
总结Summary
临床特征Clinical characteristics.
CLS的特点通常是在男性中严重到极重的智力残疾;也有不太严重的病例报道。杂合携带的女性的智力可表现为正常到严重受损。容貌在受累的特性,受累的年长男孩或成人有特征性的容貌特点。手短,软,肉质,常有手指明显过度伸展,其指骨和指甲从近侧由宽变窄 。男性身高在第三百分位数以。常见小头畸形。可能存在心脏异常,并可能导致过早死亡。刺激诱导性跌倒发作(SIDAs),即在意想不到的触觉或听觉刺激或兴奋而引发的短暂的崩溃,但无意识丧失,会出现在约20%患者中。通常SIDAs会始于童年期到青少年期。渐进性脊柱侧后凸畸形是一个长期护理中最困难的方面。寿命可能会缩短。
Coffin-Lowry syndrome (CLS) is usually characterized by severe-to-profound intellectual disability in males; less severely impaired individuals have been reported. Intellect ranges from normal to profoundly impaired in 杂合的 females. The facial appearance is characteristic in the 受累的, older male child or adult. The hands are short, soft, and fleshy, often with remarkably hyperextensible fingers that taper from wide (proximally) to narrow with small terminal phalanges and nails. Males are consistently below the third centile in height. Microcephaly is common. Cardiac abnormalities may be present and can contribute to premature death. Stimulus-induced drop attacks (SIDAs) in which unexpected tactile or auditory stimuli or excitement triggers a brief collapse but no loss of consciousness are present in approximately 20% of affected individuals. Typically SIDAs begin between mid-childhood and the teens. Progressive kyphoscoliosis is one of the most difficult aspects of long-term care. Life span may be reduced.
诊断/检测 Diagnosis/testing.
男性CLS患者的诊断是严重的发育迟缓,特征性的颅面和手部,以及影像学表现。女性携带者可能是轻度受累。RPS6KA3基因是目前为止发现的唯一的致病性变异会引起CLS的基因,其基因检测可以用来确诊但不能排除对典型的CLS的诊断。测序分析可以在约25% - 40%临床诊断的先证者中发现致病性变异。The diagnosis of CLS is established in males with severe developmental delay, characteristic craniofacial and hand findings, and radiographic findings. Carrier females may be mildly 受累的. Molecular genetic testing of RPS6KA3, the only 基因 yet published in which pathogenic variants are known to cause CLS, can be used to confirm but not to rule out the diagnosis of typical CLS. Sequence analysis identifies pathogenic variants in approximately 25%-40% of clinically diagnosed probands.
管理 Management.
对症治疗针对SIDAs的治疗药物如丙戊酸、氯硝西泮,或选择性5-羟色胺摄取抑制剂;对频繁出现SIDAs的患者可能需要使用轮椅,如果可能的话应避免惊吓。对表现出破坏性或自残行为的患者可给予利培酮。如果出现喂养困难,异常生长速度、行为问题、脊柱侧后凸畸形,肥胖,予以标准的处理。并发症的预防:预防进展的脊柱侧后凸畸形导致心肺损害。监测:定期的听力,牙科和视力检查;每年心脏检查,十岁开始加测超声心动图,每五至十年一次;针对渐进性脊柱后侧弯,定期脊柱监测;需避免的试剂/环境:有过SIDAs的患者需尽量避免受惊吓和/或跌倒。
Treatment of manifestations: SIDAs are treated with medications such as valporate, clonazepam, or selective serotonin uptake inhibitors; individuals who experience frequent SIDAs may require use of a wheelchair and should be protected, if possible, from being startled. Risperidone may be of benefit to individuals who display destructive or self-injurious behavior. Feeding difficulties, abnormal growth velocity, behavioral problems, kyphoscoliosis, and obesity, if present, are treated in a standard manner.
Prevention of secondary complications: Intervention to prevent progression of kyphoscoliosis to the point of cardio-respiratory compromise.
Surveillance: Periodic hearing, dental, and vision examinations; annual clinical cardiac examination, adding an echocardiogram by age ten years and repeating every five to ten years; regular monitoring of the spine for progressive kyphoscoliosis.
Agents/circumstances to avoid: Individuals who experience SIDAs should be protected as much as possible from being startled and/or from falls.
遗传咨询Genetic counseling.
CLS是X连锁遗传病。约70% - 80%的先证者没有CLS的家族史,20%-30%有一个以上的受累的家庭成员。女性携带者的孩子有50%的风险遗传致病性变异。遗传致病变异的男性会患病;遗传致病性变异的女性为携带者,且有出现一些发育迟缓和CLS的轻微生理体征的高风险。对于已确诊的家庭,或者连锁分析排除了X染色体携带(或有携带可能)致病性变异的家庭,可行携带者检测高风险的亲属和孕期产前检查。CLS is inherited in an X-linked manner. Approximately 70%-80% of probands have no family history of CLS, and 20%-30% have more than one additional 受累的 family member. Children of a woman known to be a 携带者 are at 50% risk of inheriting the 致病性变异. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers and at high risk for at least some developmental delay and mild physical signs of CLS. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible in families in which the pathogenic variant has been identified in an affected family member or in which linkage studies can exclude the X 染色体 that carries (or potentially carries) the pathogenic variant.
诊断Diagnosis
临床诊断Clinical Diagnosis
男性患者临床表现Affected Males
Clinical findings. 最重要的受累男性的表现如下The most important clinical signs of Coffin-Lowry syndrome (CLS) in 受累的 males are the following [Hanauer & Young 2002] (see Figure 1, Figure 2, Figure 3, Figure 4, and Figure 5):
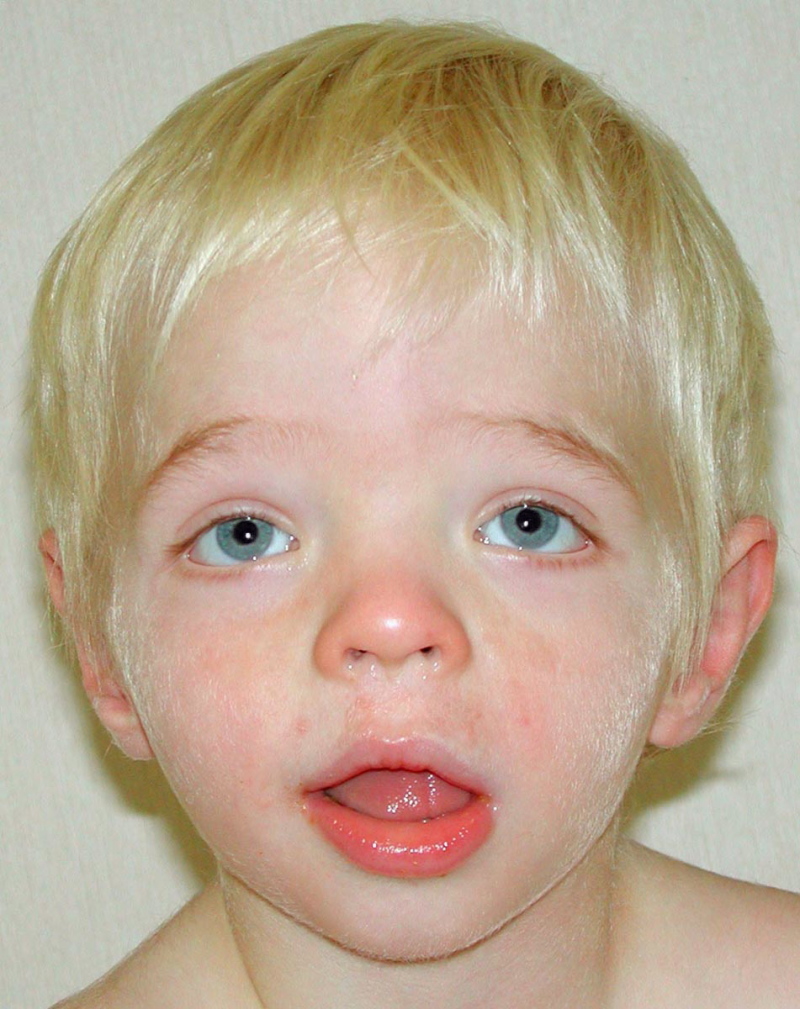
Figure 1.
图一前后位,2岁患病男孩的面部特征有双目距离远,轻度眼睑下斜,鼻短小而宽厚, 嘴唇轻度外翻唇红。 AP view of a boy age two years with CLS showing relatively fine facial features but with widely spaced eyes, mildly downslanted palpebral fissures, short nose with broad columella and thick, slightly everted vermilion of the lips. (Affected individual (more...)

Figure 2 A & B.
图2A,B前后位及侧位,同一个男孩在五岁时表现出更明显的三角形脸,粗糙度增加,以及CLS的典型面部表情(患者有已知RPS6KA3致病性变异。)AP and lateral view of the same boy at age five years showing a more triangular-shaped face, increasing coarseness, and expression of the typical facial signs of CLS. (Affected individual has a known RPS6KA3 pathogenic variant.)

Figure 3.
前后位显示一青少年表现轻度面部特征,但双眼距宽,轻度睑裂下斜,厚红双唇和小牙齿,鼻小柱宽而鼻孔正常。AP view of an adolescent showing relatively mild facial signs but with widely spaced eyes, mildly downslanted palpebral fissures, thick vermilion of the upper and lower lips, and small teeth. The columnella is broad but nares are a good size, perhaps (more...)
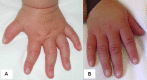
Figure 4 A & B. 图1和2中孩子的手,2岁时(A),5岁时(B)
Hand of the child illustrated in Figure 1 and Figure 2 at ages two years (A) and five years (B). (Affected individual has a known RPS6KA3 pathogenic variant.)
Figure 5
年长儿童的手,表现典型的手指变尖而软。B.患者手部更多细节差异见图3A & B. A. Hand of an older child showing classic tapering and soft appearance. B. More subtle differences seen in the hand of the individual illustrated in Figure 3. (Affected individuals have a known RPS6KA3 pathogenic variant.)
- 发育Development. 受累男性有中度到重度智力障碍,自从分子遗传学检测的出现,更多中度受累的男性被检测。Development. Affected males typically have moderate to severe intellectual disability; since the advent of 分子遗传学检测, more mildly 受累的 males are being identified [Field et al 2006].
- 颅面Craniofacial.在受累的年长男孩或成人中,面部特征为Craniofacial. In the 受累的 older male child or adult, the facial appearance is characteristic (see Figure 1, Figure 2, and Figure 3):
通常前额突出和眶上脊浓眉
通常双眼距宽,眼睑裂下斜;偶见正常眶周区与轻度内眦
特征的鼻部,包括鼻梁塌陷,钝尖,厚鼻翼及鼻中隔,导致的鼻孔小
大嘴,通常开着;厚红唇,下唇外翻
儿童期面容粗糙,并随年龄进展为更“拳击”样面容
突出的耳朵- Usually prominent forehead and supraorbital ridges with thick eyebrows
- Usually marked widely spaced eyes with downslanted palpebral fissures; occasionally, relatively normal periorbital region with mild telecanthus
- Consistent, often striking, nasal findings including depressed bridge, blunt tip, and thick alae nasi and septum, resulting in small nares
- Wide mouth, usually held open; thick vermilion of the upper and lower lips with everted vermilion of the lower lip
- Coarse facial appearance in childhood with progression to a more ‘pugilistic’ look with age
- Prominent ears
- Extremities
- 四肢
- 短,软,肉质的手,常有手指明显过度伸展,和小鱼际区短横掌
手指明显从近端宽到远端变窄,小指骨和指甲(见图4)。手上的差异有时可能很细小(见图5)。
柔软、易弯曲的手,手掌有“毛绒垫”的感觉,在肥胖的人身上可以看到。
丰满的前臂:有助于诊断年幼儿童
- 短,软,肉质的手,常有手指明显过度伸展,和小鱼际区短横掌
- Short, soft, fleshy hands, often with remarkably hyperextensible fingers, and a short horizontal palmar crease across the hypothenar area
- Soft, malleable hands with an almost 'plush-cushion' feel to the palm, as may be seen in an obese individual
- Full, fleshy forearms: a potentially useful sign in diagnosing a younger child
- Musculoskeletal
- 肌肉骨骼系统
- 鸡胸或漏斗胸
儿童期发病的脊柱侧后凸畸形往往是渐进的
- 鸡胸或漏斗胸
- Frequent pectus carinatum and/or excavatum
- Childhood onset of kyphoscoliosis that is often progressive
注:几位作者曾说过,在年幼的孩子的诊断可能是困难的。事实上,同大多数综合征相比,随着年龄的增长,CLS的面部特征变得越来越明显。然而,即使在新生儿,如果考虑CLS,其诊断是很明显的。在CLS的影像学检查是非特异性,但可能有助于确诊。Note: Several authors have stated that the diagnosis may be difficult in the young child. Indeed, more than in most syndromes, the facial characteristics of CLS become increasingly discernible with age. However, even in neonates, the diagnosis of CLS is most often apparent if considered.
Radiographic findings in CLS are nonspecific individually or as a pattern but may be helpful in confirming the diagnosis [Hanauer & Young 2002]:
- 颅骨增厚伴额窦
椎间盘狭窄,相关椎体退行性变
脊柱侧后凸畸形
狭窄骨盆
掌指pseudoepiphyses?,指骨建模差,远端指骨成簇状(掌指外形难以辅助诊断。)Thickened skull with large frontal sinuses - Anterior beaking of the vertebrae with narrow disc spaces and related degenerative vertebral changes
- Kyphoscoliosis
- Narrow pelvis
- Metacarpal pseudoepiphyses, poor modeling of the middle phalanges, and tufting of the distal phalanges (Metacarpophalangeal profiles do not appear to aid diagnosis.)
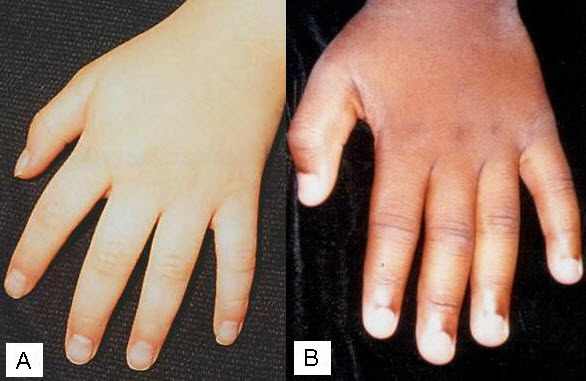
Figure 5
A & B. 年长孩子的手,表现出典型的尖细和柔软。b.图3示患者手部具体差别。A. Hand of an older child showing classic tapering and soft appearance. B. More subtle differences seen in the hand of the individual illustrated in Figure 3. (Affected individuals have a known RPS6KA3 pathogenic variant.)
- Development. 发育。男性患者通常有中度到重度智力障碍;自分子遗传学检测出现,症状较轻的男性患者被确诊。
Affected males typically have moderate to severe intellectual disability; since the advent of 分子遗传学检测, more mildly 受累的 males are being identified [Field et al 2006]. - Craniofacial. In the 受累的 older male child or adult, the facial appearance is characteristic (see Figure 1, Figure 2, and Figure 3):颅面部。在受累的年长儿童或成人,容貌特征(见图1、图2、图3):
- 通常前额突出和眶上脊浓眉
通常双眼距宽,眼睑裂下斜;偶见正常眶周区与轻度内眦
特征的鼻部,包括鼻梁塌陷,钝尖,厚鼻翼及鼻中隔,导致的鼻孔小
大嘴,通常开着;厚红唇,下唇外翻
儿童期面容粗糙,并随年龄进展为更“拳击”样面容
突出的耳朵
Usually prominent forehead and supraorbital ridges with thick eyebrows - Usually marked widely spaced eyes with downslanted palpebral fissures; occasionally, relatively normal periorbital region with mild telecanthus
- Consistent, often striking, nasal findings including depressed bridge, blunt tip, and thick alae nasi and septum, resulting in small nares
- Wide mouth, usually held open; thick vermilion of the upper and lower lips with everted vermilion of the lower lip
- Coarse facial appearance in childhood with progression to a more ‘pugilistic’ look with age
- Prominent ears
- Extremities
- 四肢
- 短,软,肉质的手,常有手指明显过度伸展,和小鱼际区短横掌
手指明显从近端宽到远端变窄,小指骨和指甲(见图4)。手上的差异有时可能很细小(见图5)。
柔软、易弯曲的手,手掌有“毛绒垫”的感觉,在肥胖的人身上可以看到。
丰满的前臂:有助于诊断年幼儿童
- 短,软,肉质的手,常有手指明显过度伸展,和小鱼际区短横掌
- Short, soft, fleshy hands, often with remarkably hyperextensible fingers, and a short horizontal palmar crease across the hypothenar area
- Soft, malleable hands with an almost 'plush-cushion' feel to the palm, as may be seen in an obese individual
- Full, fleshy forearms: a potentially useful sign in diagnosing a younger child
- Musculoskeletal
- 肌肉骨骼系统
- 鸡胸或漏斗胸
儿童期发病的脊柱侧后凸畸形往往是渐进的
- 鸡胸或漏斗胸
- Frequent pectus carinatum and/or excavatum
- Childhood onset of kyphoscoliosis that is often progressive
注:几位作者曾说过,在年幼的孩子的诊断可能是困难的。事实上,同大多数综合征相比,随着年龄的增长,CLS的面部特征变得越来越明显。然而,即使在新生儿,如果考虑CLS,其诊断是很明显的。在CLS的影像学检查是非特异性,但可能有助于确诊。
Note: Several authors have stated that the diagnosis may be difficult in the young child. Indeed, more than in most syndromes, the facial characteristics of CLS become increasingly discernible with age. However, even in neonates, the diagnosis of CLS is most often apparent if considered.
Radiographic findings in CLS are nonspecific individually or as a pattern but may be helpful in confirming the diagnosis [Hanauer & Young 2002]:
- 颅骨增厚伴额窦
椎间盘狭窄,相关椎体退行性变
脊柱侧后凸畸形
狭窄骨盆
掌指pseudoepiphyses?,指骨建模差,远端指骨成簇状(掌指外形难以辅助诊断。)Thickened skull with large frontal sinuses - Anterior beaking of the vertebrae with narrow disc spaces and related degenerative vertebral changes
- Kyphoscoliosis
- Narrow pelvis
- Metacarpal pseudoepiphyses, poor modeling of the middle phalanges, and tufting of the distal phalanges (Metacarpophalangeal profiles do not appear to aid diagnosis.)
Affected Females女性患者
发育迟缓和颅面部肢体从严重(如男性患者)到没有表型。对受累患者家属进行仔细检查可以发泄轻度面部和/或手部特征。The degree of developmental delay and craniofacial and limb changes range from severe (as seen in males) to completely absent. Careful examination of an intellectually normal female relative of an 受累的 individual may reveal mild facial and/or hand manifestations.
检测Testing
核糖体S6激酶酶测定。核糖体S6激酶酶测定,对体外培养的成纤维细胞或转化的淋巴母细胞中进行的,可在RPS6KA3致病性变异的男性患者表现酶活降低。这是一个有限的基础上的研究性检测。注:不适用于女性,因为X染色体失活导致酶活变化大。
Ribosomal S6 kinase enzyme assay. Ribosomal S6 kinase enzyme assay, performed on cultured fibroblasts or transformed lymphoblasts, may show reduced activity in males with an RPS6KA3致病性变异 [Merienne et al 1998, Delaunoy et al 2001, Zeniou et al 2002a]. This is a research test available on a limited basis.
Note: The assay is not useful in females because of the broad range of enzyme activity resulting from X-chromosome inactivation [Delaunoy et al 2001].
Molecular Genetic Testing分子遗传学检测 基因RPS6KA3(RSK2)是已知的唯一的引起CLS的致病性基因。位点异质性的证据。已证明,并不是所有临床符合CLS患者RPS6KA3 变异。然而,是否意味着CLS遗传异质性,或临床上有重叠的特征而难以鉴别,仍有待确定。还没有一个完好的CLS家系的连锁分析证明CLS存在第二个致病位点。
Gene.RPS6KA3 (also known as RSK2) is the only 基因 in which pathogenic variants are known to cause CLS.
Evidence for 位点 heterogeneity. It has been suggested that not all individuals with a clinical picture thought to be consistent with CLS have pathogenic variants in RPS6KA3 [Delaunoy et al 2001, Zeniou et al 2002b]. However, whether this finding points to true genetic heterogeneity in CLS or to inability to distinguish disorders with overlapping features on clinical grounds alone remains to be determined. There are no published linkage data from a well-described Coffin-Lowry syndrome family that would suggest a second Coffin-Lowry syndrome locus.
Clinical testing
Table 1.
Summary of Molecular Genetic Testing Used in Coffin-Lowry Syndrome
| Gene 1 | Test Method | Proportion of Probands with a Pathogenic Variant Detectable by This Method | |
|---|---|---|---|
| Affected Males | Carrier Females 2 | ||
| RPS6KA3 | Sequence analysis 3 / scanning for pathogenic variants | ~25%-40% 4, 5, 6, 7 | ~90%-95% 5, 8 |
| Deletion/重复 analysis 9 | Unknown 10 | 0% 11 | |
See Table A. Genes and Databases for 染色体位点 and protein. See Molecular Genetics for information on allelic variants.
2.到目前为止,还没有数据支持X染色体失活偏移(淋巴细胞检测)会导致到的临床变异女性携带者,或是对携带者检测有帮助。携带者的检出率是根据受试者的选择而定的。在女性的家族有一个在RPS6KA3一致病性变异受累的男性亲属,携带着检测率约为90%~95%;在疑似CLS家族中,检测率余受累男性相似。
To date there are no data to support the assumption that skewed X inactivation, as measured from lymphocytes, accounts for the observed clinical variability in 携带者 females or that it is a useful means of carrier detection. Carrier female detection rate is based on selection of subjects. In females who have a family history of an 受累的 male relative with a 致病性变异 in RPS6KA3, the carrier detection rate is approximately 90%-95%; in families with a suspected diagnosis of Coffin-Lowry syndrome the detection rate will be similar to that for affected males.
3.测序分析显示变异为良性,可能良性,意义不明,可能致病,致病。致病变异包括小的基因内缺失/插入,错义,无义突变,剪接变异,尤其是外显子或者整个基因的缺失/重复没有检测到。对于序列分析结果的解释,点击。
Sequence analysis detects variants that are benign, likely benign, of 意义不确定, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and 错义, nonsense, and 剪接位点 variants; typically, 外显子 or whole-基因 deletions/duplications are not detected. For issues to consider in interpretation of 序列分析 results, click here.
4.在序列分析之前不行PCR扩增,可以对男性患者X染色体上可能的(多)外显子或者整个基因缺失做以分析;确认还需要缺失/重复分析。
Lack of amplification by PCR prior to 序列分析 can suggest a putative (multi)外显子 or whole-基因缺失 on the X 染色体 in 受累的 males; confirmation may require additional testing by deletion/duplication analysis.
5.临床典型病例检出率;不典型病例检出率明显降低
Detection rate in clinically typical cases; detection rate in atypical cases is significantly lower [Authors, unpublished observation].
6.对男性患者PCR扩增失败的序列分析,在X染色体可以检测可能的外显子,多个外显子,和全基因缺失;确认可能需要缺失分析。序列分析不能检测男性患者外显子,多个外显子,及整个基因的重复。
Sequence analysis can detect putative 外显子, multiexon, and whole-基因 deletions on the X 染色体 in 受累的 males based on failure of amplification by PCR; confirmation may require 缺失 analysis. Sequence analysis cannot detect exon, multiexon, and whole-gene duplications in affected males.
7.
Jacquot et al [1998a], Delaunoy et al [2001], Zeniou et al [2002b], Abidi & Schwartz [unpublished] (see Molecular Genetics)
8.女性携带者,基因组DNA的序列分析不能检测一个或多个外显子,或整个X染色体关联的基因的缺失。
Sequence analysis of 基因组的 DNA cannot detect 缺失 of one or more exons or the entire X-linked基因 in a 杂合的 female.
9.序列分析检测缺失/重复,不能检测到编码和跨内含子区;可以使用的方法:定量PCR,长片段PCR、多重连接探针扩增(MLPA),和染色体芯片(CMA)。
Testing that identifies deletions/duplications not readily detectable by 序列分析 of the coding and flanking 内含子的 regions of 基因组的 DNA; included in the variety of methods that may be used are: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and 染色体芯片 (CMA) that includes this 基因/染色体 segment.
10.已发现多个PS6KA3外显子和多个外显子缺失的病例
Several cases of RPS6KA3外显子 and multiexon deletions have been described (see Table A and Molecular Genetics).
11.基因组DNA测序不能检测女性携带者X染色体上一个或多个外显子、整个基因的缺失和重复
检测策略Testing Strategy
先证者的诊断
To confirm/establish the diagnosis in a 先证者
- 男性患者,对RPS6KA3的22个外显子行双向测序,可检测单核苷酸变异和在编码区或在内含子/外显子边界存在小缺失或重复。
对受累的女性,没有以下情况的,可考虑缺失/重复分析:
序列分析发现致病性变异
一个受累的男性亲属更容易地检测致病性变异
在男性患者,表型符合CLS,序列分析没有RPS6KA3致病性变异,可以在研究基础上进行的核糖体S6激酶酶活性的测定;这个测定对女性的检测没有帮助,且只在有限的研究基础上进行。
In an 受累的 male, bidirectional sequencing of the 22 exons of RPS6KA3 should detect any single nucleotide variant and small 缺失 or 重复 present in the 编码区 or at the 内含子/外显子 boundaries.
CLS男性的特征是严重到重度的智力障碍;女性携带者智力从正常到严重受损。早期发育评估可能高估最终的预后发育。 Touraine et al[ 2002 ]没有提供细节但表示:“我们的数据表明,只要采取适当处理,大多数患者智力障碍程度仅是轻度”; Field et al [2006] 报道的家族表现为不同的和轻度体征,所涉及的成员仅有轻度症状。作者是提示一个证明RPS6KA3致病性变异的患者是在快餐店工作的。
精神神经。CLS的人通常被描述为快乐和随和,尽管也有报道自我伤害和其他行为问题。
严重的智力障碍可能无法进行详细的神经学评估。有报道包括强度和肌肉质量下降,深度腱反射下降或升高,睡眠呼吸暂停,中风,进行性痉挛,进展性截瘫,行走能力丧失。后者是由于黄韧带钙化,先天椎管狭窄。
特别值得注意的是刺激诱导的跌倒发作(SIDAs),发病年龄4-17岁之间,平均发病年龄在8.6岁。在SIDAs时,意外的触觉或听觉刺激或兴奋引发下肢60到80毫秒的肌电沉默,导致短暂的跌倒,虽然没有丧失意识。Nelson与Hahn提供的SIDAs视频说明。史蒂芬森等人从CLS基金会数据库中统计了20%(34/170)的患病率。最近,一个有CLS症状,包括刺激诱导的跌倒发作(SIDEs)的女性,有C-末端激酶结构域一致病性变异。
女性也可能受累。纳尔逊与哈恩报道的第二个患者,在六岁典型的SIDAs,之后时后短暂的肌阵挛和强直痉挛,这是伴随着增加强直肌电活动。
史蒂芬森等[ 2005 ]也强调,运动障碍的性质可能会随着年龄的增长,患者可能有不止一种类型的神经系统体征变化。表现的范围包括刺激性猝倒,过度惊骇,强直反应延长;和真正的癫痫发作。
大约5%患者有癫痫发作[史蒂芬森等人2005 ]。
女性携带者比普通人群可能有更高的精神疾病。68位女性中(22例CLS,38位杂合,和8例“受累的“姐妹),有六位(8.8%)有精神疾病诊断,表现为包括精神分裂症、躁郁症,和“精神病”。Micheli等人[ 2007 ]研究的两位女性有“精神病”,两个受累姐妹中有一位有精神分裂症。
心血管。约14%的男性患者和5%的女性患者有心血管疾病[ Hunter2002 ]。这个百分比可能被低估,因为许多CLS患者没有进行彻底的初始或进行中的心脏评估。报告包括:异常的二尖瓣、三尖瓣、主动脉阀;短腱索;心肌病(一患者有心内膜弹力纤维增生症);不明原因的充血性心力衰竭;主动脉和肺动脉扩张[Hunter2002 ]。facher等人[ 2004 ]报道,一患者有限制性心肌病。马丁内兹等人[ 2011 ]报告了一个CLS患者限制性左心室非致密性心肌病。心脏异常可能会导致过早死亡。
肌肉骨骼系统。渐进性脊柱侧后凸畸形是一种对CLS患者的长期护理中最困难的方面。确切的发病率尚不清楚,但至少有47%的男性和32%的女性受累者有渐进性脊柱侧后凸畸形[ Hunter2002 ]。发病率在一系列骨科转诊诊所中更高[Herrera Soto2007 ]。虽然没有公认的严重性的定义,很明显,病情往往随时间的进展,呼吸困难引起的脊柱侧后凸畸形可能导致过早死亡。至少两人死亡发生在手术治疗脊柱侧后凸畸形。
其他影像学看到的骨骼微小变化没有临床表现。
生长。在产前生长正常,生长障碍通常发生在产后早期。男性和严重受累的女性的身高一般低于第三百分位数,但预期会延续该曲线(?)。身高矮可能表现为不成比例的四肢短[hunter2002,Touraine et al2002 ]。而小头畸形是常见的,很多CLS患者头围正常。
牙科。牙齿异常是常见的,包括小牙,错位牙,开咬,牙发育不全,牙萌出提前或延后,和过早丧失,多种原因的牙齿过早脱落。腭高。随着年龄的增长,在年幼期颌后缩变为颌前凸。
耳聋。可能只有少数CLS患者做了视力和听力评估。然而,14/89的男性患者和1/22的女性患者报道有耳聋[Hunter 2002 ]。
听力可能揭示感音神经性听力损失。
有畸形的迷路报道,出现晚发耳聋[rosanowski et al 1998 ]。家族可能聚集出现耳聋。
视力问题。严重的视力问题似乎是罕见的,虽然有报道出现白内障、视网膜萎缩和视神经萎缩;而慢性眼睑发炎的发生率(睑缘炎)可能增加[ Hunter2002 ]。
神经影像学研究显示脑室、蛛网膜下腔,和血管周围间隙增大[Patlas et
al2003 ]。血管周围间隙是衰老的标志,与年龄和认知功能相关。胼胝体异常,包括变薄和发育不全已被多人报道 [Kondoh et al 1998, Wang et al 2006]。有患者核磁表现多发性额叶低密度影[Kondoh et al 1998]。三受累的同胞出脑脊液重点区低密度影于脑脊液[Wang et al2006];他们还显示胼胝体变薄,小脑蚓部发育不全,和轻度脑室不对称。作者认为智力残疾程度与MRI表现的严重程度有关。
凯斯勒等人[Kesler et al 2007 ]进行定量MRI和受累的男性和女性且无真空巨脑室?的低灰质和白质体积,提示早期的神经发育异常,如细胞增殖减低。最大变化区域是在小脑、颞叶和海马。后者在一个家庭中增加,另一个家庭减少;随着年龄的增加而体积增加(ρ=986,p<0=。与大脑总体积相比,胼胝体和小脑蚓部也相对增大。
在一个MRS研究中,基底神经节和侧脑室周围白质为正常[Patlas et al2003 ]。
神经病理学。尸体解剖时发现异常的回转和分层[Hunter2003 ]。
其他。在患者中有报道发现包括直肠脱垂、子宫脱垂、空肠憩室、结肠憩室伴神经节细胞减少,腘神经节、幽门狭窄、单侧肾缺如、肛门前置,面部色素增多,并扩大气管[ Hunter2002 ]。
死亡率.CLS患者寿命缩短。文献报道,4.5%的男性和13.5%的女性死亡平均年龄为20.5岁(范围:13-34)[ Hunter2002 ]。复杂的因素包括心脏异常,泡性肺气肿,呼吸道并发症,渐进性脊柱侧后凸畸形,与癫痫发作相关的吸入。Coffin[ Coffin2003 ]报告说,他的一个病人死于18.8岁时肺炎合并慢性肺病和心脏病,另一个患者死于18岁时的急性食物吸入。作者提醒CLS的患者有危及生命的中枢性和阻塞性睡眠呼吸暂停,另一名男性有慢性阻塞性和中枢性睡眠呼吸暂停的病史。
一个男性和一个女性肯定携带者死于霍奇金病。另一个携带者的母亲有肾母细胞瘤(见肾母细胞瘤的概述),和一个患者的同卵双胞胎之一死于后颅窝肿瘤[Manouvrier Hanu et al 1999 ]。
基因型-表型相关性
虽然表型和RPS6KA3致病性变异的位置和类型之间不存在很强的相关性,错义突变的患者可能有轻微症状[Delaunoy et al 2001 ]。有一个发生非综合征型智力残疾家庭(MRX19;看Genetically Related Disorders)在RPS6KA3发生错义突变,导致核糖体S6激酶酶活性降低80%,而与大多数发生致病性变异的CLS患者,造成的核糖体S6激酶酶完全失活[Merienne et al 1999 ]。这一发现表明,一些RPS6KA3变异可能引起非CLS表型或非综合征型X-连锁智力障碍。
在7位患者中,[ Harum et al2001 ]表明,智商和淋巴母细胞中RPS6KA3介导的crebtide磷酸化反应减低有相关性。
Yang等[ 2004 ]提出,由缺少RPS6KA3磷酸化-ATF4可能干扰ATF4正常调控成骨细胞分化,导致一些出现在CLS的骨性畸形,以及可能解释出现的渐进性脊柱侧后凸。
Nakamura等[ 2005 ]认为,截短变异,在N-末端激酶结构域或其上游,可能引起特异的SIDA敏感性。然而,一个出现SID的女患者,编码的C-末端的激酶结构域的区域杂合的c.1570dupa致病性变异,不符合该相关性[ Rojnueangnit et al2014]。
临床数据对受累的男性,出现或不出现RPS6KA3致病性变异,表明某些临床症状–如肉质,尖细的手指,双眼距宽,和睑裂下斜–可以帮助区分是否存在RPS6KA3突变[F Abidi & CE Schwartz, personal communication]。
命名
早期的作者使用Coffin 综合征,直到Lowry等人[ 1971 ]报道这些患者有同样的症状。
一些早期的文本和文章混淆了Coffin-Siris综合征和CLS。
患病率
CLS的患病率尚未公布。根据作者的经验,可能在1:40,000到1:50,000;这可能,但是,这可能低估了实际患病率。
Clinical Characteristics
Clinical Description
Development. Coffin-Lowry syndrome (CLS) is characterized by severe-to- profound intellectual disability in males; intellect ranges from normal to profoundly impaired in 杂合的 females. Early developmental assessments may overestimate the ultimate developmental prognosis [Hunter 2002]. Touraine et al [2002] did not provide detail but stated “our data have shown that intellectual disability is only moderate in most patients as soon as proper care is provided”; and the families reported by Field et al [2006] showed variable and mild physical signs and included members with only mild impairment. The authors are aware of a patient with a proven RPS6KA3致病性变异 who works in a fast food restaurant [C Skinner, personal communication].
Neuropsychiatric. Individuals with CLS are often described as generally happy and easygoing, although self-injury and other behavioral problems have been reported.
Detailed neurologic assessment may be hampered by the severe intellectual disability. Findings reported include loss of strength and muscle mass, both decreased and increased deep tendon reflexes, sleep apnea, stroke, progressive spasticity, and progressive paraplegia with loss of the ability to walk. The latter has been ascribed to both calcification of the ligamenta flava and 先天的 stenosis of the spinal canal [Hunter 2002].
Of particular note are stimulus-induced drop attacks (SIDAs), with onset between ages four and 17 years and a mean age at onset of 8.6 years [Nakamura et al 2005]. During a SIDA, unexpected tactile or auditory stimuli or excitement trigger a 60- to 80-millisecond electromyographic silence in the lower limbs that results in a brief collapse though no loss of consciousness [Crow et al 1998, Nakamura et al 1998, Hahn & Hanauer 2012]. Nelson & Hahn [2003] provide a video illustration of SIDAs. Stephenson et al [2005] recorded a prevalence of 20% (34/170) from the CLS Foundation database. Recently, a female with clinical features of CLS including stimulus-induced drop episodes (SIDEs) was found to have a 致病性变异 that occurred within the C-terminal kinase 结构域 of the protein [Rojnueangnit et al 2014].
Females may also be 受累的 [Fryssira et al 2002]. In the second of two individuals reported by Nelson & Hahn [2003], typical SIDAs at age six years were later replaced by brief myoclonic jerks and tonic spasms, which were accompanied by increased tonic EMG activity.
Stephenson et al [2005] have also emphasized that the nature of the movement disorder may change with age and that a single individual may have more than one type of neurologic sign. The range of manifestations include cataplexy that varies with the stimulus; hyperekplexia, a prolonged tonic reaction; and true epileptic seizures.
Epileptic seizures affect approximately 5% of individuals [Stephenson et al 2005].
Female carriers may have a higher rate of psychiatric illness than that found in the general population. Six (8.8%) of 68 women (22 females with CLS, 38 heterozygotes, and 8 '受累的' sisters) have had psychiatric diagnoses, including schizophrenia, bipolar disease, and 'psychosis' [reviewed in Hunter 2002]. One of two women studied by Micheli et al [2007] was described as having a ‘psychosis’ and one of two affected sisters reported by Wang et al [2006] as having schizophrenia.
Cardiovascular. Approximately 14% of 受累的 males and 5% of affected females have cardiovascular disease [Hunter 2002]. These percentages may be underestimates as many individuals with CLS have not had thorough initial or ongoing cardiac assessment. Reports have included: abnormalities of the mitral, tricuspid, and aortic valves; short chordae; cardiomyopathy (in one individual, with endocardial fibroelastosis); unexplained congestive heart failure; and dilatation of the aorta and of the pulmonary artery [reviewed in Hunter 2002]. An individual reported by Facher et al [2004] had a restrictive cardiomyopathy. Martinez et al [2011] reported an individual with CLS who had left ventricular non-compaction cardiomyopathy with a restrictive pattern. Cardiac anomalies may contribute to premature death.
Musculoskeletal. Progressive kyphoscoliosis is one of the most difficult aspects of the long-term care of individuals with CLS. The precise prevalence is not known, but at least 47% of 受累的 males and 32% of females have been reported to have progressive kyphoscoliosis [Hunter 2002]. The rates were higher in a series reported from an orthopedic referral clinic [Herrera-Soto et al 2007]. Although no accepted definition of severity has been adopted in published reports, it is clear that the severity often progresses over time and that respiratory compromise caused by kyphoscoliosis may contribute to premature death. At least two deaths have occurred during surgery for kyphoscoliosis.
Other minor skeletal changes that may be seen on radiographs are of no clinical consequence.
Growth. Prenatal growth is normal; growth failure usually occurs early in the postnatal period. Males and severely 受累的 females generally fall below the third centile in height but are expected to track a curve. The reduced height may reflect disproportionately short limbs [Hunter 2002, Touraine et al 2002]. While microcephaly is common, many individuals with CLS have a normal head circumference.
Dental. Dental anomalies are common and include small teeth, malpositioning, open bite, hypodontia, advanced or delayed eruption, and premature loss that appears to have more than one cause. The palate is high. With age, the retrognathia in the younger child tends to be replaced by prognathism.
Hearing loss. It is likely that only a minority of individuals with CLS have had formal assessments of vision and hearing. However, 14/89 受累的 males and 1/22 affected females have been reported to have hearing loss [Hunter 2002].
An audiogram may reveal sensorineural hearing loss.
Malformation of the labyrinth has been reported, as has late onset of hearing loss [Rosanowski et al 1998]. Clustering of hearing loss within families may occur.
Vision problems. Significant visual problems seem to be uncommon, although cataract, retinal pigment atrophy, and optic atrophy have been reported; and the incidence of chronic eyelid irritation (blepharitis) may be increased [reviewed in Hunter 2002].
Neuroimaging studies may show increased intraventricular, subarachnoid, and Virchow-Robin spaces [Patlas et al 2003]. Virchow-Robin spaces appear to be a sign of brain aging and are associated with age and cognitive function. Abnormalities of the corpus callosum including thinning and agenesis have been reported by several authors [Kondoh et al 1998, Wang et al 2006]. An individual was reported with multiple focal frontal hypodensities visible on MRI [Kondoh et al 1998]. Hypodensities attributed to focal areas of CSF were reported in three 受累的 sibs by Wang et al [2006]; they also showed thinning of the corpus callosum, vermian hypoplasia, and mild ventricular asymmetry. The authors concluded that the degree of intellectual disability correlated with the severity of the MRI findings.
Kesler et al [2007] performed quantitative MRI and demonstrated in 受累的 males and females lower gray and white matter volume without evidence of ventriculomegaly ex vacuo, suggesting an early neurodevelopmental abnormality such as reduced cellular proliferation. Areas of maximal change were the cerebellum, temporal lobes, and hippocampus. The latter was increased in one family and decreased in the other; larger volumes correlated with increasing age (rho=.986, P<0.000). The corpus callosum and cerebellar vermis were also relatively enlarged compared to total brain volume.
In a single MRS study, the basal ganglia and periventricular white matter were reported as normal [Patlas et al 2003].
Neuropathology. Abnormal gyration and lamination have been noted at autopsy [Coffin 2003].
Other. Findings reported in single individuals include rectal prolapse, uterine prolapse, jejunal diverticuli, colonic diverticuli with reduced ganglion cells, popliteal ganglion, pyloric stenosis, unilateral renal agenesis, anteriorly placed anus, increased facial pigment, and enlarged trachea [reviewed in Hunter 2002].
Mortality. Life span is reduced in some individuals with CLS. Of individuals reported in the literature, death occurred in 13.5% of males and 4.5% of females at a mean age of 20.5 (range: 13-34) years [Hunter 2002]. Complicating factors have included cardiac anomalies, panacinar emphysema, respiratory complications, progressive kyphoscoliosis, and seizure-associated aspiration. Coffin [2003] reported that one of his original patients died at age 18.8 years of pneumonia superimposed on chronic lung and heart disease, and a second individual died at age 18 years of acute food aspiration. The authors are aware of an individual with CLS who had life-threatening central and obstructive sleep apnea, and of another male who had a history of chronic obstructive and central sleep apnea who died from respiratory complications after surgery for jaw advancement.
One 受累的 male and one 肯定携带者 female died of Hodgkin disease. Another carrier mother had a Wilms tumor (see Wilms Tumor Overview), and a monozygotic twin of an affected individual died of a posterior fossa tumor [Manouvrier-Hanu et al 1999].
Genotype-Phenotype Correlations
Although no strong correlation exists between 表型 and location or type of RPS6KA3致病性变异, individuals with certain 错义 pathogenic variants may tend to have milder disease expression [Delaunoy et al 2001]. The family classified as having a form of nonsyndromic intellectual disability (MRX19; see Genetically Related Disorders) had a missense variant in RPS6KA3, which caused an 80% reduction in ribosomal S6 kinase enzyme activity, in contrast to most pathogenic variants in individuals with CLS, which cause a total loss of ribosomal S6 kinase enzyme activity [Merienne et al 1999]. This finding indicates that some RPS6KA3 variants probably give rise to non-CLS phenotypes or nonsyndromic X-linked intellectual disability.
In a sample of seven individuals, Harum et al [2001] showed a correlation between IQ and the degree of attenuation of the RPS6KA3-mediated CREBtide phosphorylation response in lymphoblasts.
Yang et al [2004] proposed that lack of phosphorylation of ATF4 by RPS6KA3 may interrupt the normal regulatory role of ATF4 in osteoblast differentiation, accounting for some of the bony anomalies seen in CLS, as well as possibly explaining the progressive nature of the kyphoscoliosis.
Nakamura et al [2005] suggested that truncating variants, either in or upstream from the N-terminal kinase 结构域, may cause a particular susceptibility to SIDAs. However, the finding of an 受累的 female with SIDAs who has a 杂合的 c.1570dupA 致病性变异 in the region encoding the C-terminal kinase domain of the protein would argue against this correlation [Rojnueangnit et al 2014].
Clinical data on a series of 受累的 males with and without a 致病性变异 in RPS6KA3 suggest that the presence of certain clinical signs – such as the fleshy, tapering fingers, widely spaced eyes, and downslanted palpebral fissures – may help distinguish those in which mutation of RPS6KA3 is present [F Abidi & CE Schwartz, personal communication].
Nomenclature
Early authors referred to Coffin syndrome until it was recognized that the individuals reported by Lowry et al [1971] had the same syndrome.
Some early texts and papers confused Coffin-Siris syndrome and CLS.
Prevalence
No estimate of the prevalence of CLS has been published. Based on the authors' experience, a rate of 1:40,000 to 1:50,000 may be reasonable; this may, however, underestimate the actual prevalence.
遗传相关(等位基因)疾病Genetically Related (Allelic) Disorders
非综合征性智力残疾。两个亲属有CLS 轻度表现,在RPS6KA3有错义变异 [ Manouvrier Hanu et al 1999 ];相似的家庭也有报道[ Field et al 2006 ]。一种非综合征性智力障碍的(MRX19)已被证明是由RPS6KA3致病性错义突变引起[ Merienne et al 1999 ];另一个致病的错义突变和轻度智力障碍患者[Delaunoy et al 2001 ]。分子遗传学诊断的出现可能有助于确定其他轻度患者的诊断。
Nonsyndromic intellectual disability. Two sibs with mild expression of CLS were reported to have 错义 pathogenic variants in RPS6KA3 [Manouvrier-Hanu et al 1999]; similar families have now been reported by Field et al [2006].
One form of nonsyndromic intellectual disability (MRX19) has been shown to be caused by an RPS6KA3 pathogenic 错义 variant [Merienne et al 1999]; another individual with a pathogenic missense variant and only mild intellectual disability was reported by Delaunoy et al [2001]. The advent of molecular genetic diagnosis may aid in confirming the diagnosis of additional mild cases.
鉴别诊断
CLS诊断年长的男孩或成人通常不难。年幼的孩子或轻度受累的女性的症状可能与其他综合征有相似。同样,年长的女孩和成年人,即使他们是先证者,可当他们完全变性出CLS综合征时,诊断就容易。
Borjeson Forssman Lehmann综合征(BFLS)是一种X-连锁隐性遗传病,有严重的智力障碍,类似CLS手部特征,短鼻子和鼻孔前倾,可能有厚间隔和小鼻孔,以及脊柱侧后凸畸形。另外有大而突出的耳朵和视力问题。BFLS患者也有极度性腺功能减退,往往有明显的乳房。女性可能有部分综合征表现。没有眼距宽、宽嘴巴和厚厚的红唇。PHF是致病突变[Lower et al 2002 ]。
而CLS与威廉姆斯综合征有面部特征相似,遗传异质性的FG综合征,和X连锁的α地中海贫血智力残疾(量)综合征(ATRX),这些疾病没有CLS出现的手特征,并各有额外的特点:
Williams syndrome还包括心血管疾病(弹性硬蛋白动脉?,外周肺动脉瓣狭窄、主动脉瓣上狭窄、高血压),结缔组织异常,智力障碍(轻微),特定的认知特征,独特的个性特征、生长发育异常、内分泌异常(高钙血症、高钙尿症、甲状腺功能减退、青春期提前)。喂养困难常导致婴儿发育不成熟。通过检荧光原位杂交(FISH)或靶向分析,超过99%患者有7q11.2邻近基因缺失。
FG综合征1型(见MED12-Related Disorders)与CLS相似:X连锁遗传,智力残疾,前额宽阔,眼距宽且眼睑裂下斜,厚红下唇,脊柱侧后凸畸形,漏斗胸,和行为特征。疾病不同的是不成比例的大头畸形;肛门异常相关的便秘;宽拇指及拇趾;突出的指尖垫;小而圆的杯状耳,往往有螺旋状卷曲[Graham et al 1998 ]。肌张力低下往往发展成关节限制。
部分缺失的胼胝体和融合的乳头体较常见。
α地中海贫血的X-连锁智力障碍综合征(ATRX)的特点是独特的面部特征,生殖器异常和严重的发育迟缓,肌张力低下,智力残疾,和α地中海贫血继发发的轻中度贫血。生殖器异常表现为尿道下裂隐睾症,到重度尿道下裂和模糊的生殖器,再到核型为46,XY患者表现的正常的女性生殖器。ATRX综合征是由ATRX致病变异引起的。
McCandless et al[ 2000 ]报道一个del(10)(q25.1q25.3)家庭,受累的患者有CLS表现。因此,对不典型或可疑诊断CLS可以做染色体分析。
Differential Diagnosis
The diagnosis of Coffin-Lowry syndrome (CLS) in the older male child or adult usually does not present a problem. The findings in a young child or more mildly 受累的 female may overlap with other syndromes. Similarly, older female children and adults, even when they are the 先证者, can be diagnosed readily when they fully express the syndrome.
Borjeson-Forssman-Lehmann syndrome (BFLS) is an X-linked recessive disorder characterized by severe intellectual disability, hand findings similar to those of CLS, short nose with anteverted nares that may have a thick septum and small nares, and kyphoscoliosis. Additional findings are large, prominent ears and visual problems. Individuals with BFLS also have extreme hypogonadism and tend to have marked gynecomastia. Females may show partial expression of the syndrome. Absent findings are marked widely spaced eyes, wide mouth, and thick vermilion of the lips. Mutation of PHF6 is causative [Lower et al 2002].
While CLS shares some facial findings with Williams syndrome, the genetically heterogeneous FG syndrome, and X-linked alpha-thalassemia intellectual disability (ATRX) syndrome, none of these disorders shows the hand changes seen in CLS, and each has additional distinguishing features:
- Williams syndrome also includes cardiovascular disease (elastin arteriopathy, peripheral pulmonary stenosis, supravalvular aortic stenosis, hypertension), connective tissue abnormalities, intellectual disability (usually mild), a specific cognitive profile, unique personality characteristics, growth abnormalities, and endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism, and early puberty). Feeding difficulties often lead to failure to thrive in infancy. More than 99% of 受累的 individuals have a 邻近基因缺失 at 7q11.2, detectable by 荧光原位杂交 (FISH) or targeted analysis.
- FG syndrome type 1 (see MED12-Related Disorders) shares with CLS: X-linked inheritance, intellectual disability, a broad forehead, widely spaced eyes with downslanted palpebral fissures, a thick vermilion of the lower lip, kyphoscoliosis, pectus excavatum, and characteristic behaviors. It is distinguished by its disproportionate macrocephaly; constipation that may be associated with anal anomalies; broad thumbs and halluces; prominent fingertip pads; and small, rounded, cupped ears that often have an overfolded superior helix [Graham et al 1998]. Hypotonia often evolves into joint restriction.
Partial absence of the corpus callosum and fused mamillary bodies are relatively common. - Alpha-thalassemia X-linked intellectual disability (ATRX) syndrome is characterized by distinctive craniofacial features, genital anomalies and severe developmental delays with hypotonia, intellectual disability, and mild-to-moderate anemia secondary to alpha-thalassemia. Genital anomalies range from hypospadias and undescended testicles to severe hypospadias and ambiguous genitalia, to normal-appearing female genitalia in individuals with a 46,XY 核型. ATRX syndrome is caused by pathogenic variants in ATRX.
McCandless et al [2000] reported a family with del(10)(q25.1q25.3) in which 受累的 members had findings suggestive of CLS. Thus, it is reasonable to obtain 染色体 studies in individuals with an atypical or doubtful diagnosis of CLS.
管理
初步诊断后的评估
对CLS患者的诊断疾病和需求的程度,建议做以下的评估:
身高、体重和头围的测量
病史和神经病学检查以评估步态、胃肠功能、癫痫或运动障碍的变化。
发育评估及干预计划制定
完整的肌肉骨骼检查,特别注意胸部和脊柱;如果临床有表现,行放射学评估
发育性、适合年龄的听力评估
牙科评价
心脏和心电图的体格检查,每十年超声心动图检查
眼科评价,包括折射和眼底检查
评估合适家庭成员的病情体征
评估家庭照顾孩子的能力,特别是如果母亲是智力受累
医学遗传学咨询
对症治疗
应该尽量提供CLS患者发展沟通技巧,参与活动和自我护理,以发展独立性。
早期干预警示SIDAs以减少刺激引发跌倒的发生:
不同的药物和剂量优化可能改善预后[O'Riordan et al 2006 ]。
抗癫痫药物(AEDs)(如丙戊酸钠、氯硝西泮,或选择5-羟色胺摄取抑制剂)又被使用[Fryssira et al2002 ],但一般无效。
苯二氮卓类药物,证明在某些情况下增加剂量有效[Touraine et al 2002, Nakamura et al 2005]。
一患者在很多药物无效,但羟丁酸钠反应良好[ Havaligi et al 2007 ]。
如果跌倒发生频率高,可能需要使用轮椅防止摔倒和受伤。
破坏性或自我伤害行为的患者利培酮有效[Valdovinos et al 2002 ]。
如果存在喂养困难,生长速度异常,肥胖,应标准评估和治疗。
治疗行为问题是标准的,需要定期重新评估。
治疗脊柱后凸是标准的,但直到成年都需要仔细再评估。
并发症再发的预防
如脊柱侧后凸畸形和狭窄的脊柱的早期诊断可能避免进展和/或干预阻止长期心血管或神经系统并发症。干预应防止脊柱侧后凸畸形进展到心肺损害,而危及生命。
同样,及早诊断某些心脏异常可能有助于防止继发性并发症或延长功能。一些CLS患者可能需要SBE(亚急性细菌性心内膜炎)预防。
注意视觉和听觉可以防止一些次要的行为改变。睑缘炎的识别和治疗可以避免揉眼和潜在的视网膜损伤。
注意牙齿卫生和牙龈疾病可以减少过早牙齿脱落的风险。
监控
以下是适当的:
听觉和视觉的定期检测
每年心脏体检,十岁以下超声心动检查。即使是正常的,鉴于心肌病发病率和发病年龄不明,超声心动应每5-10年检查一次[Massin et al 1999, Facher et al 2004]。
进展性脊柱侧后凸畸形的脊柱监测。应高度注意椎管狭窄致步态和肠/膀胱习惯改变,痛苦的表情,和局灶性神经功能变化如阵挛或腱反射异常。
同常人的常规牙科评估,但要特别注意牙齿脱落的风险
注:Hunter[ Hunter2010 ]列出类包含CLS在内的患者随访指南。
避免的试剂/环境
患CLS并有SIDA的患者应尽可能地避免受惊吓和/或跌倒。
亲属的危险评价
就高危亲属的相关检测,见Genetic Counseling。
研究中的治疗方法
对于各种疾病的临床研究信息,见ClinicalTrials.gov。注意:此疾病可能尚无临床试验。
其他
可能需要大量的社会资源来帮助CLS发育迟缓的女性患者的家庭。
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Coffin-Lowry syndrome (CLS), the following evaluations are recommended:
- Measurement of height, weight, and head circumference
- History and neurologic examination to assess for changes in gait or in bowel or bladder function and for epilepsy or movement disorder
- Developmental assessment and formulation of an intervention plan
- Complete musculoskeletal examination with particular attention to the chest and spine; radiographic assessment if clinically indicated
- Developmental, age-appropriate hearing assessment
- Dental evaluation
- Physical examination of the heart and ECG, with baseline echocardiogram by age ten years
- Ophthalmologic evaluation, including refraction and fundoscopy
- Evaluation of appropriate family members for signs of the condition
- Assessment of the family’s capacity to care for the child, especially if mother is 受累的 intellectually
- Medical genetics consultation
Treatment of Manifestations
Individuals with CLS should be provided with every opportunity to develop communication skills and to participate in activities and self-care in order to develop a degree of independence.
Awareness of SIDAs should allow early intervention to minimize the occurrence of triggering stimuli and to provide protection from falls:
- Trials with different medications and efforts to optimize the dosage may improve outcome [O'Riordan et al 2006].
- A trial of antiepileptic drugs (AEDs) (e.g., valproate, clonazepam, or selective serotonin uptake inhibitors) may be indicated [Fryssira et al 2002], although generally they are not effective.
- Benzodiazapines, sometimes in increasing doses, have proved effective in some cases [Touraine et al 2002, Nakamura et al 2005].
- In an individual who was not helped by a variety of medications, Havaligi et al [2007] reported a good response with sodium oxybate.
- If attacks occur with great frequency, use of a wheelchair may be required to prevent falling and injury.
Risperidone may be of benefit to individuals who display destructive or self-injurious behavior [Valdovinos et al 2002].
Feeding difficulties, abnormal growth velocity, and obesity, if present, should be assessed and treated in a standard manner.
Treatment of behavioral problems is standard and requires periodic reassessment.
Treatment of kyphoscoliosis is standard but requires reassessment well into adulthood.
Prevention of Secondary Complications
Early recognition of spinal problems such as kyphoscoliosis and stenosis may allow prevention of progression and/or intervention to prevent long-term cardiovascular or neurologic complications. Intervention should be directed at preventing progression of kyphoscoliosis to the point of cardio-respiratory compromise, which may be life threatening.
Similarly, early recognition of some cardiac anomalies may allow prevention of secondary complications or prolongation of adequate function. Some individuals with CLS may require SBE (subacute bacterial endocarditis) prophylaxis.
Attention to vision and hearing may prevent some secondary behavioral changes. Identification and treatment of blepharitis may prevent eye rubbing and potential retinal damage.
Attention to dental hygiene and gum disease may reduce the risk of premature tooth loss.
Surveillance
The following are appropriate:
- Periodic tests of hearing and vision
- Annual physical cardiac examination, with echocardiogram by age ten years. Even if normal, the latter should be repeated every five to ten years in light of uncertainty as to the incidence and range in age of onset of cardiomyopathy [Massin et al 1999, Facher et al 2004].
- Monitoring of the spine for the development of progressive kyphoscoliosis. There should be a high index of suspicion for narrowing of the spinal canal with attention to change in gait and bowel/bladder habits, expression of pain, and focal neurologic changes such as clonus or abnormal tendon reflexes.
- Routine dental evaluation as in the general population but with particular attention to the risk of tooth loss
Note: A table containing suggested guidelines for follow-up of individuals with CLS is provided in Hunter [2010].
Agents/Circumstances to Avoid
Individuals with CLS who experience SIDAs should be protected as much as possible from being startled and/or from falls.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for 遗传咨询 purposes.
Therapies Under Investigation
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Significant social resources may be required to support families of women with CLS and developmental delay.
遗传咨询
遗传模式
CLS是X连锁遗传。
家庭成员的风险
先证者父母
约70% - 80%的先证者没有CLS的家族史,20%—30%有一个以上的受累的家庭成员[Delaunoy2001]。散发病例的发病率高(即,家庭中有一个CLS患者)是因于遗传选择,智力障碍的女性杂合携带者[Jacquot et al1998a]。
一个受累的男性患者的父亲不会患病,或是携带致病性变异。
一个家庭有一个以上的受累的患者,男性患者的母亲是一个肯定携带者。
先证者母亲应检查CLS症状,如粗糙的五官,丰满的嘴唇,和/或尖细的手指。
如果先证者确定致病性变异,有必要对其母亲行分子遗传学检测。
先证者的同胞
先证者同胞的风险取决于母亲的携带状态。
如果该先证者的母亲有致病性变异,次怀孕50%的几率传递给子代;
遗传了致病性变异的男性同胞是受累者;女性同胞是携带者,将至少有一些发育迟缓和CLS轻度体征的高风险。
因X染色体随机失活,轻度受累的妇女可能女儿症状严重。
女性携带者表现出轻度至中度偏倚的X染色体失活,不与智商相关联[Simensen et al2002]。
在没有任何体征或智力障碍的情况下,一个没有家族史的CLS先证者的母亲可能是携带者低风险。
已经证明存在生殖嵌合。因此,即使先证者携带的致病性变异没有在母亲的DNA中发现,先证者的同胞仍有遗传该致病性变异的高风险[Jacquot et al 1998b, Horn et al 2001]。
先证者的后代
男性和严重受累的女性通常不会生育。
CLS女性有50%的机会将致病性变异遗传给孩子;遗传了致病变异的男孩将受累;女儿是携带者,一定程度表现出发育迟缓和CLS轻度体征的高风险。
先证者的其他家庭成员。如果先证者的母亲发现有一个致病性变异,她的女性家庭成员可能有携带者风险(无症状或有症状);她的男性家庭成员可能有患病风险,取决于对先证者的遗传关系。
携带者检测
高风险的女性亲属的携带者检测,需要在家庭中行致病性变异的诊断。
相关遗传咨询问题
具体咨询的问题
可能需要大量的社会资源,就生育选择和子女照顾,来帮助发育迟缓的CLS妇女及其家庭。
男性无CLS家族史携带致病性变异的患者(即,散发病例),其母亲的分子遗传学检测应慎重解释。已发现有生殖嵌合;因此,受累的子代的DNA中未检测到致病性变异,也应对该女性采取适当的提供产前检查。
计划生育
在孕前,对遗传风险的最佳测定时机,携带者检测,和是否行产前检查。
对年轻女性受累者,或携带者,或险携带者风险,提供适当的遗传咨询(包括后代和生殖选择的潜在风险)。
DNA储备是DNA的存储(通常从白细胞中提取)以备将来使用。因为检测方法和我们对基因,基因变异,疾病的理解都将逐步完善,应考虑收集受累者的DNA样本。
产前检查与胚胎植入前遗传学诊断
一旦RPS6KA3致病性变异在受累的家庭成员中鉴定,夫妻可以考虑针对CLS风险的产前检查或植入前遗传诊断为。
Genetic Counseling
Genetic counseling is the process ofproviding individuals and families with information on the nature, inheritance,and implications of genetic disorders to help them make informed medical andpersonal decisions. The following section deals with genetic risk assessment andthe use of family history and genetic testing to clarify genetic status forfamily members. This section is not meant to address all personal, cultural, orethical issues that individuals may face or to substitute for consultation witha genetics professional. —ED.
Mode of Inheritance
Coffin-Lowry syndrome (CLS) is inherited in an X-linked manner.
Risk to Family Members
Parents of a 先证者
- Approximately 70%-80% of probands have no family history of CLS, and 20%-30% have more than one 受累的 family member [Delaunoy et al 2001]. The high incidence of 单发的 cases (i.e., CLS in a single individual in a family) can be attributed to genetic selection that occurs against 杂合的 females who are intellectually disabled [Jacquot et al 1998a].
- Mothers of a 先证者 should be examined for signs of CLS such as coarse facial features, full lips, and/or tapering fingers.
Sibs of a 先证者
- As expected with random X-chromosome inactivation, a mildly 受累的 woman may have a severely affected daughter.
- Female carriers show mild-to-moderate skewing of X-chromosome inactivation that does not correlate with IQ [Simensen et al 2002].
- Germline 嵌合 has been demonstrated in this condition. Thus, even if the 致病性变异 found in the 先证者 has not been identified in the mother's DNA, sibs of the proband are still at increased risk of inheriting the pathogenic variant [Jacquot et al 1998b, Horn et al 2001].
Offspring of a 先证者
- Males and severely 受累的 females with CLS typically do not reproduce.
Other family members of a 先证者. If the mother of the proband is found to have a 致病性变异, her female family members may be at risk of being carriers (asymptomatic or symptomatic); and her male family members may be at risk of being 受累的 depending on their genetic relationship to the proband.
Carrier Detection
Carrier testing of at-risk female relatives requires prior identification of the 致病性变异 in the family.
Related Genetic Counseling Issues
Specific counseling issues
- Significant social resources may be required to support developmentally delayed women with CLS and their families with respect to reproductive choices and child care.
- Caution should be used in interpreting the results of 分子遗传学检测 of a mother of a male with no known family history of CLS (i.e., a 单发的 case) in whom a 致病性变异 has been identified. Germline 嵌合 has been observed; thus, it is appropriate to offer prenatal testing to such women even when the pathogenic variant identified in an 受累的 offspring is not detected in their DNA.
Family planning
- The optimal time for determination of genetic risk, clarification of 携带者 status, and discussion of the availability of prenatal testing is before pregnancy.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of 受累的 individuals.
Prenatal Testing and Preimplantation Genetic Diagnosis
Once the RPS6KA3致病性变异 has been identified in an 受累的 family member, prenatal testing or 植入前遗传诊断 for a pregnancy at increased risk for CLS may be an option that a couple may wish to consider.
Resources
GeneReviews staff has selected the following disease-specific and/orumbrella support organizations and/or registries for the benefit of individualswith this disorder and their families. GeneReviews is not responsible for theinformation provided by other organizations. For information on selectioncriteria, click here.
- Coffin-Lowry Syndrome FoundationPhone: 425-427-0939Email: CoffinLowry@gmail.com
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
分子遗传机制
与CLS相关的RPS6KA3(RSK2),编码生长因子调节丝氨酸/苏氨酸激酶。人类有四个密切相关的RPS6KA3(RSK)基因;每个基因有两个不相同的激酶催化结构域,两者均需要最大活性 [Yntema et al 1999, Yang et al 2004]。
RPS6KA3表达在人类胚胎脑组织中有时间和空间的限制,在妊娠九周从端脑到后脑有均匀的脑组织表达,在心室区比在皮质板有更高的水平[Guimiot et al 2004 ]。
核糖体蛋白S6激酶α-3(RPS6KA3),编码RPS6KA3蛋白,参与了许多酶活途径,包括Ras-MAPK激酶活化,蛋白激酶C和腺苷酸环化酶Harum et al 2001Pereira et al 2010]。通过MAPK /激酶通路和组蛋白H3表皮生长因子(EGF)刺激的磷酸化,它在激活G0和G1细胞周期起作用。RPS6KA3也被证明激活CREB(cAMP反应元件结合蛋白),这是参与从短期记忆到长期记忆的神经元的存活和转换[ Harum et al2001 ]。CLS患者的细胞表皮生长因子刺激的磷酸化S6,H3 [ Sassone Corsi et al 1999 ],和CREB[Harum et al2001]有缺陷的;一个或多个这些途径可能会引起一些CLS表现。
基因结构。在RPS6KA3转录本NM_004586.2由22个外显子组成;它被命名为核糖体S6激酶(别名:RSK2)。基因和蛋白质信息详细,见Table A,Gene。
良性等位基因变异。一些RPS6KA3的变异不与疾病相关[ Delaunoy et al 2001;Abidi & Schwartz, unpublished]。
致病等位基因变异。在RPS6KA3致病变异分布在整个基因,没有与特点表型聚类的证据。
在迄今为止最大的研究(250人)中,在86个不相关的家庭中发现了71个致病变异。约60%引起或预测为截短蛋白质;38%是错义突变,20%无义突变,18%剪接异常,21%片段的缺失或插入[Delaunoy et al2001 ]。
对106名疑似CLS的无关个体进行了较小的研究,发现了28种致病性变异(26%)。这28个致病变种,60%引起或预测的蛋白质截断;36%是错义,21%废话,11%错误的剪接,32%片段的缺失或插入[Abidi & Schwartz, unpublished]。
————-
对内含子变异导致的的异常剪接和LINE-1元件的内含子插入,破坏蛋白质正常功能[Zeniou et al 2002a, Martinez-Garay et al 2003, Zeniou et al 2004]。(更多信息,请参见Table A)
最近,Schneider等人[ 2013 ]已经确定了一个内含子深部的致病性变异导致蛋白异常。这一发现,证明在临床高度怀疑CLS的患者且外显子筛查阴性的病例,行RNA致病性分析。
存在的全部和部分基因重复的报道。Matsumoto等[ 2013 ]报告一家庭有轻度ID,ADHD、有关定位的癫痫,和广泛性发育障碍(PDD),检测出包括整个RPS6KA3的微重复。Marques Pereira等[ 2007 ]报告CLS患者RPS6KA3非移码串联多个外显子重复,并注意到基因中高频率Alu序列,他们认为,这可能是比较常见的情况。然而,研究有待进行。
正常基因产物。核糖体蛋白S6激酶α-3(RPS6KA3)是一种丝氨酸/苏氨酸激酶和Ras信号转导级联的一员。该蛋白被MAPK激酶磷酸化,受生长因子、胰岛素和致癌转化调节。RSK家族的成员参与细胞活动,如细胞增殖和分化。事实上,RPS6KA3的致病性变异可以导致非综合征性疾病(MRX9;看Genetically Related Disorders)以及CLS表明该基因对大脑认知功能很关键。RSK2通过PLD1调节产生胞吐所需的脂质[Zeniou-Meyer et al 2008, Zeniou-Meyer et al 2009]和调节神经递质[ Zeniou Meyeret al 2010]。
RSK2也通过介导细胞周期和DNA修复保持基因组的稳定[ Lim et al2013]。
异常基因产物。RPS6KA3致病变异导致的CLS和非综合征型XLMR。CLS患者的致病变异导致了基因产物激酶活性的丧失。然而,与MRX19相关的致病性变异,发生在基因两个激酶区之外,导致RPS6KA3活性减少到80%,表明大脑比在其他受累的器官系统对RPS6KA3活性更敏感。
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. -ED.
Table A.
Coffin-Lowry Syndrome: Genes and Databases
| Gene | Chromosome Locus | Protein | Locus-Specific Databases | HGMD | ClinVar |
|---|---|---|---|---|---|
| RPS6KA3 | Xp22-.12 | Ribosomal protein S6 kinase alpha-3 | RPS6KA3 @ LOVD | RPS6KA3 | RPS6KA3 |
Table B.
OMIM Entries for Coffin-Lowry Syndrome (View All in OMIM)
Molecular Genetic Pathogenesis
RPS6KA3(RSK2), the 基因 associated with CLS, encodes a growth factor-regulated serine/threonine kinase. Humans have four closely related RPS6KA (RSK) genes; each gene has two non-identical kinase catalytic domains, both of which are required for maximal activity [Yntema et al 1999, Yang et al 2004].
RPS6KA3 expression shows both temporal and spatial restriction in human embryogenesis, with homogeneous brain expression from the telencephalon to the rhombencephalon at nine weeks' gestation, with higher levels in the ventricular zone than in the cortical plate [Guimiot et al 2004].
Ribosomal protein S6 kinase alpha-3 (RPS6KA3), the protein encoded by RPS6KA3, is involved in kinase activation in a number of pathways including ras-MAPK, protein kinase C, and adenyl cyclase [Harum et al 2001Pereira et al 2010]. Through the MAPK/RSK pathway and the epidermal growth factor (EGF)-stimulated phosphorylation of histone H3, it appears to play a role in stimulation of the cell cycle between G0 and G1. RPS6KA3 has also been shown to activate CREB (cAMP response element binding protein), which is involved in neuronal survival and conversion from short- to long-term memory [Harum et al 2001]. Cells from individuals with CLS have shown defective EGF-stimulated phosphorylation of S6, H3 [Sassone-Corsi et al 1999], and CREB [Harum et al 2001]; and one or more of these pathways may play a role in causing some of the manifestations of CLS.
Gene structure. The RPS6KA3 transcript NM_004586.2 comprises 22 exons; it is named for ribosomal S6 kinase (alternate name: RSK2). For a detailed summary of 基因 and protein information, see Table A, Gene.
Benign allelic variants. Some variants in RPS6KA3 that are not associated with a disease 表型 have been described [Delaunoy et al 2001; Abidi & Schwartz, unpublished].
Pathogenic allelic variants. Pathogenic variants in RPS6KA3 are distributed throughout the 基因 with no evidence of clustering associated with a specific 表型.
In the largest study to date (250 individuals), 71 pathogenic variants were found in 86 unrelated families. Almost 60% caused or predicted protein truncation; 38% were 错义, 20% nonsense, 18% errors of 剪接, and 21% intragenic deletions or insertions [Delaunoy et al 2001].
A smaller study of 106 unrelated individuals with suspected CLS found 28 pathogenic variants (26%). Of the 28 pathogenic variants, 60% caused or predicted protein truncation; 36% were 错义, 21% nonsense, 11% errors of 剪接, and 32% intragenic deletions or insertions [Abidi & Schwartz, unpublished].
Pathogenic variants of 内含子的 alterations resulting in aberrant 剪接 and an intronic 插入 of a LINE-1 element that disrupts the normal function of the protein have been reported [Zeniou et al 2002a, Martinez-Garay et al 2003, Zeniou et al 2004]. (For more information, see Table A.)
Recently, Schneider et al [2013] have identified a deep 内含子的致病性变异 which results in an aberrant protein. This finding warrants analysis for pathogenic variants at the RNA level in all patients with a highly suggestive clinical diagnosis of CLS and in whom 外显子 screening has failed to detect a pathogenic variant.
Presence of full- and partial-基因 duplications has been reported. Matsumoto et al [2013] report a microduplication including the entire RPS6KA3 in a family with mild ID, ADHD, localization-related epilepsy, and pervasive developmental disorder (PDD). Marques Pereira et al [2007] report 非移码 tandem multiexon 重复 within RPS6KA3 in an individual with Coffin-Lowry syndrome, and noting the high frequency of Alu sequences within the gene, they suggest that these may be relatively common events. However, such studies have yet to be performed.
Normal 基因产物. Ribosomal protein S6 kinase alpha-3 (RPS6KA3) is a serine/threonine kinase and a member of the Ras signaling cascade. The protein is phosphorylated by MAPK kinases in response to growth factors, insulin, and oncogenic transformations. Members of the RSK family participate in cellular events such as proliferation and differentiation. The fact that a 致病性变异 in RPS6KA3 can result in nonsyndromic XLMR (MRX19; see Genetically Related Disorders) as well as CLS indicates that the gene is critical for some cognitive functions of the brain. RSK2 regulates neurite formation by phosphorylating phospholipase D1 (PLD1) [Ammar et al 2013]. Also, RSK2 mediated activation of PLD1 produces the lipids required for exocytosis [Zeniou-Meyer et al 2008, Zeniou-Meyer et al 2009] and regulates the release of neurotransmitters [Zeniou-Meyer et al 2010].
RSK2 also plays an important role in maintaining 基因组的 stability by mediating cell cycle progression and DNA repair [Lim et al 2013].
Abnormal 基因产物. Pathogenic variants in RPS6KA3 give rise to both CLS and nonsyndromic XLMR. The pathogenic variants in individuals with CLS result in the loss of kinase activity of the gene product. However, the 致病性变异 associated with MRX19 occurs outside the two kinase domains of the gene and results in a reduction to 80% RPS6KA3 activity, suggesting that the brain is more sensitive to levels of RPS6KA3 activity than are the other organ systems 受累的 in CLS.
References
Literature Cited
- Ammar MR, Humeau Y, Hanauer A, Nieswandt B, Bader MF, Vitale N. The Coffin-Lowry syndrome-associated protein RSK2 regulates neurite outgrowth through phosphorylation of phospholipase D1 (PLD1) and synthesis of phosphatidic acid. J Neurosci. 2013;33:19470鈥�9. [PubMed: 24336713]
- Coffin GS. Postmortem findings in the Coffin-Lowry Syndrome. Genet Med. 2003;5:187鈥�93. [PubMed: 12792428]
- Crow YJ, Zuberi SM, McWilliam R, Tolmie JL, Hollman A, Pohl K, Stephenson JB. "Cataplexy" and muscle ultrasound abnormalities in Coffin-Lowry syndrome. J Med Genet. 1998;35:94鈥�8. [PMC free article: PMC1051210] [PubMed: 9507386]
- Delaunoy J, Abidi F, Zeniou M, Jacquot S, Merienne K, Pannetier S, Schmitt M, Schwartz C, Hanauer A. Mutations in the X-linked RSK2 gene (RPS6KA3) in patients with Coffin- Lowry syndrome. Hum Mutat. 2001;17:103鈥�16. [PubMed: 11180593]
- Facher JJ, Regier EJ, Jacobs GH, Siwik E, Delaunoy JP, Robin NH. Cardiomyopathy in Coffin-Lowry syndrome. Am J Med Genet A. 2004;128A:176鈥�8. [PubMed: 15214012]
- Field M, Tarpey P, Boyle J, Edkins S, Goodship J, Luo Y, Moon J, Teague J, Stratton MR, Futreal PA, Wooster R, Raymond FL, Turner G. Mutations in the RSK2(RPS6KA3) gene cause Coffin-Lowry syndrome and nonsyndromic X-linked mental retardation. Clin Genet. 2006;70:509鈥�15. [PMC free article: PMC2714973] [PubMed: 17100996]
- Fryssira H, Kountoupi S, Delaunoy JP, Thomaidis L. A female with Coffin-Lowry syndrome and "cataplexy". Genet Couns. 2002;13:405鈥�9. [PubMed: 12558110]
- Graham JM Jr, Tackels D, Dibbern K, Superneau D, Rogers C, Corning K, Schwartz CE. FG syndrome: report of three new families with linkage to Xq12-q22.1. Am J Med Genet. 1998;80:145鈥�56. [PubMed: 9805132]
- Guimiot F, Delezoide AL, Hanauer A, Simonneau M. Expression of the RSK2 gene during early human development. Gene Expr Patterns. 2004;4:111鈥�4. [PubMed: 14678837]
- Hanauer A, Young ID. Coffin-Lowry syndrome: clinical and molecular features. J Med Genet. 2002;39:705鈥�13. [PMC free article: PMC1734994] [PubMed: 12362025]
- Hahn JS, Hanauer A. Stimulus-induced drop episodes in Coffin-Lowry syndrome. Eur J Med Genet. 2012;55:335鈥�7. [PubMed: 22490425]
- Harum KH, Alemi L, Johnston MV. Cognitive impairment in Coffin-Lowry syndrome correlates with reduced RSK2 activation. Neurology. 2001;56:207鈥�14. [PubMed: 11160957]
- Havaligi N, Matadeen-Ali C, Khurana DS, Marks H, Kothare SV. Treatment of drop attacks in Coffin-Lowry syndrome with the use of sodium oxybate. Pediatr Neurol. 2007;37:373鈥�4. [PubMed: 17950427]
- Herrera-Soto JA, Santiago-Cornier A, Segal LS, Ramirez N, Tamai J. The musculoskeletal manifestations of the Coffin-Lowry syndrome. J Pediatr Orthop. 2007;27:85鈥�9. [PubMed: 17195803]
- Horn D, Delaunoy JP, Kunze J. Prenatal diagnosis in Coffin-Lowry syndrome demonstrates germinal mosaicism confirmed by mutation analysis. Prenat Diagn. 2001;21:881鈥�4. [PubMed: 11746134]
- Hunter AG. Coffin-Lowry syndrome. In: Cassidy S, Allanson J, eds. Management of Genetic Syndromes. 3 ed. Hoboken, NJ: Wiley-Liss; 2010:127-38.
- Hunter AG. Coffin-Lowry syndrome: a 20-year follow-up and review of long-term outcomes. Am J Med Genet. 2002;111:345鈥�55. [PubMed: 12210291]
- Jacquot S, Merienne K, De Cesare D, Pannetier S, Mandel JL, Sassone-Corsi P, Hanauer A. Mutation analysis of the RSK2 gene in Coffin-Lowry patients: extensive allelic heterogeneity and a high rate of de novo mutations. Am J Hum Genet. 1998a;63:1631鈥�40. [PMC free article: PMC1377634] [PubMed: 9837815]
- Jacquot S, Merienne K, Pannetier S, Blumenfeld S, Schinzel A, Hanauer A. Germline mosaicism in Coffin-Lowry syndrome. Eur J Hum Genet. 1998b;6:578鈥�82. [PubMed: 9887375]
- Kesler SR, Simensen RJ, Voeller K, Abidi F, Stevenson RE, Schwartz CE, Reiss AL. Altered neurodevelopment associated with mutations of RSK2: a morphometric MRI study of Coffin-Lowry syndrome. Neurogenetics. 2007;8:143鈥�7. [PMC free article: PMC3055244] [PubMed: 17318637]
- Kondoh T, Matsumoto T, Ochi M, Sukegawa K, Tsuji Y. New radiological finding by magnetic resonance imaging examination of the brain in Coffin-Lowry syndrome. J Hum Genet. 1998;43:59鈥�61. [PubMed: 9610000]
- Lim HC, Xie L, Zhang W, Li R, Chen ZC, Wu GZ, Cui SS, Tan EK, Zeng L. Ribosomal S6 Kinase 2 (RSK2) maintains genomic stability by activating the Atm/p53-dependent DNA damage pathway. PLoS One. 2013;8:e74334. [PMC free article: PMC3781089] [PubMed: 24086335]
- Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, Schelley S, Hoyme HE, White SM, Delatycki MB, Lampe AK, Clayton-Smith J, Stewart H, van Ravenswaay CM, de Vries BB, Cox B, Grompe M, Ross S, Thomas P, Mulley JC, Gecz J. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet. 2002;32:661鈥�5. [PubMed: 12415272]
- Lowry B, Miller JR, Fraser FC. A new dominant gene mental retardation syndrome. Association with small stature, tapering fingers, characteristic facies, and possible hydrocephalus. Am J Dis Child. 1971;121:496鈥�500. [PubMed: 5581017]
- Manouvrier-Hanu S, Amiel J, Jacquot S, Merienne K, Moerman A, Coeslier A, Labarriere F, Vallee L, Croquette MF, Hanauer A. Unreported RSK2 missense mutation in two male sibs with an unusually mild form of Coffin-Lowry syndrome. J Med Genet. 1999;36:775鈥�8. [PMC free article: PMC1734232] [PubMed: 10528858]
- Marques Pereira P, Heron D, Hanauer A. The first large duplication of the RSK2 gene identified in a Coffin-Lowry syndrome patient. Hum Genet. 2007;122:541鈥�3. [PubMed: 17717706]
- Martinez-Garay I, Ballesta MJ, Oltra S, Orellana C, Palomeque A, Molto MD, Prieto F, Martinez F. Intronic L1 insertion and F268S, novel mutations in RPS6KA3 (RSK2) causing Coffin-Lowry syndrome. Clin Genet. 2003;64:491鈥�6. [PubMed: 14986828]
- Martinez HR, Niu MC, Sutton VR, Pignatelli R, Vatta M, Jefferies JL. Coffin-Lowry syndrome and left ventricular noncompaction cardiomyopathy with a restrictive pattern. Am J Med Genet A. 2011;155A:3030鈥�4. [PubMed: 22009732]
- Massin MM, Radermecker MA, Verloes A, Jacquot S, Grenade T. Cardiac involvement in Coffin-Lowry syndrome. Acta Paediatr. 1999;88:468鈥�70. [PubMed: 10342551]
- Matsumoto A, Kuwajima M, Miyake K, Kojima K, Nakashima N, Jimbo EF, Kubota T, Momoi MY, Yamagata T. An Xp22.12 microduplication including RPS6KA3 identified in a family with variably affected intellectual and behavioral disabilities. J Hum Genet. 2013;58:755鈥�7. [PubMed: 23985797]
- McCandless SE, Schwartz S, Morrison S, Garlapati K, Robin NH. Adult with an interstitial deletion of chromosome 10 [del(10)(q25. 1q25.3)]: overlap with Coffin-Lowry syndrome. Am J Med Genet. 2000;95:93鈥�8. [PubMed: 11078556]
- Merienne K, Jacquot S, Pannetier S, Zeniou M, Bankier A, Gecz J, Mandel JL, Mulley J, Sassone-Corsi P, Hanauer A. A missense mutation in RPS6KA3 (RSK2) responsible for non-specific mental retardation. Nat Genet. 1999;22:13鈥�4. [PubMed: 10319851]
- Merienne K, Jacquot S, Trivier E, Pannetier S, Rossi A, Scott C, Schinzel A, Castellan C, Kress W, Hanauer A. Rapid immunoblot and kinase assay tests for a syndromal form of X-linked mental retardation: Coffin-Lowry syndrome. J Med Genet. 1998;35:890鈥�4. [PMC free article: PMC1051479] [PubMed: 9832033]
- Micheli V, Sestini S, Parri V, Fichera M, Romano C, Ariani F, Longo I, Mari F, Bruttini M, Renieri A, Meloni I. RSK2 enzymatic assay as a second level diagnostic tool in Coffin-Lowry syndrome. Clin Chim Acta. 2007;384:35鈥�40. [PubMed: 17586481]
- Nakamura M, Yamagata T, Momoi MY, Yamazaki T. Drop episodes in Coffin-Lowry syndrome: exaggerated startle responses treated with clonazepam. Pediatr Neurol. 1998;19:148鈥�50. [PubMed: 9744638]
- Nakamura M, Yamagata T, Mori M, Momoi MY. RSK2 gene mutations in Coffin-Lowry syndrome with drop episodes. Brain Dev. 2005;27:114鈥�7. [PubMed: 15668050]
- Nelson GB, Hahn JS. Stimulus-induced drop episodes in Coffin-Lowry syndrome. Pediatrics. 2003;111:e197鈥�202. [PubMed: 12612271]
- O'Riordan S, Patton M, Schon F. Treatment of drop episodes in Coffin-Lowry syndrome. J Neurol. 2006;253:109鈥�10. [PubMed: 16021355]
- Patlas M, Joseph A, Cohen JE, Gomori JM. MRI and MRS of Coffin-Lowry syndrome: a case report. Neurol Res. 2003;25:285鈥�6. [PubMed: 12739239]
- Pereira PM, Schneider A, Pannetier S, Heron D, Hanauer A. Coffin-Lowry syndrome. Eur J Hum Genet. 2010;18:627鈥�33. [PMC free article: PMC2987346] [PubMed: 19888300]
- Rojnueangnit K, Jones JR, Basehore MJ, Robin NH. Classic phenotype of Coffin-Lowry syndrome in a female with stimulus-induced drop episodes and a genotype with preserved N-terminal kinase domain. Am J Med Genet A. 2014;164A:516鈥�21. [PubMed: 24311527]
- Rosanowski F, Hoppe U, Proschel U, Eysholdt U. Late-onset sensorineural hearing loss in Coffin-Lowry syndrome. ORL J Otorhinolaryngol Relat Spec. 1998;60:224鈥�6. [PubMed: 9646311]
- Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886鈥�91. [PubMed: 10436156]
- Schneider A, Maas SM, Hennekam RC, Hanauer A. Identification of the first deep intronic mutation in the RPS6KA3 gene in a patient with a severe form of Coffin-Lowry syndrome. Eur J Med Genet. 2013;56:150鈥�2. [PubMed: 23261961]
- Simensen RJ, Abidi F, Collins JS, Schwartz CE, Stevenson RE. Cognitive function in Coffin-Lowry syndrome. Clin Genet. 2002;61:299鈥�304. [PubMed: 12030896]
- Stephenson JB, Hoffman MC, Russell AJ, Falconer J, Beach RC, Tolmie JL, McWilliam RC, Zuberi SM. The movement disorders of Coffin-Lowry syndrome. Brain Dev. 2005;27:108鈥�13. [PubMed: 15668049]
- Touraine R-L, Zeniou M, Hanauer A. A syndromic form of X-linked mental retardation: the Coffin-Lowry syndrome. Eur J Pediatr. 2002;161:179鈥�87. [PubMed: 12014383]
- Valdovinos MG, Napolitano DA, Zarcone JR, Hellings JA, Williams DC, Schroeder SR. Multimodal evaluation of risperidone for destructive behavior: functional analysis, direct observations, rating scales, and psychiatric impressions. Exp Clin Psychopharmacol. 2002;10:268鈥�75. [PubMed: 12233987]
- Wang Y, Martinez JE, Wilson GL, He XY, Tuck-Muller CM, Maertens P, Wertelecki W, Chen TJ. A novel RSK2 (RPS6KA3) gene mutation associated with abnormal brain MRI findings in a family with Coffin-Lowry syndrome. Am J Med Genet A. 2006;140:1274鈥�9. [PubMed: 16691578]
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387鈥�98. [PubMed: 15109498]
- Yntema HG, van den Helm B, Kissing J, van Duijnhoven G, Poppelaars F, Chelly J, Moraine C, Fryns JP, Hamel BC, Heilbronner H, Pander HJ, Brunner HG, Ropers HH, Cremers FP, van Bokhoven H. A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics. 1999;62:332鈥�43. [PubMed: 10644430]
- Zeniou M, Ding T, Trivier E, Hanauer A. Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin-Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum Mol Genet. 2002a;11:2929鈥�40. [PubMed: 12393804]
- Zeniou M, Gattoni R, Hanauer A, Stevenin J. Delineation of the mechanisms of aberrant splicing caused by two unusual intronic mutations in the RSK2 gene involved in Coffin-Lowry syndrome. Nucleic Acids Res. 2004;32:1214鈥�23. [PMC free article: PMC373406] [PubMed: 14973203]
- Zeniou M, Pannetier S, Fryns JP, Hanauer A. Unusual splice-site mutations in the RSK2 gene and suggestion of genetic heterogeneity in Coffin-Lowry syndrome. Am J Hum Genet. 2002b;70:1421鈥�33. [PMC free article: PMC379129] [PubMed: 11992250]
- Zeniou-Meyer M, Liu Y, Béglé A, Olanich ME, Hanauer A, Becherer U, Rettig J, Bader MF, Vitale N. The Coffin-Lowry syndrome-associated protein RSK2 is implicated in calcium-regulated exocytosis through the regulation of PLD1. Proc Natl Acad Sci U S A. 2008;105:8434鈥�9. [PMC free article: PMC2448854] [PubMed: 18550821]
- Zeniou-Meyer M, Béglé A, Bader MF, Vitale N. The Coffin-Lowry syndrome-associated protein RSK2 controls neuroendocrine secretion through the regulation of phospholipase D1 at the exocytotic sites. Ann N Y Acad Sci. 2009;1152:201鈥�8. [PubMed: 19161391]
- Zeniou-Meyer M, Gambino F, Ammar MR, Humeau Y, Vitale N. The Coffin-Lowry syndrome-associated protein RSK2 and neurosecretion. Cell Mol Neurobiol. 2010;30:1401鈥�6. [PubMed: 21061166]
Chapter Notes
Author History
Fatima E Abidi, PhD (2002-present)
R Curtis Rogers, MD (2014-present)
Alisdair GW Hunter, MD; University of Ottawa (2002-2014)
Charles E Schwartz, PhD; Greenwood Genetics Center (2002-2009)
Revision History
- 27 March 2014 (me) Comprehensive update posted live
- 15 January 2009 (me) Comprehensive update posted live
- 6 August 2007 (cd) Revision: deletion/duplication analysis available clinically
- 31 August 2006 (me) Comprehensive update posted to live Web site
- 27 December 2004 (cd) Revision: change in 分子遗传学检测 availability
- 28 June 2004 (me) Comprehensive update posted to live Web site
- 16 July 2002 (me) Review posted to live Web site
- 24 January 2002 (ah) Original submission