
概述
临床特征。
Ehlers -Danlos综合征(EDS),经典型是以皮肤过度伸张,伤口愈合异常和关节超过度活动为特征的结缔组织病变。它包括两个亚型(EDS I型和EDS II型),现在被认为是形成一个连续的临床表现。皮肤光滑,触感柔软,弹性大,即,它在释放后容易延伸并且回弹(不像松弛的,冗余的皮肤)。 皮肤脆弱,如同晓得创伤之后,特别是在压力点(膝盖,肘部)和易于创伤的区域(胫骨,前额,下巴)上真皮分裂所表现出来的一样。 伤口愈合慢,创伤愈合后具有明显的瘢痕伸展。 关节活动过度并发症,如肩胛骨,髌骨,髋关节,半径和锁骨脱位,通常自发恢复,或容易缓解。 其他特征包括运动发育延迟,疲劳和肌肉痉挛以及容易瘀伤,低血压。 较不常见的发现包括二尖瓣和三尖瓣脱垂,主动脉根部扩张和大动脉自发性破裂。
诊断/检测。
经典型EDS诊断由家族史和临床表现确定。V型胶原链的定量和定性研究通常在确诊中没有作用。 至少有50%的具有典型EDS的个体在COL5A1或COL5A2(编码V型胶原的基因)中具有可鉴定的致病变异; 然而,因为在临床上明确定义的组中没有进行COL5A1和COL5A2的前瞻性分子研究,这个数字可能是低估的。
管理。
治疗表现:患有低血压和运动发育延迟的 儿童受益于物理治疗。 非负重运动促进肌肉力量和协调性。 抗炎药可缓解关节疼痛。 伴有低血压,关节不稳定性和慢性疼痛的患者可能需要相应地调整生活方式。 皮肤伤口需在没有张力的情况下封闭,最好是两层。 对于其他伤口,可以大量使用深针迹; 皮肤针迹保留的时间需延长, 并且仔细地粘贴相邻皮肤的边界以防止疤痕的拉伸。 心血管问题以标准方式进行治疗。
预防原发性表现:皮肤脆弱的幼儿可以在前额,膝盖和胫骨上佩戴保护垫或绷带,以避免皮肤撕裂。 年龄较大的孩子可以在活动期间穿足球垫或滑雪袜。 抗坏血酸(维生素C)可减少瘀伤。
监控:当主动脉扩张和/或二尖瓣脱垂存在时,每年超声心动图监测。
禁忌事项:乙酰水杨酸,关节的负重运动。
遗传咨询。
EDS,经典类型以常染色体显性方式遗传。 据估计,约50%的受影响致病性变变异遗传自父母,约50%的受影响个体携带新发突变。 受影响个体的每个孩子有50%的可能性遗传致病性变异。 在含有携带致病性变异患者的家庭中,对于高风险的孕妇进行产前检测具有中的意义。
诊断
临床诊断
典型Ehlers-Danlos综合征(EDS)的诊断是通过家族史和临床检查建立的。诊断标准在1997年Villefranche会议(由Ehlers-Danlos基金会[美国]和Ehlers-Danlos支持组织[英国]发起)的由医疗咨询小组制定 [Beighton et al 1998] (全文;pdf)。
前三主要诊断标准相结合,诊断经典型EDS具有较高的特异性。 存在一个或多个次要标准有助于诊断经典型EDS,但不足以建立诊断。
经典类型EDS的主要诊断标准
-
皮肤过度伸展性。 皮肤过度伸展(见 图 1)应在中性部位进行测试(一个不受机械力或无瘢痕形成部位),如前臂的手腕表面。 通过拉起皮肤来测量,直到感觉到阻力。 在幼儿中,由于皮下脂肪丰富,皮肤过度伸张性难以评估。
-
扩大萎缩性瘢痕。(见图 2)(组织脆弱的表现)
-
阳性家族史

Figure 1.
Skin hyperextensibility
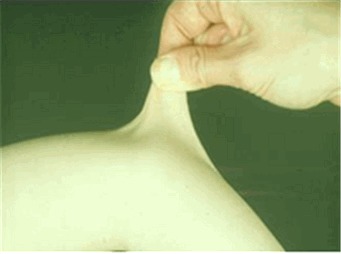
Figure 2.
Widened atrophic scars
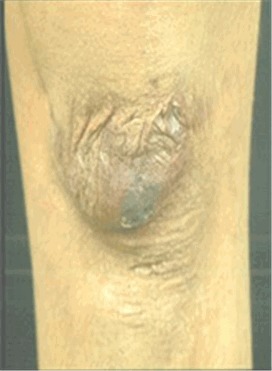
Figure 3.
Passive flexion of thumbs to the forearm: manifestation of joint hypermobility
表 1。
关节过度活动的Beighton's 标准
| 关节/发现 | 阴性 | 单侧 | 双侧 |
|---|---|---|---|
| 五指被动背屈> 90° | 0 | 1 | 2 |
| 拇指被动屈曲到前臂 | 0 | 1 | 2 |
| 肘屈>10° | 0 | 1 | 2 |
| 膝盖过度伸展>10° | 0 | 1 | 2 |
| 膝盖向前弯曲完全伸展,手掌可平放在在地板上 | 0 | 1 | |
-
总分≥5定义为关节过度活动。
经典类型EDS的最低诊断标准
-
光滑,柔软的皮肤
-
Molluscoid 假性肿瘤:肉质,与压力点如肘部和膝盖的疤痕相关的病变。
-
皮下球状体:小,囊肿样,硬粒状结节,可以在皮下组织上自由移动,在腿部和双臂的骨骼突起处。大约三分之一受影响个体都会产生,数量众多,感觉像米粒一样坚硬。 X射线显示具有半透明芯的外钙化层。 球状体代表失去血液供应,变纤维化和钙化的皮下脂肪球。
-
关节过度运动并发症(如扭伤,脱位/半脱位,扁平足)
-
肌张力低下,运动发育迟缓
-
容易挫伤
-
组织表现出伸展性和脆弱性(例如,疝气疝,儿童肛门脱垂,宫颈功能不全)
-
手术并发症(术后疝气)
检查
电子显微镜皮肤活检 在EDS,经典类型中通常呈现胶原纤维断裂,主要表现为胶原纤维的畸形 [Hausser & Anton-Lamprecht 1994]。然而,这些发现不是EDS特有的,因此不能诊断。 此外,如果皮肤活检不是全厚度,那么在网状真皮的中心部分通常最明显的超微结构变化可能会被错过。
真皮成纤维细胞的生物化学检测。对 皮肤来源的成纤维细胞进行胶原蛋白分析,以获得I,III和V型胶原蛋白进行电泳分析。在SDS-聚丙烯酰胺凝胶电泳上标记和分析胶原蛋白。与对照样品相 比,异常蛋白在凝胶上的迁移不同。由于V型胶原蛋白由低水平的成纤维细胞合成,所以电泳迁移率的改变不太可重现,这是常规诊断评估的无效方法。 然而,该测试有助于排除在临床鉴别诊断困难的个体中的EDS的其他亚型(例如,血管型,脊柱侧凸型,关节肉芽肿和皮肤痉挛类型)。 由于在编码I型胶原的胶原链的COL1A1中存在精氨酸突变为胱氨酸,因此很少检测到I型胶原的异常电泳图[Nuytinck et al 2000, Malfait et al 2007]。
分子遗传学检测
基因。 在大多数受影响家庭(≥50%)中,在编码V型胶原的基因COL5A1和COL5A2中可鉴定出致病性变异。 然而,由于在临床上明确定义的患者组中没有进行COL5A1和COL5A2的前瞻性分子生物学研究,所以这个数字可能低估了携带两个基因致病变异的经典EDS的个体的实际比例。
基因异质性的证据。COL1A1 致病性变异,p.Arg134Cys,已在两个散发的经典型EDS儿童中检出 [Nuytinck et al 2000]。随后在三个散发的年轻动脉瘤患者检出了相同的变异位点,这些患者也表现出薄而可伸张的皮肤,容易瘀伤,伤口愈合异常[Malfait et al 2007; Malfait and De Paepe, personal observation]。然而,COL1A1的突变不是经典EDS的主要原因 [Malfait et al 2005]。
临床检测
-
测序分析。 大约50%的经典EDS的个体在COL5A1或COL5A2中具有可鉴定的致病变异。大约30-40%的典型EDS的个体中,存在COL5A1无效等位变异 [Malfait et al 2005]。
-
缺失/重复分析。 目前没有报道涉及COL5A1或COL5A2的缺失或重复导致典型EDS,所以这类检测的意义尚不清楚。
-
COL5A1 无效等位基因检测。COL5A1无效等位基因检测可确定个体gDNA中是否存在若干COL5A1多态性杂合子,然后在cDNA水平确定两个等位基因是否被表达。 如果两个COL5A1等位基因中只有一个存在于cDNA中,则认为缺失的等位基因为空。 由于该测试检查了gDNA和cDNA,因此COL5A1无效等位基因检测需要培养的皮肤成纤维细胞 [Malfait et al 2005]。
表 2。
用于Ehlers-Danlos综合征的经典型分子遗传测试
| 基因 1 | 该基因突变致病的比例 | 检测方法 | 检测到等位基因变异 2 |
|---|---|---|---|
| COL5A1 | 46% 3 | 测序分析 4 | 序列变异 |
| 缺失/重复分析 5 | 外显子和全基因组缺失/重复 6 | ||
| COL5A2 | 4% 3 | 测序分析 4 | 序列变异 |
| 缺失/重复分析 5 | 外显子和全基因组缺失/重复 6 |
- 1.
-
染色体位置和蛋白参考 表 A. 基因和数据库。
-
等位突变体信息参考 分子遗传学。
- 3.
- 4.
-
通过序列分析检测到的致病性变异的实例包括小的基因缺失/插入和错义突变,无义突变和剪接位点变异,通常,没有检测到外显子或全基因缺失/重复。 对于解释序列分析结果时要考虑的问题,点击 这儿。
- 5.
-
通过基因组DNA的编码区和侧翼内含子区域的序列分析不能检测出外显子或全基因缺失/重复,可以使用的其他的方法包括:定量PCR,long-range PCR,多重连接依赖性探针扩增(MLPA)和包括该基因/染色体片段的染色体微阵列(CMA)。
- 6.
-
COL5A1或COL5A2的缺失或重复引起经典型Ehlers-Danlos综合症的案例尚未报道。(注意:根据定义,删除/重复分析表明通过基因组DNA的序列分析不可识别的重排。)
检测特点。有关测试特征的信息,包括灵敏度和特异性参考 临床实用基因卡 [Mayer et al 2013]。
检测策略
先证者证实/确定诊断。 经典EDS的分子遗传学检测由编码序列包含大量外显子(COL5A1中为66个,COL5A2中为52个)以及致病变异的广泛分布而复杂化。 怀疑经典EDS的临床诊断时,我们推荐以下评估:
-
进行序列分析。 通过在gDNA或cDNA上对COL5A1和COL5A2进行Sanger测序,建议首先对gDNA进行COL5A1序列分析,因为大多数具有经典EDS的个体在该基因中具有独特的致病性变体,导致引入早期终止密码子和无义介导的mRNA衰变。 当没有发现COL5A1致病变异时,应进行COL5A2的序列分析。
-
COL5A1无效等位基因检测和生化检测。如果COL5A1和COL5A2两者的序列分析不能识别具有经典EDS表型的人的致病原因,则建议进行皮肤活检以进行COL5A1无效等位基因检测和生物化学检测。
产前诊断和植入前遗传诊断(PGD) 对于风险妊娠需要事先鉴定家族中的致病变异。
临床特点
临床描述
Ehlers-Danlos 综合征(EDS)是一种结缔组织疾病,其特征在于皮肤过度伸展性,伤口愈合异常和关节过度活动。 以前,依据表型严重程度不同,分为两种亚型,EDS I型和EDS型II,现在研究表明两者构成一个连续的临床发现。
皮肤
-
皮肤过度伸展是EDS的基本特征之一,特别是经典的EDS。 皮肤容易伸展,并在释放后快速恢复(不像松弛,多余的皮肤,如皮肤松弛)。
-
皮肤光滑,触感柔软。
-
皮肤脆弱,类似小的创伤之后,特别是在压力点(膝盖,肘部)和易于创伤的区域(胫骨,前额,下巴)真皮分裂的表现。 皮肤脆性可能导致皮肤或粘膜缝合切口开裂。
-
伤口愈合延迟,创伤愈合后的明显的瘢痕伸展,疤痕变大,带有“卷烟纸”状外观。
-
经典EDS中的其他皮肤特征:
组织脆弱。在多个器官中观察到广泛组织可扩展性和脆性的表现:
-
怀孕期间宫颈功能不全
-
腹股沟和脐疝
-
疝和切口疝
-
儿童早期复发性直肠脱垂
关节
-
并发症,包括肩关节脱位,髌骨,手指,髋关节,桡骨和锁骨可能发生的并发症,通常可以自发恢复或由受影响的个人轻松缓解。尽管正常的骨骼射线照相术,一些具有经典EDS的人尽管骨骼射线结果正常,也可能会经历慢性关节和肢体疼痛。
-
与关节过度活动有关的其他问题包括关节不稳定,脚畸形,如先天性马蹄足或扁平足,颞下颌关节功能障碍,关节积液和骨关节炎[Hagberg et al 2004, De Coster et al 2005a, De Coster et al 2005b]。
神经功能。 可能发生原发性肌张力减低症,并可能导致运动发育迟缓,移动问题和轻度运动障碍,疲劳和肌肉抽筋相对频繁。 据报道,脑脊液泄漏很少导致经典EDS患者的姿势性低血压和头痛 [Schievink et al 2004]。
容易挫伤。 容易瘀伤是一种常见的症状,并且表现为自发性瘀斑,经常在相同的区域重复出现,并引起皮肤特征性的褐色变色,特别是在暴露的区域如胫骨和膝盖。尽管血凝状态正常,但仍存在延长出血的趋势(例如,刷牙之后)。
心血管
-
结构性心脏畸形在经典EDS中不常见。
-
二尖瓣脱垂,鲜有三尖瓣脱垂。 二尖瓣脱垂的诊断标准应更严格。
-
主动脉根部扩张可能比以前想象的更常见 [Wenstrup et al 2002]。最近的一项回顾性研究显示,50例经典EDS患者中,有3例(6%)第一次超声心动图显示主动脉扩张,平均年龄为16岁。 然而,扩张往往没有什么临床后果,二尖瓣脱垂也不会产生严重的后果,两者都很少需要外科手术 [Atzinger et al 2011]。
-
大动脉自发性破裂以及颅内动脉瘤和动静脉瘘可能发生严重的经典EDS的罕见个体中。
妊娠 经典EDS的女性怀孕,新生儿以及母亲都面临风险。 总体而言,这些并发症比正常人群概率更高, 然而缺少研究支持,难以定量受影响个体的每个并发症的发病率:
-
当母亲以及胎儿都受影响时,最严重的可能会发生膜过早破裂和早产儿。
-
由于低血压,如果婴儿受到影响,臀位呈现较为频繁,并可能导致新生儿臀部或肩膀脱位。
-
在受影响的女性中,可能会发生会阴部皮肤撕裂,分娩后会出现外阴切口扩张和子宫和/或膀胱脱垂。
基因型 - 表型相关性
检出COL5A1或COL5A2致病变异的患者数目相对较少。 尽管表型的严重程度可能有一些差异,但至今没有发现基因型/表型相关性。 尤其是与具有结构变异的个体及不携带突变的个体相比,具有COL5A1无效变体的个体,在严重性方面没有差异。
-
编码proα1(V)型胶原链氨基末端区域的COL5A1中的致病变异可能与经典EDS略有差异的表型相关。
-
α1(V)链的氨基末端前肽中的杂合突变p.Gly530Ser可以进行疾病修饰,并且在纯合状态下引起疾病 [Giunta & Steinmann 2000, Giunta et al 2002]。
-
最近研究表明,在proα1(V)胶原链的氨基末端区域内的特定剪接位点变异可导致经典的EDS样表型,其仅具有轻微的皮肤受累(不存在特征性萎缩性瘢痕形成),但是具有严重的脊柱后凸和视网膜脱落 [Symoens et al 2011]。
外显率
家族间和家族内表型严重程度的变异性很大。
在具有非功能性(即无效)COL5A1等位基因变异的一些家族中,受影响的成员可具有轻度的经典EDS表型,而其他家族成员可具有严重表型[Malfait & De Paepe 2005]。
预期
疾病无法预测。
命名法
1997年Villefranche 关于EDS的会议 [Beighton et al 1998], 以前的EDS I型和II型现在被重新分类为EDS,经典型。
流行病学
EDS I型的患病率估计为1:20,000 [Byers 2001],然而,有些患有这种疾病的病人,以前被列为EDS II型的人,很可能没有关注,因此未被发现。
遗传相关(等位基因)疾病
没有其他表型与COL5A1或COL5A2的突变相关。
鉴别诊断
在容易发生瘀伤,关节活动过度和/或慢性关节脱位的个体中,应考虑其他形式的Ehlers-Danlos综合征(EDS)。 临床发现与经典类型的EDS重叠的疾病包括:
Ehlers-Danlos 综合征, 超动力型 (EDS type III)。在这种形式中,关节过度活动是主要的表现。皮肤通常柔软,可能轻度可过度伸展。常见半脱位和脱位,它们可能自发发生或由小创伤引起,伴随急性疼痛。常见退行性关节病, 与急性脱臼或晚期骨关节炎相关的慢性疼痛是严重的并发症,并且可能造成身体和心理上的功能障碍。 容易的瘀伤,但萎缩性瘢痕形成则是典型EDS的特征。临床上主要表现为关节过度活动,并且发现皮肤异常,如皮肤过度伸展性和光滑柔软,存在萎缩性瘢痕,提示经典EDS的诊断。
EDS,超动力型的诊断完全基于临床评估和家族史。 在大多数具有超动力型EDS的个体中,突变致病的基因是未知的和未被研究的 [Malfait et al 2006a]。 在TNXB(编码腱生蛋白X的基因)半倍剂量不足和杂合错义突变与少部分超动力型EDS有关(参考腱生蛋白X缺陷)[Zweers et al 2003, Zweers et al 2005]。已有报道,一个超动力型EDS家族中检出COL3A1变异。 呈常染色体显性遗传。
腱生蛋白X缺乏症。 在具有常染色体隐性EDS表型的个体中已经鉴定了TNXB纯合致病变异,其特征在于轻度关节过度活动,皮肤过度伸展性和容易瘀伤,但没有萎缩性瘢痕形成 [Schalkwijk et al 2001, Lindor & Bristow 2005]。对于相同致病性变异杂合子携带者,特别是女性,似乎具有EDS超动力表型。
家族性关节过度活动综合征,.和其他发现过度活动的综合征,与经典EDS都表现出关节的过度活动,但不存在皮肤过度伸张性和萎缩性瘢痕。
Ehlers-Danlos综合征,血管型 (EDS type IV) 其特征是薄而半透明的皮肤,容易挫伤,特殊的面部特征,动脉,肠和/或子宫脆性大。 受影响的个体有动脉破裂,动脉瘤和/或夹层的风险,怀孕期间容易发生胃肠穿孔/破裂,子宫破裂。 四分之一的血管型EDS患者,20岁以上患有重大医疗问题,40岁以上者超过80%。 死亡中位数年龄为48岁。
血管型EDS的诊断是基于临床表型,并通过生物化学和/或分子遗传测试证实。受影响个体的生物化学研究证明培养的真皮成纤维细胞的III型前胶原具有异常的电泳迁移率和异常分泌的效率。分子遗传检测用于鉴定COL3A1中的致病性变异。常染色体显性方式遗传。
Ehlers-Danlos综合征, 早衰型 是一种罕见的常染色体隐性遗传病,其特征除了典型的EDS表现,还包括皱纹相,卷曲和细毛,少量眉毛和睫毛以及牙周炎。它是由B4GALT7(编码β-1,4-半乳糖基转移酶7的基因)中的纯合致病变异引起的。
Ehlers-Danlos 综合征,脊柱侧凸型 (以前称为EDS型VI)是一种广泛的结缔组织病,其特征在于脊柱后凸,关节松弛,肌张力减低,以及在某些个体中,眼球的脆弱性。智力正常,寿命可能是正常的,但是如果脊柱后凸严重,受影响的个体有不同程度的的动脉破裂和呼吸道受损的风险。
PLOD1的突变导致酶前胶原 - 赖氨酸2-氧戊二酸5-加氧酶1(PLOD1:赖氨酰羟化酶1)的活性不足,从而引起脊柱侧凸型EDS。 脊柱侧凸型EDS的诊断可依赖于通过HPLC测量的尿液中脱氧吡啶啉与吡啶啉啉交联的比例增加,具有高度敏感和特异性,也可通过皮肤成纤维细胞中赖氨酰羟化酶活性的测定和PLOD1的分子遗传检测进行诊断。 以常染色体隐性遗传方式遗传。
Ehlers-Danlos综合征,关节痛型 (以前称为VIIA和B型) 由先天性双侧髋关节脱位和严重的关节过度活动等症状进行区分。通常存在组织脆性(包括萎缩性瘢痕)和皮肤过度伸展性,严重程度从轻度到严重。 它是由COL1A1或COL1A2的突变引起的,分别导致编码I型胶原(EDS VIIA)或α2链(EDS VIIB)的mRNA的外显子6的缺失。以常染色体显性遗传方式遗传。
Ehlers-Danlos综合征,皮肤痉挛型 (以前称为EDS型VIIC) 特点是皮肤脆弱,松弛,下垂,冗长。 其他不同的特征包括是延迟囟门的闭合,特殊面容,眼睑水肿,蓝色巩膜,脐疝,短手指和身材矮小。 这种疾病是由前胶原N-蛋白酶的活性不足引起的,前蛋白酶N蛋白酶是在前胶原I,II和III中切除N-末端前肽的酶[Malfait et al 2005]。 以常染色体隐性遗传方式遗传。
Ehlers-Danlos综合征,心脏瓣膜型 其特征在于关节过度活动,皮肤过度伸张性,有时萎缩性瘢痕形成,以及心脏瓣膜缺损。 COL1A2中纯合子或复合杂合突变引起的I型胶原的proα2(I)链的缺失是致病原因[Schwarze et al 2004, Malfait et al 2006b]。 以常染色体隐性遗传方式遗传。
经典样EDS具有动脉破裂倾向。 已经在多个个体中鉴定了I型胶原的proα1(I)链中的一种精氨酸至半胱氨酸(Arg-to-Cys)变异(p.Arg134Cys),此类个体都具有典型EDS的表现,包括皮肤过度扩张性, 容易发生瘀伤,萎缩性瘢痕形成,以及关节活动过度以及成年动脉破裂的倾向[Nuytinck et al 2000, Malfait et al 2007]。 另外两种proα1(I)R-to-C突变(p.Arg396Cys和p.Arg915Cys)也与动脉破裂有关,但受影响的个体没有EDS样皮肤特征 [Malfait et al 2007]。 此外,在呈现EDS /成骨不全重叠表型的家族中报道了proα1(I)-Arg888Cys突变 [Cabral et al 2007],并且proα1(I)-Arg836Cys突变被证明与常染色体显性的Caffey病 相关 [Gensure et al 2005]。
Ehlers-Danlos综合征和脑室周围结节异位症。已报道在少数脑室周围异位症(以癫痫发作为特征的神经元迁移障碍和围绕脑侧脑室的神经细胞集合体)和EDS的特征的患者中,检测出了FLNA中的致病性变异 [Gómez-Garre et al 2006]。参考 X连锁心室异位症。
Ehlers-Danlos综合征脊柱侧凸型 其特征在于皮肤薄,容易瘀伤,具有挛缩趋势的小关节的过度运动,具有蓝色巩膜的突出的眼睛,具有细皱纹的手掌,臀部肌肉的萎缩以及尖的手指。 骨骼上,身材矮小,骨质减少,体宽。 编码膜结合锌转运蛋白SLC39A13的SLC39A13的突变是致病原因[Giunta et al 2008]。以常染色体隐性遗传方式遗传。
RIN2综合征 (又称MACS综合征) 表现为进行性脊柱侧凸,进行性面部粗化,牙龈肥大,稀疏的头发,以及皮肤和关节的膨胀。 它是由RIN2突变导致的,该基因编码Ras和Rab相互作用因子2,其作为GTP酶Rab5的鸟嘌呤核苷酸交换因子(GEF),参与早期内吞作用 [Basel-Vanagaite et al 2009, Syx et al 2010]。 以常染色体隐性遗传方式遗传。
Ehlers-Danlos综合征肌肉型 其特征在于颅面畸形,可伸缩的薄皮肤,萎缩性瘢痕,容易瘀伤,小关节过度活动,手掌细小和渐尖的手指,远端关节先天性挛缩,脊柱侧凸,进行性肌张力减退和可变的胃肠道和泌尿生殖器受累。 由编码皮肤素4磺基转移酶-1的CHST14突变引起,其涉及硫酸皮肤素的生物合成。 遗传方式是常染色体隐性遗传 [Malfait et al 2010, Miyake et al 2010]。
经典EDS与其他结缔组织疾病(包括以下几类)部分表型重叠,这些疾病可通过其他独特的临床特征来区分:
-
马凡综合征,由FBN1的突变引起,涉及眼,骨骼和心血管系统的广泛的临床表现。晶状体异位是一个标志性的特征约占60%。 近视,视网膜脱离,青光眼和早期白内障形成。 骨骼过度生长导致长肢体,骨折畸形和关节松弛,常见脊柱侧凸。 心血管表现包括主动脉扩张,主动脉夹层,二尖瓣脱垂伴有或没有反流,三尖瓣脱垂和近端肺动脉扩大。 马凡综合征是基于家族史的临床诊断和多器官系统特征性发现的观察,诊断标准已经建立。 常染色体显性方式遗传。
-
Occipital horn 综合征(OHS)(参考 ATP7A相关铜运输障碍) 其特征在于“枕骨角”,在斜方肌和胸锁乳突肌附着到枕骨的位置处的特征性楔形钙化。 临床上可以通过颅骨X光触及或观察。OHS患者也有松弛的皮肤和关节,膀胱憩室,腹股沟疝气和血管曲折,没有特别容易的瘀伤或脆弱的皮肤, 血清铜浓度和血清铜蓝蛋白浓度低。ATP7A的突变是致病原因,X连锁遗传。
管理
对于并发症和管理的详细信息,参考 Wenstrup & Hoechstetter [2004]。
初步诊断后评估
为了确定被诊断患有经典型Ehlers-Danlos综合征(EDS)患者的疾病程度,建议进行以下评估:
-
皮肤临床检查评估皮肤过敏性,萎缩性瘢痕和瘀伤以及经典EDS的其他表现
-
使用Beighton得分评估关节过度活动
-
评估婴儿和儿童的低血压和运动发育
-
对10岁以下个体进行基线超声心动图测量主动脉直径
-
如果存在严重的容易瘀伤,则评估凝血因子
表现处理
在患有低血压和延迟运动发育的儿童中,物理治疗很重要。
非负重肌肉运动,如游泳,有助于促进肌肉发育和协调。
患有肌张力低下症和伴有慢性疼痛的关节不稳定的个体可能需要相应调整生活方式和选择专业治疗。改善情绪和心理治疗可能有助于提高接受度和应对能力。
皮肤伤口应无张力关闭,最好是两层。 深层针脚应保证松散。皮肤针迹比正常情况保留更久,通常情况下,相邻皮肤用胶带进行额外的固定可以帮助防止疤痕的拉伸。
关于治疗关节松弛和脱位的建议,参考 EDS, 超动力型。(注意:关节稳定手术效果可能不佳,或只是暂时的改善。)
抗炎药可能有助于缓解关节疼痛。
长期慢性疼痛可能需要精神咨询。
心血管问题应以标准方式处理。
预防主要表现
具有明显的皮肤脆性的年幼的孩子,可以在前额,膝盖和胫骨上佩戴保护垫或绷带,以避免皮肤撕裂。 活跃的大龄儿童在活动期间可以穿足球鞋垫或长筒袜。
关于预防关节松弛和脱位的的建议,请参考 EDS,超动力型:管理,预防主要表现。
抗坏血酸(维生素C)可以减少瘀伤,但对皮肤过度伸张性,萎缩性瘢痕形成和关节过度活动的主要表现没有影响。 一般来说,建议每天使用两克的成人剂量,儿童剂量按比例减少,没有限制。
预防继发并发症
关于预防关节松弛和脱位继发并发症的建议,请参考 EDS,超动力型:管理,预防继发并发症。
监控
如果成人超声心动图无异常,则无需追踪超声心动图。 (因为关于主动脉扩张进展的纵向数据不可用,所以没有具体正常主动脉直径的个体随访的建议。)
如果存在主动脉扩张或二尖瓣脱垂等异常情况,则需要每年超声心动图检查。
注意事项
应避免以下情况:
-
重关节劳损(身体接触的运动,格斗运动,足球,跑步)运动
-
乙酰水杨酸(阿司匹林)
风险亲属评估
涉及与危险亲属遗传咨询有关的问题,请参考遗传咨询。
怀孕管理
由于皮肤撕裂,产后出血和子宫和/或膀胱脱垂的风险增加,建议在怀孕期间和产后期对女性进行监测。
抗坏血酸(维生素C)可以减轻瘀伤(参考 预防主要表现)。一般来说,建议每天使用两克的成人剂量,然而,怀孕第三孕期没有关于推荐剂量的严格指标。
妊娠早期如发生风险,需对孕妇进行监测。
调查治疗
可搜索 ClinicalTrials.gov 获取关于各种疾病和病症的临床研究的信息。 注意:这种疾病可能没有临床试验。
遗传咨询
遗传咨询的内容是向患者及其家庭提供该病的性质、遗传方式及其可能造成的影响方面的信息,帮助他们做出基于足够的背景知识,以及符合个人情况的决 定。接着几个段落是涉及遗传风险评估,根据家族史和遗传学检测来确定家庭成员的遗传状态。这一段的目的并不是为了解决所有患者可能面临的个人、文化或者伦 理的问题,或者企图替代遗传学专业人员的咨询工作。 —ED.
遗传方式
经典型 Ehlers-Danlos综合征(EDS)以常染色体显性遗传遗传。
家庭成员的风险
先证者父母
-
据估计,约50%的受影响个体是遗传了受影响的父母的致病性变异,约50%的受影响个体携带新发突变。
-
携带新发突变的先证者的父母应通过皮肤检查来评估,特别注意伤口愈合是否延迟,是否容易瘀伤,关节高度活动或复发性脱位以及慢性关节疼痛。 如果在先证者中鉴定出致病性变异,则需在父母中进行分子遗传检测。
注意:虽然诊断为经典EDS的患者中约有50%都具有受影响的父母,但由于家族成员无法识别家族史,家族史可能标记为阴性。
先证者的同胞
-
先证者同胞的风险取决于先证者父母的遗传状况。
-
如果先证者的父母受到影响,同胞的风险为50%。
-
当父母临床不受影响时,先证者同胞的风险很低。
-
虽然没有胚系嵌合体情况的报道,但在少数情况下仍然存在理论上的可能。
先证者的后代。经典EDS患者的每个孩子有50%的机会遗传致病性变异。
先证者的其他家庭成员
-
其他家庭成员的风险取决于先证者父母的遗传状况。
-
如果父母受到影响或携带致病性变异,其家人有风险。
相关遗传咨询问题
表型预测。 由于家族存在临床变异性,难以预测携带致病性变异的家族成员的表型。
具有新发致病变异家庭的意见。当常染色体显性遗传病患者的两亲均无致病性变异或病理证据时,可能突变发生在先证者中, 然而,父母嵌合的频率是未知的。 还可能存在包括代孕(例如辅助生殖)或未公开收养的其他解释。
家庭计划
-
怀孕之前是确定遗传风险的和讨论产前检测的最佳时间。
-
适当的向受影响或有风险的年轻成人提供遗传咨询(包括讨论潜在的后代和生殖选择风险)。
DNA 库 存储DNA(通常从白细胞中提取)以备日后使用。 因为测试方法和我们对基因,等位基因变异和疾病的理解在将来可能会有所改善,所以受影响的个体可以对DNA进行存储。
产前检测
如果已经在受影响的家庭成员中鉴定出致病性变异,那么可以通过提供目标基因检测或基因组合检测的临床实验室对高危产妇进行产前检测。
对于不影响智力或寿命的疾病(如经典EDS)产前测试的请求并不常见。 在医疗专业人员和家庭内可能存在使用产前检测的差异,特别是如果为了妊娠终止而不是早期诊断而进行检查。 虽然大多数中心会考虑关于产前检测的决定是父母的选择,但是对这些问题的讨论是适当的。
植入前遗传诊断(PGD) 可能是携带致病性变异一些家庭的选择。
资源
为了该患者及其家庭成员的方便,GeneReviews的员工已经选择了下述的针对该病的或者包括其他GeneReviews疾病的患者支持组组和患者注册组织。 GeneReviews不为其他组织提供的信息承担责任,选择这些组织的标准,及获取详细信息,请 点击这儿。
-
Association Francaise des Syndrome d'Ehlers Danlos34 rue L茅on JoulinTurns 37 000FranceEmail: contact@afsed.com
-
Ehlers-Danlos SocietyP.O. Box 87463Montgomery Village MD 20886Phone: 410-670-7577Email: info@ehlers-danlos.com
-
Ehlers-Danlos Support GroupPO Box 337Aldershot Surrey GU12 6WZUnited KingdomPhone: 01252 690940Email: director@ehlers-danlos.org
-
Ehlers-Danlos Syndrome Network C.A.R.E.S. FoundationPO Box 66Muskego WI 53150Phone: 262-514-2851Email: EDSLynnCARES@gmail.com
-
National Library of Medicine Genetics Home Reference
-
Medline Plus
-
National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC)Phone: 800-334-8571 ext 24640Email: gentac-registry@rti.org
分子遗传学
分子遗传学和OMIM表中的信息可能与GeneReview中的其他信息不同:表可能包含更多最新信息。-ED。
表 A。
Ehlers-Danlos综合征,经典类型:基因和数据库
| Gene | Chromosome Locus | Protein | Locus Specific | HGMD |
|---|---|---|---|---|
| COL5A1 | 9q34 |
Collagen alpha-1(V) chain | Ehlers-Danlos Syndrome Variant Database (COL5A1) | COL5A1 |
| COL5A2 | 2q32 |
Collagen alpha-2(V) chain | Ehlers-Danlos Syndrome Variant Database (COL5A2) | COL5A2 |
-
数据来自以下标准参考文献:基因参考 HGNC;染色体位点,基因座名称,临界区,互补群参考 OMIM;蛋白参考 UniProt。 有关提供链接的数据库(Locus Specific,HGMD)的描述,点击 这儿。
表 B.
经典类型的Ehlers-Danlos综合征的OMIM条目 (View All in OMIM)
| 120190 | COLLAGEN, TYPE V, ALPHA-2; COL5A2 |
| 120215 | COLLAGEN, TYPE V, ALPHA-1; COL5A1 |
| 130000 | EHLERS-DANLOS SYNDROME, CLASSIC TYPE |
COL5A1
基因结构。COL5A1 DNA全长超过150 kb,cDNA包含66个外显子。 有关基因和蛋白质信息的详细信息参考 表 A,基因。
致病变异。已经在COL5A1和COL5A2中鉴定出了几种类型的致病变异:
-
最常见的分子缺陷类型是COL5A1 mRNA的单倍剂量不足。 约40%的经典EDS的个体携带无义或移码突变,导致COL5A1基因功能缺失[Schwarze et al 2000, Wenstrup et al 2000, Schwarze et al 2001, Malfait et al 2005]。 引入终止密码子的无义突变,移码或剪接位点突变是产生非功能性COL5A1等位基因的主要原因。无义介导的突变可通过多种机制导致mRNA的衰变,结果可能是产生大约一半的正常V型胶原的量。
-
在大约10~15个典型EDS的患者中,已经证明了COL5A1的结构变异具有显性负相作用。在一小部分个体中,一种变异影响V型胶原蛋白的结构完整性,导致功能缺陷型V型胶原蛋白(显性负性变体)的产生。这些结构变异最常见的是导致外显子跳跃的剪接位点突变 [Burrows et al 1998, Malfait et al 2005] 和可导致胶原分子的三螺旋区域中的甘氨酸改变的单核苷酸变异 [Giunta & Steinmann 2000, Malfait et al 2005]。还已经鉴定了COL5A1中一个特定的单核苷酸变体,其将α1(V)胶原链的C末端前肽中的高度保守的半胱氨酸残基改变为丝氨酸 (p.Cys1639Ser) (NM_000093.3:c.4916G>C)。与以纤维胶原基因突变为特征的其他病症相反,甘氨酸改变类型的致病变异数量较少。
-
α1(V)链的氨基末端前肽中的p.Gly530Ser (NM_000093.3: c.1588G>A) 改变可能在杂合状态下存在疾病修饰作用,并且在纯合状态下致病 [Giunta & Steinmann 2000, Giunta et al 2002]。
正常基因产物。 胶原α1(V)链(V型胶原链)。V型胶原蛋白是广泛分布在各种组织中的定量小纤维状胶原蛋白。主要以皮肤,骨骼和肌腱中的[α1(V)] 2α2(V)异源三聚体形式存在。 它通过其非常大的氨基末端前肽形成具有I型胶原的异型原纤维,并调节这些原纤维的直径。 最近的数据表明V型胶原蛋白在多种组织中控制胶原原纤维组装 [Wenstrup et al 2004]。
异常基因产物。 α1(V)或α2(V)链的三重螺旋结构域中的错义变异可能具有显性负相作用,突变形式可以干扰正常等位基因编码的正常蛋白质的作用。 COL5A1或mRNA产物中的过早引入终止密码子,可能会改变正常的胶原纤维的产生。
COL5A2
基因结构。COL5A2 全长67 kb,cDNA包含51个外显子,有关基因和蛋白质的详细信息请参考 表 A,基因。
致病性等位变异。COL5A2中的结构性致病变异已在少数经典EDS患者中被证实,这些结构变异最常见的是导致外显子跳跃的剪接位点变异 [Michalickova et al 1998, Malfait et al 2005] 和导致胶原分子的三重螺旋区域中的甘氨酸改变的单核苷酸变异 [Richards et al 1998]。
正常基因产物。胶原α2(V)链(V型胶原链)。V型胶原蛋白是广泛分布在各种组织中的定量小纤维状胶原蛋白。它主要以皮肤,骨骼和腱中的[α1(V)] 2,α2(V)异源三聚体存在。 它通过其非常大的氨基末端前肽形成具有I型胶原的异型原纤维,并调节这些原纤维的直径。
异常基因产物。α1(V)或α2(V)链的三重螺旋结构域中的错义变异可能具有显性负相作用,也就是说,突变形式可以干扰正常等位基因编码的正常蛋白质的作用。
参考文献
指南/共识声明
-
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Available online. 1998. Accessed 9-16-15. [PubMed: 9557891]
文献引用
-
Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011;158:826 - 830.e1. [PubMed: 21193204]
-
Basel-Vanagaite L, Sarig O, Hershkovitz D, Fuchs-Telem D, Rapaport D, Gat A, Isman G, Shirazi I, Shohat M, Enk CD, Birk E, Kohlhase J, Matysiak-Scholze U, Maya I, Knopf C, Peffekoven A, Hennies HC, Bergman R, Horowitz M, Ishida-Yamamoto A, Sprecher E. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am J Hum Genet. 2009;85:254 - 63. [PMC free article: PMC2725231] [PubMed: 19631308]
-
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers- Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31 - 7. [PubMed: 9557891]
-
Burrows NP, Nicholls AC, Richards AJ, Luccarini C, Harrison JB, Yates JR, Pope FM. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am J Hum Genet. 1998;63:390 - 8. [PMC free article: PMC1377290] [PubMed: 9683580]
-
Byers PH. Disorders of collagen biosynthesis and structure. In: Scriver, Beaudet, Sly, Valle, eds. The Metabolic and Molecular Bases of Inherited Disease. 2 ed. Edinburgh, UK: Churchill Livingstone; 2001:1065-81.
-
Cabral WA, Makareeva E, Letocha AD, Scribanu N, Fertala A, Steplewski A, Keene DR, Persikov AV, Leikin S, Marini JC. Y-position cysteine substitution in type I collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers-Danlos syndrome phenotype. Hum Mutat. 2007;28:396 - 405. [PubMed: 17206620]
-
De Coster PJ, Martens LC, De Paepe A. Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med. 2005a;34:298 - 307. [PubMed: 15817074]
-
De Coster PJ, Van den Berghe LI, Martens LC. Generalized joint hypermobility and temporomandibular disorders: inherited connective tissue disease as a model with maximum expression. J Orofac Pain. 2005b;19:47 - 57. [PubMed: 15779539]
-
Gensure RC, Mäkitie O, Barclay C, Chan C, Depalma SR, Bastepe M, Abuzahra H, Couper R, Mundlos S, Sillence D, Ala Kokko L, Seidman JG, Cole WG, Jüppner H. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest. 2005;115:1250 - 7. [PMC free article: PMC1087158] [PubMed: 15864348]
-
Giunta C, Elçioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, Steinmann B. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome--an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet. 2008;82:1290 - 305. [PMC free article: PMC2427271] [PubMed: 18513683]
-
Giunta C, Nuytinck L, Raghunath M, Hausser I, De Paepe A, Steinmann B. Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers-Danlos syndrome. Am J Med Genet. 2002;109:284 - 90. [PubMed: 11992482]
-
Giunta C, Steinmann B. Compound heterozygosity for a disease-causing G1489E [correction of G1489D] and disease-modifying G530S substitution in COL5A1 of a patient with the classical type of Ehlers-Danlos syndrome: an explanation of intrafamilial variability? Am J Med Genet. 2000;90:72 - 9. [PubMed: 10602121]
-
Gómez-Garre P, Seijo M, Gutiérrez-Delicado E, Castro del Río M, de la Torre C, Gómez-Abad C, Morales-Corraliza J, Puig M, Serratosa JM. Ehlers-Danlos syndrome and periventricular nodular heterotopia in a Spanish family with a single FLNA mutation. J Med Genet. 2006;43:232 - 7. [PMC free article: PMC2563248] [PubMed: 15994863]
-
Hagberg C, Berglund B, Korpe L, Andersson-Norinder J. Ehlers-Danlos Syndrome (EDS) focusing on oral symptoms: a questionnaire study. Orthod Craniofac Res. 2004;7:178 - 85. [PubMed: 15359504]
-
Hausser I, Anton-Lamprecht I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet. 1994;93:394 - 407. [PubMed: 8168810]
-
Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A. 2005;135:75 - 80. [PubMed: 15793839]
-
Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A. The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum Mutat. 2005;25:28 - 37. [PubMed: 15580559]
-
Malfait F, De Paepe A. Molecular genetics in classic Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2005;139C:17 - 23. [PubMed: 16278879]
-
Malfait F, Hakim AJ, De Paepe A, Grahame R. The genetic basis of joint hypermobility syndromes. Rheumatology. 2006a;45:502 - 7. [PubMed: 16418200]
-
Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. Total absence of the alpha2(I) chain of collagen type I is a rare cause of Ehlers-Danlos Syndrome hyermobility type. J Med Genet. 2006b;43:e36. [PMC free article: PMC2564565] [PubMed: 16816023]
-
Malfait F, Symoens S, De Backer J, Hermanns-Lê T, Sakalihasan N, Lapière CM, Coucke P, De Paepe A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387 - 95. [PubMed: 17211858]
-
Malfait F, Syx D, Vlummens P, Symoens S, Nampoothiri S, Hermanns-Lê T, Van Laer L, De Paepe A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum Mutat. 2010;31:1233 - 9. [PubMed: 20842734]
-
Mayer K, Kennerknecht I, Steinmann B. Clinical utility gene card for: Ehlers-Danlos syndrome types I-VII and variants - update 2012. Eur J Hum Genet. 2013;21(1) [PMC free article: PMC3533317] [PubMed: 22892533]
-
Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG. Mutations of the alpha2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet. 1998;7:249 - 55. [PubMed: 9425231]
-
Miyake N, Kosho T, Mizumoto S, Furuichi T, Hatamochi A, Nagashima Y, Arai E, Takahashi K, Kawamura R, Wakui K, Takahashi J, Kato H, Yasui H, Ishida T, Ohashi H, Nishimura G, Shiina M, Saitsu H, Tsurusaki Y, Doi H, Fukushima Y, Ikegawa S, Yamada S, Sugahara K, Matsumoto N. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum Mutat. 2010;31:966 - 74. [PubMed: 20533528]
-
Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet. 2000;66:1398 - 402. [PMC free article: PMC1288203] [PubMed: 10739762]
-
Richards AJ, Martin S, Nicholls AC, Harrison JB, Pope FM, Burrows NP. A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II. J Med Genet. 1998;35:846 - 8. [PMC free article: PMC1051462] [PubMed: 9783710]
-
Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167 - 75. [PubMed: 11642233]
-
Schievink WI, Gordon OK, Tourje J. Connective tissue disorders with spontaneous spinal cerebrospinal fluid leaks and intracranial hypotension: a prospective study. Neurosurgery. 2004;54:65 - 70. [PubMed: 14683542]
-
Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH. Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II). Am J Hum Genet. 2000;66:1757 - 65. [PMC free article: PMC1378060] [PubMed: 10796876]
-
Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, Byers PH. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004;74:917 - 30. [PMC free article: PMC1181985] [PubMed: 15077201]
-
Schwarze U, Schievink WI, Petty E, Jaff MR, Babovic-Vuksanovic D, Cherry KJ, Pepin M, Byers PH. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989 - 1001. [PMC free article: PMC1274375] [PubMed: 11577371]
-
Symoens S, Malfait F, Vlummens P, Hermanns-Lê T, Syx D, De Paepe A. A novel splice variant in the N-propeptide of COL5A1 causes an EDS phenotype with severe kyphoscoliosis and eye involvement. PLoS One. 2011;6:e20121. [PMC free article: PMC3096658] [PubMed: 21611149]
-
Syx D, Malfait F, Van Laer L, Hellemans J, Hermanns-Lê T, Willaert A, Benmansour A, De Paepe A, Verloes A. The RIN2 syndrome: a new autosomal recessive connective tissue disorder caused by deficiency of Ras and Rab interactor 2 (RIN2). Hum Genet. 2010;128:79 - 88. [PubMed: 20424861]
-
Wenstrup RJ, Hoechstetter LB. Ehlers-Danlos syndromes. In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. 2 ed. New York, NY: John Wiley & Sons; 2004:211-24.
-
Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331 - 7. [PubMed: 15383546]
-
Wenstrup RJ, Florer JB, Willing MC, Giunta C, Steinmann B, Young F, Susic M, Cole WG. COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet. 2000;66:1766 - 76. [PMC free article: PMC1378044] [PubMed: 10777716]
-
Wenstrup RJ, Meyer RA, Lyle JS, Hoechstetter L, Rose PS, Levy HP, Francomano CA. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med. 2002;4:112 - 7. [PubMed: 12180144]
-
Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214 - 7. [PMC free article: PMC1180584] [PubMed: 12865992]
-
Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet. 2005;67:330 - 4. [PubMed: 15733269]
章节注释
修订记录
-
18 August 2011 (me) Comprehensive update posted live
-
11 May 2010 (cd) Revision: changes in testing
-
24 July 2008 (me) Comprehensive update posted live
-
10 April 2006 (me) Comprehensive update posted to live Web site
-
29 October 2003 (ca) Review posted to live Web site
-
20 June 2003 (rw, ad) Original submission