
摘要
临床特征.
贝克-维德曼综合征(Beckwith-Wiedemann syndrome,BWS)是一种以新生儿低血糖、巨大儿、大舌癌、偏侧发育过度、脐膨出、胚胎肿瘤(如肾母细胞瘤、肝母细胞瘤、神经母细胞瘤和横纹肌肉瘤)、内脏肿大、肾上腺皮质细胞瘤、肾脏异常(如髓质发育不良、肾髓质增生、髓质海棉肾、肾肥大)和耳皱/窝为特征的生长障碍。
BWS被认为是一种临床表现,受累的个体可能有许多这写特征,也可能只有一两个临床特征,早逝可能发生于早产、低血糖、心肌病、大舌咽炎或肿瘤等并发症。然而,先前报道的20%的死亡率很可能是高估,因为人们对这种疾病有了更好的认识,同时也增加了治疗方案。巨舌症和巨大儿通常出现在出生时,但有可能产后发病。增长率在7-8岁左右减慢。偏侧发育过度可能影响身体的阶段区域或选定的器官和组织。
诊断/检测.
基于临床评估的BWS的临床诊断可由分子/细胞遗传的 检测来确立。细胞遗传的 染色体 11p15 异常在1%或更少的 受累的 个体中被发现。分子遗传学检测可以识别BWS患者的表观遗传和染色体11p15 基因组的 改变:
CDKN1C的序列分析在大约40%的家族性 病例中识别出一种 杂合的 母系遗传致病性变异 变异,并且 5%-10% 的病例没有BWS家族史。
管理.
对各种症状的处理:治疗低血糖以减少中枢神经系统并发症的风险,脐突出腹壁修补术,气管内插管治疗气道受损,并使用专门的乳头或鼻饲喂养来管理大舌咽炎造成的喂养困难。大舌咽炎的儿童在婴儿期或幼儿期可能会受益于减舌手术和言语治疗。手术可在青春期早期进行,以平衡偏侧发育过度的腿部长度的显著差异;颅面手术可能有利于面部偏侧发育过度的患者。肿瘤采用标准儿科肿瘤学治疗方案。肾钙质沉着症和其他肾脏表现应由儿科肾病专家评估和治疗。将有胃肠道结构异常的儿童转诊至相关专家;标准管理心脏问题;标准干预发育迟缓儿童。
预防继发性并发症: 及时评估和标准治疗可疑泌尿道感染,以防止继发性肾损伤。
监测: 低血糖监测,尤其是新生儿期;每三个月进行一次胚胎肿瘤筛查,直到8岁;在出生后的头四年每两至三个月监测一次血清甲胎蛋白(AFP)浓度,以早期发现肝母细胞瘤。每年为年龄在8岁至青春期中期的受累的个体进行一次肾超声检查,以鉴别肾钙质沉着症或髓质海绵肾患者;考虑每年或每年两次测量尿钙/肌酐比值。
遗传咨询.
贝克-维德曼综合征(Beckwith-Wiedemann syndrome,BWS)与11p15.5染色体上两个印记的区域的基因 转录调控异常有关。大多数BWS患者都有正常的染色体研究或核型。约85%的BWS患者没有家族史,约有15%的家族史与同源 常染色体显性遗传 一致。辅助生殖技术(ART)孕育的不孕不育父母的孩子可能有更高的 印记 障碍风险,包括BWS。识别导致BWS的潜在遗传机制可以更好的估计再发风险。在一般人群中对孕妇进行产前筛查,以确定诊断BWS的发现,可能会考虑染色体分析,染色体芯片,和/或 分子遗传学检测。通过对遗传性染色体异常家系的染色体分析或对已明确BWS机制的家系进行分子遗传学检测,可以进行特定的遗传学检查。
诊断
推测性结论
贝克-维德曼综合征 (BWS)目前尚无公认的临床诊断标准。
对于有下列一项或多项主要和/或次要发现的个体,应该怀疑存在贝克-维德曼综合征(BWS)。
与BWS相关的主要发现
- 巨大儿(传统定义为体重和身长/身高>97百分位)
- 巨舌症
- 偏侧发育过度(身体一个或多个区域的不对称过度生长)
- 脐膨出(亦称脐外疝)或脐疝
- 儿童期胚胎肿瘤(例如,Wilms 肿瘤,肝母细胞瘤,神经母细胞瘤,横纹肌肉瘤)
- 内脏肿大,涉及一个或多个腹腔内器官,包括肝、脾、肾上腺和/或胰腺
- 胎儿肾上腺皮质细胞肿大(病理)
- 肾脏异常,包括结构异常、肾肥大、肾钙质沉着症和/或髓质海棉肾的后期发育
- 前线形耳叶折痕和或后螺旋耳孔
- 胎盘间充质发育不良 [Wilson et al 2008]
- 腭裂 (BWS中罕见)
- 心肌病(BWS中罕见)
- 阳性家族史(≥1个家族成员,有临床诊断BWS或提示BWS的病史或特征)
与BWS相关的次要发现
- 妊娠相关发现包括羊水过多和早产
- 新生儿低血糖
- 血管病变包括单纯痣(通常出现在额头、眉毛和/或颈部后部)或血管瘤(皮肤或皮肤外)
- 面部特征包括中面后缩和眶下折痕
- 心脏结构异常或心脏肿大
- 直肠纵裂
- 骨龄超前(常见于过度生长/内分泌失调)
建立诊断
BWS的诊断建立在 先证者 有以下情况:
BWS与 染色体 11p15.5 (又称 BWS 关键区域)两个 印记的 区域的 基因 转录调控异常有关。调节可能被多种机制中的任何一种破坏;这里给出了已知致病机制的简化描述,以澄清 遗传测试 描述的测试通道。有关该区域基因表达调控的详细描述,请参见分子遗传学发病机制 。
BWS 关键区域 包括两个区域: 印记 中心1 (IC1) 结构域 1 中的 IGF2和 H19 的表达;印记中心2(IC2)调控CDKN1C,KCNQ10T1,和 KCNQ1在2区域中的表达 (Figure 1)。基因组印记是一种基因的两个等位基因 的DNA发生差异修饰,使每个基因通常只表达一个亲本 等位基因 [Barlow 1994]。参看 Figure 1a, IC1和IC2的差异甲基化与未受影响的个体的父系和母系等位基因上特定基因的表达有关。
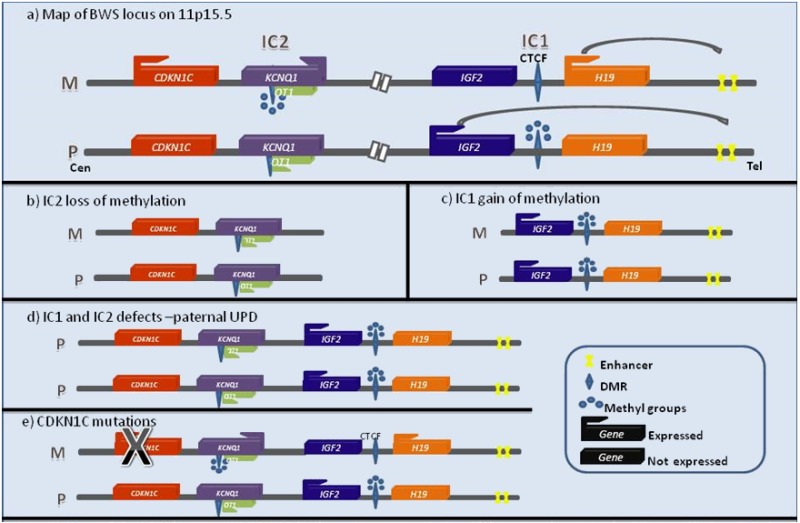
注解: IC1 和 IC2 有时分别被称为差异甲基化区域DMR1 和 DMR2。
在超过80%的BWS患者中,基因检测可以检测到五种突变中的一种 [Weksberg et al 2003, Weksberg et al 2005]:
- 下面四个分子改变的原理图显示在 Figure 1:
- 以下基因改变未在 Figure 1表示:
- 涉及 染色体 11p15.5 的基因组变异,包括细胞遗传学可见的复制、倒置或易位,或包括11p15.5的微复制或微缺失的拷贝数变异。
注解: 甲基化改变可能与上述任何主要 基因组的 变异有关,除了母体CDKN1C 等位基因上的致病变异 [Niemitz et al 2004, Sparago et al 2004, Prawitt et al 2005, Baskin et al 2014]。
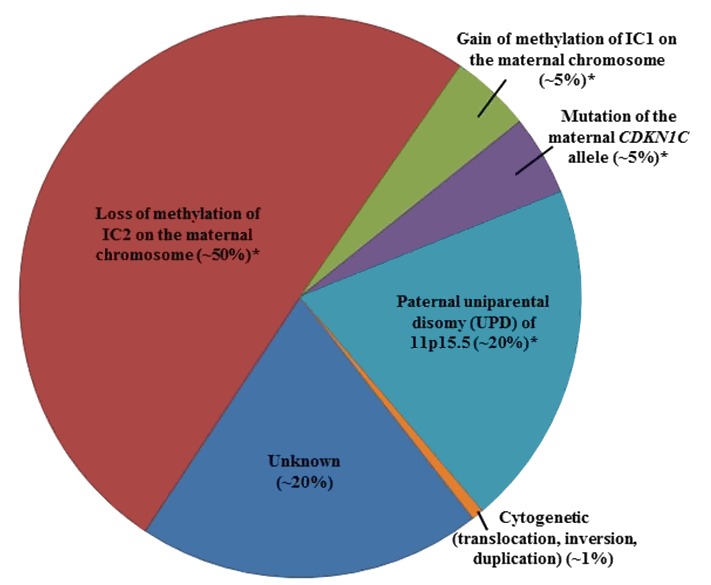
Figure 2 通过遗传机制总结了BWS个体的百分比。
基因检测
基因检测方法包括DNA甲基化 研究,单-基因 检测,11p15.5(序列)的拷贝数变异, 染色体芯片, 核型,,和包括BWS 关键区域基因的多基因面板 的使用:qazi
- IC1 和 IC2 DNA 甲基化 研究 应同时进行。
- IC1 和 IC2的甲基化改变提示 单亲二体性.
- 单-基因 测试. 家族性 病例、BWS和中线异常(腭裂、后窝异常、脐突出或尿道下裂[Gardiner et al 2012, Brioude et al 2015])患者,或者在临床怀疑BWS存在但未检测到 染色体 11p15.5 细胞遗传的 异常、拷贝数变异、 甲基化 异常或UPD的个体应考虑对CDKN1C进行序列分析,然后对CDKN1C进行基因靶向 缺失/重复分析 。
- 染色体微阵列(CMA) 利用寡核苷酸阵列或SNP 基因分型 阵列,可以检测出 先证者 中的 缺失 或 重复 。在一个有智力缺陷的序列中,CMA可能被考虑是第一位的。删除大小取决于使用的微阵列类型和11p15.5区域的探针密度 [Keren et al 2013, Baskin et al 2014, Russo et al 2016]. SNP 阵列 分析还可以检测阶段性父系单亲二体性。
Table 1.
贝克-维德曼综合征的遗传检测
| 试验方法 | 致病变异/变异检测 | 检测到BWS变异的比例1 |
|---|---|---|
| 甲基化分析 2 | IC2甲基化 缺失 (母系) | 50% 3 |
| IC1 甲基化 获得 (母系) | 5% 3 | |
| IC2 甲基化 缺失和CI1 甲基化 获得 (父系 UPD) | 20% 3 | |
| 序列分析 / 基因-靶向 删除/重复分析4. 5 | 杂合母系 CDKN1C 致病变异 | 5% 无BWS家族史 6 |
| ~40% 有BWS家族史 6 | ||
| 核型 | 11p15.5细胞遗传 重复,倒位,或 易位 | <1% 7 |
| 微阵列 (基于SNP) | 微缺失,微重复,父系UPD 8 | ~ 9% 9 |
- 1.
按 基因/位点、 表型、人群和/或检测方法分类的 受累的人群在符合BWS的临床诊断标准的人群中的比例。注解:不同人群的频率可能不同 [Sasaki et al 2007]。
- 2.
- 3.
- 4.
- 5.
- 6.
CDKN1C测序的检出率因家族史而异 [Hatada et al 1997, Lee et al 1997, O'Keefe et al 1997, Lam et al 1999, Algar et al 2000, Li et al 2001, Brioude et al 2015]。
- 7.
- 8.
- 9.
临床特征. 有关测试特征的敏感性和特异性信息参看 临床实用基因卡[Eggermann et al 2014] 。
临床特征
临床描述
贝克-维德曼综合征(BWS)是一种以新生儿低血糖、巨大儿、巨舌、偏侧发育过度、脐突出、胚胎肿瘤(如肾母细胞瘤、肝母细胞瘤、神经母细胞瘤、横纹肌肉瘤)、内脏肿大、肾上腺皮质巨细胞症、肾脏异常(如髓质发育不良、肾钙质沉着、髓质海绵肾、肾巨细胞症)和耳朵折痕/为主要特征的生长障碍。BWS被认为是一种临床范围, 受累的 个体可能具有许多或只有一两个临床特征。
贝克-维德曼综合征(BWS)具体的个体临床表现在已发表的报告中差异很大,部分原因是测量偏倚。然而,以下特征显然是 表型 的一部分。
产前和围产期. 羊水过多、早产、巨大儿的发生率可高达50%。其它共同特征包括脐带增长和胎盘增大,胎盘重量平均是正常孕龄胎盘重量的两倍。据报道,胎盘间充质发育不良的婴儿后来发现有BWS的特征[Wilson et al 2008]。
患有BWS的婴儿死亡率增加主要原因是早产、大舌癌、低血糖以及极少出现的心肌病等并发症。然而,鉴于最近在本病识别和治疗方面的改善,先前报告的20%的死亡率可能是被高估的。
代谢异常. 新生儿低血糖有很好的文献记载,发生于50%的BWS患儿 [Mussa et al 2016a]。如果未被发现或未经治疗,它会有极大风险造成生长发育后遗症。低血糖多为轻度、短暂性低血糖;然而,在更严重的情况下,低血糖会持续。偶尔会观察到低血糖延迟发作(即,在出生后的第一个月)。
其它不常见的内分泌/代谢/血液学表现包括甲状腺功能减退、高脂血症/高胆固醇血症和红细胞增多症。
即使没有肾脏异常,BWS患儿也会出现高尿钙。在超声检查中,22%的BWS患者表现出肾结石病,而在一般人群中发病率为7%-10%[Goldman et al 2003]。
生长. 巨舌(约~90%)和巨大儿(约~50%)通常出现在出生时,但也观察到出生后出现这两种症状 [Chitayat et al 1990, Brioude et al 2013, Ibrahim et al 2014, Mussa et al 2016b]。虽然大多数BWS患者在儿童早期发育迅速,但身高通常保持在正常范围的上限。生长速度通常在七岁到八岁左右出现减缓。
偏侧发育过度*一般在出生时就能被察觉,但随着孩子的生长或多或少会变得明显。偏侧发育过度可能会影响身体的节段性区域或选定的器官组织。当涉及多个节段时,偏侧发育过度可能局限于身体的一侧(同侧)或累及身体的另一侧(对侧)[Hoyme et al 1998]。
*注解:偏侧发育过度是指细胞增殖异常,导致细胞不对成过度生长;在BWS中, 偏侧发育过度是指细胞数量增加,取代了细胞肥大一词,即细胞大小增加。
肿瘤. 患有BWS的儿童因肿瘤儿死亡的风险增加,特别是Wilms 肿瘤和肝母细胞瘤,还有神经母细胞瘤、肾上腺皮质癌、横纹肌肉瘤。也曾见到各种各样的其它肿瘤,包括恶性和良性 [Cohen 2005]。BWS患儿发生肿瘤的风险估计为7.5%,风险范围估计在4%到21%之间 [Cohen 2005, Tan & Amor 2006, Mussa et al 2016a]。肿瘤形成的风险增加似乎集中在生命的前8年。8岁以上受累的个体发生肿瘤虽不常见,但已有报告。
表型较轻的儿童(例如,巨舌症和脐疝)可能患有体细胞型BWS,而且(与一般人群相比)患BWS相关肿瘤的风险可能增加。这在一定程度上是因为BWS相关的分子改变可能是镶嵌型的;也就是说,许多与BWS相关的细胞可能存在于肿瘤发生的“危险”器官(例如,肝脏或肾脏),而不是存在于影响临床表现的组织中。因此,在评价BWS表型谱中临床特征最轻的儿童时,应考虑使用基因检测来确认诊断,怀疑指数应较高。
其它器官系统
- 前腹壁缺损包括脐膨出、脐疝、和直肠纵隔是常见的。
- 关于BWS 心血管疾病的许多信息都是道听途说。大约20% 受累的个体存在心脏肥大[Pettenati et al 1986]。如果在婴儿期进行胸部X光检查,通常会发现心脏肥大,但通常不需要特殊治疗即可解决。
- 心肌病曾被报道,但很罕见。
- 肾异常 包括髓质发育不良、重复收集系统、肾钙化、髓质海绵肾、囊性改变、憩室、和肾巨细胞症 [Choyke et al 1998, Borer et al 1999, Mussa et al 2012]。
- 听力损失 很少报道于BWS患者,听力损失要么是感觉神经性的[Kantaputra et al 2013],要么是由镫骨固定引起的传导性听力损失[Hopsu et al 2003]。
注解:虽然BWS患儿的父母偶尔会对听力损失和低张力症表示关注,但很难确定这些问题和其他问题在BWS患者中的发生率是否高于一般人群的发生率。
生长发育 在BWS患儿中通常是正常的,除非有染色体异常、脑畸形、缺氧史或未治疗的低血糖。神经行为问题,如自闭症谱系障碍,在父母报告确定的BWS患儿中报告的频率增加。然而还需要进行其他研究,包括正式的神经发育评估,以评估BWS中此类问题的发生率。
成年期. 儿童期后,预后一般良好。然而,并发症-包括肾髓质发育不良和男性不育症-可能会发生。这些问题可能与特定的分子亚型有关[Greer et al 2008]。
表型相关性的分子机制研究
虽然分子机制的一般表型相关性如下所示,但任何BWS个体的具体临床结果都不能通过分子改变来准确预测。BWS个体中剩余的变异可能是由于 体细胞嵌合 、遗传背景和/或其他未知因素造成的。
- 当11p15的UPD或IC1甲基化增加时,Wilms肿瘤和肝母细胞瘤的风险最高。
- IC2 甲基化缺失与较低的肿瘤发生风险有关,且迄今报道的肿瘤不包括Wilms肿瘤。
- 母系CDKN1C 等位基因的基因内变异与以下因素有关:
- 少数神经母细胞瘤病例 [Bliek et al 2001, Weksberg et al 2001, DeBaun et all 2002, Rump et al 2005, Alsultan et al 2008, Kuroiwa et al 2009];
- 神经节母细胞瘤、急性淋巴细胞白血病、儿童神经母细胞瘤和成人黑色素瘤的单个病例[Brioude et al 2015]。
注解: 鉴于白血病和黑色素瘤在没有BWS的人群中的发生率较高,很难确定这些疾病的发生是两种不相关单一疾病的的巧合,还是恶性肿瘤确实与BWS有关。
- 偏侧发育过度 通常与父系11p15的UPD 嵌合 有关,但也见于IC2或IC1发生分子改变的个体[DeBaun et al 2002, Shuman et al 2002, Enklaar et al 2006, Ibrahim et al 2014, Mussa et al 2016b]。
- 阳性家族史 与CDKN1C 杂合的致病变异、IC1位点缺失或IC2位点 重复(很少)有关[Weksberg & Shuman 2004, Cooper et al 2005, Prawitt et al 2005, Enklaar et al 2006, Percesepe et al 2008, Scott et al 2008b, Bliek et al 2009]。
- 脐膨出主要与IC2改变或母系来源的CDKN1C 等位基因 杂合的 致病性变异 有关 [Ibrahim et al 2014, Brioude et al 2015, Mussa et al 2015, Mussa et al 2016a]。
- 生育能力低下似乎都与因IC2 甲基化 缺失导致的婴儿BWS发病率增加有关,无论是否使用辅助生殖技术(ART)[DeBaun et al 2003, Gicquel et al 2003, Maher et al 2003a, Maher et al 2003b, Halliday et al 2004]。
外显度
如果考虑到 印记的 区域的起源效应,家族性病例的外显度较高。例如,一个人可能遗传了一种CDKN1C 致病性变异,但没有BWS的特征,因为CDKN1C致病变异实在父系遗传的等位基因上,通常不表达(例如,致病性变异通过正常 印记表达时沉默)。
命名
BWS最初被称为EMG,是根据 脐膨出(exomphalos)、巨舌(macroglossia)、和巨人症(gigantism)三种临床表现来命名的。
流行率
已报道的1:10,000 [Mussa et al 2013]到1:13,700 [Thorburn et al 1970]的患病率可能被低估了,因为存在一些表型较轻的未确诊个体。
BWS 在多种种族人群中均有报道,男性和女性发病率相等。
遗传相关(等位基因)疾病
11p15位点的分子改变包括IC2位点甲基化缺失、IC1位点甲基化增加 [Martin et al 2005]和已在 明显孤立的偏侧发育过度个体中得到报道的11p15父系单亲二体性[Shuman et al 2002]。一些明显孤立的偏侧发育过度病例代表了WBS临床谱的轻度末端;然而,其他孤立的偏侧发育过度可能是由体细胞嵌合引起的其他分子改变。
孤立的Wilms 肿瘤可能与染色体11p15的结构改变有关,包括IC1高甲基化,11p15的父系单亲二体性、11p15基因组的异常,包括缺失和插入[Scott et al 2008a]。
父系IC1甲基化 缺失的体细胞嵌合与罗素-西尔弗综合征 Russell-Silver syndrome(RSS) 和/或孤立的 偏侧发育过度有关[Zeschnigk et al 2008, Eggermann 2009, Eggermann et al 2015]。
在一个患有罗素-西尔弗综合征(RSS)的家族中,有一种母系遗传的CDKN1C致病性变异导致细胞环素依赖酶抑制剂1C的稳定性增强,[Brioude et al 2013],而母系遗传功能获得性杂合的CDKN1C致病性变异导致IMAGe 综合征[Arboleda et al 2012, Milani et al 2014]。
鉴别诊断
过度发育. 贝克-维德曼综合征(BWS)常被认为是儿童发育过度的鉴别诊断。值得注意的是,目前尚未分类的过度发育综合征需要与BWS进行区分,患有BWS和发育迟缓的儿童, 染色体 研究正常,无缺氧或低血糖史,需要考虑发育迟缓的其他原因,如果存在结构性或心脏传导缺陷,鉴别诊断应包括Simpson-Golabi-Behmel综合征和Costello综合征。
Table 2.
过度发育紊乱在贝克-维德曼综合征鉴别诊断中的应用
| 紊乱 | 基因 | MOI | 相同的临床特征 | 不同的临床特征 |
|---|---|---|---|---|
| Simpson-Golabi-Behmel 综合征1型 | GPC3 GPC4 | XL |
|
|
| Perlman 综合征 (OMIM) | DIS3L2 | AR |
|
|
| Costello 综合征 | HRAS | AD 2 |
|
|
| Sotos 综合征 | NSD1 | AD 2 |
|
|
| 嵌合全基因组父系单亲同二体 4 | Multiple genes | Sporadic |
|
|
偏侧发育过度或节段性增生可能是一种孤立的发现,也可能与其他综合征有关,如Proteus 综合征, PTEN 错构瘤综合征, Klippel-Trenaunay-Weber 综合征 (OMIM), 和 1型神经纤维瘤病[Hoyme et al 1998]。不对称,如脸或胸部,应评估排除斜头畸形和胸壁畸形。
管理
初步诊断后的评估
为确定被诊断为贝克-维德曼综合征(BWS)的患者的患病程度和需求,建议进行以下评估:
- 巨舌症患者气道充分性评估
- 如果巨舌症引起明显的喂养困难,由喂养专家进行评估
- 新生儿低血糖的评估由儿科内分泌学专家评估低血糖是否持续超过出生最初几天。
- 腹部超声检查,以评估器官肿大、结构异常和肿瘤
- 在手术前或怀疑有心脏异常时进行心脏综合评估,包括心电图和超声心动图
- 初步诊断时用甲胎蛋白测定法评估肝母细胞瘤。利用特定年龄类别的正常范围来指导结果解释是很重要的,尤其是在非常年幼的婴儿中。
- 咨询临床遗传学家和/或遗传顾问
以下是合适的:
- 及时治疗低血糖,降低中枢神经系统并发症的风险。由于低血糖的发病有时会延迟几天,甚至几个月,所以应该告知父母低血糖的症状,以便它们能够寻求适当的医疗照顾。
- 腹壁修补术治疗出生后不久的脐膨出。一般来说,这种手术的耐受性是很好的。
- 巨舌症处理的困难:
- 预期气管插管困难[Kimura et al 2008]
- 评估呼吸功能,可能包括睡眠研究,以解决对潜在睡眠呼吸暂停的担忧
- 使用特殊乳头解决喂养困难,如建议腭裂患儿使用较长的乳头,或很少短期使用鼻饲管喂养
- 由熟悉BWS自然史的整形外科医生、正畸医生和言语病理学专家组成的颅面小组进行随访。随着时间的推移,舌头的生长速度会减慢,而下巴的生长速度会加快,以适应舌头的扩张。一些儿童受益于舌复位手术;然而,手术复位通常影响舌头的长度而不是厚度;残留的整形和语言问题可能需要进一步的评估/治疗[Tomlinson et al 2007]。
- 如儿童/青少年后期有需要,可进行牙齿矫正治疗
- 言语困难评估
- 腭裂的治疗遵循标准程序
- 如面部偏侧增生严重,应转介颅面外科医生
- 如果偏侧发育过度导致腿部长度有差异,请咨询骨科医生。在青春早期可能需要手术来闭合较长的腿生长板,以平衡最终的腿长。
- 肿瘤的治疗遵循标准的儿科肿瘤学建议
- 在一些BWS患者中,肾脏发育异常与钙排泄和沉积增加有关(即肾钙质沉着症)。在肾脏超声检查有钙沉积证据的个体中,高钙尿症的评估和肾脏CT扫描可能会有所帮助。
- 如果发现尿钙升高和/或肾结构异常,请转诊儿科肾病专家
- 将患有胃肠道结构异常的儿童转介给相关专家
- 标准规程下处理心脏问题
- 标准干预措施,如婴儿刺激计划、职业和物理治疗以及针对发育迟缓儿童的个体化教育方案
预防继发性并发症
及时评估和标准治疗可疑尿路感染是预防继发性肾损害的适当方法。
监测
以下是适当的方法:
- 检测低血糖,特别是在新生儿期
- 胚胎肿瘤的筛查,传统上涉及以下方面(见另注解):
- 在出生后前四年每两至三个月测一次甲胎蛋白(AFP)浓度,以早期发现肝母细胞瘤[Clericuzio & Martin 2009]。在BWS患儿出生后第一年,AFP血清浓度可能升高[Everman et al 2000]。 如果AFP升高,影像学检查未见可疑病变,可随访测定血清AFP浓度,一月后进行肝功能基线检查,已确定血清AFP浓度随时间变化的趋势。如果浓度没有下降,就应该对潜在的肿瘤进行彻底的搜索[Clericuzio et al 2003]。
注解: (1)有人提出根据检测到的分子改变修改肿瘤检测指南。Scott et al [2006] 认为BWS患儿和IC2改变患儿不需要Wilms 肿瘤筛查。Brioude et al [2013] 提出,BWS患儿在IC2甲基化缺失时,应该在临床诊断时进行超声评估,只有内脏肿大或“严重”偏侧发育过度时才继续超声检测;否侧,建议单独进行临床检查。Mussa et al [2016b] 怀疑检测超声和IC2甲基化缺失个体的“肿瘤标志物”的理由,但随后在意大利BWS科学委员会的指导方针中提出,在不久的将来,临床实践中的肿瘤筛查将停止。(2) 虽然建议对神经母细胞瘤进行定期X胸片、尿VMA和VHA筛查,但由于其检出率低,尚未纳入常用筛查方案。
- 对八岁至青春期中期的患者进行一年一次的肾超声检查,以确定那些需要进一步评估的发现,如肾钙质沉着和髓质海棉肾病。如有阳性结果,应转介肾科医生作进一步评估及跟进。由于成人疾病的自然史尚未被评估,可能存在没有早期发现的成人肾脏疾病。因此,应考虑成年期的定期肾评估。
- 因为在超声检查结果正常的BWS患者中可能存在异常,考虑从确立BWS诊断时开始,每年或每两年测尿钙/肌酐比值 [Goldman et al 2003]
- 把筛查发展为日常托儿工作的一部分
评估有风险的亲属
适当的做法是评估BWS患者的新生儿同胞,以便尽早确定哪些人将从预防措施中受益。
评估可以包括:
- 即使在产前检查没有明显临床表现的情况下,仍要检测高危新生儿亲属的低血糖;
- 考虑到共享胎儿的可能性和由此导致的体细胞嵌合,强烈考虑对单卵双生的双胞胎进行肿瘤检测,明确哪一个是BWS患儿。
正在调查中的疗法
搜索 ClinicalTrials.gov以获取各种疾病和条件的临床研究信息。注解:可能还没有这种疾病的临床试验。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、遗传和影响的信息,以帮助他们做出知情的医疗和个人决定的过程。下一节讨论遗传风险评估以及利用家族史和遗传检测来澄清家庭成员的遗传状况。本节并不是为了解决个体可能面临的所有个人、文化或道德问题,也不是用来代替与遗传学专业人员进行咨询的。 —ED.
正常染色体先证者对家庭成员的风险
先证者的父母
- 大多数BWS患者没有受累的父母。
- 对患有BWS且无已知BWS家族史的儿童的父母进行评估,建议包括一份医学史和一份家族史,重点关注儿童早期BWS相关的医学问题,婴儿和儿童的照片可能有用。在成年期,体格检查的价值可能有限,但耳坑/皱纹可能仍然存在。这种折痕在一般人群中也并不少见。
- 与潜在微缺失或微重复无关的甲基化改变通常是不可遗传的,因此对亲本的检测可能会得到正常的甲基化结果。
先证者的亲属. 患BWS的儿童对其他亲属的风险取决于BWS的遗传基础(Table 3)。 大多数家庭的再发风险 小于1%,然而,某些病因涉及高达50%的复发风险。
Table 3.
基于家族史和分子机制的BWS先证者对亲属的危险
| 家族史 | BWS分子机制 | 先证者对亲属的风险 |
|---|---|---|
| 阴性 (正常核型) (~85%) | 母系染色体IC2甲基化缺失(50%) | 在没有基因组的异常的情况下(例如,如微缺失/微重复[经MS-MLPA证实]或在另一个位点如NLRP2发生病原性变异的情况下,这一比例非常低) |
| 未知 (~20%) | 未知但经验上较低 1 | |
| 11p15.5的父系单亲二体性 (~20%) | 非常低,因为这个区域的UPD似乎来自于合子后的体细胞重组 | |
| IC1在母系遗传染色体 上甲基化增加(5%) | 在没有基因组的异常的情况下非常低(经由MS-MLPA证实) | |
| CDKN1C 致病性变异 (~5%) | 母系遗传的几个例子,CDKN1C 致病性变异从一个临床上未受影响的母亲到她受累的子代报告 2, 3 这类家庭的复发风险可能高达50% | |
| 微缺失,微重复(~9%) | ≤50%。当基因组的导致甲基化改变时,即使没有阳性家族史,也应考虑对父母双方进行分子检测。 4, 5 如果一个亲本具有相同的基因组的异常,必须考虑亲本效应,因为再发风险可能高达50%。 | |
| 阳性 (正常 核型) (~15%) | 先证者CDKN1C 致病性变异 (40%) | 如果母亲有CDKN1C 致病性变异,则先证者亲属的风险为50%。 如果父亲有CDKN1C 致病性变异,亲属的风险会增加,但确切数字还有待进一步的经验研究。 如果双亲都没有CDKN1C 致病性变异,先证者已确诊,亲属的风险很低;但是,有胚系嵌合的可能。 |
| 未在先证者中发现CDKN1C 致病性变异 (~60%) | ≤50% | |
| 微缺失,微重复 (~9%) | ≤50%。当基因组的异常导致甲基化改变时,即使没有阳性家族史,也应考虑对父母双方进行分子检测。 4, 5 如果双亲之一具有相同的基因组的异常,必须考虑亲本效应,因为再发风险可能高达50%。 |
- 1.
这些个体中的一部分,尤其是那些有偏侧发育过度的个体,可能有UPD11p15嵌合现象,但由于取样组织的变异程度较低,可能没有被发现。应考虑检测其他组织(例如,来自较大一侧的皮肤)。
- 2.
- 3.
出乎意料的是,还报告了一例来自临床未受影响的父亲的CDKN1C 致病性变异的父系遗传[Lee et al 1997]。
- 4.
- 5.
在未检测到CDKN1C 致病性变异且核型正常的家族性病例中,应考虑检测可遗传微缺失/微重复[Niemitz et al 2004, Sparago et al 2004, Prawitt et al 2005]。
先证者的后代
Table 4.
基于家族史和分子机制,BWS先证者对后代的影响
先证者的其他家族成员。 其他家族成员的风险取决于分子病因。
先证者染色体异常对家族成员的影响
先证者的父母. 先证者的父母有染色体结构平衡或不平衡风险,可能拥有一个平衡的染色体重排,应该提供染色体分析。
- 亲属的风险取决于父母细胞遗传的发现。
染色体异常的先证者的后代. 如果先证者是女性,风险可能高达50%。对于男性先证者来说,这种风险很低。
Table 5.
BWS和细胞遗传学异常的先证者对亲属和后代的风险
相关基因咨询问题
有关为早期诊断和治疗目的而评估高危亲属的信息,请参阅管理、风险亲属评估。
家庭计划
- 确定遗传基因风险和产前遗传咨询的最佳时间是怀孕前。关于检测高危无症状家庭成员遗传状况的决定最好在怀孕前做出。
与辅助生殖技术相关的(ART)相关的可能印记风险. 资料表明,生育力低下/ART与印记障碍之间可能存在联系[DeBaun et al 2002, Maher et al 2003a, Maher et al 2003b]。最近 DeBaun et al [2003], Gicquel et al [2003], Maher et al [2003a], 和 Halliday et al [2004] 等人报道的资料表明,ART可能倾向于对中心粒 印记的 11p15 位点 IC2 的印记改变,并且,因此可能增加不孕不育夫妇和/或正在接受抗逆转录病毒治疗的女性的子女患BWS的发病率。与ART程序相比,这些印记缺陷在造成生育力低下的潜在问题方面起着什么样的作用还不清楚。
着床前的胚胎发育阶段是印记维持的关键时期。虽然没有明确的ART技术被证实会增加贝克-维德曼综合征(Beckwith-Wiedemann syndrome,BWS)的风险,与ART有关的许多程序可能会影响 印记 ,包括卵巢卵泡刺激方案、生物技术、配子成熟阶段、培养基和胚胎移植时机。
与ART相关的Angelman 综合征的报道表明,早期发育中的表观遗传错误并不局限于贝克-维德曼综合征(Beckwith-Wiedemann syndrome,BWS)。
这些资料虽然具有回溯性,但强调有必要对ART技术出生后的孩子进行随访,并进行更大的前瞻性研究,以澄清 印记 错误风险显著增加是否与ART有关,如果是这样的话,这一发现是否与ART程序有关,还是与父母潜在的不孕有关[DeBaun et al 2003, Gicquel et al 2003, Gosden et al 2003, Maher et al 2003a, Weksberg et al 2003, Schieve et al 2004, Weksberg et al 2005]。
同卵双生. 与BWS不一致的同卵双生(通常是女性)也被证实与皮肤成纤维细胞中IC2甲基化缺失不一致,但在血细胞中却不同程度的一致,这可能是共享胎儿循环的结果[Weksberg et al 2002]。男性同卵双生子的观察频率要低得多,并显示了更多的分子发现,包括11p15的UDP和IC1的甲基化。由于在这些双胞胎中没有复发的报告,再发风险是未知的。
DNA 库 被用来存储DNA,(通常从白细胞中提取),以备将来使用。由于检测方法和我们对基因、等位基因突变、和疾病的理解可能会在未来有所改善,因此应考虑将受累的个体的DNA储存起来。
产前检测
阳性家族史. 对于有一个患有BWS孩子并且对 产前诊断 感兴趣的家庭来说,有许多选择。一些家庭可能考虑产前检查来管理怀孕和分娩;另一些家庭则可能考虑终止妊娠。
如果在先证者中发现了细胞遗传的或分子遗传异常,则对处于危险中的妊娠进行适当的检测是合理的。
基因改变,包括11p15或致病性CDKN1C变异的重复/缺失,可通过分析CVS或羊膜穿刺术获得的胎儿DNA来检测。然而,从羊水中提取的DNA目前被认为是评估胎儿 甲基化 状态最可靠的组织来源,尽管也有假阴性的结果报道[Eggermann et al 2015]。培养的羊水细胞可能表现出不代表胎儿情况的克隆结果,类似于与体细胞嵌合有关的出生后检测色问题。同样,通过CVS获得的用于产前甲基化状态检测的组织可能不会产生可靠的结果;据报道,存在假阳性检测[Eggermann et al 2015]。后一个问题反应了早期胎盘发育中甲基化建立的时机。应就表观遗传变异产前检测的潜在局限性进行遗传咨询[Eggermann et al 2015]。
对于所有妊娠期BWS风险增加的孕妇,不论其遗传机制是否已知:
- 孕妇血清甲胎蛋白(AFP)浓度在妊娠16周时可能升高。
- 可在妊娠19~20周和孕25~32周进行超声检查,以评估妊娠中晚期的生长参数,并检测腹壁缺陷、器官肿大、肾异常、腭裂、心脏异常和巨舌症。在一份报告中,对10至14周间的妊娠进行超声检查,发现胎儿有颈项透明层增厚和脐膨出,后来发现胎儿有BWS [Souka et al 1998]。
注解: (1) 如果超声检查不能显示胎儿生长畸形或异常,考虑到临床表现的多变性,BWS复发的残余风险仍然存在。(2) 即使没有明显的临床发现和产前检查结果,也应对新生儿进行低血糖检测。
阴性家族史. 在没有BWS家族史但超声检查发现明显孤立的脐膨出的异常妊娠中,应考虑进行其他调查 [Porter et al 2009, Wilkins-Haug et al 2009]包括:
- 连续超声检查评估胎儿生长发育及检测BWS的其他异常特征
注解: 妊娠年龄表示为月经周,从上一次正常月经的第一天算起,或者用超声测量。如果对BWS有很高的怀疑指数,就可以进行分子遗传测试。
资源
GeneReviews工作人员选择了以下针对特定疾病和/或伞式支持组织和/或登记册,以造福于患有这种疾病的个人及其家人。 GeneReviews 不负责其他组织提供的信息。有关选择标准的信息。请单击此处。
- 贝克-维德曼国际儿童基金会
- My46 特质剖析图
- 国家医学遗传学图书馆主页参考资料
分子遗传学
分子遗传学和OMIM表中的信息可能与基因审查的其他部分不同,表格可能包含更多的最新信息。 - ED.
Table A.
贝克-维德曼综合征(Beckwith-Wiedemann Syndrome,BWS): 基因和数据库
Table B.
贝克-维德曼综合征(Beckwith-Wiedemann Syndrome,BWS)的OMIM条目(在OMIM中查看所有)
| 103280 | H19, IMPRINTED MATERNALLY EXPRESSED NONCODING TRANSCRIPT; H19 |
| 130650 | BECKWITH-WIEDEMANN SYNDROME; BWS |
| 147470 | INSULIN-LIKE GROWTH FACTOR II; IGF2 |
| 600856 | CYCLIN-DEPENDENT KINASE INHIBITOR 1C; CDKN1C |
| 604115 | KCNQ1-OVERLAPPING TRANSCRIPT 1; KCNQ1OT1 |
| 607542 | POTASSIUM CHANNEL, VOLTAGE-GATED, KQT-LIKE SUBFAMILY, MEMBER 1; KCNQ1 |
| 616186 | H19/IGF2-IMPRINTING CONTROL REGION |
分子遗传学发病机制
印记是一种表观遗传过程,通过对一个基因的两个等位基因 的DNA进行差异修饰,使每一个基因通常只表达一个亲本等位基因 [Barlow 1994]。 印记基因聚集在基因组的不同区域, 印记中心控制着一组紧密相连的印记的基因通过胚系在传代过程中的复位 [Nicholls 1994]。
基因/位点. 一些候选的印记的基因包括生长因子和肿瘤抑制基因映射到11p15区域已是有牵连的。印记中心(IC1和IC2)通过大染色体区域控制 基因表达。该区域许多不同的分子改变与贝克-维德曼综合征(Beckwith-Wiedemann syndrome,BWS)有关。
与BWS有关的印记的 基因表达的改变. 在BWS 关键区域 中有两个印记区域(见 Figure 1a).
结构域1是端粒,并且含有 印记的 基因H19和IGF2。H19是一种非编码、非翻译的RNA,可作为肿瘤抑制因子。IGF2是一种有效的胎儿生长因子。H19和IGF2是相互表达的印记基因,H19为母系表达,IGF2为父系表达。该 结构域 的表达由H19上游的一个 印记 中心调控,称为印记中心(IC1)(也称为差异甲基化区域1[DMR1])。IC1通常在父系 等位基因 上甲基化,在母系等位基因上不甲基化。转录调控是通过将锌指绝缘体蛋白CTCF与IC1内的一致序列结合而实现的。CTCF只与非甲基化序列(母系等位基因)结合,并不干扰下游增强子与IGF2启动子的相互作用 [Hark et al 2000]。
结构域2位于着丝粒,含有CDKN1C、KCNQ1、和KCNQ1OT1 印记的基因。该 结构域 的调控由印记中心2(IC2)(以前称为差异甲基化区域2[DMR2])控制。IC2位于KCNQ1内含子10中。IC2 [Smilinich et al 1999]含有KCNQ1OT1的启动子—一种具有潜在调节功能的非编码RNA [Pandey et al 2008]。虽然这一区域的确切调控机制尚不清楚,但已知的是,母源染色体上IC2甲基化缺失会导致正常父系双等位基因的表现出 KCNQ1OT1 的表达。此外,研究表明,BWS患者和母系染色体上IC2 甲基化 缺失降低了CDKN1C的表达 [Diaz-Meyer et al 2003]。
印记中心 (IC) 是DNA的一个区域,它可以在cis中调控相邻 印记的 基因的长距离表达。ICS通常以差异DNA甲基化和差异组蛋白修饰为特征,也可以称为印记控制区域(ICRs)或差异甲基化区域(DMRs)。
- IC2 是控制KCNQ1OT1、KCNQ1和CDKN1C等多种等位基因的着丝粒印记中心。它也可称为ICR2、 DMR2、或 KvDMR1。它通常在母系等位基因上甲基化,在父系等位基因上不甲基化(例如,差异甲基化)。
- H19. 这种母系表达的基因编码了一种具有生物活性的非编码mRNA,这种信使RNA可作为肿瘤抑制因子发挥作用。大约50%的BWS患者在其组织中表现出双等位基因的 IGF2表达,表现为IGF2和H19的非耦联表达;也就是说,大多数保留了H19的正常母系单等位基因表达。较不常见的是,BWS患者体内H19表达或甲基化的变化[Joyce et al 1997, Sparago et al 2004]。
- CDKN1C 编码蛋白p57KIP2,p57KIP2 是细胞周期依赖性激酶抑制因子家族成员,对细胞增殖起负调节作用。该基因既是一个可能的抑癌基因,也是一个潜在的负调控胎儿生长的基因。这两种功能以及该基因在母体中的优先表达(对父系等位基因转录的不完全抑制)均表明该基因是BWS的候选基因。约有5% 受累的个体报告了该基因的致病变异。CDKN1C致病性变异多见于脑膨出、腭裂和家族史阳性的个体。然而,目前并非所有的BWS垂直传播都可以归因于CDKN1C 的致病变异 [Hatada et al 1997, Lee et al 1997]。
- KCNQ1. KCNQ1编码的蛋白构成钾通道的一部分,并且至少与两种心律失常综合征, Romano-Ward 综合征 和 Jervell and Lange-Nielsen 综合征有关。该基因在大多数组织(不包括心脏)中呈母系表达,有四个选择性转录本,其中2个未翻译。
- KCNQ1OT1 是一种反义转录本,起源于KCNQ1内含子10。50%BWS患者KCNQ1OT1的5'差异甲基化 启动子区(IC2)发生印记丢失 [Bliek et al 2001, Weksberg et al 2001]。
其它印记的基因.PHLDA2 (又称IPL、HLDA2、或 BWR1C) 和SLC22A18 (又称TSSC5、 BWR1A、或 ITM)是11p15区域的印记基因[Qian et al 1997, Dao et al 1998]。这两种基因在胎儿生命中均表现出优先的母系表达,且均位于CDKN1C的着丝粒。虽然这两种基因都与BWS无直接相关,但两者都被假定具有负性的生长调节功能。最近,PHLDA2已被证明对正常的胎盘/胎儿发育非常重要[Dória et al 2010, Tunster et al 2010]。
该区域基因表达量对小鼠胎儿生长的调控有重要意义。在小鼠模型中上调Igf2表达和下调Cdkn1c (p57Kip2)导致类似BWS的表型[Caspary et al 1999]。
参考文献
指导方针/共同声明
- Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Available online. 2001. Accessed 8-8-16. [PMC free article: PMC3111049] [PubMed: 11388763]
引用文献
- Algar E, Brickell S, Deeble G, Amor D, Smith P. Analysis of CDKN1C in Beckwith Wiedemann syndrome. Hum Mutat. 2000;15:497 - 508. [PubMed: 10862080]
- Alsultan A, Lovell MA, Hayes KL, Allshouse MJ, Garrington TP. Simultaneous occurrence of right adrenocortical tumor and left adrenal neuroblastoma in an infant with Beckwith-Wiedemann syndrome. Pediatr Blood Cancer. 2008;51:695 - 8. [PubMed: 18668518]
- Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, Délot EC, Rodriguez-Fernandez IA, Braslavsky D, Bergadá I, Dell'Angelica EC, Nelson SF, Martinez-Agosto JA, Achermann JC, Vilain E. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44:788 - 92. [PMC free article: PMC3386373] [PubMed: 22634751]
- Barlow DP. Imprinting: a gamete's point of view. Trends Genet. 1994;10:194 - 9. [PubMed: 7864936]
- Baskin B, Choufani S, Chen YA, Shuman C, Parkinson N, Lemyre E, Micheil Innes A, Stavropoulos DJ, Ray PN, Weksberg R. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum Genet. 2014;133:321 - 30. [PubMed: 24154661]
- Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, Munnich A, Gicquel C, Cormier-Daire V, Colleaux L. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet. 2004;74:715 - 20. [PMC free article: PMC1181947] [PubMed: 14997421]
- Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268 - 73. [PubMed: 9781906]
- Bliek J, Maas SM, Ruijter JM, Hennekam RC, Alders M, Westerveld A, Mannens MM. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum Mol Genet. 2001;10:467 - 76. [PubMed: 11181570]
- Bliek J, Verde G, Callaway J, Maas SM, De Crescenzo A, Sparago A, Cerrato F, Russo S, Ferraiuolo S, Rinaldi MM, Fischetto R, Lalatta F, Giordano L, Ferrari P, Cubellis MV, Larizza L, Temple IK, Mannens MM, Mackay DJ, Riccio A. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2009;17:611 - 9. [PMC free article: PMC2986258] [PubMed: 19092779]
- Borer JG, Kaefer M, Barnewolt CE, Elias ER, Hobbs N, Retik AB, Peters CA. Renal findings on radiological followup of patients with Beckwith-Wiedemann syndrome. J Urol. 1999;161:235 - 9. [PubMed: 10037413]
- Brioude F, Netchine I, Praz F, Le Jule M, Calmel C, Lacombe D, Edery P, Catala M, Odent S, Isidor B, Lyonnet S, Sigaudy S, Leheup B, Audebert-Bellanger S, Burglen L, Giuliano F, Alessandri JL, Cormier-Daire V, Laffargue F, Blesson S, Coupier I, Lespinasse J, Blanchet P, Boute O, Baumann C, Polak M, Doray B, Verloes A, Viot G, Le Bouc Y, Rossignol S. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Hum Mutat. 2015;36:894 - 902. [PubMed: 26077438]
- Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, Thibaud N, Faussat AM, Tauber M, Le Bouc Y, Netchine I. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet. 2013;50:823 - 30. [PubMed: 24065356]
- Caspary T, Cleary MA, Perlman EJ, Zhang P, Elledge SJ, Tilghman SM. Oppositely imprinted genes p57(Kip2) and igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev. 1999;13:3115 - 24. [PMC free article: PMC317182] [PubMed: 10601037]
- Chitayat D, Rothchild A, Ling E, Friedman JM, Couch RM, Yong SL, Baldwin VJ, Hall JG. Apparent postnatal onset of some manifestations of the Wiedemann-Beckwith syndrome. Am J Med Genet. 1990;36:434 - 9. [PubMed: 2389800]
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:343 - 54. [PubMed: 20803657]
- Choyke PL, Siegel MJ, Oz O, Sotelo-Avila C, DeBaun MR. Nonmalignant renal disease in pediatric patients with Beckwith-Wiedemann syndrome. AJR Am J Roentgenol. 1998;171:733 - 7. [PubMed: 9725306]
- Clericuzio CL, Chen E, McNeil DE, O'Connor T, Zackai EH, Medne L, Tomlinson G, DeBaun M. Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr. 2003;143:270 - 2. [PubMed: 12970646]
- Clericuzio CL, Martin RA. Diagnostic criteria and tumor screening for individuals with isolated hemiphyperplasia. Genet Med. 2009;11:220 - 2. [PMC free article: PMC3111026] [PubMed: 19367194]
- Cohen MM Jr. Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr Dev Pathol. 2005;8:287 - 304. [PubMed: 16010495]
- Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, Bowdin SC, Riccio A, Sebastio G, Bliek J, Schofield PN, Reik W, Macdonald F, Maher ER. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2005;13:1025 - 32. [PubMed: 15999116]
- Dao D, Frank D, Qian N, O'Keefe D, Vosatka RJ, Walsh CP, Tycko B. IMPT1, an imprinted gene similar to polyspecific transporter and multi-drug resistance genes. Hum Mol Genet. 1998;7:597 - 608. [PubMed: 9499412]
- DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72:156 - 60. [PMC free article: PMC378620] [PubMed: 12439823]
- DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70:604 - 11. [PMC free article: PMC384940] [PubMed: 11813134]
- Diaz-Meyer N, Day CD, Khatod K, Maher ER, Cooper W, Reik W, Junien C, Graham G, Algar E, Der Kaloustian VM, Higgins MJ. Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J Med Genet. 2003;40:797 - 801. [PMC free article: PMC1735305] [PubMed: 14627666]
- Dória S, Sousa M, Fernandes S, Ramalho C, Brandão O, Matias A, Barros A, Carvalho F. Gene expression pattern of IGF2, PHLDA2, PEG10 and CDKN1C imprinted genes in spontaneous miscarriages or fetal deaths. Epigenetics. 2010;5:444 - 50. [PubMed: 20484977]
- Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res. 2009;71 Suppl 2:30 - 5. [PubMed: 19407494]
- Eggermann T, Algar E, Lapunzina P, Mackay D, Maher ER, Mannens M, Netchine I, Prawitt D, Riccio A, Temple IK, Weksberg R. Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur J Hum Genet. 2014;22(3) [PMC free article: PMC3925261] [PubMed: 23820480]
- Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol Med. 2015;21:77 - 87. [PubMed: 25547535]
- Enklaar T, Zabel BU, Prawitt D. Beckwith–Wiedemann syndrome: multiple molecular mechanisms. Expert Rev Mol Med. 2006;8:1 - 19. [PubMed: 16842655]
- Everman DB, Shuman C, Dzolganovski B, O'riordan MA, Weksberg R, Robin NH. Serum alpha-fetoprotein levels in Beckwith-Wiedemann syndrome. J Pediatr. 2000;137:123 - 7. [PubMed: 10891834]
- Gardiner K, Chitayat D, Choufani S, Shuman C, Blaser S, Terespolsky D, Farrell S, Reiss R, Wodak S, Pu S, Ray PN, Baskin B, Weksberg R. Brain abnormalities in patients with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2012;158A:1388 - 94. [PubMed: 22585446]
- Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72:1338 - 41. [PMC free article: PMC1180288] [PubMed: 12772698]
- Goldman M, Shuman C, Weksberg R, Rosenblum ND. Hypercalciuria in Beckwith-Wiedemann syndrome. J Pediatr. 2003;142:206 - 8. [PubMed: 12584548]
- Gosden R, Trasler J, Lucifero D, Faddy M. Rare congenital disorders, imprinted genes, and assisted reproductive technology. Lancet. 2003;361:1975 - 7. [PubMed: 12801753]
- Greer KJ, Kirkpatrick SJ, Weksberg R, Pauli RM. Beckwith-Wiedemann syndrome in adults: observations from one family and recommendations for care. Am J Med Genet A. 2008;146A:1707 - 12. [PubMed: 18546283]
- Halliday J, Oke K, Breheny S, Algar E, J, Amor D. Beckwith–Wiedemann syndrome and IVF: a case – control study. Am J Hum Genet. 2004;75:526 - 8. [PMC free article: PMC1182036] [PubMed: 15284956]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486 - 9. [PubMed: 10839547]
- Hatada I, Nabetani A, Morisaki H, Xin Z, Ohishi S, Tonoki H, Niikawa N, Inoue M, Komoto Y, Okada A, Steichen E, Ohashi H, Fukushima Y, Nakayama M, Mukai T. New p57KIP2 mutations in Beckwith-Wiedemann syndrome. Hum Genet. 1997;100:681 - 3. [PubMed: 9341892]
- Hopsu E, Aarnisalo A, Pitkaranta A. Progressive stapedial fixation in Beckwith-Wiedemann syndrome. Arch Otolaryngol Head Neck Surg. 2003;129:1131 - 4. [PubMed: 14568801]
- Hoyme HE, Seaver LH, Jones KL, Procopio F, Crooks W, Feingold M. Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. 1998;79:274 - 8. [PubMed: 9781907]
- Ibrahim A, Kirby G, Hardy C, Dias RP, Tee L, Lim D, Berg J, MacDonald F, Nightingale P, Maher ER. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics. 2014 Jun 4;6(1):11. [PMC free article: PMC4064264] [PubMed: 24982696]
- Inbar-Feigenberg M, Choufani S, Cytrynbaum C, Chen YA, Steele L, Shuman C, Ray PN, Weksberg R. Mosaicism for genome-wide paternal uniparental disomy with features of multiple imprinting disorders: diagnostic and management issues. Am J Med Genet A. 2013 Jan;161A(1):13 - 20. [PubMed: 23239666]
- Joyce JA, Lam WK, Catchpoole DJ, Jenks P, Reik W, Maher ER, Schofield PN. Imprinting of IGF2 and H19: lack of reciprocity in sporadic Beckwith- Wiedemann syndrome. Hum Mol Genet. 1997;6:1543 - 8. [PubMed: 9285792]
- Kalish JM, Conlin LK, Bhatti TR, Dubbs HA, Harris MC, Izumi K, Mostoufi-Moab S, Mulchandani S, Saitta S, States LJ, Swarr DT, Wilkens AB, Zackai EH, Zelley K, Bartolomei MS, Nichols KE, Palladino AA, Spinner NB, Deardorff MA. Clinical features of three girls with mosaic genome-wide paternal uniparental isodisomy. Am J Med Genet A. 2013 Aug;161A(8):1929 - 39. [PMC free article: PMC4082120] [PubMed: 23804593]
- Kaltenbach S, Capri Y, Rossignol S, Denjoy I, Soudée S, Aboura A, Baumann C, Verloes A. Beckwith-Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin Genet. 2013 Jul;84(1):78 - 81. [PubMed: 23061425]
- Kantaputra PN, Sittiwangkul R, Sonsuwan N, Romanelli V, Tenorio J, Lapunzina P. A novel mutation in CDKN1C in sibs with Beckwith-Wiedemann syndrome and cleft palate, sensorineural hearing loss, and supernumerary flexion creases. Am J Med Genet A. 2013 Jan;161A(1):192 - 7. [PubMed: 23197429]
- Keren B, Chantot-Bastaraud S, Brioude F, Mach C, Fonteneau E, Azzi S, Depienne C, Brice A, Netchine I, Le Bouc Y, Siffroi JP, Rossignol S. SNP arrays in Beckwith-Wiedemann syndrome: an improved diagnostic strategy. Eur J Med Genet. 2013 Oct;56(10):546 - 50. [PubMed: 23892181]
- Kimura Y, Yasuhiro K, Kimura S. Anesthetic management of two cases of Beckwith-Wiedemann syndrome. J Anesth. 2008;22:93 - 95. [PubMed: 18306025]
- Kuroiwa M, Sakamoto J, Shimada A, Suzuki N, Hirato J, Park MJ, Sotomatsu M, Hayashi Y. Manifestation of alveolar rhabdomyosarcoma as primary cutaneous lesions in a neonate with Beckwith-Wiedemann syndrome. J Pediatr Surg. 2009;44:e31 - 5. [PubMed: 19302842]
- Lam WW, Hatada I, Ohishi S, Mukai T, Joyce JA, Cole TR, Donnai D, Reik W, Schofield PN, Maher ER. Analysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype- phenotype correlation. J Med Genet. 1999;36:518 - 23. [PMC free article: PMC1734395] [PubMed: 10424811]
- Lee MP, DeBaun M, Randhawa G, Reichard BA, Elledge SJ, Feinberg AP. Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am J Hum Genet. 1997;61:304 - 9. [PMC free article: PMC1715913] [PubMed: 9311734]
- Lew JM, Fei YL, Aleck K, Blencowe BJ, Weksberg R, Sadowski PD. CDKN1C mutation in Wiedemann-Beckwith syndrome patients reduces RNA splicing efficiency and identifies a splicing enhancer. Am J Med Genet A. 2004;127A:268 - 76. [PubMed: 15150778]
- Li M, Squire J, Shuman C, Fei YL, Atkin J, Pauli R, Smith A, Nishikawa J, Chitayat D, Weksberg R. Imprinting status of 11p15 genes in Beckwith-Wiedemann syndrome patients with CDKN1C mutations. Genomics. 2001;74:370 - 6. [PubMed: 11414765]
- Li M, Squire JA, Weksberg R. Molecular genetics of Wiedemann-Beckwith syndrome. Am J Med Genet. 1998;79:253 - 9. [PubMed: 9781904]
- Maher ER, Afnan M, Barratt CL. Epigenetic risks related to assisted reproductive technologies: epigenetics, imprinting, ART and icebergs? Hum Reprod. 2003a;18:2508 - 11. [PubMed: 14645164]
- Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, Macdonald F, Sampson JR, Barratt CL, Reik W, Hawkins MM. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet. 2003b;40:62 - 4. [PMC free article: PMC1735252] [PubMed: 12525545]
- Martin RA, Grange DK, Zehnbauer B, Debaun MR. LIT1 and H19 methylation defects in isolated hemihyperplasia. Am J Med Genet A. 2005;134A:129 - 31. [PubMed: 15651076]
- Milani D, Pezzani L, Tabano S, Miozzo M. Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet. 2014 Sep 16;7:169 - 75. [PMC free article: PMC4173641] [PubMed: 25258553]
- Mussa A, Di Candia S, Russo S, Catania S, De Pellegrin M, Di Luzio L, Ferrari M, Tortora C, Meazzini MC, Brusati R, Milani D, Zampino G, Montirosso R, Riccio A, Selicorni A, Cocchi G, Ferrero GB. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur J Med Genet. 2016a Jan;59(1):52 - 64. [PubMed: 26592461]
- Mussa A, Peruzzi L, Chiesa N, De Crescenzo A, Russo S, Melis D, Tarani L, Baldassarre G, Larizza L, Riccio A, Silengo M, Ferrero GB. Nephrological findings and genotype-phenotype correlation in Beckwith-Wiedemann syndrome. Pediatr Nephrol. 2012 Mar;27(3):397 - 406. [PubMed: 22015620]
- Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, Richiardi L, Larizza L, Silengo MC, Riccio A, Ferrero GB. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am J Med Genet A. 2013 Oct;161A(10):2481 - 6. [PubMed: 23918458]
- Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, Macchiaiolo M, Molinatto C, Baldassarre G, Mariani M, Tarani L, Bedeschi MF, Milani D, Melis D, Bartuli A, Cubellis MV, Selicorni A, Cirillo Silengo M, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016b Feb;24(2):183 - 90. [PMC free article: PMC4717210] [PubMed: 25898929]
- Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet. 2015 Jul 3; Epub ahead of print. [PubMed: 26138266]
- Nicholls RD. New insights reveal complex mechanisms involved in genomic imprinting. Am J Hum Genet. 1994;54:733 - 40. [editorial; comment] [PMC free article: PMC1918270] [PubMed: 8178814]
- Niemitz EL, DeBaun MR, Fallon J, Murakami K, Kugoh H, Oshimura M, Feinberg AP. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am J Hum Genet. 2004;75:844 - 9. [PMC free article: PMC1182113] [PubMed: 15372379]
- O'Keefe D, Dao D, Zhao L, Sanderson R, Warburton D, Weiss L, Anyane-Yeboa K, Tycko B. Coding mutations in p57KIP2 are present in some cases of Beckwith-Wiedemann syndrome but are rare or absent in Wilms tumors. Am J Hum Genet. 1997;61:295 - 303. [PMC free article: PMC1715902] [PubMed: 9311733]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232 - 46. [PubMed: 18951091]
- Percesepe A, Bertucci E, Ferrari P, Lugli L, Ferrari F, Mazza V, Forabosco A. Familial Beckwith-Wiedemann syndrome due to CDKN1C mutation manifesting with recurring omphalocele. Prenat Diagn. 2008;28:447 - 9. [PubMed: 18395877]
- Pettenati MJ, Haines JL, Higgins RR, Wappner RS, Palmer CG, Weaver DD. Wiedemann-Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet. 1986;74:143 - 54. [PubMed: 3770742]
- Porter A, Benson CB, Hawley P, Wilkins-Haug L. Outcome of fetuses with a prenatal ultrasound diagnosis of isolated omphalocele. Prenat Diagn. 2009;29:668 - 73. [PubMed: 19367563]
- Prawitt D, Enklaar T, Gartner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, Schmidtke P, Reutzel D, Fees S, Lucito R, Korzon M, Brozek I, Limon J, Housman DE, Pelletier J, Zabel B. Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc Natl Acad Sci U S A. 2005;102:4085 - 90. [PMC free article: PMC554791] [PubMed: 15743916]
- Qian N, Frank D, O'Keefe D, Dao D, Zhao L, Yuan L, Wang Q, Keating M, Walsh C, Tycko B. The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum Mol Genet. 1997;6:2021 - 9. [PubMed: 9328465]
- Rump P, Zeegers MP, van Essen AJ. Tumor risk in Beckwith–Wiedemann syndrome: a review and meta-analysis. Am J Med Genet. 2005;136:95 - 104. [PubMed: 15887271]
- Russo S, Calzari L, Mussa A, Mainini E, Cassina M, Di Candia S, Clementi M, Guzzetti S, Tabano S, Miozzo M, Sirchia S, Finelli P, Prontera P, Maitz S, Sorge G, Calcagno A, Maghnie M, Divizia MT, Melis D, Manfredini E, Ferrero GB, Pecile V, Larizza L. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin Epigenetics. 2016 Mar 1;8:23. [PMC free article: PMC4772365] [PubMed: 26933465]
- Sasaki K, Soejima H, Higashimoto K, Yatsuki H, Ohashi H, Yakabe S, Joh K, Niikawa N, Mukai T. Japanese and North American/European patients with Beckwith-Wiedemann syndrome have different frequencies of some epigenetic and genetic alterations. Eur J Hum Genet. 2007;15:1205 - 10. [PubMed: 17700627]
- Schieve LA, Rasmussen SA, Buck GM, Schendel DE, Reynolds MA, Wright VC. Are children born after assisted reproductive technology at increased risk for adverse health outcomes? Obstet Gynecol. 2004;103:1154 - 63. [PubMed: 15172847]
- Scott RH, Douglas J, Baskcomb L, Nygren AO, Birch JM, Cole TR, Cormier-Daire V, Eastwood DM, Garcia-Minaur S, Lupunzina P, Tatton-Brown K, Bliek J, Maher ER, Rahman N. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J Med Genet. 2008b;45:106 - 13. [PubMed: 18245390]
- Scott RH, Walker L, Olsen ØE, Levitt G, Kenney I, Maher E, Owens CM, Pritchard-Jones K, Craft A, Rahman N. Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child. 2006 Dec;91(12):995 - 9. [PMC free article: PMC2083016] [PubMed: 16857697]
- Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, Craft A, Gerrard M, Kohler JA, Levitt GA, Picton S, Pizer B, Ronghe MD, Williams D., Factors Associated with Childhood Tumours (FACT) Collaboration. Cook JA, Pujol P, Maher ER, Birch JM, Stiller CA, Pritchard-Jones K, Rahman N. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. 2008a Nov;40(11):1329 - 34. [PubMed: 18836444]
- Shuman C, Steele L, Fei YL, Ray PN, Zackai E, Parisi M, Squire J, Weksberg R (2002) Paternal uniparental disomy of 11p15 is associated with isolated hemihyperplasia and expands Beckwith-Wiedemann syndrome spectrum. Am J Hum Genet 71:A1800, 477.
- Slavotinek A, Gaunt L, Donnai D. Paternally inherited duplications of 11p15.5 and Beckwith-Wiedemann syndrome. J Med Genet. 1997;34:819 - 26. [PMC free article: PMC1051088] [PubMed: 9350814]
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB, Higgins MJ. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A. 1999;96:8064 - 9. [PMC free article: PMC22188] [PubMed: 10393948]
- Smith AC, Rubin T, Shuman C, Estabrooks L, Aylsworth AS, McDonald MT, Steele L, Ray PN, Weksberg R. New chromosome 11p15 epigenotypes identified in male monozygotic twins with Beckwith–Wiedemann syndrome. Cytogenet Genome Res. 2006;113:313 - 7. [PubMed: 16575195]
- Souka AP, Snijders RJ, Novakov A, Soares W, Nicolaides KH. Defects and syndromes in chromosomally normal fetuses with increased nuchal translucency thickness at 10-14 weeks of gestation. Ultrasound Obstet Gynecol. 1998;11:391 - 400. [PubMed: 9674084]
- Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36:958 - 60. [PubMed: 15314640]
- Tan TY, Amor DJ. Tumour surveillance in Beckwith-Wiedemann syndrome and hemihyperplasia: a critical review of the evidence and suggested guidelines for local practice. J Paediatr Child Health. 2006;42:486 - 90. [PubMed: 16925531]
- Thorburn MJ, Wright ES, Miller CG, Smith-Read EH. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child. 1970 Apr;119(4):316 - 21. [PubMed: 5434588]
- Tomlinson JK, Morse SA, Bernard SP, Greensmith AL, Meara JG. Long-term outcomes of surgical tongue reduction in Beckwith–Wiedemann syndrome. Plast Reconstr Surg. 2007;119:992 - 1002. [PubMed: 17312506]
- Tunster SJ, Tycko B, John RM. The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol Cell Biol. 2010;30:295 - 306. [PMC free article: PMC2798284] [PubMed: 19884348]
- van Eeghen AM, van Gelderen I, Hennekam RC. Costello syndrome: report and review. Am J Med Genet. 1999;82:187 - 93. [PubMed: 9934987]
- Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, Steele L, Cameron J, Smith A, Ambus I, Li M, Ray PN, Sadowski P, Squire J. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989 - 3000. [PubMed: 11751681]
- Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, Stockley TL, Best L, Chitayat D, Olney A, Ives E, Schneider A, Bestor TH, Li M, Sadowski P, Squire J. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet. 2002;11:1317 - 25. [PubMed: 12019213]
- Weksberg R, Shuman C, Smith AC. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:12 - 23. [PubMed: 16010676]
- Weksberg R, Shuman C. Beckwith-Wiedemann syndrome and hemihypertrophy In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. 2 ed. Hoboken, New Jersey: John Wiley & Sons; 2004:101-16.
- Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet. 2003;12(Spec No 1):R61 - 8. [PubMed: 12668598]
- Wilkins-Haug L, Porter A, Hawley P, Benson CB. Isolated fetal omphalocele, Beckwith-Wiedemann syndrome, and assisted reproductive technologies. Birth Defects Res A Clin Mol Teratol. 2009;85:58 - 62. [PubMed: 19107956]
- Wilson M, Peters G, Bennetts B, McGillivray G, Wu ZH, Poon C, Algar E. The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: Two new reports. Am J Med Genet. 2008;146A:137 - 148. [PubMed: 18033734]
- Zarate YA, Mena R, Martin LJ, Steele P, Tinkle BT, Hopkin RJ. Experience with hemihyperplasia and Beckwith-Wiedemann syndrome surveillance protocol. Am J Med Genet A. 2009;149A:1691 - 7. [PubMed: 19610116]
- Zeschnigk M, Albrecht B, Buiting K, Kanber D, Eggermann T, Binder G, Gromoll J, Prott EC, Seland S, Horsthemke B. IGF2/H19 hypomethylation in Silver-Russell syndrome and isolated hemihypoplasia. Eur J Hum Genet. 2008;16:328 - 34. [PubMed: 18159214]
推荐阅读
- Bliek J, Gicquel C, Maas S, Gaston V, Le Bouc Y, Mannens M. Epigenotyping as a toll for the predicition of tumor risk and tumor type in patients with Beckwith–Wiedemann syndrome (BWS). J Pediatr. 2004;145:796 - 9. [PubMed: 15580204]
- Du M, Zhou W, Beatty LG, Weksberg R, Sadowski PD. The KCNQ1OT1 promoter, a key regulator of genomic imprinting in human chromosome 11p15.5. Genomics. 2004;84:288 - 300. [PubMed: 15233993]
- Nicholls RD. The impact of genomic imprinting for neurobehavioral and developmental disorders. J Clin Invest. 2000;105:413 - 8. [PMC free article: PMC289176] [PubMed: 10683369]
- Schwienbacher C, Sabbioni S, Campi M, Veronese A, Bernardi G, Menegatti A, Hatada I, Mukai T, Ohashi H, Barbanti-Brodano G, Croce CM, Negrini M. Transcriptional map of 170-kb region at chromosome 11p15.5: identification and mutational analysis of the BWR1A gene reveals the presence of mutations in tumor samples. Proc Natl Acad Sci U S A. 1998;95:3873 - 8. [PMC free article: PMC19930] [PubMed: 9520460]
章节注释
致谢
Sanaa Choufani, Adam Smith, Khadine Wiltshire
作者背景
J Bruce Beckwith, MD (2000-present)
Cheryl Shuman, MS, CGC (2000-present)
Adam C Smith, PhD; The Hospital for Sick Children (2000-2016)
Rosanna Weksberg, MD, PhD, FRCPC, FCCMG, FACMG (2000-present)
修订记录
- 2016年8月11日(ma)全面即时更新
- 2010年12月14日(me)全面即时更新
- 2015年9月8日8(me)全面即时更新并发布到网站
- 2033年4月10日(tk)全面即时更新并发布到网站
- 2000年3月3日(me)回顾并发布到网站
- 1999年7月28日(cs)原始意见