
摘要
临床特征.
强直性肌营养不良2型(DM2)以肌强直(占90%受累的个体)和肌肉功能障碍(无力、疼痛和僵硬)(82%)为特征,较少见心脏传导缺陷、虹膜后囊下白内障、胰岛素不敏感性2型糖尿病和睾丸功能衰竭。 尽管肌强直(不自主的肌肉收缩并延迟放松)在生命第一个十年被报道过,但通常在第三个十年发病,最常见的是波动性或阵发性肌肉疼痛,可使颈部屈肌和手指屈肌虚弱无力。随后,肘伸肌和髋屈肌及伸肌出现无力。面部无力和踝背屈肌无力较少见。肌强直罕见引起严重症状。
Diagnosis/testing诊断/检测.
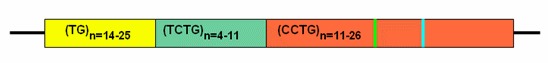
CNBP(ZNF9)是唯一已知突变导致强直性营养不良2型的 基因 。内含子包含一个复杂的重复序列(TG)n(TCTG)n(CCTG)n。 CCTG重复序列的扩增导致DM2。 在扩增的等位基因中CCTG的重复次数从约75到11000以上,平均约5000次重复。通过常规PCR、Southern blot 分析和PCR重复引物检测技术相结合,CNBP CCTG扩增的检出率超过99%。
管理.
对症治疗: 踝足矫形器、轮椅或其他辅助设备(视虚弱情况而定);心律失常患者放置除颤器;摘除损害视力的白内障;睾酮替代疗法治疗男性性腺功能低下。肌强直很少需要治疗。 日常体育锻炼可能有助于保持肌力和耐力,并控制缓解肌肉骨骼疼痛。在控制缓解疼痛治疗中取得一定成效的药物包括美西律,加巴喷丁,非甾体类抗炎药(NSAIDS),低剂量甲状腺替代品,低剂量类固醇(例如,5 mg泼尼松隔日给药)和三环类抗抑郁药。
预防继发并发症: 麻醉风险可能增加,因此建议在手术前后评估心脏和呼吸功能。及时治疗甲状腺功能减退症以减少继发性虚弱。
监测: 每年进行心电图和超声心动图或可能的心脏磁共振检查,以检测/监测心脏传导缺陷和心肌病;每年测量空腹血糖浓度和糖化血红蛋白水平;每隔几年对男性进行性腺功能减退的检查。
避免接触药物/环境: 与增加肌无力有关的降胆固醇药物。
遗传咨询.
强直性肌营养不良2型为常染色体显性遗传。 迄今为止,所有已通过分子遗传学检测 进行评估的生物学亲本个体,其父母中均有一位携带CNBP 扩增;de novo 尚未有报道。携带CNBP扩增个体的子代均有50%的概率遗传该变异。 无论是显性等位基因 的大小,还是单个样本中可检测到的不同扩增的总数,都不能预测疾病的严重程度、发病年龄或临床表现。如果在受累的父母中发现CNBP扩增,则可对风险增加的妊娠进行产前检测。
诊断
临床诊断
有以下临床表现应怀疑有强直性肌营养不良2型:
- 肌无力早期表现为临床手部运动测验示颈部屈肌和手指屈肌无力,后期肌无力通常累及臀部肌肉,发生在爬楼梯和从椅子上站起来时
- 肌强直(持续性肌肉收缩),可表现为紧握性肌强直(无法快速松开紧握的拳头),最早出现于生命的第一个十年,叩击性肌强直(用叩诊锤敲击肌肉后持续收缩)或电性肌强直(在肌电图上观察到重复自发放电)。DM2患者的肌强直并不总是可以通过肌电图检测到 [Dabby et al 2011].
- 后囊下白内障在直接检眼镜上表现为非特异性的空泡和混浊,或在裂隙灯检查中表现为病理性的后囊下红色和绿色虹膜混浊
- 心脏传导缺陷或进行性心肌病,前者可通过常规心电图诊断为房室或各种室内传导缺陷,后者可通过超声心动图上诊断为扩张型心肌病
- 低丙种球蛋白血症,定义为血清蛋白电泳的低γ蛋白组分或免疫蛋白电泳的低免疫球蛋白G或免疫球蛋白M含量,该症发生在75%的1型和2型强直性肌营养不良的成人中,与任何临床异常无关
- 胰岛素敏感性不佳,尽管血清胰岛素浓度正常或升高,但在临床上可能表现为葡萄糖耐量试验中葡萄糖正常化受损,并且易导致高血糖和糖尿病
- 男性原发性性腺功能衰竭,表现为血清睾酮浓度低、血清FSH浓度升高、少精症和不育
检测
肌肉活检. 肌肉病理学包括萎缩性肌纤维、散在重度萎缩纤维致密肌核、纤维明显增生中央核[Day et al 1999, Day et al 2003, Schoser et al 2004c],所有这些均可发生在强直性肌营养不良1型(DM1)和DM2型中,因此不能用来鉴别二者:
- 1型纤维萎缩是先天性DM1患者的一个常见特征,与DM2不同。.
- 在患有DM2的个体中观察到了优先型2型纤维萎缩 [Vihola et al 2003, Schoser et al 2004c, Pisani et al 2008, Giagnacovo et al 2012].
分子遗传学检测
基因.CNBP基因(原名ZNF9)是编码细胞核酸结合蛋白(锌指蛋白9),是唯一已知突变导致DM2的基因。CNBP内含子1包含一个复杂的重复序列(TG)n(TCTG)n(CCTG)n。CCTG重复序列扩增导致DM2 [Liquori et al 2001].
等位基因大小
正常等位基因. 所有三个重复域(TG、TCTG和CCTG)以一个复杂序列存在于所有正常和致病等位基因上;此外,正常等位基因中的CCTG重复域通常包含一个或多个四核苷酸阻断(TCTG或GCTG)[Liquori et al 2003] (参见 Figure 1). 正常等位基因中的(TG)n(TCTG)n(CCTG)n复杂重复序列的总长度在104~176个碱基对之间。由于TG和TCTG重复序列具有高度多态性,用测序以外的方法确定等位基因的大小以总碱基对长度报告,而不是以确定的CCTG重复次数报告。.
24个正常等位基因序列分析 [Liquori et al 2001, Liquori et al 2003] 显示:- 连续的CCTG重复次数最多是9次(下面讨论的单个案例除外);参见可变正常等位基因.
- 整个正常的CCTG重复序列,包括任何GCTC和TCTG阻断,范围为11-26个四核苷酸序列重复.
- 在85%的未受累个体中,复杂的重复序列全长在两个等位基因上明显不同,因此在PCR扩增分析中也有所不同.
- 可变的正常等位基因(也称为中间型或前突变等位基因). 在177-372bp(相当于约27-74个CCTG重复)的大小范围内没有CNBP等位基因的报道;此外,目前尚不清楚具体特定大小范围或具体序列特征的正常等位基因会更易于扩增至致病范围. 序列中断常发生在正常等位基因CCTG序列中,而致病性CCTG扩增序列中未发现. 正常等位基因缺乏中断可能会增加不稳定性,并使这些重复域在后代中易于扩增;尽管这种不稳定性已被其他疾病证实,但在DM2患者中尚未观察到. 然而,同这一假设相符的报道显示,在一个未受累个体的等位基因中,20个连续的CCTG重复序列出现在与所有受累个体相同的单倍体上 [Liquori et al 2003]. 即使该个体的重复序列在传递给下一代时没有扩增,但是序列缺乏中断可能会使连续的CCTG重复序列在后代中易于扩增. 由于尚未证实大的正常CNBP等位基因重复可以扩增到致病范围,因此177-372bp范围内的等位基因被更准确地称为边缘扩增而不是前突变.
- 完全 外显率 (或者异常) 等位基因. 在已测序的较小致病性等位基因中,仅复杂重复序列的CCTG部分显示出扩增。在不能精确测序的大的扩增中,大于372 bp的等位基因(相当于75个CCTG重复)似乎导致DM2完全外显.致病性等位基因的大小范围从372 bp到44,000 bp以上(相当于约75-11,000个CCTG重复),平均约20,000 bp(相当于约5,000个重复).
CCTG重复序列显示:- 体细胞不稳定性. CCTG重复序列大小随年龄增长而增加。超过25%的受累个体在外周血中检测到两个或两个以上的CCTG扩增。CCTG重复序列大小的体细胞异质性使其难以确认致病阈值;例如,在一个等位基因上CCTG重复扩增最短的受累个体(约75个CCTG重复或300个bp),也有一个非常大的超过11000个CCTG重复或44000个bp扩增的等位基因;其中的的一个或两个扩增等位基因都可能是致病的[Liquori et al 2001, Day et al 2003].
- 代际不稳定性. 在传递至下一代时,CNBP重复序列长度有时会显著减少,这种显著差异与父母性别无关;然而,重复序列长度的 体细胞嵌合 和年龄依赖性使这一观察结果的解释复杂化 [Day et al 2003].
临床检测
- 靶向变异分析. 由于CCTG扩增的范围较大且存在 体细胞嵌合 (由于CCTG扩增的体细胞不稳定性导致等位基因长度变化),因此异常CNBP等位基因的检测变得更为复杂. 通过应用常规 PCR, Southern blot分析和“PCR重复引物分析”相结合的诊断测试方法,CNBP CCTG扩增的检测率可提高到99%以上[Day et al 2003]. 2012年,发表了有关1型和2型强直性肌营养不良分子诊断的最佳实践指南和建议. 参见 Kamsteeg et al [2012] (full text).
强直性肌营养不良2型分子遗传学检测概述
| 基因 1 | 检测方法 | 检出变异 2 | 该检测方法检出率 3 |
|---|---|---|---|
| CNBP | 靶向变异分析 4 | 内含子 1的CCTG 四核苷酸重复扩增 | 99% 5 |
| 序列分析 6 | 序列变异 | 无案例报道 7 |
1. 染色体 位点和蛋白质信息请参阅Table A. Genes and Databases.
2. 等位基因变异参见Molecular Genetics .
3. 该检测方法检测致病性 基因变异的检出率.
- 4.
可能包括:
(a) 常规 PCR,可检测正常大小的等位基因,但不能检测有异常的等位基因,因为它不能在扩增中放大;
(b) Southern blot分析,可检测到约80%的扩增;
(c) PCR重复引物分析,有助于检测CCTG重复扩增。该检测方法中引物与延伸的CCTG重复序列相邻,其中以涂片方法对不同重复扩增大小的等位基因进行差异检测,而对照等位基因则以离散带方法进行检测。用内部探针检测PCR重复引物的产物,以确保必要的特异性.5.- 5.
致病变异检出率因检测方法不同。当常规 PCR分析, Southern blot 分析和PCR重复引物分析均使用时,致病变异检出率大于99%.
- 6.
- 7.
由于侧翼TG和TCTG重复次数的变化以及CCTG重复域内中断次数的变化,精确的CCTG重复数只能通过序列分析确定。即使对一些扩增等位基因进行测序以表明只有部分CCTG重复序列在受累个体中扩增是必要的,但通过测序确定特定的CCTG重复长度不用于诊断目的,因为大多数扩增数过长,无法进行有效的序列分析.
结果解释
检测策略
明确/建立先证者诊断。如果常规 PCR分析在15%的 纯合性正常个体和所有受累的个体中,只检测到一个 等位基因,有必要进行Southern blot 分析和PCR重复引物分析以确定该个体是否为纯合子。因为正常大小的等位基因或者既有正常大小的等位基因又有扩增的等位基因大小较大而无法通过PCR扩增 [Liquori et al 2001].
对有患病风险亲属进行携带者筛查前提是家系中已检出致病性变异。
产前诊断和植入前遗传诊断 (PGD) 在家系中检出已知致病性变异时可施行。
临床特征
临床表现
强直性肌营养不良2型(DM2)是以肌强直(90%)和肌肉功能障碍(无力、疼痛和僵硬)(82%)为特征的多系统疾病,以及一些看似无关的临床特征,包括:心脏传导缺陷(19%)、随年龄增长增加的虹膜后囊下白内障(36%-78%),一组特定的内分泌变化包括胰岛素不敏感(25%-75%,随年龄增长而增加)和睾丸功能障碍(29%-65%)。
尽管在生命的第一个十年有肌强直的报道,但DM2患者的症状通常出现在第三个十年,最常见的症状是肌肉无力和疼痛[Day et al 1999, Day et al 2003, Udd et al 2011]. 注意,与 myotonic dystrophy type 1(DM1)不同,DM1在成年期可表现为退行性疾病或具有不同严重的 先天 特征,而DM2与发育异常无关,因此不会引起严重的儿童期症状[Udd等人2011] [Udd et al 2011]. 任何患有DM2 受累的的家庭成员中都没有发育缺陷,这是两种DM类型之间可靠且具临床意义的显著差异。
肌肉功能障碍. DM2患者常因肌肉无力、疼痛和肌强直就诊 [Ricker et al 1994, Moxley 1996, Day et al 1999, Ricker 1999, Ricker et al 1999, Thornton 1999, Harper 2001, Day et al 2003]. 在疾病的最早阶段 受累的肌肉部位是颈部屈肌和手指屈肌。 随后,在肘部伸肌以及髋屈肌和伸肌中出现无力症状。30%的人在50岁以后会出现臀肌无力。
面部肌肉无力和踝背屈肌无力也可能发生,但较少见。
肌强直,即肌肉过度兴奋引起的不自主肌肉收缩和放松延迟,几乎发生在所有DM2患者中,但很少导致严重的临床病症。
大多数 受累的 的个体均有波动性或阵发性肌肉疼痛的报道,该疼痛会使人虚弱无力 [George et al 2004, Auvinen et al 2008, Tieleman et al 2011, Suokas et al 2012]
多系统功能. 早在生命的第二个十年可于裂隙灯观察下发现虹膜后囊下白内障。报道的白内障摘除年龄范围为28至74岁[Day et al 2003]。
尽管DM2患者的心脏受累程度比DM1患者轻 [Meola et al 2002; Sansone et al 2013], 但DM2与房室传导和脑室内传导缺陷、心律失常、心肌病和猝死有关[Nguyen et al 1988, Colleran et al 1997, Merino et al 1998, Philips et al 1998, Day et al 2003, Schoser et al 2004b]. 一项研究表明DM2与最初报道估计相比,与左心室功能障碍更为相关 [Wahbi et al 2009].
麻醉并发症在DM2患者中未见报道,其发生率可能低于DM1, DM1患者术中及术后心律失常、通气抑制和气道护理不当被认为是造成严重发病率和死亡率的可能原因 [Kirzinger et al 2010, Weingarten et al 2010].
DM2患者的内分泌异常包括胰岛素不敏感型2型糖尿病和导致男性不育的睾丸功能障碍 [Day et al 2003, Savkur et al 2004].
DM2患者,和DM1患者一样,低丙种球蛋白血症的发生率较高,IgG和IgM均低于正常水平,但未观察到相关临床问题。
报道的DM2患者中枢神经系统异常包括MRI明显的白质改变和PET扫描显示额叶和颞叶的脑血流明显减少[Hund et al 1997, Meola et al 1999, Franc et al 2012]. 这些解剖学上改变似乎对认知、行为和性格有一定的影响,虽然与DM1不同,但DM2与智力障碍无关 [Meola et al 2002, Meola et al 2003].
在DM2患者的大量案例报道和个体报道中,观察到一组睡眠障碍,包括白天嗜睡,失眠,REM行为障碍和不宁腿综合征 [Day et al 1999, Bhat et al 2012, Chokroverty et al 2012, Shepard et al 2012].
胃肠道并发症在DM2中较为常见,可能包括便秘、吞咽困难和腹痛 [Tieleman et al 2008]
患有DM2的女性在怀孕期间症状可能会恶化[Day et al 1999, Newman et al 1999, Rudnik-Schöneborn et al 2006]. 羊水过多是DM1的一个公认特征,但在DM2患者中未见报道。
致癌风险. 一些案例报道显示,患有强直性肌营养不良患者的癌症发病率增加 。最近的回顾性研究表明,肌强直性营养不良2型 受累的个体似乎患癌症的整体风险更高。DM2受累个体的癌症可能涉及结肠、大脑、甲状腺、胰腺、卵巢和子宫内膜 [Gadalla et al 2011, Win et al 2012].
基因型-表型关联
CCTG重复序列的大小与肌无力的发病年龄或其他疾病严重程度无显著相关性(如白内障摘除年龄)。观察发现在两个CNBP等位基因中均带有CCTG重复扩增的个体与他们杂合的同胞及父母的表型特征相同,这进一步表明CCTG重复数不会改变临床进程 [Schoser et al 2004a].
在测量重复大小时,重复大小与DM2个体的年龄之间存在相关性,表明重复长度随年龄增加而增加[Schneider et al 2000, Liquori et al 2001, Day et al 2003].
外显率
疾病 的外显率 既反映了个体对症状的敏感性,也反映了医生正确识别和解释疾病迹象的能力。随着 受累的 家庭和医生对DM2临床特征的认识日益加深,外显率接近100%。
经验丰富的调查员研究. 在一项由经验丰富的调查员对234位18岁或18岁以上的成年人进行的研究中,除一名DM2扩增个体外,其他人均被正确归为 受累的个体 (一名50岁男性被误认为未受影响,可能是由于检查不完全所致)。
家族史提示以下内容:
- 未经检测的杂合子携带者在生命的第五和第六个十年中未被诊断,或经常被误诊为“风湿病”、纤维肌痛、类风湿关节炎、炎症性肌病、非典型运动神经元病或代谢性肌病。
- 除骨骼肌特征外的疾病因素,如早发性白内障、睾丸衰竭和心律失常,通常被认为是DM2的表现,却不能够被识别 [Day et al 2003].
预期
据报道,在参与DM2最初特征鉴定的家庭中,临床特征逐代恶化[Schneider et al 2000, Day et al 2003]. 数据表明,这是由遗传早现(连续几代人发病倾向于更早的年龄和/或更严重的临床表现)引起的,而不是由于偏见确认(研究中无意纳入更多受累的年轻一代家庭成员)。
- 此外,疾病严重程度与CCTG重复长度之间缺乏相关性,强调了重复长度的代际变化并不会造成疾病恶化。
命名法
国际强直性肌营养不良联盟(IDMC)和人类在线孟德尔遗传(OMIM)均认为DM2和近端强直性肌病(PROMM)是指相同的情况。患有PROMM的个体最初被描述为具有DM1的一些特征,但没有DMPK 三核苷酸重复扩增的典型特征。 DM2与PROMM最初认为在临床上有所区别,因为PROMM和DM2家庭成员的临床描述存在明显差异。 然而,现表明大多数患有PROMM的家庭成员在DM2患者身上观察到了CNBP扩增的典型特征。
注意:如果致病变异未知,术语PROMM有时仍用于指代临床表型。 但是,当通过CNBP的分子遗传学检测建立诊断时,更精确的术语DM2是可取的。
尽管已提出存在多系统肌强直营养不良的其他遗传因素,但尚未确认。 国际强直性肌营养不良联盟已经同意,任何新发现的多系统肌强直营养不良将依次命名为强直性肌营养不良的形式。
一个被认为患有强直性肌营养不良3型(DM3)的家庭[Le Ber et al 2004] 后来被证实患有佩吉特氏病和 家族性 包涵体肌炎的不寻常表现[Udd et al 2006],也被称为包涵体肌病伴佩吉特病和额颞叶痴呆(IBMPFTD),由VCP突变引起。(参见 Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia.)
流行病学
强直性肌营养不良是最常见的成人型肌营养不良,估计影响约1/8000的总人口。DM1和DM2引起的强直性肌营养不良的比例尚不清楚。由于临床严重程度的不同,DM2的发病率仍不清楚。
不同人群的患病率似乎有所不同,然而很少有确切的人口统计学研究。 在德国和波兰以及德国或波兰血统的个体中,DM2的患病率较高 [Udd et al 2003]。 在芬兰,DM2发病率(1:1830)的发病率高于DM1 [Suominen et al 2011]。 DM2在阿富汗和斯里兰卡已有报道,但在中国、日本或撒哈拉以南非洲尚无报道。
遗传相关(等位基因)疾病
除此GeneReview中讨论的表型外,没有其他 表型 与CNBP突变相关。
鉴别诊断
多系统强直性肌病. 目前为止,造成强直性肌营养不良表型的唯一确认原因是DMPK(myotonic dystrophy type 1,DM1)的3'端非翻译区CTG扩增或CNBP(DM2) 内含子 1的CCTG扩增。 这两种类型的强直性肌营养不良的明确诊断依赖于 分子遗传学检测。
虽然常规的临床评估可以可靠地识别强直性肌营养不良,但单凭临床标准无法可靠地区分DM1和DM2的成人发病类型。 患有DM1和DM2个体的白内障难以进行区分。DM1和DM2之间最明显的区别是,在DM1患者中有报道过畸形足、新生儿无力和呼吸功能不全、智力障碍、颅面畸形、儿童肌张力低下和肌无力,而DM2患者尚无此类报道。 此外,可能由于 先天的 影响,成人DM1无力和肌强直表现程度比DM2更大;成人DM1患者更具明显的面部特征和阴茎无力、肌肉萎缩、心脏受累和包括中枢性嗜睡的中枢神经系统异常 [Meola et al 2002, Ranum & Day 2002, Ranum & Day 2004, Day & Ranum 2005].
在发现一些家庭成员有CNBP CCTG扩增,被证实确诊为DM2后 ,先前关于多系统强直性肌病与DM1 位点或DM2基因座无关的报道(即“非DM1,非DM2的PROMM病例”)已被撤回,[Day et al 2003],尽管如此,仍有其他遗传因素可导致DM2。
患有新型多系统强直性肌病(“ DM3”)的家庭成员 [Le Ber et al 2004]与DM1和DM2(白内障和肌强直)具有某些共同特征,但在神经系统异常方面有明显不同(运动神经元疾病和海绵状脑病) ); 尽管该家庭成员中的 表型最初反映为强直性肌营养不良,该家族现已被划分为由VCP突变引起的生理病理上不同于DM的佩吉特病和 家族性包涵体肌炎。 (See Inclusion Body Myopathy with Paget Disease of Bone and/or Frontotemporal Dementia.)
远端肌病. 其他主要鉴别诊断是远端肌病 (参见 Table 2).
- Udd 远端肌病 Udd肌病的特点是踝关节背屈无力,35岁以后无法行走。 疾病进展缓慢,肌无力局限于胫骨前肌。 10 至 20年后,长趾伸肌出现临床症状,导致足下垂和行走笨拙。编码肌联蛋白的TTN 基因 突变导致Udd肌病。 [Hackman et al 2002].
- Nonaka肌病 通常于第二或第三十年在腿前腔室和足趾伸肌开始受累。足下垂和跨域步态在12至15年后可进一步发展为无法行走。这与由GNE [Nishino et al 2002]突变引起的保留股四头肌肌病相同。参见 GNE-Related Myopathy.
- Zaspopathy 迟发性远端型肌病的特征是通常于40岁之后发作踝关节背屈无力,随后缓慢发展到手指和手腕伸肌以及手部固有肌。最后,近端腿部肌肉受累。该疾病由LDB3(也称为ZASP)突变所导致 [Griggs et al 2007]. 参见 Myofibrillar Myopathy.
- MPD3是报道于芬兰的一种显性远端肌病,在一个单亲家庭中,有些受累的 个体影响上肢,有些影响下肢;之后,上肢和下肢均受累 [Haravuori et al 2004].
- 成人早发型Miyoshi肌病始于腿部后腔室,表现为爬楼梯和脚趾行走困难,并发展到其他远端和近端肌肉,如LGMD2B(参见 Dysferlinopathy)。血清CK浓度通常是正常值的50倍以上。
- Welander远端肌病有时可能发生在小腿前室肌,而不是通常发生在手和手指伸肌 [von Tell et al 2002]。一般情况下, 受累的个体40岁后出现食指伸肌无力,随后缓慢进展到其他手指伸肌和小腿前后肌。
Table 2.
远端肌病
| 疾病名称 | 平均发病年龄(岁) | 起初受累肌群 | 血清肌酸激酶浓度 | 肌肉活检 | 基因(基因座)1 |
|---|---|---|---|---|---|
| 常染色体显性遗传 | |||||
| Welander远端肌病 | >40 | 上肢远端(手指和手腕伸肌) | 正常或轻微增加 | 镶边空泡 | (2p13) |
| Udd distal myopathy | >35 | 腿部前室 | ± 镶边空泡 | TTN | |
| Zaspopathy (Markesbery-Griggs远端型肌病) | >40 | 空泡和肌原纤维肌病 | LDB3 (也称为 ZASP) | ||
| Distal myotilinopathy | >40 | 腿后部> 腿前 | 轻微增加 | 空泡和肌原纤维 | MYOT |
| Laing early-onset distal myopathy (MPD1) | <20 | 腿部前室和颈部屈肌 | 正常至中度增加(少见) | 胫骨前肌1型纤维萎缩;近端肌肉不均衡 | MYH7 |
| 远端肌病伴咽喉和声带受累 (MPD2) | 35-60 | 手脚不对称;言语障碍 | 1-8 倍 | 镶边空泡 | MATR3 |
| 远端肌病伴弓形足和反射消失 | 15-50 | 小腿前后;言语障碍和吞咽困难 | 2-6 倍 | 营养不良, 镶边空泡 | (19p13) |
| 新型芬兰远端肌病(MPD3) | >30 | 手或小腿前部 | 1-4 倍 | 营养不良, 镶边空泡,嗜酸性包涵体 | (8p22-q11 and 12q13-q22) |
| 常染色体隐性遗传 | |||||
| Nonaka early-adult-onset distal myopathy | 15-20 | 腿部前室 | <10 倍 | 镶边空泡 | GNE |
| Miyoshi early-adult-onset myopathy | 腿部后室 | >10 倍 | 肌病性改变 | DYSF | |
Udd & Griggs [2001]
1. 基因座信息仅在 基因不明确时给出
肌强直. 电性肌强直可在多种情况下发生,但多个家庭成员中的肌强直限制DM或非营养不良性肌强直的诊断可能性,其后者是由氯离子和钠离子通道基因突变引起的,可导致myotonia congenita、 hyperkalemic periodic paralysis和高钾型周期性麻痹。这些情况与肌营养不良或DM1和DM2的多系统特征无关,因此可以根据临床情况加以区分。
其他. 有时,DM2患者会被误诊为患有非典型运动神经元疾病 [Rotondo et al 2005],炎性肌病、纤维肌痛、类风湿关节炎或代谢性肌病。
管理
初步诊断后的评估
为确认诊断为强直性肌营养不良2型(DM2)个体的疾病严重程度,建议进行以下评估:
- 肌力与功能状态的常规临床评估
- 由熟悉DM虹膜后囊下白内障的眼科医生进行检查,确定基线
- 初步心脏评估至少包括:
- 建立心电图基线记录以先后比较
- 如果患者有症状或在常规心电图上提示明显的心律或传导异常应进行动态心电图监测或有创性电生理检查
- 由于有心肌病风险,考虑超声心动图和必要时心脏MRI [Spengos et al 2012]
- 基础的血清学检验,包括空腹血脂水平、血糖和糖化血红蛋白浓度,以评估胰岛素不敏感和糖尿病的证据
- 测定青春期后男性的血清睾酮浓度和FSH,以评估性腺功能
- 甲状腺研究。虽然甲状腺功能不全与DM2致病性变异没有明确的因果关系,但任何原因引起的甲状腺功能减退都与DM2患者肌无力和症状增加有关[Sansone et al 2000, Day & Ranum 2005].
- 血清CK、转氨酶(AST、ALT)、γ-谷氨酰转移酶(GGT)活性测定。血清AST、ALT和GGT活性在DM2患者中经常升高,但目前尚不清楚这些异常水平是肝细胞性还是肌源性。测定DM2引起的转氨酶基线和GGT活性异常有助于避免不必要的肝脏检查。
- 血清蛋白电泳和免疫蛋白电泳建立一个基线,因为在DM2患者中,由于IgG和IgM水平较低,γ分数经常降低。尽管这些变化与临床问题无关,但测定DM2患者的异常免疫球蛋白水平可确定个体基线值,避免对将来低丙种球蛋白血症研究的错误判读。
对症治疗
DM2治疗指南已发布. 参见 Udd et al [2011] (full text).
随疾病进展,物理治疗师、职业治疗师或理疗学家可以帮助确认是否需要踝足矫形器、轮椅或其他辅助设备 [Johnson et al 1995].
常规体力活动可能有益于DM2患者保持肌力和耐力,并有助于控制肌肉骨骼疼痛。
肌强直通常是轻微的,很少需要治疗 [Ricker 1999],尽管在某些DM2患者中,使用美西律可有效地控制某些肌强直,有助于控制肌肉疼痛。
药物和联合用药治疗疼痛的效果各不相同。没有一种药物一直有效;已取得一些成功应用的药物包括美西律、加巴喷丁、非甾体抗炎药(NSAIDS)、低剂量甲状腺替代品、低剂量类固醇(如5毫克强的松隔日给药)和三环类抗抑郁药。小剂量麻醉性镇痛药作为全面疼痛管理计划的一部分,可能所帮助,但也可能导致药物耐受性和剂量依赖。
Due to the increased risk for cardiomyopathy, echocardiography and possibly cardiac MRI should be considered.强烈建议有心脏症状或心电图提示心律失常的患者咨询心脏病专家,因为致命性的心律失常可能发生在在其他症状之前。应通过心电图、动态心电图监测和超声心动图评估晕厥、心悸和其他潜在心源性症状。可能需要对心脏进行更高级的有创性电生理测试 [Florek et al 1990, Hawley et al 1991]。由于心肌病风险的增加,应考虑超声心动图和必要时心脏MRI检查。
除颤器置入的价值在有明显心律不齐的DM2患者中越来越明显,但是起搏器/除颤器在无症状患者中的作用尚待确定 [Schoser et al 2004b]
如果白内障损害视力则可以摘除。与较典型的老年核性白内障相比,直接检眼镜甚至裂隙灯检查会低估DM2患者白内障的功能意义,因为视力的改变取决于位置,而不仅仅是囊下混浊的数量。
睾酮替代疗法对有性腺功能减退症状的男性是有益的。
(Zelnorm™).DM2的直接胃肠道表现尚不明确,但一些患者抱怨餐后腹痛、腹胀、便秘和腹泻。与 myotonic dystrophy type 1(DM1)一样,一些个体对促进胃动力药物如胃复安(Reglan)™) 和替加色罗(Zelnorm™)有反应.
预防继发性并发症
DM2患者可能会增加麻醉风险; 因此,建议在手术前后仔细评估心脏和呼吸功能 [Veyckemans & Scholtes 2013].
DM2患者的无力增加程度与甲状腺功能减退有关;因此,如果治疗甲状腺功能减退,一些力量可能会恢复。
监测
每年心电图和超声心动图或必要时心脏MRI检查可提示无症状和进展性心脏传导缺陷及心肌病。
一些中心甚至在没有心脏症状的情况下也会进行24小时动态心电图监测。
每年应测定空腹血糖浓度和糖化血红蛋白水平。
如果男性变得越来越疲劳或性欲减少,应该对其进行性腺功能减退测试,并且应该每隔几年进行一次测试,即使没有症状,看看他们是否从替代疗法中受益。
药物禁忌
无力的增加程度与某些降胆固醇药物的使用有关。在这种情况下,如果禁服他汀类降胆固醇药物,一些力量可以得到恢复。
注意:并不是所有DM2患者对他汀类药物都有不良反应,因此诊断为DM2并不是使用这些药物的绝对禁忌症。
亲属患病风险评估
涉及与遗传咨询目的有关的高危亲属检测请参阅 Genetic Counseling .
在研治疗
检索 ClinicalTrials.gov 以获得有关各种疾病和状况的临床研究信息。注意:可能没有针对该疾病的临床试验。
遗传咨询
遗传咨询是一个给患者及家属提供关于遗传性疾病本质、遗传特性以及影响并帮助他们做出知情的医疗决定的过程。下列段落描述遗传风险的评估以及根据家族史和基因检测判断家族成员遗传状态。本段落描述不适用于解决患者实际面对的个人、文化或伦理问题,也不能代替专业的遗传咨询。—ED.
遗传模式
强直性肌营养不良2型的遗传模式是 常染色体显性遗传 .
家庭成员风险
先证者父母
- 迄今为止,所有先证者的亲生父母都经过 分子遗传学检测 均发现其中一方具有CNBP扩增。
- 先证者 de novo 尚未有过报道。
- CNBP扩增显示出同一家族中各代之间的大小差异。 通常,重复序列的大小在传递给后代时似乎会缩小,然后随着 受累的个体年龄的增长而增加(请参见 Related Genetic Counseling Issues,体细胞嵌合)。 没有母亲或父亲的其中一方更倾向于缩短或扩增。
先证者同胞
先证者后代
- 每个携带CNBP扩增个体的子代都有50%的概率遗传到该变异。
- CNBP CCTG重复序列大小从一代传给下一代时倾向于缩短,随着 受累的个体年龄的增长而增大。
先证者家系其他成员. 其他家庭成员的风险取决于先证者父母的遗传状态。如果父母其中一个是 受累的,他或她的家庭成员就有风险。
遗传咨询相关问题
家系中有明显de novo(新发)突变注意事项。当患有常染色体显性遗传 疾病的先证者的父母均没有导致该疾病的突变或临床证据时,先证者很可能是新发突变。 但是,也可能存在非医学解释,包括 非生物学父亲或产妇(例如,辅助生殖)或未公开收养。
所测重复序列大小与疾病严重程度之间没有相关性;因此,从分子遗传学检测结果无法预测发病年龄或临床病程。
体细胞 嵌合.CNBP CCTG重复扩增是高度不稳定的,并且随着 受累的 个体年龄的增长而趋于增大。在同一时间或不同年龄从受累个体提取的不同外周血样本上进行多项检测结果可能在扩增大小上有所不同。此外,在单个外周血样本中,通过 Southern blot 分析,可能检测到不止一个扩增 等位基因 的大小。一个显性等位基因的大小和单个样本中可检测到的不同扩增的总数都不能预测疾病的严重程度、发病年龄或临床症状。
有风险的无症状成人检测. 通过Molecular Genetic Testing(分子遗传学检测)中提及的检测方法,可以对有风险的无症状成人进行检测。通过检测可以确认个体是否携带有CNBP扩增,从而确认个体患病风险。重复序列的大小不能预测发病年龄、严重程度或临床症状。在对有风险的家庭成员检测之前,应先对 受累的的家庭成员进行检测,以确认家系中是否携带有CNBP扩增先证者。风险筛查应结合遗传咨询 进行,以确保个体意识到分子遗传学检测的局限性以及与风险筛查相关的可能性风险。
对18岁以下的无症状风险个体进行检测. 不建议在没有已知的有效治疗预防该疾病或改善预后的成人发病情况下,对18岁以下无症状的风险个体进行检测。 有症状的18岁以下的个体通常会建立明确的诊断。 另请参见国家遗传咨询师协会关于未成年人成年发作情况的基因检测的立场声明,以及美国儿科学会和美国医学遗传学和基因组学的 policy statement(政策声明):儿童基因检测和筛查中的道德和政策问题。
18岁以下高危人群的检测。对18岁以下的无症状高危人群的检测不推荐用于没有已知有效治疗预防疾病或改善预后的成人发病情况。18岁以下有症状的个人通常从确定特定诊断中获益。另见国家遗传咨询师协会关于未成年人成人发病情况遗传检测的position statement(立场声明),以及美国儿科学会和美国医学遗传学和基因组学学院的政策声明:儿童遗传检测和筛查中的伦理和政策问题。
家庭计划
- 确定遗传风险、探讨产前检测的最佳时机是怀孕前。
DNA库 DNA 库用于存储DNA(通常为提取自白细胞DNA)已备将来之需,我们对基因、等位基因突变的理解、对疾病的认识以及检测方法都将提升,应考虑对患者 受累的个体DNA进行保存。
产前检测
如果在一个 受累的家庭成员中确认CNBP致病性扩增变异,高危妊娠孕妇可以从提供针对该疾病/基因检测或者 定制产前检测致病变异的临床实验室进行产前检测。
对不影响智力的典型成人发病情况(如DM2)的产前诊断需求并不常见。在应用产前检测方面,医学专业人员和家庭内部可能存在不同的看法,特别是在考虑产前检测是为了终止妊娠而不是进行早期诊断的情况下。尽管大多数中心会考虑由父母决定是否进行产前检测,但对这些问题的讨论是合理的。
植入前遗传学诊断 (PGD) 对于某些检出致病性扩增的家系可选。
资源
GeneReviews 工作人员已经筛选了以下专科疾病和患者帮扶组织,注册登记能使患者及其家庭获益。GeneReviews对该类组织提供的信息不负责,信息的筛选标准,参见此处here
- My46 Trait Profile
- Myotonic Dystrophy: Making an Informed Choice About Genetic TestingBooklet providing information about Myotonic Dystrophy and genetic testing (PDF file)University of Washington Medical Center, Medical Genetics and NeurologySeattle WA
- National Library of Medicine Genetics Home Reference
- Muscular Dystrophy Association - USA (MDA)222 South Riverside PlazaSuite 1500Chicago IL 60606Phone: 800-572-1717Email: mda@mdausa.org
- Muscular Dystrophy UK61A Great Suffolk StreetLondon SE1 0BUUnited KingdomPhone: 0800 652 6352 (toll-free); 020 7803 4800Email: info@musculardystrophyuk.org
- Myotonic Dystrophy Family Registry (MDFR)Phone: 602-435-7496Email: coordinator@myotonicregistry.org
- National Registry of Myotonic Dystrophy and FSHD Patients and Family MembersNational Registry of Myotonic Dystrophy and FSHD601 Elmwood AvenueBox 673Rochester NY 14642Phone: 888-925-4302Fax: 585-273-1255Email: dystrophy_registry@urmc.rochester.edu
分子遗传学
分子遗传学检测的信息和OMIM相关列表可能同其它GeneReview信息会有不同。(可能有更新原因)—ED.
Table A.
强直性肌营养不良2型: 基因和数据库
| 基因 | 染色体位置 | 蛋白 | 基因座特异性 | HGMD |
|---|---|---|---|---|
| CNBP | 3q21 | Cellular nucleic acid-binding protein | CNBP database | CNBP |
Table B.
OMIM 收录强直性肌营养不良2型词条 (View All in OMIM)
分子遗传致病基础
强直性肌营养不良1型(DM1)和强直性肌营养不良2型(DM2)的临床和分子相似性强调表明,含有重复扩增的未翻译RNAs是两种疾病共同的病理特征。
DM1和DM2的发病机理可以通过功能获得性RNA机制来解释,分别为CUG和CCUG重复,改变细胞功能,包括不同基因的选择性 剪接 [Tapscott & Thornton 2001, Ranum & Day 2002, Ranum & Day 2004, Day & Ranum 2005]. DM1和DM2都有含有异常扩增 等位基因RNA的RNA团簇,与多种形式的盲肌样RNA结合蛋白(MBNL、MBLL和MBXL)共同定位 [Mankodi et al 2001, Fardaei et al 2002].
盲肌和RNA结合蛋白CUG-BP(由CNBP编码)失调后会改变各种下游基因的基因 剪接。增加的CUG-BP RNA导致心肌肌钙蛋白T,胰岛素受体和氯离子通道的错接,可能分别导致心脏受累,胰岛素不敏感和肌强直[Philips et al 1998, Mankodi et al 2001, Savkur et al 2001]. DM1和DM2的胰岛素受体剪接异常的下游效应与胰岛素不敏感相关[Savkur et al 2004]. 敲除盲肌蛋白导致DM1和DM2的肌强直、白内障和肌病特征[Kanadia et al 2003].
基因结构. CNBP(原名ZNF9) CCTG重复序列是一个复杂重复序列的一部分,其整体结构为(TG)n(TCTG)n(CCTG)n。 在正常的CNBP等位基因中,CCTG重复序列包含核苷酸中断。 已知最长的正常CNBP 等位基因,重复序列全长为176 bp,包括26个CCTG重复和两个中断 [Liquori et al 2001]. 这样的核苷酸中断与导致其他核苷酸重复疾病基因的正常长度等位基因相似(spinocerebellar ataxia type 1 和 FMR1-related disorders) [Chung et al 1993, Kunst & Warren 1994].
CNBP广泛表达。 它与DM1 CTG扩增的3'非翻译区域内的DM1 位点上的任何基因,包括肌营养不良蛋白激酶基因(DMPK),没有任何功能相似性。同样,DM2区域内的任何基因与DM1区域内的基因都没有相似性。两个区域缺乏相似基因进一步表明,致病突变导致致病性RNA扩增,而不是基因表达或基因产物的改变。
致病性变异. DM2是由超过75个CCTG四核苷酸重复扩增(重复全长大于372 bp)的单一突变机理引起的。扩增重复长度范围从372 bp到44,000 bp以上(相当于75到> 11,000个重复数),由于重复域的高度不稳定性和明显的 体细胞嵌合,尚未确认准确的致病性阈值。重复扩增最小的 受累的的个体,也有第二次大扩增,两者或其一的致病性仍是未知的。在DM2中扩增的CCTG重复序列位于CNBP的内含子1中,转录为RNA,但未翻译成蛋白质。
正常 基因产物.CNBP编码带有177个氨基酸的19463-dalton细胞核酸结合蛋白 (NP_003409.1) [Pellizzoni et al 1997, Pellizzoni et al 1998].
异常 基因产物. 在DM2中扩增的CCTG重复序列被转录为RNA,但未翻译为蛋白质。没有证据表明DM2中CNBP单倍剂量不足 [Margolis et al 2006].
参考文献
已发表的指南和共识
- Kamsteeg EJ, Kress W, Catalli C, Hertz JM, Witsch-Baumgartner M, Buckley MF, van Engelen BGM, Schwartz M, Scheffer H (2012) Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet. 20:1203-8. Available online. Accessed 2-25-15. [PMC free article: PMC3499739] [PubMed: 22643181]
- Udd B, Meola G, Krahe R, Wansink DG, Bassez G, Kress W, Schoser B, Moxley R (2011) Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3–5 December 2010, Naarden, The Netherlands. Neuromuscul Disord. 21:443-50. Available online. Accessed 2-25-15. [PubMed: 21543227]
引用文献
- Auvinen S, Suominen T, Hannonen P, Bachinski LL, Krahe R, Udd B. Myotonic dystrophy type 2 found in two of sixty-three persons diagnosed as having fibromyalgia. Arthritis Rheum. 2008;58:3627 - 31. [PMC free article: PMC2585600] [PubMed: 18975316]
- Bhat S, Sander HW, Grewal RP, Chokroverty S. Sleep disordered breathing and other sleep dysfunction in myotonic dystrophy type 2. Sleep Med. 2012;13:1207 - 8. [PubMed: 22959494]
- Chokroverty S, Bhat S, Rosen D, Farheen A. REM behavior disorder in myotonic dystrophy type 2. Neurology. 2012;78:2004. [PubMed: 22689737]
- Chung MY, Ranum LP, Duvick LA, Servadio A, Zoghbi HY, Orr HT. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet. 1993;5:254 - 8. [PubMed: 8275090]
- Colleran JA, Hawley RJ, Pinnow EE, Kokkinos PF, Fletcher RD. Value of the electrocardiogram in determining cardiac events and mortality in myotonic dystrophy. Am J Cardiol. 1997;80:1494 - 7. [PubMed: 9399734]
- Day JW, Ranum LP. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr Neurol Neurosci Rep. 2005;5:55 - 9. [PubMed: 15676109]
- Dabby R, Sadeh M, Herman O, Leibou L, Kremer E, Mordechai S, Watemberg N, Frand J. Clinical, electrophysiologic and pathologic findings in 10 patients with myotonic dystrophy 2. Isr Med Assoc J. 2011;13:745 - 7. [PubMed: 22332444]
- Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657 - 64. [PubMed: 12601109]
- Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul Disord. 1999;9:19 - 27. [PubMed: 10063831]
- Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805 - 14. [PubMed: 11929853]
- Florek RC, Triffon DW, Mann DE, Ringel SP, Reiter MJ. Electrocardiographic abnormalities in patients with myotonic dystrophy. West J Med. 1990;153:24 - 7. [PMC free article: PMC1002461] [PubMed: 2202157]
- Franc DT, Muetzel RL, Robinson PR, Rodriguez CP, Dalton JC, Naughton CE, Mueller BA, Wozniak JR, Lim KO, Day JW. Cerebral and muscle MRI abnormalities in myotonic dystrophy. Neuromuscul Disord. 2012;22:483 - 91. [PMC free article: PMC3350604] [PubMed: 22290140]
- Gadalla SM, Lund M, Pfeiffer RM, Gørtz S, Mueller CM, Moxley RT III, Kristinsson SY, Björkholm M, Shebl FM, Hilbert JE, Landgren O, Wohlfahrt J, Melbye M, Greene MH. Cancer risk among patients with myotonic muscular dystrophy. JAMA. 2011;306:2480 - 6. [PMC free article: PMC3286183] [PubMed: 22166607]
- George A, Schneider-Gold C, Zier S, Reiners K, Sommer C. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol. 2004;61:1938 - 42. [PubMed: 15596616]
- Giagnacovo M, Malatesta M, Cardani R, Meola G, Pellicciari C. Nuclear ribonucleoprotein-containing foci increase in size in non-dividing cells from patients with myotonic dystrophy type 2. Histochem Cell Biol. 2012;138:699 - 707. [PubMed: 22706481]
- Griggs R, Vihola A, Hackman P, Talvinen K, Haravuori H, Faulkner G, Eymard B, Richard I, Selcen D, Engel A, Carpen O, Udd B. Zaspopathy in a large classic late-onset distal myopathy family. Brain. 2007;130:1477 - 84. [PubMed: 17337483]
- Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492 - 500. [PMC free article: PMC379188] [PubMed: 12145747]
- Haravuori H, Siitonen HA, Mahjneh I, Hackman P, Lahti L, Somer H, Peltonen L, Kestilä M, Udd B. Linkage to two separate loci in a family with a novel distal myopathy phenotype (MPD3). Neuromuscul Disord. 2004;14:183 - 7. [PubMed: 15036327]
- Harper PS. Myotonic Dystrophy. London, UK: WB Saunders; 2001.
- Hawley RJ, Milner MR, Gottdiener JS, Cohen A. Myotonic heart disease: a clinical follow-up. Neurology. 1991;41:259 - 62. [PubMed: 1992371]
- Hund E, Jansen O, Koch MC, Ricker K, Fogel W, Niedermaier N, Otto M, Kuhn E, Meinck HM. Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology. 1997;48:33 - 7. [PubMed: 9008490]
- Johnson ER, Abresch RT, Carter GT, Kilmer DD, Fowler WM, Sigford BJ, Wanlass RL. Profiles of neuromuscular diseases. Myotonic dystrophy. Am J Phys Med Rehabil. 1995;74:S104 - 16. [PubMed: 7576418]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978 - 80. [PubMed: 14671308]
- Kamsteeg E-J, Kress W, Catalli C, Hertz JM, Witsch-Baumgartner M, Buckley MF, van Engelen Baziel GM, Schwartz M, Scheffer H. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet. 2012;20:1203 - 8. [PMC free article: PMC3499739] [PubMed: 22643181]
- Kirzinger L, Schmidt A, Kornblum C, Schneider-Gold C, Kress W, Schoser B. Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. Eur J Neurol. 2010 Jun 1;17:842 - 5. [PubMed: 20100232]
- Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853 - 61. [PubMed: 7911740]
- Le Ber I, Martinez M, Campion D, Laquerriere A, Betard C, Bassez G, Girard C, Saugier-Veber P, Raux G, Sergeant N, Magnier P, Maisonobe T, Eymard B, Duyckaerts C, Delacourte A, Frebourg T, Hannequin D. A non-DM1, non-DM2 multisystem myotonic disorder with frontotemporal dementia: phenotype and suggestive mapping of the DM3 locus to chromosome 15q21-24. Brain. 2004;127:1979 - 92. [PubMed: 15215218]
- Liquori CL, Ikeda Y, Weatherspoon M, Ricker K, Schoser BG, Dalton JC, Day JW, Ranum LP. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet. 2003;73:849 - 62. [PMC free article: PMC1180607] [PubMed: 14505273]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864 - 7. [PubMed: 11486088]
- Mankodi A, Takahashi M, Beck C, Cannon S, Thornton CA. Myotonia is associated with loss of transmembrane chloride conductance and aberrant splicing of Clcn1, the skeletal muscle chloride channel, in a transgenic model of myotonic dystrophy (DM1). Am J Hum Genet. 2001;69:A211.
- Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LP. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet. 2006;15:1808 - 15. [PubMed: 16624843]
- Meola G, Sansone V, Marinou K, Cotelli M, Moxley RT, Thornton CA, De Ambroggi L. Proximal myotonic myopathy: a syndrome with a favourable prognosis? J Neurol Sci. 2002;193:89 - 96. [PubMed: 11790388]
- Meola G, Sansone V, Perani D, Colleluori A, Cappa S, Cotelli M, Fazio F, Thornton CA, Moxley RT. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology. 1999;53:1042 - 50. [PubMed: 10496264]
- Meola G, Sansone V, Perani D, Scarone S, Cappa S, Dragoni C, Cattaneo E, Cotelli M, Gobbo C, Fazio F, Siciliano G, Mancuso M, Vitelli E, Zhang S, Krahe R, Moxley RT. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord. 2003;13:813 - 21. [PubMed: 14678804]
- Merino JL, Carmona JR, Fernandez-Lozano I, Peinado R, Basterra N, Sobrino JA. Mechanisms of sustained ventricular tachycardia in myotonic dystrophy: implications for catheter ablation. Circulation. 1998;98:541 - 6. [PubMed: 9714111]
- Moxley RT 3rd. Proximal myotonic myopathy: mini-review of a recently delineated clinical disorder. Neuromuscul Disord. 1996;6:87 - 93. [PubMed: 8664567]
- Newman B, Meola G, O'Donovan DG, Schapira AH, Kingston H. Proximal myotonic myopathy (PROMM) presenting as myotonia during pregnancy. Neuromuscul Disord. 1999;9:144 - 9. [PubMed: 10382907]
- Nguyen HH, Wolfe JT, Holmes DR, Edwards WD. Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol. 1988;11:662 - 71. [PubMed: 3278037]
- Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, Ohi T, Nishimiya J, Sunohara N, Ciafaloni E, Kawai M, Aoki M, Nonaka I. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689 - 93. [PubMed: 12473753]
- Pellizzoni L, Lotti F, Maras B, Pierandrei-Amaldi P. Cellular nucleic acid binding protein binds a conserved region of the 5' UTR of Xenopus laevis ribosomal protein mRNAs. J Mol Biol. 1997;267:264 - 75. [PubMed: 9096224]
- Pellizzoni L, Lotti F, Rutjes SA, Pierandrei-Amaldi P. Involvement of the Xenopus laevis Ro60 autoantigen in the alternative interaction of La and CNBP proteins with the 5'UTR of L4 ribosomal protein mRNA. J Mol Biol. 1998;281:593 - 608. [PubMed: 9710533]
- Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737 - 41. [PubMed: 9563950]
- Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, Bernardi G, Angelini C, Massa R. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve. 2008;38:1405 - 11. [PubMed: 18816606]
- Ranum LP, Day JW. Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr Neurol Neurosci Rep. 2002;2:465 - 70. [PubMed: 12169228]
- Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004;74:793 - 804. [PMC free article: PMC1181975] [PubMed: 15065017]
- Ricker K. Myotonic dystrophy and proximal myotonic myophathy. J Neurol. 1999;246:334 - 8. [PubMed: 10399862]
- Ricker K, Grimm T, Koch MC, Schneider C, Kress W, Reimers CD, Schulte-Mattler W, Mueller-Myhsok B, Toyka KV, Mueller CR. Linkage of proximal myotonic myopathy to chromosome 3q. Neurology. 1999;52:170 - 1. [PubMed: 9921867]
- Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley RT. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448 - 52. [PubMed: 8058147]
- Rotondo G, Sansone V, Cardani R, Mancinelli E, Krahe R, Stangalini D, Meola G. Proximal myotonic dystrophy mimicking progressive muscular atrophy. Eur J Neurol. 2005;12:160 - 1. [PubMed: 15679706]
- Rudnik-Schöneborn S, Schneider-Gold C, Raabe U, Kress W, Zerres K, Schoser BG. Outcome and effect of pregnancy in myotonic dystrophy type 2. Neurology. 2006;66:579 - 80. [PubMed: 16505316]
- Sansone VA, Brigonzi E, Schoser B, Villani S, Gaeta M, De Ambroggi G, Bandera F, De Ambroggi L, Meola G. The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): Long-term outcomes. Int J Cardiol. 2013;168:1147 - 53. [PubMed: 23266299]
- Sansone V, Griggs RC, Moxley RT. Hypothyroidism unmasking proximal myotonic myopathy. Neuromuscul Disord. 2000;10:165 - 72. [PubMed: 10734262]
- Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40 - 7. [PubMed: 11528389]
- Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LP, Day JW. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74:1309 - 13. [PMC free article: PMC1182097] [PubMed: 15114529]
- Schneider C, Ziegler A, Ricker K, Grimm T, Kress W, Reimers CD, Meinck H, Reiners K, Toyka KV. Proximal myotonic myopathy: evidence for anticipation in families with linkage to chromosome 3q. Neurology. 2000;55:383 - 8. [PubMed: 10932272]
- Schoser BG, Kress W, Walter MC, Halliger-Keller B, Lochmuller H, Ricker K. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain. 2004a;127:1868 - 77. [PubMed: 15231584]
- Schoser BG, Ricker K, Schneider-Gold C, Hengstenberg C, Durre J, Bultmann B, Kress W, Day JW, Ranum LP. Sudden cardiac death in myotonic dystrophy type 2. Neurology. 2004b;63:2402 - 4. [PubMed: 15623712]
- Schoser BG, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC, Pongratz DE, Toyka KV, Lochmuller H, Ricker K. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve. 2004c;29:275 - 81. [PubMed: 14755494]
- Shepard P, Lam EM, St Louis EK, Dominik J. Sleep disturbances in myotonic dystrophy type 2. Eur Neurol. 2012;68:377 - 80. [PMC free article: PMC4259012] [PubMed: 23108384]
- Spengos K, Gialafos E, Vassilopoulou S, Toulas P, Manta P. Delayed contrast enhancement on cardiac MRI unmasks subclinical cardiomyopathy in a case of myotonic dystrophy type 2. Hellenic J Cardiol. 2012;53:324 - 6. [PubMed: 22796821]
- Suokas KI, Haanpaa M, Kautiainen H, Udd B, Hietaharju AJ. Pain in patients with myotonic dystrophy type 2: a postal survey in finland. Muscle Nerve. 2012;45:70 - 4. [PubMed: 22190310]
- Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, Peltonen L, Krahe R, Udd B. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19:776 - 82. [PMC free article: PMC3137497] [PubMed: 21364698]
- Tapscott SJ, Thornton CA. Biomedicine. Reconstructing myotonic dystrophy. Science. 2001;293:816 - 7. [PubMed: 11486078]
- Thornton C. The myotonic dystrophies. Semin Neurol. 1999;19:25 - 33. [PubMed: 10711986]
- Tieleman AA, Jenks KM, Kalkman JS, Borm G, van Engelen BG. High disease impact of myotonic dystrophy type 2 on physical and mental functioning. J Neurol. 2011;258:1820 - 6. [PMC free article: PMC3184219] [PubMed: 21461958]
- Tieleman AA, van Vliet J, Jansen JB, van der Kooi AJ, Borm GF, van Engelen BG. Gastrointestinal involvement is frequent in Myotonic Dystrophy type 2. Neuromuscul Disord. 2008;18:646 - 9. [PubMed: 18602828]
- Udd B, Griggs R. Distal myopathies. Curr Opin Neurol. 2001;14:561 - 6. [PubMed: 11562566]
- Udd B, Meola G, Krahe R, Wansink DG, Bassez G, Kress W, Schoser B, Moxley R. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul Disord. 2011;21:443 - 50. [PubMed: 21543227]
- Udd B, Meola G, Krahe R, Thornton C, Ranum L, Day J, Bassez G, Ricker K. Report of the 115th ENMC workshop: DM2/PROMM and other myotonic dystrophies. 3rd Workshop, 14-16 February 2003, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:589 - 96. [PubMed: 12921797]
- Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, Kress W, Schoser B, Moxley R. 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006;16:403 - 13. [PubMed: 16684600]
- Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth. 2013;23:794 - 803. [PubMed: 23384336]
- Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, Mancinelli E, Rouche A, Hogrel JY, Laforet P, Maisonobe T, Pellissier JF, Krahe R, Eymard B, Udd B. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854 - 7. [PubMed: 12796551]
- von Tell D, Somer H, Udd B, Edström L, Borg K, Ahlberg G. Welander distal myopathy outside the Swedish population: phenotype and genotype. Neuromuscul Disord. 2002;12:544 - 7. [PubMed: 12117477]
- Wahbi K, Meune C, Bécane HM, Laforêt P, Bassez G, Lazarus A, Radvanyi-Hoffman H, Eymard B, Duboc D. Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: a case control study. Neuromuscul Disord. 2009;19:468 - 72. [PubMed: 19481939]
- Weihl CC, Dalal S, Pestronk A, Hanson PI. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet. 2006;15:189 - 99. [PubMed: 16321991]
- Weingarten TN, Hofer RE, Milone M, Sprung J. Anesthesia and myotonic dystrophy type 2: a case series. Can J Anaesth. 2010;57:248 - 55. [PubMed: 20077169]
- Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc. 2012;87:130 - 5. [PMC free article: PMC3498332] [PubMed: 22237010]
推荐阅读
- Chaudhry SP, Frishman WH. Myotonic dystrophies and the heart. Cardiol Rev. 2012;20:1 - 3. [PubMed: 22143278]
- Johnson NE, Heatwole CR. Myotonic dystrophy: from bench to bedside. Semin Neurol. 2012;32:246 - 54. [PubMed: 23117949]
- Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891 - 905. [PubMed: 22995693]
章节注释
修订记录
- 3 July 2013 (me) Comprehensive update posted live
- 23 April 2007 (jwd) Revision: Allele sizes; to provide information to aid clinicians in interpreting test reports
- 21 September 2006 (me) Review posted to live Web site
- 14 June 2004 (jwd) Original submission