15860307]
- Ajayi A, Yu X, Lindberg S, Langel U, Ström AL. Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7 (SCA7) model. BMC Neurosci. 2012;13:86. [PMC free article: PMC3412756] [PubMed: 22827889]
- Aleman TS, Cideciyan AV, Volpe NJ, Stevanin G, Brice A, Jacobson SG. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res. 2002;74:737-45. [PubMed: 12126946]
- Andrew SE, Goldberg YP, Hayden MR. Rethinking genotype and phenotype correlations in polyglutamine expansion disorders. Hum Mol Genet. 1997;6:2005-10. [PubMed: 9328463]
- Ansorge O, Giunti P, Michalik A, Van Broeckhoven C, Harding B, Wood N, Scaravilli F. Ataxin-7 aggregation and ubiquitination in infantile SCA7 with 180 CAG repeats. Ann Neurol. 2004;56:448-52. [PubMed: 15349877]
- Anttinen A, Nikoskelainen E, Marttila RJ, Grenman R, Falck B, Aarnisalo E, Kalimo H. Familial olivopontocerebellar atrophy with macular degeneration: a separate entity among the olivopontocerebellar atrophies. Acta Neurol Scand. 1986;73:180-90. [PubMed: 3705927]
- Babovic-Vuksanovic D, Snow K, Patterson MC, Michels VV. Spinocerebellar ataxia type 2 (SCA 2) in an infant with extreme CAG repeat expansion. Am J Med Genet. 1998;79:383-7. [PubMed: 9779806]
- Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY. Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology. 1998;51:1081-6. [PubMed: 9781533]
- Bloch M, Hayden MR. Opinion: predictive testing for Huntington disease in childhood: challenges and implications. Am J Hum Genet. 1990;46:1-4. [PMC free article: PMC1683548] [PubMed: 2136787]
- Cancel G, Duyckaerts C, Holmberg M, Zander C, Yvert G, Lebre AS, Ruberg M, Faucheux B, Agid Y, Hirsch E, Brice A. Distribution of ataxin-7 in normal human brain and retina. Brain. 2000;123:2519-30. [PubMed: 11099453]
- Carpenter S, Schumacher GA. Familial infantile cerebellar atrophy associated with retinal degeneration. Arch Neurol. 1966;14:82-94. [PubMed: 5900234]
- Chen YC, Gatchel JR, Lewis RW, Mao CA, Grant PA, Zoghbi HY, Dent SY. Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model. Hum Mol Genet. 2012;21:394-405. [PMC free article: PMC3276287] [PubMed: 22002997]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302-11. [PubMed: 16936724]
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65-70. [PubMed: 9288099]
- David G, Dürr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, Holmberg M, Yahyaoui M, Hentati F, Chkili T, Agid Y, Brice A. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum Mol Genet. 1998;7:165-70. [PubMed: 9425222]
- Del-Favero J, Krols L, Michalik A, Theuns J, Löfgren A, Goossens D, Wehnert A, Van den Bossche D, Van Zand K, Backhovens H, van Regenmorter N, Martin JJ, Van Broeckhoven C. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA type II) caused by CAG triplet repeat expansion. Hum Mol Genet. 1998;7:177-86. [PubMed: 9425224]
- Enevoldson TP, Sanders MD, Harding AE. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain. 1994;117:445-60. [PubMed: 8032856]
- Filla A, Mariotti C, Caruso G, Coppola G, Cocozza S, Castaldo I, Calabrese O, Salvatore E, De Michele G, Riggio MC, Pareyson D, Gellera C, Di Donato S. Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol. 2000;44:31-6. [PubMed: 10894992]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG Jr, Warren ST, Oostra BA, Nelson DL, Thomas Caskey C. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047-58. [PubMed: 1760838]
- Furrer SA, Mohanachandran MS, Waldherr SM, Chang C, Damian VA, Sopher BL, Garden GA, La Spada AR. Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous bergmann glia degeneration. J Neurosci. 2011;31:16269-78. [PMC free article: PMC3256125] [PubMed: 22072678]
- Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, Damian VA, Garden GA, La Spada AR. Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. 2013;22:890-903. [PMC free article: PMC3561911] [PubMed: 23197655]
- Garden GA, La Spada AR. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7:138-49. [PMC free article: PMC4195584] [PubMed: 18418675]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897-905. [PubMed: 12077187]
- Giunti P, Stevanin G, Worth PF, David G, Brice A, Wood NW. Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet. 1999;64:1594-603. [PMC free article: PMC1377902] [PubMed: 10330346]
- Gouw LG, Castañeda MA, McKenna CK, Digre KB, Pulst SM, Perlman S, Lee MS, Gomez C, Fischbeck K, Gagnon D, Storey E, Bird T, Jeri FR, Ptácek LJ. Analysis of the dynamic mutation in the SCA7 gene shows marked parental effects on CAG repeat transmission. Hum Mol Genet. 1998;7:525-32. [PubMed: 9467013]
- Gouw LG, Digre KB, Harris CP, Haines JH, Ptacek LJ. Autosomal dominant cerebellar ataxia with retinal degeneration: clinical, neuropathologic, and genetic analysis of a large kindred. Neurology. 1994;44:1441-7. [PubMed: 8058146]
- Gu W, Wang Y, Liu X, Zhou B, Zhou Y, Wang G. Molecular and clinical study of spinocerebellar ataxia type 7 in Chinese kindreds. Arch Neurol. 2000;57:1513-8. [PubMed: 11030806]
- Han Y, Deng B, Liu M, Jiang J, Wu S, Guan Y. Clinical and genetic study of a Chinese family with spinocerebellar ataxia type 7. Neurol India. 2010a;58:622-6. [PubMed: 20739808]
- Han Y, Yu L, Zheng HM, Guan YT. Clinical and genetic study of spinocerebellar ataxia type 7 in East Asian population. Chin Med J (Engl) 2010b;123:2274-8. [PubMed: 20819679]
- Harper PS, Clarke A. Should we test children for "adult" genetic diseases? Lancet. 1990;335:1205-6. [PubMed: 1971046]
- Havener WH. Cerebellar-macular abiotrophy. AMA Arch Ophthalmol. 1951;45:40-3. [PubMed: 14789289]
- Hugosson T, Gränse L, Ponjavic V, Andréasson S. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet. 2009;30:1-6. [PubMed: 19172503]
- Illarioshkin SN, Slominsky PA, Ovchinnikov IV, Markova ED, Miklina NI, Klyushnikov SA, Shadrina M, Vereshchagin NV, Limborskaya SA, Ivanova-Smolenskaya IA. Spinocerebellar ataxia type 1 in Russia. J Neurol. 1996;243:506-10. [PubMed: 8836939]
- Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, Duyckaerts C, Brice A, Dejean A, Sittler A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet. 2010;19:181-95. [PubMed: 19843541]
- Jardim LB, Silveira I, Pereira ML, Ferro A, Alonso I, do Céu Moreira M, Mendonça P, Ferreirinha F, Sequeiros J, Giugliani R. A survey of spinocerebellar ataxia in South Brazil - 66 new cases with Machado-Joseph disease, SCA7, SCA8, or unidentified disease-causing mutations. J Neurol. 2001;248:870-6. [PubMed: 11697524]
- Kim JY, Park SS, Joo SI, Kim JM, Jeon BS. Molecular analysis of Spinocerebellar ataxias in Koreans: frequencies and reference ranges of SCA1, SCA2, SCA3, SCA6, and SCA7. Mol Cells. 2001;12:336-41. [PubMed: 11804332]
- Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62-73. [PubMed: 15895563]
- Konigsmark BW, Weiner LP. The olivopontocerebellar atrophies: a review. Medicine (Baltimore) 1970;49:227-41. [PubMed: 4910986]
- Koob MD, Benzow KA, Bird TD, Day JW, Moseley ML, Ranum LP. Rapid cloning of expanded trinucleotide repeat sequences from genomic DNA. Nat Genet. 1998;18:72-5. [PubMed: 9425905]
- Lebre AS, Stevanin G, Brice A. Spinocerebellar ataxia 7 (SCA7). In: Pulst SM, ed. Genetics of Movement Disorders. San Diego, CA: Academic Press. 2003:85-94.
- Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, Szliwowski H, Evrard P, Ceuterick C, Tassignon MJ, Smet-Dieleman H, Willems PJ. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol. 1994;88:277-86. [PubMed: 7839819]
- Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12:2-15. [PubMed: 14571264]
- Michalik A, Van Broeckhoven C. Pathogenesis of polyglutamine disorders: aggregation revisited. Hum Mol Genet. 2003;12(Spec No 2):R173-86. [PubMed: 14504263]
- Miller RC, Tewari A, Miller JA, Garbern J, Van Stavern GP. Neuro-ophthalmologic features of spinocerebellar ataxia type 7. J Neuroophthalmol. 2009;29:180-6. [PubMed: 19726938]
- Mittal U, Roy S, Jain S, Srivastava AK, Mukerji M. Post-zygotic de novo trinucleotide repeat expansion at spinocerebellar ataxia type 7 locus: evidence from an Indian family. J Hum Genet. 2005;50:155-7. [PubMed: 15750685]
- Mookerjee S, Papanikolaou T, Guyenet SJ, Sampath V, Lin A, Vitelli C, DeGiacomo F, Sopher BL, Chen SF, La Spada AR, Ellerby LM. Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J Neurosci. 2009;29:15134-44. [PMC free article: PMC2907146] [PubMed: 19955365]
- Nakamura Y, Tagawa K, Oka T, Sasabe T, Ito H, Shiwaku H, La Spada AR, Okazawa H. Ataxin-7 associates with microtubules and stabilizes the cytoskeletal network. Hum Mol Genet. 2012;21:1099-110. [PMC free article: PMC3277310] [PubMed: 22100762]
- Nardacchione A, Orsi L, Brusco A, Franco A, Grosso E, Dragone E, Mortara P, Schiffer D, De Marchi M. Definition of the smallest pathological CAG expansion in SCA7. Clin Genet. 1999;56:232-4. [PubMed: 10563484]
- Rüb U, Brunt ER, Seidel K, Gierga K, Mooy CM, Kettner M, Van Broeckhoven C, Bechmann I, La Spada AR, Schöls L, den Dunnen W, de Vos RA, Deller T. Spinocerebellar ataxia type 7 (SCA7): widespread brain damage in an adult-onset patient with progressive visual impairments in comparison with an adult-onset patient without visual impairments. Neuropathol Appl Neurobiol. 2008;34:155-68. [PubMed: 17971076]
- Sasaki H, Yabe I, Yamashita I, Tashiro K. Prevalence of triplet repeat expansion in ataxia patients from Hokkaido, the northernmost island of Japan. J Neurol Sci. 2000;175:45-51. [PubMed: 10785256]
- Scholefield J, Greenberg LJ, Weinberg MS, Arbuthnot PB, Abdelgany A, Wood MJ. Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One. 2009;4:e7232. [PMC free article: PMC2747278] [PubMed: 19789634]
- Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rüb U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1-21. [PubMed: 22684686]
- Sokolovsky N, Cook A, Hunt H, Giunti P, Cipolotti L. A preliminary characterisation of cognition and social cognition in spinocerebellar ataxia types 2, 1, and 7. Behav Neurol. 2010;23:17-29. [PMC free article: PMC5434399] [PubMed: 20714058]
- Sopher BL, Ladd PD, Pineda VV, Libby RT, Sunkin SM, Hurley JB, Thienes CP, Gaasterland T, Filippova GN, La Spada AR. CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron. 2011;70:1071-84. [PMC free article: PMC3139428] [PubMed: 21689595]
- Stevanin G, Giunti P, Belal GD, Dürr A, Ruberg M, Wood N, Brice A. De novo expansion of intermediate alleles in spinocerebellar ataxia 7. Hum Mol Genet. 1998;7:1809-13. [PubMed: 9736784]
- Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ, Forrest SM, Biros I, Nicholson GA. Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet. 2000;95:351-7. [PubMed: 11186889]
- Tang B, Liu C, Shen L, Dai H, Pan Q, Jing L, Ouyang S, Xia J. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol. 2000;57:540-4. [PubMed: 10768629]
- Thurtell MJ, Fraser JA, Bala E, Tomsak RL, Biousse V, Leigh RJ, Newman NJ. Two patients with spinocerebellar ataxia type 7 presenting with profound binocular visual loss yet minimal ophthalmoscopic findings. J Neuroophthalmol. 2009;29:187-91. [PMC free article: PMC2987707] [PubMed: 19726939]
- To KW, Adamian M, Jakobiec FA, Berson EL. Olivopontocerebellar atrophy with retinal degeneration. An electroretinographic and histopathologic investigation. Ophthalmology. 1993;100:15-23. [PubMed: 8433819]
- Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Yves Agid Y, Brice A, Mandel J. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403-6. [PubMed: 7477379]
- Tsai HF, Liu CS, Leu TM, Wen FC, Lin SJ, Liu CC, Yang DK, Li C, Hsieh M. Analysis of trinucleotide repeats in different SCA loci in spinocerebellar ataxia patients and in normal population of Taiwan. Acta Neurol Scand. 2004;109:355-60. [PubMed: 15080863]
- van de Warrenburg BP, Frenken CW, Ausems MG, Kleefstra T, Sinke RJ, Knoers NV, Kremer HP. Striking anticipation in spinocerebellar ataxia type 7: the infantile phenotype. J Neurol. 2001;248:911-4. [PubMed: 11697534]
- Weiner LP, Konigsmark BW, Stoll J Jr, Magladery JW. Hereditary olivopontocerebellar atrophy with retinal degeneration. Report of a family through six generations. Arch Neurol. 1967;16:364-76. [PubMed: 6021917]
- Yvert G, Lindenberg KS, Picaud S, Landwehrmeyer GB, Sahel JA, Mandel JL. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet. 2000;9:2491-506. [PubMed: 11030754]
推荐书目
- Gouw LG, Kaplan CD, Haines JH, Digre KB, Rutledge SL, Matilla A, Leppert M, Zoghbi HY, Ptácek LJ. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat Genet. 1995;10:89-93. [PubMed: 7647799]
- Zoghbi HY, Orr HT. Spinocerebellar ataxias. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chapter 226. McGraw-Hill. Available online. Accessed 5-4-16.
章节注释
作者履历
Thomas D Bird, MD; University of Washington (2007-2012)
Gwenn Garden, MD, PhD (2012-present)
Launce G-C Gouw, MD, PhD; University of Utah School of Medicine (1998-2007)
Albert R La Spada, MD, PhD; University of California, San Diego (2007-2012)
Roberta A Pagon, MD, University of Washington (2007-2012)
Louis J Ptacek, MD; University of California at San Francisco (1998-2007)
Revision History
- 20 December 2012 (me) Comprehensive update posted live
- 6 September 2007 (tb) Revision: Natural History (Clinical Description)
- 9 February 2007 (me) Comprehensive update posted to live Web site
- 11 December 2003 (me) Comprehensive update posted to live Web site
- 20 June 2001 (me) Comprehensive update posted to live Web site
- 27 August 1998 (pb) Review posted to live Web site
- 1 June 1998 (lg) Original submission